

Abstract
CD1d-restricted T (natural killer T; NKT) cells are important for controlling herpesvirus infections. Interestingly, herpes simplex virus (HSV) can down-regulate CD1d-mediated activation of NKT cells. We have previously shown that the Thr322 residue in the cytoplasmic tail of human CD1d is important for its intracellular trafficking and functional expression. We proposed that the phosphorylation of T322 is a signal for CD1d lysosomal targeting and subsequent degradation. In the current study, we generated dual mutants by substituting the T322 and S323 residues of wild-type (WT) CD1d with Ala (non-phosphorylatable) or Asp (mimicking phosphorylation) and ectopically expressed them in human embryonic kidney 293 cells. We found that the surface expression levels of the CD1d mutants was in this order: T322AS323A > WT > T322A > S323A > S323D > T322D > T322DS323D. Our results therefore suggest that mimicking the phosphorylation of both T322 and S323 has a cumulative negative effect on the functional expression of CD1d. As previously reported, we also found that upon an HSV infection, antigen presentation by WT CD1d is reduced and the CD1d molecule is degraded. Interestingly, the T322A/S323A double mutation inhibited CD1d degradation and rescued CD1d-mediated antigen presentation following an HSV-1 infection. This suggests that the T322/S323 dyad may be phosphorylated, which then targets CD1d for lysosomal degradation post-infection as a means of immune evasion, explaining (at least in part) the reduced antigen presentation observed. Hence, our findings strongly suggest that T322 and S323 form a dual residue motif that can regulate the functional expression of CD1d during a viral infection.
Keywords: antigen presentation, innate immunity, signalling, trafficking, viruses
Introduction
Herpes simplex virus (HSV) belongs to the Alpha Herpesviridae family.1 It is one of the best known viruses, due to its worldwide distribution and high frequency of infection in the human population. There are two types of HSV: HSV-1 and HSV-2. HSV-1 is mainly responsible for oral and ocular lesions, whereas HSV-2 causes genital and anal lesions. After an acute infection with HSV, the virus may persist in the body in a quiescent state called latency.2 Latent HSV-1 has been found in neural tissues, such as the vagus nerve and dorsal sensory nerve root ganglia.1 Reactivation of latent HSV-1 in brain tissue often results in encephalitis and has also been reported to be associated with the occurrence of Alzheimer's disease.3 Despite the clinical introduction of the antiviral drug acyclovir in 1980,4 herpes encephalitis remains a significant life-threatening disease with potential long-term neurological side-effects.5
Both the innate and adaptive immune systems are important for controlling a viral infection in a host. HSV has evolved many ways to evade the host immune response. Some HSV-encoded viral proteins, such as γ34.5, ICP0 and US11, counteract the host's antiviral response induced by interferons.6–8 The HSV-1-encoded molecule ICP47 suppresses MHC class I-mediated antigen presentation to cytotoxic T lymphocytes by binding to the transporter associated with antigen processing (TAP) and prevents peptide translocation into the endoplasmic reticulum for loading onto MHC class I molecules.9–13 An HSV-1 infection also decreases the level of invariant chain (Ii) in lymphoid cells and thereby impairs peptide loading onto MHC class II molecules.14 The HSV protein gB binds to HLA-DR and causes the release of HLA-DR into the exosome pathway.14,15
CD1d is an MHC class I-like molecule that mediates lipid antigen presentation to natural killer T (NKT) cells.16 Activated NKT cells secrete both T helper type 1 (Th1) and Th2 cytokines, linking the innate and adaptive immune responses.17 We and others have shown that the CD1d/NKT cell interaction plays critical roles in anti-tumour and anti-microbial immune defence.18–21 NKT cells have been reported to be particularly important in HSV clearance. NKT cell-deficient mice are less able to clear HSV-1 or HSV-2,22,23 although there have been reports that conflict with these observations.24 Nonetheless, in humans, control of other α-herpesviruses is dependent upon the CD1d/NKT cell system. For example, an 11-year-old girl who died after vaccination with an attenuated strain of varicella virus, exhibited a specific dysfunction and deficiency in the NKT cell population.25 Additionally, it was recently reported that a 6-year-old boy, who had a deficiency in NKT cells and diminished CD1d expression, developed a life-threatening infection with the vaccine strain of varicella zoster virus.26 This further indicates a link between a deficiency of the NKT cell/CD1d system and increased susceptibility to herpesvirus infections.
CD1d molecules are expressed predominantly on professional antigen-presenting cells, such as dendritic cells, B cells and macrophages.16 Upon infection of CD1d+ cells by many viruses, including varicella virus, vesicular stomatitis virus and HIV, CD1d molecules are reduced on the cell surface.27–29 Infections by HSV, like those by other viruses, also suppress CD1d-mediated activation of NKT cells, probably by inhibiting the transport of newly synthesized CD1d molecules to the cell surface.30–32
We have previously reported that a threonine-based (Thr322) signal in the cytoplasmic tail of human CD1d is important for its functional expression.33 In this study, we found that the Thr322 and Ser323 residues in the human CD1d cytoplasmic tail form a dyad that controls its surface expression and function. Upon an HSV-1 infection, a non-phosphorylatable mutant of CD1d (T322A/S323A) activates NKT cells better than the wild-type (WT) molecule does, strongly suggesting that phosphorylation of this dual-residue motif is involved in HSV-1-mediated immuno-evasion.
Materials and methods
CD1d and mutant constructs
HEK293-CD1d cells have been previously described.33 The CD1d mutants were generated by site-directed mutagenesis of the WT human CD1d (hCD1d) cDNA in the pcDNA3.1-neo vector (Invitrogen, Carlsbad, CA). Forward and reverse primers generating CD1d cytoplasmic tail mutants are shown in Table 1. CD25/CD1d fusion proteins (containing the CD1d transmembrane domain and cytoplasmic tails) were generated as previously reported.33 The same primers shown in Table 1 were used to generate mutations in the cytoplasmic tail of the CD25/CD1d fusion constructs.
Table 1.
Primers used for site-directed mutagenesis
| Primers | DNA sequence (5′–3′) |
|---|---|
| T322A forward | CATTGTGGGCTTTGCCTCCCGGTTTAAG |
| T322A reverse | CTTAAACCGGGAGGCAAAGCCCACAATG |
| S323A forward | TGTGGGCTTTACCGCCCGGTTTAAGAGGC |
| S323A reverse | GCCTCTTAAACCGGGCGGTAAAGCCCACA |
| T322A/S323A forward | CATTGTGGGCTTTGCCGCCCGGTTTAAG |
| T322A/S323A reverse | CTTAAACCGGGCGGCAAAGCCCACAATG |
| S323D forward | TGTGGGCTTTACCGACCGGTTTAAGAGGC |
| S323D reverse | GCCTCTTAAACCGGTCGGTAAAGCCCACA |
| T322D forward | CATTGTGGGCTTTGACTCCCGGTTTAAG |
| T322D reverse | CTTAAACCGGGAGTCAAAGCCCACAATG |
| T322D/S323D forward | CATTGTGGGCTTTGACGATCGGTTTAAG |
| T322D/S323D reverse | CTTAAACCGATCGTCAAAGCCCACAATG |
Cell culture and viral infections
C1R.CD1d cells (kindly provided by S. Balk, Harvard University, Cambridge, MA) were cultured in RPMI-1640 supplemented with 10% fetal bovine serum and 2 mm l-glutamine. HeLa.CD1d cells (obtained from J. Blum, Indiana University, Indianapolis, IN with permission from P. Cresswell, Yale University, New Haven, CT) and HEK293 cells (kindly provided by Prof. Philip Cohen, MRC Protein Phosphorylation Unit, University of Dundee, UK) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2 mm l-glutamine. HEK293 cells were transfected with the pcDNA3.1-neo-based vectors containing cDNAs encoding WT CD1d, the indicated cytoplasmic tail mutants, or the CD25/CD1d fusion constructs as previously described.33
Cells were infected with the HSV-1 F strain WT virus, or US3 and UL13 double-deficient HSV-1 (ΔUS3/ΔUL13, kindly provided by B. Roizman, University of Chicago, Chicago, IL) for the indicated periods of time at 37°.
Antibodies and related reagents
The human CD1d-specific monoclonal antibody (mAb) clone 51.1 (for immunoprecipitation) was purchased from eBiosciences (San Diego, CA). Phycoerythrin (PE) -conjugated and purified human CD1d-specific mAb clone 42.1, as well as FITC-labelled mAbs against human LAMP-1 and CD25, were purchased from BD Biosciences (San Diego, CA). A pan-HSV-1- and HSV-2-specific mAb (clone 4F10.3) was purchased from Millipore (Temecula, CA). A PE-labelled goat anti-mouse immunoglobulin antiserum was obtained from Dako (Carpenteria, CA). A Texas Red-conjugated donkey anti-rabbit immunoglobulin antiserum was from Jackson ImmunoResearch Inc. (West Grove, PA). A Texas Red-conjugated goat anti-mouse immunoglobulin antiserum and Hoechst stain were purchased from Molecular Probes (Portland, OR). The HB95 hybridoma (pan-HLA class I-specific mAb) was a kind gift from J. Yewdell and J. Bennink (Laboratory of Viral Diseases, NIAID, NIH). The anti-CD1d free heavy chain-specific mAb (C3D5; for immunoprecipitation and Western blot analysis) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The CD1d-binding lipid α-galactosylceramide (α-GalCer) was synthesized as described elsewhere34 or purchased from Alexis Biochemicals (San Diego, CA). Recombinant human interleukin-2 (IL-2), IL-4 and granulocyte–macrophage colony-stimulating factor (GM-CSF) were from PeproTech, Inc. (Rocky Hill, NJ), whereas antibody pairs for the human IL-4 and GM-CSF ELISAs (described below) were obtained from BD-Biosciences and Biolegend (San Diego, CA), respectively. Antibodies specific for phospho- and total-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p38 were from Cell Signaling Technology (Danvers, MA). A GAPDH-specific antibody was also obtained from Cell Signaling Technology. Methyl-β-cyclodextrin was from Sigma-Aldrich (St Louis, MO).
Flow cytometry
Aliquots of cells were washed three times in PBS and then fixed in 1% paraformaldehyde. For surface staining, the cells were washed three times in Hanks' balanced salt solution (HBSS)/BSA and incubated with the appropriate mAb followed by a PE-conjugated rabbit anti-mouse immunoglobulin antiserum. For intracellular staining, the cells were permeabilized in HBSS/BSA with 0·1% saponin and then incubated with the appropriate mAb followed by a PE-conjugated rabbit anti-mouse immunoglobulin antiserum in the presence of saponin. Analysis was performed by flow cytometry as previously described.15
NKT cell stimulation assays
Human NKT cells were isolated from human peripheral blood and expanded ex vivo,33 HEK293-CD1d (WT and mutants; 1 × 105 cells/well) were incubated with human NKT cells at an effector to target (E : T) cell ratio of 1 : 1 in 7·5 ng/ml of recombinant hIL-2, in the presence or absence of the indicated concentrations of α-GalCer for 48 hr. Secreted IL-4 and GM-CSF levels were measured by ELISA.
For virus infections, HEK293-CD1d cells were infected with WT or mutant HSV-1 at the indicated multiplicity of infection and incubation time. The cells were then fixed in 0·05% of paraformaldehyde and co-cultured with human NKT cells (E : T = 1 : 1, 1 × 105 cells/well) for 48 hr. The supernatants were harvested for the measurement of IL-4 and GM-CSF by ELISA.
Confocal microscopy
Staining for confocal microscopy was performed as described previously.27 Briefly, HEK293-CD1d cells were plated in sterile glass-bottom 35-mm dishes coated with collagen (MatTek, Ashland, MA). After the cells became 50–80% confluent, they were washed, fixed in 4% paraformaldehyde and then permeabilized with 0·1% saponin in HBSS containing 0·1% BSA. The cells were stained with an anti-human LAMP-1 mAb followed by a Texas Red-conjugated donkey anti-mouse immunoglobulin antiserum. After blocking the free reactive sites with normal mouse serum (Sigma-Aldrich), the cells were incubated with an FITC-conjugated anti-CD25 antibody. For nuclear staining, cells were overlayed in HBSS/BSA with 0·1% saponin-containing Hoechst (1 : 2000) for 5 min. Just before confocal analysis, the cells were placed in mounting medium [10 mm Tris–HCl (pH 8·5), 2% DABCO]. The cells were viewed on an Olympus FV1000-MPE Confocal/Multiphoton microscope modified for one-photon microscopy using an oil immersion lens at 60 ×.
Recycling assay
The recycling assay was performed as previously described.30 Briefly, 5 × 106 HEK293-CD1d WT and T322A/S323A cells were infected with WT or mutant HSV-1 (multiplicity of infection = 2) for 4 hr. One hour before harvest, the cells were treated with cyclohexamide (25 μg/ml) for 1 hr at 37°, then washed in HBSS/BSA. Unlabelled 42.1 mAb was added to block surface CD1d (10 μg/ml in 500 μl, 30 min on ice). The cells were washed in Iscove's modified Dulbecco's medium (IMDM) and resuspended in 1 ml of IMDM containing cyclohexamide for various chasing times at 37° as indicated. At each time-point, a 100-μl aliquot was placed in a well of a 96-well plate on ice. All cells were stained with a PE-labelled CD1d-specific 42.1 mAb. An aliquot not blocked by the 42.1 mAb was used as a positive control. Cells were washed in ice-cold PBS and fixed in 1% paraformaldehyde before flow cytometry analysis.
Statistical analysis
Graphs were generated and statistics were calculated using GraphPad Prism 5 (GraphPad Software, La Jolla, CA). The mean of triplicates of a representative assay is shown with error bars representing the SEM. A one-way analysis of variance with Bonferroni's post-test or Student's t-test was used as appropriate. A P-value < 0·05 was considered significant.
Results
An HSV-1 infection inhibits CD1d-mediated activation of NKT cells in different types of cells
A previous report showed that an HSV-1 infection of CD1d-expressing HeLa cells inhibits CD1d-mediated antigen presentation to NKT cells.30 To confirm that observation in our laboratory, we infected CD1d-expressing HEK293 (human embryonic kidney) cells, C1R (B lymphoblast) cells and HeLa cells with HSV-1 for 4–6 hr. The infectivity of HSV-1 in these cells was shown by flow cytometry using an HSV-specific antibody (Fig. 1a). An HSV-1 infection caused minimal changes on surface CD1d expression on these cells (Fig. 1b). Consistent with that previous report,30 our data also showed a reduction in antigen presentation by CD1d (Fig. 1c). Hence, our results confirm that HSV-1 can down-regulate antigen presentation by CD1d, and it can do so in a variety of cell types.
Figure 1.
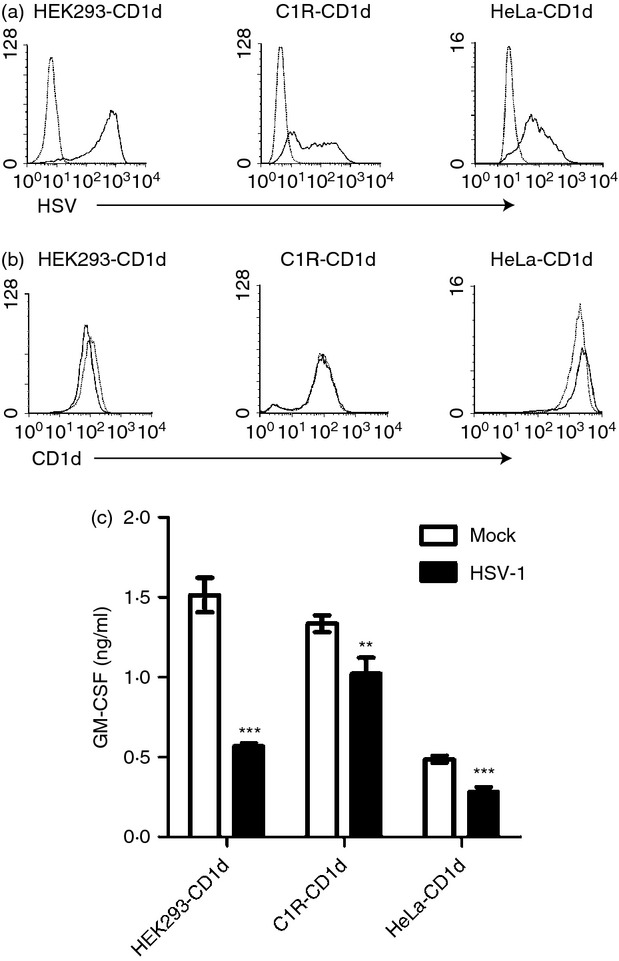
A herpes simplex virus type 1 (HSV-1) infection inhibits CD1d-mediated antigen presentation to NKT cells in a variety of cell lines. (a) Human CD1d-expressing HEK293, C1R and HeLa cells were mock-treated (dotted line) or infected with HSV-1 [solid line, multiplicity of infection (MOI) = 2 for HEK293, MOI = 10 for C1R and HeLa] for 4 hr. The cells were fixed, permeabilized with 0·1% saponin and stained with an anti-HSV antibody (clone 4F10.3) and analysed by flow cytometry. (b) The same mock-treated (dotted line) or HSV-1-infected (solid line) cells were also fixed and stained with the CD1d-specific 42.1 monoclonal antibody (mAb) and analysed by flow cytometry. (c) Aliquots of cells used in (a) and (b) were fixed and co-cultured with human NKT cells (105/well, E:T = 1 : 1) for 48 hr. The supernatants were analysed for granulocyte–macrophage colony-stimulating factor (GM-CSF) production by ELISA. The data shown are representative of three independent experiments. **P < 0·01; ***P < 0·001.
Substitution of the human CD1d cytoplasmic tail residues T322 and S323 with Asp causes a cumulative negative effect on its cell surface expression
It has previously been shown that an HSV-1 infection impairs CD1d recycling and down-regulates CD1d-mediated antigen presentation.30 We have reported that one amino acid residue in the cytoplasmic tail of CD1d, Thr322, is important for surface expression of the CD1d-β2M heterodimer, the functional form of CD1d, but not the free CD1d heavy chain (HC).33 We hypothesized that an HSV-1 infection may cause some modifications to the cytoplasmic tail of CD1d, resulting in the observed reduced recycling rate and inhibition of CD1d-mediated antigen presentation.
Substitution of another CD1d cytoplasmic tail residue (Ser323) with Asp (S323D, Fig. 2a), mimics a phosphorylated form.35 This mutation also reduces the surface expression of the CD1d-β2M complex, but has a minimal effect on CD1d HC expression (Fig. 2b). When both T322 and S323 were substituted with Asp (T322D/S323D, Fig. 2a), the surface expression level of the CD1d-β2M complex was reduced to a level below that observed with either of the single amino acid mutants alone (Fig. 2b). Similar to what we have shown with a tail-deleted mutant of CD1d (TD-14),33 the T322D/S323D mutant expressed CD1d HC at a level similar to WT CD1d (Fig. 2b), suggesting that the T322D/S323D mutation only affects the intracellular trafficking of CD1d, and not protein synthesis. As expected (and as we have previously shown),33 cells expressing the single T322D or S323D CD1d mutations showed a reduced ability to present endogenous lipid antigens to NKT cells compared with WT CD1d (Fig. 2c). Not surprisingly, the CD1d T322D/S323D double mutant-expressing cells were even less able to activate NKT cells (Fig. 2c), potentially because of lower cell surface CD1d (Fig. 2b). However, adding the CD1d-specific glycolipid ligand α-galactosylceramide (α-GalCer) exogenously,36 T322D, S323D and T322D/S323D expressing cells were all able to activate NKT cells at WT levels (Fig. 2d). These results suggest that these mutations in the cytoplasmic tail of CD1d altered its ability to present endogenous (but not exogenous) lipid antigens. Moreover, they suggest a cumulative negative effect on surface expression and function of CD1d by phosphorylation of both the T322 and S323 residues.
Figure 2.
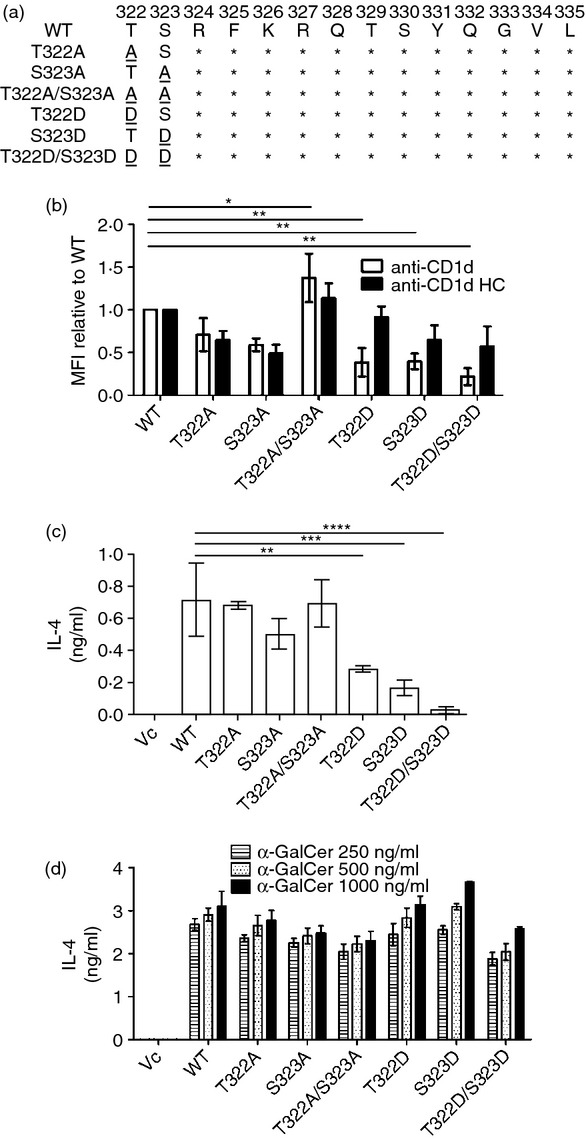
T322D and S323D mutations in the human CD1d cytoplasmic tail cause a cumulative negative effect on its cell surface expression. (a) Amino acid sequence of the cytoplasmic tail mutants used in the experiments. Wild-type (WT). (b) CD1d WT- and mutant-expressing HEK293 cells were fixed and stained with the CD1d-β2m-specific monoclonal antibody (mAb) 42.1 (white bars), followed by a phycoerythrin-conjugated anti-mouse immunoglobulin antiserum. For the CD1d heavy chain (HC)-specific mAb C3D5 (black bars), the cells were permeabilized with 0·1% saponin following fixation. Analysis was by flow cytometry. The data are displayed as the mean fluorescence intensity (MFI) relative to WT (WT = 1). *P < 0·05; **P < 0·01. CD1d WT and the indicated CD1d tail mutant-expressing cells were co-cultured with human NKT cells (E:T = 1 : 1) for 48 hr in the presence of vehicle (c) or the indicated concentrations of α-GalCer (d). Interleukin-4 (IL-4) production in the supernatants was measured by ELISA.
The T322D/S323D mutation impairs the surface expression of a CD25/CD1d fusion protein
As another means to assess whether phosphorylation of the CD1d T322 and S323 cytoplasmic tail residues could be important for its surface expression, we used a different model: a chimeric construct consisting of the human CD25 extracellular domain fused with the CD1d transmembrane domain and cytoplasmic tail (CD25/CD1d fusion).33 We mutated the T322D, S323D and T322D/S323D residues and generated stably transfected HEK293 cells expressing these individual CD1d mutants or the WT CD1d cytoplasmic tail. Using confocal microscopic analysis, we found that neither the T322D nor S323D mutants inhibited the surface expression of CD25. Only the T322D/S323D double mutant limited expression of CD25 on the cell surface (Fig. 3). This confirmed the cumulative negative effect on WT CD1d surface expression observed when both T322 and S323 were mutated to Asp (Fig. 2). Therefore, these data are consistent with the idea that the phosphorylation of the T322/S323 dyad impairs cell surface expression of human CD1d.
Figure 3.
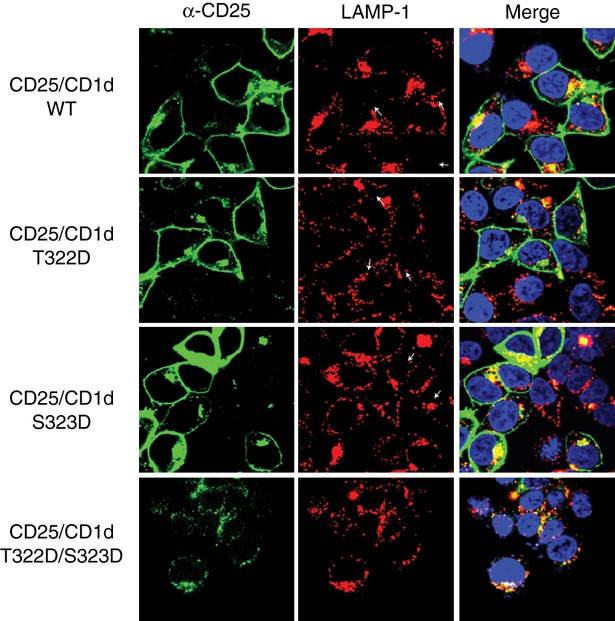
Substitution of the human CD1d cytoplasmic tail residues T322 and S323 with Asp inhibits cell surface expression of a CD25/CD1d chimeric protein. HEK293 cells were transfected with the CD25 extracellular domain fused to wild-type (WT) CD1d, or the T322D, S323D and T322D/S323D CD1d cytoplasmic tail mutants. The cells were stained with an anti-CD25 (green) and anti-LAMP-1 (red) monoclonal antibody (mAb), and Hoechst (blue). Analysis was by confocal microscopy.
Substitution of T322 and S323 to Ala inhibits the down-regulation of CD1d-mediated antigen presentation caused by an HSV-1 infection
In contrast to what was observed with the T322D/S323D double mutant, substitution of both T322 and S323 with Ala (T322A/S323A, Fig. 2a) significantly increased the surface expression of CD1d (Fig. 2b), but did not change its co-colocalization with the lysosomal marker LAMP-1.33 Substituting T322 or S323 to Ala individually did not increase CD1d surface expression (Fig. 2b), or change its intracellular distribution.33 These CD1d mutant-expressing cells activated NKT cells at WT levels (Fig. 2c,d).
It has been shown that an HSV-1 infection reduces CD1d surface expression and antigen presentation by human CD1d.30,31 Because the T322D/S323D mutation caused a negative effect on the functional expression of CD1d, we hypothesized that an HSV-1 infection may result in phosphorylation of the T322 and S323 residues of CD1d and impair its function. As an indirect means to test this hypothesis, HEK293 cells expressing WT or non-phosphorylatable mutants of CD1d (T322A, S323A and T322A/S323A), were infected with HSV-1 and co-cultured with human NKT cells. Interestingly, the decrease in CD1d-mediated antigen presentation by HSV-1-infected cells expressing the T322A/S323A double mutant was less compared with cells expressing WT CD1d, or the individual cytoplasmic tail mutants, T322A and S323A (Fig. 4). This suggested that phosphorylation of the T322/S323 dyad is probably critical for the inhibition of antigen presentation by CD1d following an HSV-1 infection. It should be noted that HSV-1 showed similar infectivity in HEK293 cells, whether they expressed WT or mutant CD1d molecules (see Supplementary material, Fig. S1a). The 4 hr infection caused little change in the surface expression of CD1d, regardless of which mutant CD1d molecules the HEK293 cells were expressing (see Supplementary material, Fig. S1b).
Figure 4.
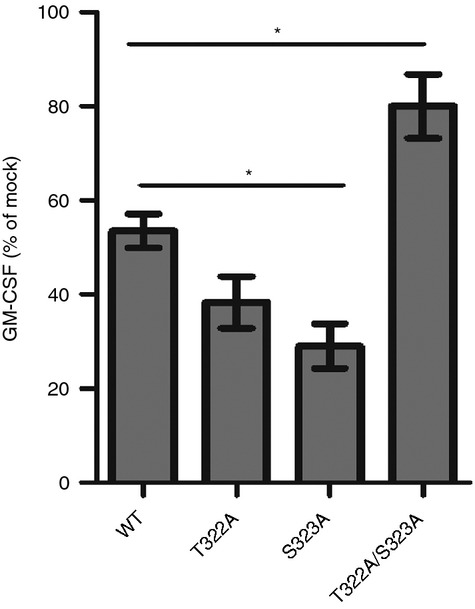
The CD1d T322A/S323A double mutant is more resistant to the down-regulation of CD1d-mediated antigen presentation caused by a herpes simplex virus type 1 (HSV-1) infection than wild-type (WT) CD1d. HEK293 cells expressing WT CD1d, or the T322A, S323A and T322A/S323A mutants were infected with HSV-1 at a multiplicity of infection (MOI) of 2 for 4 hr. The cells were then fixed with 0·05% paraformaldehyde and co-cultured with human NKT cells (105/well, E:T = 1 : 1) for 48 hr. The supernatants were analysed for granulocyte–macrophage colony-stimulating factor (GM-CSF) production by ELISA. The data are shown as the percentage of mock-treated cells (mock = 100%). *P < 0·05.
The T322A/S323A double mutant of CD1d recycles faster than WT following an HSV-1 infection
As previously reported, an HSV-1 infection causes redistribution of CD1d to lysosome-like structures and reduces the surface expression of CD1d.30 In line with that report, we found that an HSV-1 infection decreased the surface expression of both WT CD1d and the T322A/S323A double mutant 18 hr post-infection (Fig. 5a). We also found that an HSV-1 infection reduced the level of CD1d HC in both WT CD1d and the T322A/S323A mutant (Fig. 5a). There was less of a reduction of the CD1d HC in the T322A/S323A mutant compared with WT CD1d, but the difference was not significant (Fig. 5b). This suggests that protein synthesis of the T322A/S323A mutant is similarly inhibited as is WT CD1d following an HSV-1 infection. However, the intracellular level of the CD1d-β2M heterodimer in the double mutant-expressing cells was significantly less affected by an HSV-1 infection compared with those expressing WT CD1d molecules (Fig. 5a,b). The results suggest that the T322A/S323A double mutant is probably more resistant to lysosomal degradation following an HSV-1 infection. We next tested whether the T322A/S323A double mutant has a longer half-life than WT molecules post-HSV-1 infection. Surprisingly, the CD1d T322A/S323A double mutant molecules detected as either CD1d HC (see Supplementary material, Fig. S2) or CD1d-β2M heterodimeric forms (data not shown), had a similar half-life as WT CD1d molecules.
Figure 5.
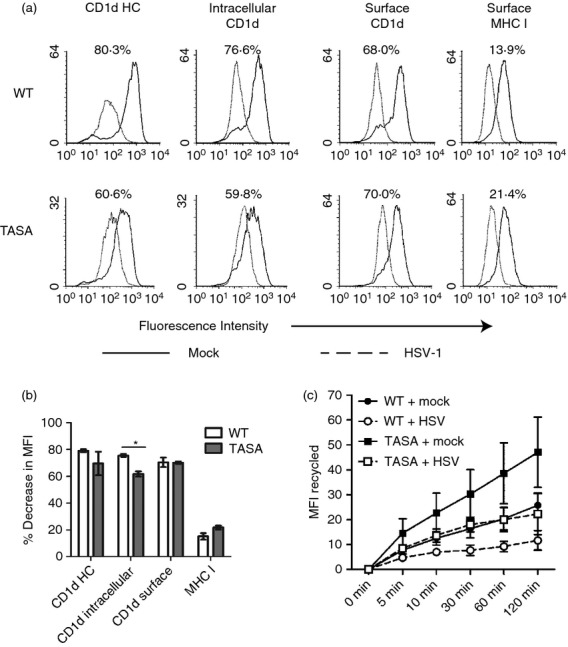
The T322A/S323A double mutant of CD1d recycles faster than wild-type (WT) following a herpes simplex virus type 1 (HSV-1) infection. (a) HEK293 cells expressing WT CD1d or a T322A/S323A double mutant were mock- (solid line) or HSV-1-infected (dotted line, multiplicity of infection = 2) for 18 hr. The cells were then fixed and stained for intracellular CD1d HC, intracellular CD1d-β2m complex, surface CD1d-β2m and surface MHC class I as indicated. The per cent decrease in mean fluorescence intensity (MFI) post-infection is shown and calculated using the following formula: [decrease in MFI = 100% × (MFIvirus−MFImock)/MFImock]; where MFImock: MFI from mock-treated cells; MFIvirus: MFI from virus-infected cells. The percentage decrease in MFI after virus infection is shown in (b). *P < 0·05. (c) HEK293 cells expressing WT CD1d (circles) or a T322A/S323A double mutant (squares) were mock- (black symbols) or HSV-1-infected (white symbols) at a multiplicity of infection of 2 for 4 hr. The level of recycled cell surface CD1d is shown and was calculated by subtracting the MFI at each time point from time 0. The data shown are combined from three independent experiments.
An HSV-1 infection also inhibits intracellular CD1d from recycling back to the cell surface,30 which is critical for CD1d-mediated antigen presentation.37 The T322A/S323A CD1d double mutant recycled faster than WT CD1d in the absence of an HSV-1 infection (Fig. 5c). Although an HSV-1 infection reduced the recycling rate of both WT CD1d and the T322A/S323A mutant, the T322A/S323A double mutant still recycled more quickly than WT CD1d-expressing cells infected with HSV-1. In fact, the recycling was at a similar level as mock-treated WT CD1d (Fig. 5c). The faster recycling rate in the CD1d T322A/S323A mutant may explain why it is resistant to the down-regulation of CD1d-mediated antigen presentation caused by an HSV-1 infection.
A US3/UL13 double-deficient HSV-1 (ΔUS3/ΔUL13) inhibits CD1d-mediated antigen presentation at a level similar to the WT virus
Because it was likely that the phosphorylation of the CD1d cytoplasmic tail T322 and S323 residues following an HSV-1 infection was responsible for the decrease in CD1d-mediated antigen presentation post-infection, it was important to determine if virus-encoded serine/threonine protein kinases played a role in this observation. This was particularly important, because cells expressing the non-phosphorylatable mutant of CD1d (T322A/S323A) are less sensitive to the HSV-1-mediated down-regulation of antigen presentation by CD1d. There are two well-characterized viral serine/threonine protein kinases in HSV-1: US3 and UL13.38 Hence, HEK293-CD1d cells were infected with WT HSV-1 or the ΔUS3/ΔUL13 virus for 4 hr. The ΔUS3/ΔUL13 virus, which had similar infectivity as the WT virus (Fig. 6a), inhibited CD1d-mediated antigen presentation to the same degree as the WT virus post-infection (Fig. 6c). There was a minimal change in surface expression of CD1d following infection with either WT HSV-1 or the ΔUS3/ΔUL13 mutant virus (Fig. 6b). Our data suggest that the US3 or UL13 viral serine/threonine kinases do not affect CD1d-mediated antigen presentation following an HSV-1 infection, and neither T322 nor S323 is likely to be phosphorylated by these HSV-1-encoded serine/threonine kinases. Consistent with this observation, when hydrogen peroxide treatment or serum starvation was used to induce oxidative stress in HEK293-CD1d cells, we found that antigen presentation by the CD1d T322A/S323A double mutant was much higher than that observed with WT CD1d (see Supplementary material, Fig. S3). Hence, we believe that it is likely that a host serine/threonine kinase(s), activated by stress (e.g. following an HSV-1 infection), is responsible for phosphorylating T322 and S323 (directly or indirectly).
Figure 6.
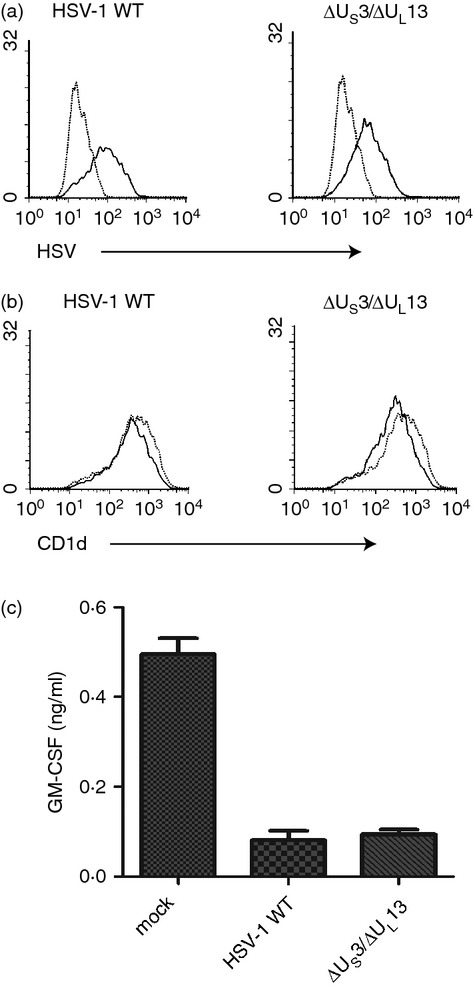
A US3/UL13 double-deficient herpes simplex virus type 1 (HSV-1; ΔUS3/ΔUL13) inhibits CD1d-mediated antigen presentation at a similar level as the wild-type (WT) virus. (a) HEK293-CD1d cells were mock-treated (dotted line) or infected with HSV-1 (solid line, multiplicity of infection = 2) for 4 hr. The cells were fixed, permeabilized and stained with an anti-HSV monoclonal antibody (mAb; clone 4F10.3) and analysed by flow cytometry. (b) Aliquots of the same mock-treated (dotted line) or HSV-1-infected (solid line) cells were stained with a CD1d-specific mAb and cell surface expression was analysed by flow cytometry. (c) Aliquots of cells also used for (a) and (b) were fixed and co-cultured with human NKT cells (105/well, E : T = 1 : 1) for 48 hr. granulocyte–macrophage colony-stimulating factor production into the supernatants was measured by ELISA.
Discussion
A deficiency in NKT cells and diminished CD1d expression were reported to be associated with a life-threatening varicella zoster virus infection in humans.26 In this study, we found that the T322 and S323 residues in the CD1d cytoplasmic tail are critical in the HSV-1-mediated reduction in antigen presentation by CD1d. Via strong (albeit indirect) evidence, we believe this effect is probably a result of the phosphorylation of these residues. We found that substitution of T322 and S323 with Asp, a regularly used method to mimic phosphorylation,35 impairs the functional expression of CD1d [ref. 33 and this report]. In the context of an HSV-1 infection, the non-phosphorylatable T322A/S323A mutant was more resistant to the HSV-1-dependent down-regulation of antigen presentation by CD1d. We attempted to detect the phosphorylation of T322 or S323 in CD1d by Western blot analysis using Phospho-Ser/Thr-specific antibodies and by using [32P] in vivo labelling. By neither approach were we able to observe any phosphorylation of CD1d (data not shown). This was not entirely surprising, because it is likely that the phosphorylation of T322 and S323 is transient and so very difficult to detect. Furthermore, we have previously reported that the likely phosphorylation of T322 is a signal for lysosomal degradation of CD1d.33 Consistent with this, Rao et al.39 proposed that the HSV-1 viral kinase US3 is important for the down-regulation of CD1d-mediated antigen presentation; they also did not directly observe phosphorylation of CD1d when US3 was over-expressed. Overall, our data strongly support our hypothesis that the HSV-1-dependent down-regulation of CD1d-mediated antigen presentation is due, at least in part, to phosphorylation of the T322/S323 dual-residue motif in the CD1d cytoplasmic tail.
Because the phosphorylation of T322 or S323 was never directly detected, we cannot rule out other possibilities of why the T322A/S323A mutant expresses higher surface levels of CD1d and becomes resistant to HSV-1-mediated down-regulation of antigen presentation. Our data suggest that the presence of hydrophobic residues at these two positions is important for its surface expression. Indeed, it has been reported that some G protein-coupled receptors contain hydrophobic motifs in the membrane-proximal C-terminal tail that are important for their transport from the endoplasmic reticulum to the Golgi.40,41 Although the T322/S323 motif in CD1d does not resemble any of those hydrophobic motifs in G protein-coupled receptors, it is possible that hydrophobic or neutral amino acids at these two positions are necessary for binding to some chaperones for transporting CD1d to the cell surface. When the hydrophobic residues are absent (e.g. tail-deleted CD1d, TD-14),33 or become negatively charged (e.g. T322D/S323D), it is likely that these chaperones could not transport CD1d to the cell surface, so the surface expression of CD1d is impeded. We are actively in the process of identifying those potential chaperones.
Relevant to what is discussed above, another possible mechanism by which T322A/S323A mutant is more resistant to viral-mediated down-regulation of antigen presentation is that it may have a stronger association with lipid rafts compared with WT CD1d. Several reports demonstrate that murine CD1d (mCD1d) localizes to specialized membrane microdomains called lipid rafts.42,43 Infection of macrophages with the protozoan parasite Leishmania donovani, changes the distribution of mCD1d to non-lipid raft regions and inhibits antigen presentation by mCD1d.44 To test the hypothesis of whether T322A/S323A mutant has stronger association with lipid rafts, we treated CD1d WT and T322A/S323A mutant with different concentration of methyl-β-cyclodextrin (MBCD), an inhibitor of lipid rafts. We found that MBCD similarly inhibits CD1d-mediated antigen presentation in both WT and T322A/S323A CD1d (see Supplementary material, Fig. S4), suggesting that the T322A/S323A mutant is not more associated with lipid rafts than WT CD1d.
The YXXZ motif of human CD1d is important for its endocytosis.45 Substituting Y to A (Y331A) or deleting YXXZ (TD-6) blocks CD1d from internalization and causes CD1d accumulation at the cell surface; hence, the Y331A and TD-6 mutants express higher surface levels of CD1d than WT.33 The T322A/S323A mutant, which also expresses CD1d at a higher level on the cell surface, has a similar internalization rate as the WT CD1d (data not shown). In contrast to T322A/S323A, the TD-6 and Y331A mutants are less effective at activating NKT cells, compared with WT CD1d.33 On the other hand, the intracellular CD1d-β2M complex in Y331A mutant-expressing cells (but not in the T322A/S323A mutant), was down-regulated to a similar degree as WT CD1d-expressing cells upon an HSV-1 infection (see Supplementary material, Fig. S5). Our data suggest that the resistance of the T322A/S323A mutant to HSV-1-mediated down-regulation of CD1d-mediated antigen presentation is not merely because of its high surface expression, but rather its faster recycling rate.
Another question concerns the Ser/Thr kinase that catalyses the phosphorylation of T322 and S323 following an HSV-1 infection. We are not aware of any existing Ser/Thr kinase consensus motif that includes these two residues in CD1d. However, there are two well-characterized HSV-1-encoded Ser/Thr protein kinases: US3 and UL13.38 Rao et al.39 reported that US3 is important for inhibiting CD1d-mediated antigen presentation by HSV-1. But phosphorylation of CD1d was never detected when US3 was over-expressed, suggesting that US3 does not directly phosphorylate the CD1d cytoplasmic tail.39 It is unlikely that US3 phosphorylates the cytoplasmic tail of CD1d, because we found that a US3-deficient HSV-1 inhibited CD1d-mediated antigen presentation to the same degree as a US3-rescued virus (data not shown). The difference between the two observations is probably due to different cell lines (HEK293 vs HeLa) and/or different infection conditions (4 hr versus 12 hr post-infection). We also found that a UL13-deficient HSV-1 was no different from WT HSV-1 in inhibiting CD1d function (data not shown), suggesting that UL13 is not the Ser/Thr kinase that phosphorylates the CD1d T322 and/or S323 residues. We speculate that the stress induced by a viral infection activates a host Ser/Thr kinase(s) that is responsible for phosphorylating these residues in CD1d. In line with this idea, when hydrogen peroxide treatment or serum starvation was used to induce oxidative stress in HEK293-CD1d cells, we found that antigen presentation by the CD1d T322A/S323A double mutant was much higher than that observed with WT CD1d (Fig. S3). Hence, we believe it is likely that a host serine/threonine kinase(s), activated by stress (e.g. following an HSV-1 infection), is responsible for phosphorylating T322 and S323 (directly or indirectly). Potential candidates for these host Ser/Thr kinases include the mitogen-activated kinases (MAPK), p38, ERK and JNK. An HSV-1 infection does activate p38 and JNK, but not ERK (see Supplementary material, Fig. S6). We have previously shown that p38 and JNK (Liu et al., manuscript in preparation) are negative regulators of CD1d-mediated antigen presentation, whereas ERK promotes antigen presentation by CD1d.27,29 Therefore, those data in our previous reports are consistent with the observations reported here.
In conclusion, we have identified a dual-residue motif in the cytoplasmic tail of human CD1d that is important for its functional expression upon an HSV-1 infection. Another study (Liu et al., manuscript in preparation) suggests that this motif is most likely involved with binding to the cytoskeletal network, which is likely to be modified after a viral infection or stress. Activation of host Thr/Ser kinases after an HSV-1 infection may cause phosphorylation of the T322/S323 dual-residue motif of CD1d and thereby down-regulate CD1d-mediated antigen presentation (Fig. 7). Our current studies are focused on how those changes in the cytoskeletal network can modulate CD1d-mediated antigen presentation and, as a result, impact the host's innate anti-viral immune response.
Figure 7.
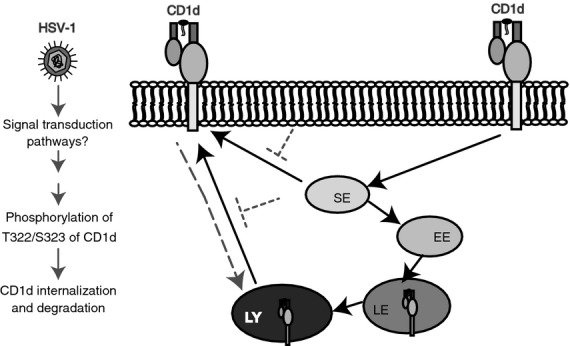
Model illustrating how a herpes simplex virus type 1 (HSV-1) infection inhibits CD1d-mediated antigen presentation. The solid arrows show normal CD1d trafficking.33 The dotted arrows indicate CD1d trafficking upon an HSV-1 infection. The virus infection likely activates host Thr/Ser kinases, causing the phosphorylation of the CD1d T322/S323 residues. The phosphorylation of T322/S323 results in lysosomal degradation and a reduced CD1d recycling rate. SE, sorting endosome; EE, early endosome; LE, late endosome; LY, lysosomes.
Acknowledgments
The authors would like to thank Drs B. Roizman (University of Chicago), S. Balk (Harvard University), P. Cresswell (Yale University), J. Blum (Indiana University) and P. Cohen (University of Dundee, UK) for critical reagents, Drs M. Harrington and J. Blum (Indiana University) for helpful discussions, as well as the Indiana Center for Biological Microscopy and the Flow Cytometry Resource Facility, Indiana University School of Medicine. This work was supported by National Institutes of Health grants R01 AI46455 and P01 AI056097 (to RRB), and NSF CHE-0194682 from the National Science Foundation (to JGH).
Disclosures
The authors have no financial conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. HEK293 cells expressing wild-type (WT) CD1d, or the T322A, S323A and T322A/S323A mutants were mock-infected (black line) or infected with herpes simplex virus type 1 (HSV-1; red line) at a multiplicity of infection of 2 for 4 hr.
Figure S2. Wild-type (WT) CD1d and the CD1d T322A/S323A double mutant have similar half-lives following an herpes simplex virus type 1 (HSV-1) infection.
Figure S3. The T322A/S323A CD1d double mutant is more resistant to the inhibition of CD1d-mediated antigen presentation caused by oxidative stress than wild-type (WT) CD1d.
Figure S4. The lipid-raft inhibitor methyl-β-cyclodextrin reduces CD1d-mediated antigen presentation in both wild-type (WT) and T322A/S323A CD1d-expressing cells.
Figure S5. The intracellular CD1d-β2m complex in T322A/S323A double mutant-expressing cells is less reduced than wild-type (WT) CD1d following a herpes simplex virus type 1 (HSV-1) infection.
Figure S6. A herpes simplex virus type 1 (HSV-1) infection activates Jun-N-terminal kinase (JNK) and p38, but not extracellular signal-regulated kinase 1/2 (ERK1/2). HEK293-CD1d cells were mock-infected or infected with HSV-1 (multiplicity of infection = 5) for 4 hr.
References
- 1.Perng GC, Jones C. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis. 2010;2010:262415. doi: 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–21. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 3.Letenneur L, Peres K, Fleury H, et al. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer's disease: a population-based cohort study. PLoS One. 2008;3:e3637. doi: 10.1371/journal.pone.0003637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teare EL, Clements MR. Acyclovir for suspected systemic herpes infections. Lancet. 1980;1:42. doi: 10.1016/s0140-6736(80)90577-2. [DOI] [PubMed] [Google Scholar]
- 5.Sellner J, Trinka E. Seizures and epilepsy in herpes simplex virus encephalitis: current concepts and future directions of pathogenesis and management. J Neurol. 2012;259:2019–30. doi: 10.1007/s00415-012-6494-6. [DOI] [PubMed] [Google Scholar]
- 6.Leib DA. Counteraction of interferon-induced antiviral responses by herpes simplex viruses. Curr Top Microbiol Immunol. 2002;269:171–85. doi: 10.1007/978-3-642-59421-2_11. [DOI] [PubMed] [Google Scholar]
- 7.Koelle DM, Corey L. Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med. 2008;59:381–95. doi: 10.1146/annurev.med.59.061606.095540. [DOI] [PubMed] [Google Scholar]
- 8.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol. 2004;78:1675–84. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 1994;77:525–35. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 10.Fruh K, Ahn K, Djaballah H, et al. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375:415–8. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 11.Hill A, Jugovic P, York I, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–5. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 12.Tomazin R, Hill AB, Jugovic P, et al. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 1996;15:3256–66. [PMC free article] [PubMed] [Google Scholar]
- 13.Ahn K, Meyer TH, Uebel S, et al. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 1996;15:3247–55. [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann J, Eis-Hubinger AM, Koch N. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J Immunol. 2003;171:3075–83. doi: 10.4049/jimmunol.171.6.3075. [DOI] [PubMed] [Google Scholar]
- 15.Temme S, Eis-Hubinger AM, McLellan AD, Koch N. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J Immunol. 2010;184:236–43. doi: 10.4049/jimmunol.0902192. [DOI] [PubMed] [Google Scholar]
- 16.Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–90. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 17.Yoshimoto T, Paul WE. CD4pos, NK1.1pos T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J Exp Med. 1994;179:1285–95. doi: 10.1084/jem.179.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasquier B, Yin L, Fondaneche MC, et al. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med. 2005;201:695–701. doi: 10.1084/jem.20042432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Santo C, Salio M, Masri SH, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. 2008;118:4036–48. doi: 10.1172/JCI36264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terabe M, Swann J, Ambrosino E, et al. A nonclassical non-Vα14Jα18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med. 2005;202:1627–33. doi: 10.1084/jem.20051381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renukaradhya GJ, Khan MA, Vieira M, Du W, Gervay-Hague J, Brutkiewicz RR. Type I NKT cells protect (and Type II NKT cells suppress) the host's innate antitumor immune response to a B cell lymphoma. Blood. 2008;111:5637–45. doi: 10.1182/blood-2007-05-092866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grubor-Bauk B, Simmons A, Mayrhofer G, Speck PG. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant Vα14-Jα281 TCR. J Immunol. 2003;170:1430–4. doi: 10.4049/jimmunol.170.3.1430. [DOI] [PubMed] [Google Scholar]
- 23.Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol. 2003;77:10168–71. doi: 10.1128/JVI.77.18.10168-10171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cornish AL, Keating R, Kyparissoudis K, Smyth MJ, Carbone FR, Godfrey DI. NKT cells are not critical for HSV-1 disease resolution. Immunol Cell Biol. 2006;84:13–9. doi: 10.1111/j.1440-1711.2005.01396.x. [DOI] [PubMed] [Google Scholar]
- 25.Levy O, Orange JS, Hibberd P, et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J Infect Dis. 2003;188:948–53. doi: 10.1086/378503. [DOI] [PubMed] [Google Scholar]
- 26.Banovic T, Yanilla M, Simmons R, et al. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J Infect Dis. 2011;204:1893–901. doi: 10.1093/infdis/jir660. [DOI] [PubMed] [Google Scholar]
- 27.Renukaradhya GJ, Khan MA, Shaji D, Brutkiewicz RR. Vesicular stomatitis virus matrix protein impairs CD1d-mediated antigen presentation through activation of the p38 MAPK pathway. J Virol. 2008;82:12535–42. doi: 10.1128/JVI.00881-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho S, Knox KS, Kohli LM, et al. Impaired cell surface expression of human CD1d by the formation of an HIV-1 Nef/CD1d complex. Virology. 2005;337:242–52. doi: 10.1016/j.virol.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 29.Renukaradhya GJ, Webb TJ, Khan MA, et al. Virus-induced inhibition of CD1d1-mediated antigen presentation: reciprocal regulation by p38 and ERK. J Immunol. 2005;175:4301–8. doi: 10.4049/jimmunol.175.7.4301. [DOI] [PubMed] [Google Scholar]
- 30.Yuan W, Dasgupta A, Cresswell P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol. 2006;7:835–42. doi: 10.1038/ni1364. [DOI] [PubMed] [Google Scholar]
- 31.Raftery MJ, Winau F, Kaufmann SH, Schaible UE, Schonrich G. CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J Immunol. 2006;177:6207–14. doi: 10.4049/jimmunol.177.9.6207. [DOI] [PubMed] [Google Scholar]
- 32.Bosnjak L, Sahlstrom P, Paquin-Proulx D, Leeansyah E, Moll M, Sandberg JK. Contact-dependent interference with invariant NKT cell activation by herpes simplex virus-infected cells. J Immunol. 2012;188:6216–24. doi: 10.4049/jimmunol.1100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Shaji D, Cho S, Du W, Gervay-Hague J, Brutkiewicz RR. A threonine-based targeting signal in the human CD1d cytoplasmic tail controls its functional expression. J Immunol. 2010;184:4973–81. doi: 10.4049/jimmunol.0901448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du W, Gervay-Hague J. Efficient synthesis of α-galactosyl ceramide analogues using glycosyl iodide donors. Org Lett. 2005;7:2063–5. doi: 10.1021/ol050659f. [DOI] [PubMed] [Google Scholar]
- 35.Sakaguchi M, Miyazaki M, Takaishi M, et al. S100C/A11 is a key mediator of Ca2+-induced growth inhibition of human epidermal keratinocytes. J Cell Biol. 2003;163:825–35. doi: 10.1083/jcb.200304017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brutkiewicz RR. CD1d ligands: the good, the bad, and the ugly. J Immunol. 2006;177:769–75. doi: 10.4049/jimmunol.177.2.769. [DOI] [PubMed] [Google Scholar]
- 37.Moody DB, Porcelli SA. Intracellular pathways of CD1 antigen presentation. Nat Rev Immunol. 2003;3:11–22. doi: 10.1038/nri979. [DOI] [PubMed] [Google Scholar]
- 38.Smith-Donald BA, Roizman B. The interaction of herpes simplex virus 1 regulatory protein ICP22 with the cdc25C phosphatase is enabled in vitro by viral protein kinases US3 and UL13. J Virol. 2008;82:4533–43. doi: 10.1128/JVI.02022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao P, Pham HT, Kulkarni A, et al. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J Virol. 2011;85:8093–104. doi: 10.1128/JVI.02689-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bermak JC, Li M, Bullock C, Zhou QY. Regulation of transport of the dopamine D1 receptor by a new membrane-associated ER protein. Nat Cell Biol. 2001;3:492–8. doi: 10.1038/35074561. [DOI] [PubMed] [Google Scholar]
- 41.Robert J, Clauser E, Petit PX, Ventura MA. A novel C-terminal motif is necessary for the export of the vasopressin V1b/V3 receptor to the plasma membrane. J Biol Chem. 2005;280:2300–8. doi: 10.1074/jbc.M410655200. [DOI] [PubMed] [Google Scholar]
- 42.Park YK, Lee JW, Ko YG, Hong S, Park SH. Lipid rafts are required for efficient signal transduction by CD1d. Biochem Biophys Res Commun. 2005;327:1143–54. doi: 10.1016/j.bbrc.2004.12.121. [DOI] [PubMed] [Google Scholar]
- 43.Lang GA, Maltsev SD, Besra GS, Lang ML. Presentation of α-galactosylceramide by murine CD1d to natural killer T cells is facilitated by plasma membrane glycolipid rafts. Immunology. 2004;112:386–96. doi: 10.1111/j.1365-2567.2004.01896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karmakar S, Paul J, De T. Leishmania donovani glycosphingolipid facilitates antigen presentation by inducing relocation of CD1d into lipid rafts in infected macrophages. Eur J Immunol. 2011;41:1376–87. doi: 10.1002/eji.201040981. [DOI] [PubMed] [Google Scholar]
- 45.Rodionov DG, Nordeng TW, Pedersen K, Balk SP, Bakke O. A critical tyrosine residue in the cytoplasmic tail is important for CD1d internalization but not for its basolateral sorting in MDCK cells. J Immunol. 1999;162:1488–95. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.