

Abstract
Autologous haematopoietic stem cell transplantation (HSCT) for relapsing–remitting multiple sclerosis is a potentially curative treatment, which can give rise to long-term disease remission. However, the mode of action is not yet fully understood. The aim of the study was to evaluate similarities and differences of the CD4+ T-cell populations between HSCT-treated patients (n = 12) and healthy controls (n = 9). Phenotyping of memory T cells, regulatory T (Treg) cells and T helper type 1 (Th1) and type 17 (Th17) cells was performed. Further, T-cell reactivity to a tentative antigen, myelin oligodendrocyte glycoprotein, was investigated in these patient populations. Patients treated with natalizumab (n = 15) were included as a comparative group. White blood cells were analysed with flow cytometry and T-cell culture supernatants were analysed with magnetic bead panel immunoassays. HSCT-treated patients had similar levels of Treg cells and of Th1 and Th17 cells as healthy subjects, whereas natalizumab-treated patients had lower frequencies of Treg cells, and higher frequencies of Th1 and Th17 cells. Cells from HSCT-treated patients cultured with overlapping peptides from myelin oligodendrocyte glycoprotein produced more transforming growth factor-β1 than natalizumab-treated patients, which suggests a suppressive response. Conversely, T cells from natalizumab-treated patients cultured with those peptides produced more interleukin-17 (IL-17), IL-1 and IL-10, indicating a Th17 response. In conclusion, we demonstrate circumstantial evidence for the removal of autoreactive T-cell clones as well as development of tolerance after HSCT. These results parallel the long-term disease remission seen after HSCT.
Keywords: haematopoietic stem cell transplantation, multiple sclerosis, natalizumab, neuroimmunology
Introduction
Autologous haematopoietic stem cell transplantation (HSCT) has been performed for multiple sclerosis (MS) since 1995.1 The aim is to achieve long-term disease remission through short-lasting lymphoablation followed by rebuilding of a new immune system. Initially this procedure was reserved for patients with treatment-resistant progressive forms of MS, largely with disappointing results. Eventually, HSCT was tried for relapsing–remitting MS (RRMS) and it has become clear that HSCT can be a very effective and possibly curative treatment for RRMS.2,3 Most patients remain free from disease activity, even with long follow up. At our centre we have seen patients with remission up to 9 years after HSCT; others have reported similar outcomes.4–6
The mode of action is not fully understood, and several mechanisms probably contribute to the effect. We know that HSCT causes a profound renewal of the immune system and not just long-lasting immune suppression,7 and at least part of the effect is probably related to removal of autoreactive clones. Nevertheless, some autoreactive cells probably escape the treatment and remain after HSCT, which was demonstrated in a recent study.8 Such autoreactive cells must be kept in control to maintain remission, and a possible mechanism is restoration of tolerance to self-antigens. In this setting, tolerance can be achieved through massive exposure to myelin degradation products in the developing immune system after HSCT and lack of co-stimulation during antigen presentation. The developing immune system has low levels of co-stimulatory molecules such as CD289 and the presence of myelin peptides in the absence of co-stimulation could give rise to clonal anergy and tolerance through myelin-specific natural T regulatory (Treg) cells or induction of Treg type 1 cells.
In a recent study of HSCT in experimental autoimmune encephalomyelitis, non-myeloablative conditioning without stem cell transplantation was not sufficient for the induction of long-term remission, whereas with transplantation long-term remission could be achieved.10 This suggests that the haematopoietic stem cells themselves are important for the establishment of tolerance.
The aim of the present study was to evaluate similarities and differences of the CD4+ T-cell populations between HSCT-treated patients and healthy controls. More specifically, we performed phenotyping of memory T cells, Treg cells, T helper type 1 (Th1) cells and Th17 cells. In addition, we also investigated a novel marker of a subset of Treg cells previously not investigated in MS: the transcription factor Helios. Helios is a member of the Ikaros family and binds to the FoxP3 promoter. It has been suggested as a marker of natural Treg (nTreg) cells developed in the thymus as opposed to peripherally induced Treg (pTreg) cells.11,12 T-cell reactivity to a tentative antigen: myelin oligodendrocyte glycoprotein (MOG) was investigated in CD4+ T cells.
Patients were treated with HSCT up to 7 years before the study, and therefore no pre-HSCT blood samples were available. In lieu, patients treated with natalizumab (NZB) were included as a contrasting group with immunologically highly active MS. NZB inhibits extravasation of T cells and causes a sequestration of pathological T cells in the peripheral circulation without activation of the T cells.13
Methods
Ethics statement
The study was approved by the local ethics committee of Uppsala University (DNr 2006:089). All participants provided written informed consent.
Subjects
In total 36 individuals were included: 12 patients who had undergone HSCT as rescue therapy for aggressive RRMS, 15 NZB-treated patients with RRMS and nine healthy controls. HSCT was performed with the BEAM/ATG protocol (bis-chloroethylnitrosourea, etoposide, cytosine-arabinoside, melphalan with addition of anti-thymocyte globulin), which is considered to be a medium-intensity protocol. Peripheral haematopoietic stem cells were mobilized with cyclophosphamide and granulocyte colony-stimulating factor. No ex vivo graft manipulation was performed. NZB-treated patients received infusions every month, and the total number of infusions received ranged from 9 to 49. All patients were treated at the Department of Neurology at Uppsala University Hospital; blood donors served as healthy controls.
The HSCT-treated patients had a severe disease course with an expanded disability disease scale score of up to 9·0 pre-treatment and in several cases > 20 gadolinium-enhancing lesions. The annualized relapse rate was on average 6·5 with an increased number of relapses in the year before HSCT, reaching annualized relapse rate > 10 in some cases. The clinical characteristics of some of the patients treated with HSCT as well as the clinical effects have been reported previously.2 NZB-treated patients had a fairly aggressive disease with an annualized relapse rate of 2·0 during the pre-treatment course. The characteristics of patients and controls are summarized in Table 1. A more detailed description of the HSCT-treated patients is available in the Supplementary material (Table S1).
Table 1.
Demographic data and clinical characteristics of the included subjects
| Control | HSCT | Natalizumab | |
|---|---|---|---|
| n | 9 | 12 | 15 |
| Male/female | 3/6 | 4/8 | 4/11 |
| Age (years) | 27 (24–37) | 32 (16–41) | 39 (18–67) |
| Total disease duration (months) | n/a | 94 (44–170) | 140 (17–393) |
| Disease duration after commencement of treatment (months) | n/a | 53 (3–84) | 24 (9–49) |
| Number of relapses in the year before treatment start | n/a | 6·9 | 2·0 |
| Annualized relapse rate after treatment | n/a | 0·06 | 0·1 |
HSCT, haematopoietic stem cell transplant; n/a, not applicable.
Data for age, disease duration and duration of treatment are given as means (range). For HSCT-treated patients, duration of treatment equals time since HSCT.
Cell isolation
White blood cells were isolated by incubation of heparin blood with a lysing buffer (cat. no. 555899; BD Biosciences, San Jose, CA) according to the manufacturer's protocol. The cells were washed with PBS and then cryopreserved at − 80°.
Short-term recall assay of MOG-reactive T cells
A total of 1·5 × 106 white blood cells were incubated at 37° for 4 hr with 1 μg of a 15-mer peptide mix (in total 29 different peptides) of 11 overlapping amino acids from the MOG (JPT Peptide Technologies, Berlin, Germany). The complete sequence of MOG can be found in the Supplementary material (Appendix S1). This was followed by a 5-hr incubation with brefeldin A (cat. no. 347688; BD Biosciences). Intracellular staining for flow cytometry was performed using a Cytofix/Cytoperm™ kit (cat. no. 554714; BD Biosciences).
Long-term recall assay of MOG-reactive T cells
A total of 106 cells were labelled with CFSE using the CellTrace™ CFSE Cell Proliferation Kit (cat. no. C34554; Invitrogen, Carlsbad, CA). Labelled cells were divided into three groups: unstimulated cells; cells stimulated with 100 IU/ml human recombinant interleukin-2 (IL-2; R&D Systems Inc., Minneapolis, MN) and 0·3 μg MOG peptide mix; and cells stimulated with 100 IU/ml IL-2 and 2 μg/ml OKT-3 Orthoclone (anti-CD3) antibody (Cilag Ag Int., Zug, Switzerland). The cells were kept in culture for 7 days, and additional medium supplemented with IL-2 was added every other day. At endpoint, cells were analysed with flow cytometry. Supernatants were analysed using magnetic bead panel immunoassays.
Flow cytometry
A total of 105 unstimulated white blood cells were stained with antibodies against surface markers for 15 min at 4°. Intracellular staining for FoxP3 and Helios was performed with 5 × 105 unstimulated cells for 30 min at 4° using a FOXP3 Fix/Perm Buffer set according to the manufacturer's protocol (cat. no. 421403; BioLegend, San Diego, CA). The combinations of antibodies used are specified in the Supplementary material (Appendix S2).
Memory T cells were defined as CD4+ CD45RO+ T cells. Th1 cells were defined as CD4+ T cells producing interferon-γ (IFN-γ), and Th17 cells were defined as CD4+ T cells producing IL-17. Treg cells were defined as CD4+ FoxP3+ T cells, nTreg cells were defined as CD4+ FoxP3+ Helios+ T cells and pTreg cells were defined as CD4+ FoxP3+ Helios– T cells.
Cells were acquired on a FACS Calibur or a FACS CantoII cytometer (BD Biosciences) and data were analysed using flowjo version 9.3.3 (Tree Star Inc., Ashland, OR). All data analyses were performed at the end of the study to minimize potential temporal artefacts. Further, all analyses were performed by an investigator who was blinded to the subject's identity to minimize bias. A quality control was performed after data collection, namely, proliferative ability after polyclonal stimulation. If T cells did not respond to polyclonal stimulation in the long-term recall assay, all results from the long-term recall assay of those subjects were discarded and not included in the analysis. The numbers listed in the results reflect the samples that passed the quality control criteria and that were included in the analysis.
Magnetic bead panel immunoassay
Supernatants from the long-term recall assay were analysed on a Magpix (Luminex, Austin, TX). For analysis of IL-1, IL-6, IL-10, IL-17 and IFN-γ, a Milliplex MAG human cytokine/chemokine magnetic bead panel immunoassay (Merck Millipore, Billerica, MA) was used. For analysis of transforming growth factor-β (TGF-β), a Bio-Plex Pro TGF-β 3-plex assay (Bio-Rad, Hercules, CA) was used.
Statistical analysis
All statistical analyses were done with graphpad prism 5·0 (GraphPad Software, La Jolla, CA). For establishing statistical significance between the groups the Kruskal–Wallis test was used and Dunn's test was used for the post hoc analysis. A two-tailed P value of < 0·05 was considered significant. All described differences are statistically significant unless otherwise stated. Statistical significances are indicated by *P < 0·05 and ***P < 0·001 in the figures.
Results
HSCT-treated patients display circulating Treg cell levels comparable to healthy controls
Frequencies of memory T cells, Treg cells and Th1 and Th17 cells were investigated (Table 2). A trend towards a difference in percentage of CD4+ CD45RO+ T cells was observed between the three groups: the HSCT-treated group had most, followed by the NZB group, and healthy controls had the lowest numbers of CD4+ CD45RO+ T cells. HSCT-treated patients and healthy controls had similar levels of CD4+ FoxP3+ T cells, CD4+ Helios+ T cells as well as nTreg and pTreg cells. NZB-treated patients, on the other hand, had fewer CD4+ FoxP3+ T cells than the other two groups. This difference was due to a lower level of nTreg cells, whereas the frequencies of pTreg cells were similar in the three groups. CD4+ Helios+ T cells were fewer in the NZB-treated group than the HSCT-treated patients, and a similar trend could be observed towards controls. The gating strategy and FACS plots from a typical HSCT-treated patient are seen in Supplementary material, Fig. S1.
Table 2.
Frequencies of memory T cells and regulatory T cells
| Control | HSCT | Natalizumab | P-value | |
|---|---|---|---|---|
| CD4 | 55·8 (50·4–66·4) | 44·1 (33·3–53·5) | 64·5 (49·3–69·2) | 0·003 |
| CD45RO | 30·3 (21·2–45·0) | 49·1 (35·6–65·0) | 37·1 (29·3–46·7) | 0·06 |
| FoxP3+ | 2·93 (2·47–3·75) | 3·00 (2·76–3·39) | 1·87 (1·60–2·64) | 0·006 |
| Helios+ | 7·74 (6·07–9·27) | 8·84 (7·33–10·7) | 5·98 (4·30–7·82) | 0·04 |
| pTreg | 1·20 (0·90–1·45) | 1·33 (1·16–1·91) | 1·18 (0·76–1·58) | NS |
| nTreg | 1·89 (1·15–2·59) | 1·54 (1·22–1·85) | 0·84 (0·45–1·13) | 0·001 |
| pTreg/nTreg | 0·55 (0·42–1·06) | 0·74 (0·62–1·57) | 1·19 (0·81–2·99) | 0·03 |
HSCT, haematopoietic stem cell transplant; nTreg, natural regulatory T cells; pTreg, peripheral regulatory T cells.
Medians (interquartile range). CD4+ is a percentage of CD3+ cells. All other numbers are percentages of total CD3+ CD4+ cells. pTreg is defined as CD3+ CD4+ Helios– FoxP3+. nTreg is defined as CD3+ CD4+ Helios+ FoxP3+. P-values are given for the Kruskal–Wallis test.
HSCT patients have normal frequencies of Th1 and Th17 cells
In the short-term recall assay we investigated ex vivo MOG-specific T-cell responses. The presence of MOG peptides did not seem to have an impact on the production of IFN-γ or IL-17 in CD4+ T cells (see Supplementary material, Fig. S2); however, baseline cytokine production varied between the groups. HSCT-treated patients and healthy controls had similar levels of Th1 cells, but a trend towards a higher production of IFN-γ was observed in the NZB-treated group. Similarly, HSCT-treated patients and healthy controls had equal numbers of Th17 cells, whereas NZB-treated patients had an increased level of Th17 cells (Fig. 1).
Figure 1.
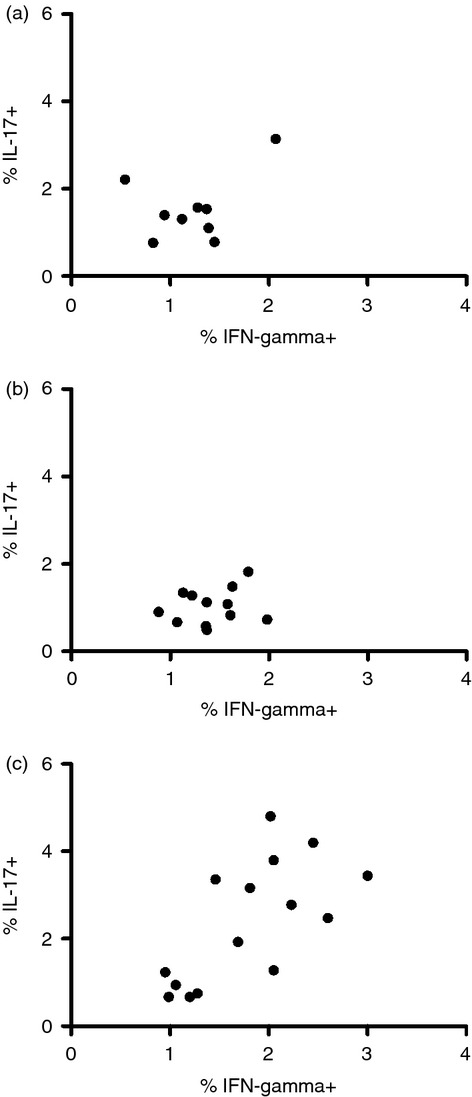
Interleukin-17 (IL-17) and interferon-γ (IFN-γ) production in a short-term recall assay using myelin oligodendrocyte glycoprotein (MOG) peptide stimulation. Frequencies of CD4+ T cells producing IL-17 and IFN-γ after MOG peptide stimulation from (a) healthy controls, (b) haematopoietic stem cell transplant-treated patients and (c) natalizumab-treated multiple sclerosis patients.
T central memory cells are less frequent among MOG-stimulated T cells from HSCT-treated patients compared with those treated with NZB
In the long-term recall assay we investigated if co-culture with MOG peptides could affect the proliferation of T cells and if the proliferating cells were memory T cells. No statistically significant differences in proliferation among the groups could be seen after stimulation with MOG peptide and IL-2 (Fig. 2). Some individual patients had a high level of responsive cells but they were too few to allow any meaningful conclusions.
Figure 2.
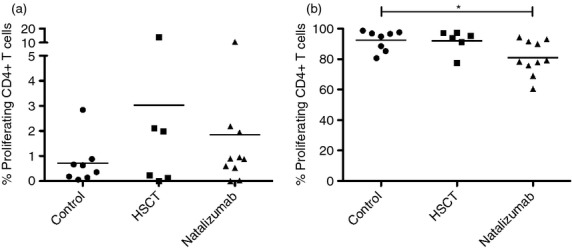
Proliferation of CD4+ T cells in a long-term recall assay using myelin oligodendrocyte glycoprotein (MOG) peptide stimulation and polyclonal stimulation. Proliferation of CD4+ T cells subjected to a long-term recall assay measured with CFSE. (a) Cells cultured with MOG and interleukin-2 and (b) cells cultured with OKT-3 and interleukin-2, i.e. polyclonal stimulation. HSCT, haematopoietic stem cell transplant.
T cells were then cultured with anti-CD3 (OKT-3), i.e. polyclonal stimulation, and no difference in proliferation could be seen between HSCT-treated patients and healthy controls. However, CD4+ T cells from NZB-treated patients proliferated less than the other groups after polyclonal stimulation.
CD4+ T cells stimulated with OKT-3 and IL-2 (Fig. 3) expressed high levels of CD45RO and CCR7 in all groups but activated CD4+ T cells (as evidenced by an increase in size) from NZB-treated patients stimulated with MOG peptide and IL-2 expressed more CD45RO than the other groups. Double-positive CD45RO+ CCR7+ CD4+ T cells were much more frequent in the NZB-treated group. The gating strategy and FACS plots from a typical NZB-treated patient are seen in Supplementary material, Fig. S3.
Figure 3.
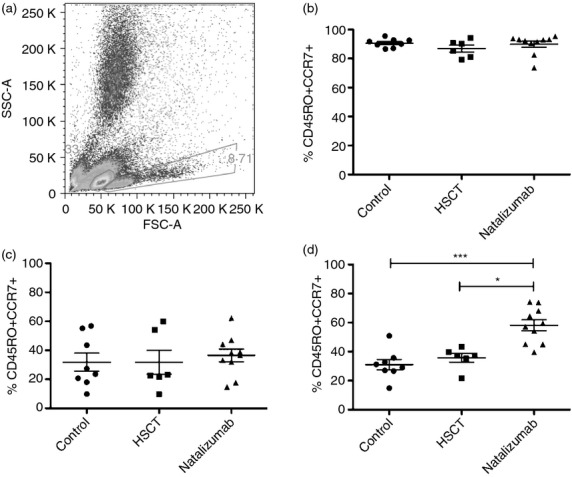
Frequencies of CD45RO and CCR7 in CD4+ T cells cells in a long-term recall assay using myelin oligodendrocyte glycoprotein (MOG) peptide stimulation and polyclonal stimulation. (a) Ungated white blood cells from stimulation with MOG and interleukin-2. The gate on the left contains unactivated T cells and the gate on the right contains activated T cells. (b) Polyclonal stimulation with OKT-3 and interleukin-2, CD4+ T cells from the ‘activated’ gate. T cells acquire a T central memory phenotype, indicated by high expression of CD45RO and CCR7. (c) MOG stimulation of CD4+ T cells, cells from the ‘unactivated’ gate. T cells from all groups have similar frequencies of T central memory cells. (d) CD4+ T cells from the ‘activated’ gate. T cells from natalizumab-treated patients predominantly are of a T central memory phenotype. HSCT, haematopoietic stem cell transplant
Stimulated T cells from HSCT patients produce less IL-17 than controls
In the analysis of supernatants from the long-term recall stimulation assay, some numerical differences of interest were observed, but no statistically significant differences could be demonstrated between the groups (Table 3, and Supplementary material, Table S2). After MOG stimulation the Th17-related cytokine IL-17 could not be detected in the HSCT-treated group, whereas it was detected in three of seven controls and four of seven NZB-treated patients. Production of the Th1-related cytokine IFN-γ was lower in the HSCT-treated group, than in the other two groups. Production of TGF-β1 was higher after MOG stimulation in the HSCT-treated group than in the NZB-treated group; this relation reversed after OKT-3 stimulation, when mean TGF-β1 production was 27% lower in the HSCT-treated group and 35% higher in the NZB-treated group (Fig. 4).
Table 3.
Magnetic bead panel immunoassay of cytokines from supernatants of T cells cultures with myelin oligodendrocyte glycoprotein in long-term recall assay
| Control | HSCT | Natalizumab | ||||
|---|---|---|---|---|---|---|
| Detected | Mean value (pg/ml) | Detected | Mean value (pg/ml) | Detected | Mean value (pg/ml) | |
| IFN-γ | 7/7 | 17·09 (±29·46) | 6/6 | 5·28(± 2·73) | 7/7 | 9·41 (± 8·90) |
| IL-1β | 7/7 | 2·12 (± 0·41) | 6/6 | 1·90 (± 0·46) | 7/7 | 2·69 (± 0·94) |
| IL-6 | 3/7 | 6·80 (± 7·48) | 3/6 | 8·64 (± 3·71) | 4/7 | 9·51 (± 7·76) |
| IL-10 | 7/7 | 5·84 (± 3·17) | 6/6 | 3·08 (± 0·71) | 7/7 | 5·15 (1·62) |
| IL-17 | 3/7 | 3·66 (± 0·85) | 0/6 | n/a | 4/7 | 4·42 (1·75) |
| TGF-β1 | 3/7 | 33443 (± 15977) | 6/6 | 32849 (± 16296) | 7/7 | 20954 (± 7562) |
| TGF-β2 | 7/7 | 468 (± 42·1) | 6/6 | 469 (± 48·9) | 7/7 | 428 (± 53·2) |
| TGF-β3 | 7/7 | 32 (± 8·83) | 6/6 | 32 (± 4·52) | 7/7 | 27 (± 4·63) |
HSCT, haematopoietic stem cell transplant; IFN-γ, interferon-γ; IL-1β, interleukin-1β; n/a, not applicable; TGF-β1, transforming growth factor-β1.
Means (SD).
Differences are not statistically significant.
Figure 4.
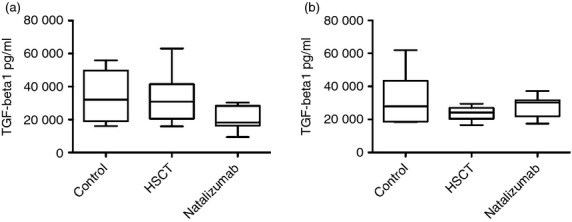
Magnetic bead panel immunoassay of transofrming growth factor-β1 (TGF-β1) from supernatants of T-cell cultures in long-term recall assay with myelin oligodendrocyte glycoprotein (MOG) peptide stimulation and polyclonal stimulation. Bars represent medians. Whiskers represent minimum to maximum. Differences are not statistically significant. (a) TGF-β1 in supernatants of T-cell cultures with MOG peptide stimulation. (b) TGF-β1 in supernatants of T-cell cultures with OKT-3 and interleukin-2, i.e. polyclonal stimulation. HSCT, haematopoietic stem cell transplant.
Discussion
In this study, some of the aspects of tolerance in the immune system after HSCT for aggressive MS were explored. HSCT-treated patients were compared with healthy controls and NZB-treated patients. An obvious weakness of this study is that the HSCT-treated patients were not compared with themselves before and after HSCT. Patients had been treated with HSCT on average for more than 4 years and up to 7 years before this study, making a comparative approach difficult for practical reasons. At the point of HSCT most of our patients had cataclysmic MS with several severe relapses in the year before HSCT. Such patients are exceedingly rare, even at a tertiary centre. With today's ‘treat early’ paradigm, untreated patients with active MS are few and far between and a group of untreated patients with highly active MS should not be found today.
Natalizumab is a monoclonal antibody binding to integrin α4/βl, preventing extravasation of leucocytes into the central nervous system.14 It has until recently been considered a second-line drug, used for patients with break-through disease on first-line therapy. As it is non-cytotoxic, it was reported that pro-inflammatory T cells from NZB-treated patients are sequestered in the peripheral circulation13 and that NZB does not stimulate proliferation of T cells in vitro;13,15 as a result we decided to include NZB-treated patients as a comparative group and to ensure that our methods of evaluating central nervous system reactivity was appropriate.
CD45 exists in two complementary isoforms: CD45RA and CD45RO. CD45RA is a marker of naive cells recently issued from the thymus, whereas CD45RO is a marker of memory cells.16 Conflicting data exist regarding the expression of CD45RO in MS.7,17 In the larger series17 the frequency of CD4+ CD45RO+ T cells was 34·8% in healthy controls and somewhat higher, c. 45% in MS patients. Our data from healthy controls and NZB-treated patients adhere well to the data from this study. The highest expression of CD45RO was seen in HSCT-treated patients and most in the two patients who underwent HSCT within 2 years before this study. This seems counterintuitive, because HSCT has been proposed to reset the immune system. However, it is well-known that the CD4+ CD45RA+ subset is profoundly reduced after HSCT and may take up to 2 years to recover.7,9 The expansion of the CD45RO+ subset of CD4+ T cells has been considered to be the result of proliferation of surviving T cells in response to homeostatic mechanisms. In a lymphopenic setting, low-affinity self and microbial flora-derived antigens may induce proliferation of mature cells in the periphery.18
FoxP3 is a transcription factor and a marker of the Treg cell subset of CD4+ T cells. Conflicting data exist regarding frequency and function of Treg cells in MS. Others have reported unchanged frequencies of Treg cells after 1 month of treatment with NZB,19 non-significant increase after 12 months of treatment with NZB,20 or non-significant reduction15 after 1 month of treatment. Helios is a transcription factor of the Ikaros family, suggested to be a marker of nTreg cells. The nTreg cells are believed to be educated in the thymus and are more stable than pTreg cells, which are induced in the inflammatory response and probably converted from effector T cells.
In our study HSCT-treated patients and controls had similar levels of FoxP3+ and Helios+ CD4+ T cells, they also had similar levels of pTreg and nTreg cells. In contrast, NZB-treated MS patients had fewer CD4+ FoxP3+ T cells and fewer CD4+ Helios+ T cells (Table 2). This difference in Treg cell frequency was the result of a selective decrease of nTreg cells, because levels of pTreg cells were similar between all groups. The pTreg cell pool is continuously replenished with short-lived pTreg cells converted from effector T cells.11 Hence, the number of pTreg cells would increase with the number of effector T cells. It is well known that the absolute number of T cells increases with NZB treatment. The nTreg cells are not dependent on the T-cell pool in the same way as pTreg cells and the lower frequencies in nTreg cells observed in the NZB-treated group could represent the original population of nTreg cells. Further studies on Helios in MS may elucidate on this.
In the short-term recall assay to detect MOG-reactive CD4 T cells, the frequency of Th17 cells was similar in the HSCT-treated group and healthy controls (Fig. 1). In the NZB-treated group the frequency was higher, but this phenomenon was not a result of MOG reactivity, but rather reflected an elevated baseline. A similar trend was noted for IFN-γ. Interleukin-17 is a well-known marker for Th17 cells, which have been associated with disease activity in MS as well as in experimental autoimmune encephalomyelitis.21 Interferon-γ is produced by Th1 cells, which have also been associated with disease activity in MS.22 When combining the analyses of these two CD4+ T-cell populations, signs of immunological disease activity were demonstrated in 10 out of 15 NZB-treated patients, whereas no Th1 or Th17 activation could be seen in the HSCT-treated group or controls. This lends support to the supposition that HSCT causes a removal of autoreactive T-cell clones.
As the T cells did not seem to respond to MOG antigen in the short-term recall assay, cells were stimulated in a traditional 1-week recall assay. In this assay, a polyclonal stimulation with OKT-3 was added as well. T cells from all groups proliferated poorly when stimulated with MOG and IL-2. Polyclonally stimulated T cells from HSCT patients and controls proliferated vigorously, but CD4+ T cells from MS patients treated with NZB proliferated less (Fig. 2b), in concordance with earlier studies on MS patients.23
As no effect of MOG on proliferation could be demonstrated, activated T cells were examined. Such T cells from HSCT-treated patients and controls expressed relatively low levels of CD45RO and CCR7 (Fig. 3d), most likely representing unspecific activation by IL-2. Activated T cells from the NZB-treated group expressed high levels of CD45RO and CCR7, suggesting activation of a T central memory phenotype.24 This implies that MOG was recognized by a group of T cells in the NZB-treated group, even though we failed to demonstrate a MOG-specific proliferation. Myelin reactivity in CD4+ memory T cells has been described in humans with MS.25 Again, our observation lends support to the supposition that HSCT causes a removal of autoreactive T-cell clones, whereas they persist during NZB treatment.
Haematopoietic stem cell transplantation does not seem to impede T-cell activation in general. We saw no difference between T cells from HSCT-treated patients and healthy controls in the ability to proliferate and produce IFN-γ after polyclonal stimulation (Table S1). When stimulated with MOG and IL-2, however, T cells from HSCT-treated patients produced less IFN-γ than T cells from both healthy controls and NZB-treated patients. This is consistent with data from a recent study by Darlington et al.,8 in which patients were treated with a high-intensity protocol, the frequency of CD4+ T cells producing IFN-γ in response to myelin basic protein, proteolipid protein and MOG was markedly diminished 12 months after HSCT. Similar to Darlington et al.,8 we noticed that CD4+ T cells produced less IL-17 after polyclonal stimulation. Production of IL-17 was overall very low in the MOG stimulation, and we failed to detect IL-17 production in about half of the controls and natalizumab-treated patients. In HSCT-treated patients we could not detect it at all, again suggesting that reduction in IL-17 production may be important for the therapeutic effect of HSCT.
In addition, T cells from HSCT-treated patients and healthy controls produced similar levels of TGF-β1, whereas T cells from NZB-treated patients produced about 50% less (Fig. 4). When stimulated polyclonally, T cells from HSCT-treated patients produced less than during peptide stimulation, healthy controls produced similar levels, and NZB-treated patients produced more TGF-β1. Even if these differences were non-significant, taken together it seems reasonable to suggest that Treg cells from HSCT-treated patients recognize MOG, produce TGF-β1 and inhibit the Th17 response seen in MS. This idea deserves to be further explored.
Haematopoietic stem cell transplantation and NZB are both viable treatment options for aggressive MS. In our patients both had a strong effect on the annualized relapse rate and most patients were clinically stable. In patients treated with HSCT, we could not demonstrate any of the pathological changes commonly associated with MS, such as increased frequencies of Th17 cells. MOG is abundantly found in oligodendrocytes, thought to be the prime target of the autoimmune attack in MS. We have demonstrated that T cells from HSCT-treated patients exhibit signs of tolerance in the presence of MOG in contrast to T central memory cells from NZB-treated patients that were activated by MOG. Far from being proof that HSCT might entail a cure for MS, these results offer support to this idea. NZB on the other hand seems to do nothing to alter the way the immune system works, as exemplified by the clinical rebound activity seen after cessation of NZB therapy, which in some cases can be severe.26 Although the issue of progressive multifocal leucoencephalopathy and the risks associated with NZB treatment are now well recognized, it remains to be seen if HSCT can be performed in a way that is safe, and at least one phase III study is underway to address this question (ClinicalTrials no. NCT00273364).
Acknowledgments
The authors would like to thank Emma Svensson, who was very helpful with the magnetic bead panel immunoassays. The study was supported by an unrestricted grant from MerckSerono Corporation; a donation by Lars Tenerz; Uppsala University Hospital, the Swedish Research Council and the Medical Faculty at Uppsala University.
Disclosures
Joachim Burman has received travel support and/or lecture honoraria from Almirall, BiogenIdec, Genzyme/SanofiAventis and MerckSerono; has received an unconditional research grant from MerckSerono. Jan Fagius has received travel support and/or lecture honoraria from BiogenIdec, Genzyme/SanofiAventis and MerckSerono; has received an unconditional research grant from BiogenIdec. Moa Fransson, Thomas Tötterman, Sara Mangsbo and Angelica Loskog have no disclosures.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Short-term recall assay using myelin oligodendrocyte glycoprotein peptide stimulation.
Figure S2. Gating strategy and example of FACS plots from a typical haematopoietic stem cell transplant-treated patient.
Figure S3. Gating strategy and example of FACS plots from a typical natalizumab-treated patient.
Appendix S1. Myelin oligodendrocyte glycoprotein protein sequence.
Appendix S2. Antibodies used.
Table S1. Information on individual haematopoietic stem cell transplant-treated patients.
Table S2. Magnetic bead panel immunoassay of cytokines from supernatants of T cells cultures with OKT-3 in long-term recall assay.
References
- 1.Fassas A, Anagnostopoulos A, Kazis A, Kpainas K, Sakellari I, Kimiskidis V, Tsompanakou A. Peripheral blood stem cell transplantation in the treatment of progressive multiple sclerosis: first results of a pilot study. Bone Marrow Transplant. 1997;20:631–8. doi: 10.1038/sj.bmt.1700944. [DOI] [PubMed] [Google Scholar]
- 2.Fagius J, Lundgren J, Öberg G. Early highly aggressive MS successfully treated by hematopoietic stem cell transplantation. Mult Scler. 2008;15:229–37. doi: 10.1177/1352458508096875. [DOI] [PubMed] [Google Scholar]
- 3.Burt R, Loh Y, Cohen B, et al. Autologous non-myeloablative haemopoietic stem cell transplantation in relapsing-remitting multiple sclerosis: a phase I/II study. Lancet Neurol. 2009;8:244–53. doi: 10.1016/S1474-4422(09)70017-1. [DOI] [PubMed] [Google Scholar]
- 4.Krasulova E, Trneny M, Kozak T, et al. High-dose immunoablation with autologous haematopoietic stem cell transplantation in aggressive multiple sclerosis: a single centre 10-year experience. Mult Scler. 2010;16:685–93. doi: 10.1177/1352458510364538. [DOI] [PubMed] [Google Scholar]
- 5.Fassas A, Kimiskidis VK, Sakellari I, Kapinas K, Anagnostopoulos A, Tsimourtou V, Sotirakoglou K, Kazis A. Long-term results of stem cell transplantation for MS: a single-center experience. Neurology. 2011;76:1066–70. doi: 10.1212/WNL.0b013e318211c537. [DOI] [PubMed] [Google Scholar]
- 6.Mancardi G, Sormani M, Di Gioia M, et al. Autologous haematopoietic stem cell transplantation with an intermediate intensity conditioning regimen in multiple sclerosis: the Italian multi-centre experience. Mult Scler. 2012;18:835–42. doi: 10.1177/1352458511429320. [DOI] [PubMed] [Google Scholar]
- 7.Muraro P, Douek D, Packer A, et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. 2005;201:805–16. doi: 10.1084/jem.20041679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darlington PJ, Touil T, Doucet JS, et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol. 2013;73:341–54. doi: 10.1002/ana.23784. [DOI] [PubMed] [Google Scholar]
- 9.Guillaume T, Rubinstein DB, Symann M. Immune reconstitution and immunotherapy after autologous hematopoietic stem cell transplantation. Blood. 1998;92:1471–90. [PubMed] [Google Scholar]
- 10.Meng L, Ouyang J, Zhang H, Wen Y, Chen J, Zhou J. Treatment of an autoimmune encephalomyelitis mouse model with nonmyeloablative conditioning and syngeneic bone marrow transplantation. Restor Neurol Neurosci. 2011;29:177–85. doi: 10.3233/RNN-2011-0590. [DOI] [PubMed] [Google Scholar]
- 11.Getnet D, Grosso JF, Goldberg MV, et al. A role for the transcription factor Helios in human CD4+ CD25+ regulatory T cells. Mol Immunol. 2010;47:1595–600. doi: 10.1016/j.molimm.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–41. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kivisakk P, Healy BC, Viglietta V, Quintana FJ, Hootstein MA, Weiner HL, Khoury SJ. Natalizumab treatment is associated with peripheral sequestration of proinflammatory T cells. Neurology. 2009;72:1922–30. doi: 10.1212/WNL.0b013e3181a8266f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yednock T, Cannon C, Fritz L, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–6. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 15.Ramos-Cejudo J, Oreja-Guevara C, Stark Aroeira L, Rodriguez de Antonio L, Chamorro B, Diez-Tejedor E. Treatment with natalizumab in relapsing–remitting multiple sclerosis patients induces changes in inflammatory mechanism. J Clin Immunol. 2011;31:623–31. doi: 10.1007/s10875-011-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michie CA, McLean A, Alcock C, Beverley PC. Lifespan of human lymphocyte subsets defined by CD45 isoforms. Nature. 1992;360:264–5. doi: 10.1038/360264a0. [DOI] [PubMed] [Google Scholar]
- 17.Mikulkova Z, Praksova P, Stourac P, Bednarik J, Michalek J. Imbalance in T-cell and cytokine profiles in patients with relapsing-remitting multiple sclerosis. J Neurol Sci. 2011;300:135–41. doi: 10.1016/j.jns.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 18.Muraro PA, Douek DC. Renewing the T cell repertoire to arrest autoimmune aggression. Trends Immunol. 2006;27:61–7. doi: 10.1016/j.it.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Stenner MP, Waschbisch A, Buck D, Doerck S, Einsele H, Toyka KV, Wiendl H. Effects of natalizumab treatment on Foxp3+ T regulatory cells. PLoS One. 2008;3:e3319. doi: 10.1371/journal.pone.0003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frisullo G, Iorio R, Plantone D, Marti A, Nociti V, Patanella AK, Batocchi AP. CD4+ T-bet+, CD4+ pSTAT3+ and CD8+ T-bet+ T cells accumulate in peripheral blood during NZB treatment. Mult Scler. 2010;17:556–66. doi: 10.1177/1352458510392263. [DOI] [PubMed] [Google Scholar]
- 21.Segal BM. Th17 cells in autoimmune demyelinating disease. Semin Immunopathol. 2010;32:71–7. doi: 10.1007/s00281-009-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 23.Fransson ME, Liljenfeldt LSE, Fagius J, Tötterman TH, Loskog ASI. The T-cell pool is anergized in patients with multiple sclerosis in remission. Immunology. 2009;126:92–101. doi: 10.1111/j.1365-2567.2008.02881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 25.Venken K, Hellings N, Hensen K, Rummens J-L, Stinissen P. Memory CD4+ CD127high T cells from patients with multiple sclerosis produce IL-17 in response to myelin antigens. J Neuroimmunol. 2010;226:185–91. doi: 10.1016/j.jneuroim.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 26.Miravalle A, Jensen R, Kinkel RP. Immune reconstitution inflammatory syndrome in patients with multiple sclerosis following cessation of natalizumab therapy. Arch Neurol. 2010;68:186–91. doi: 10.1001/archneurol.2010.257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.