

Abstract
Diffuse large B-cell lymphoma (DLBCL), the most common form of non-Hodgkin's lymphoma (NHL) diagnosed in the USA, consists of at least two distinct subtypes: germinal centre B (GCB) and activated B-cell (ABC). Decreased MHC class II (MHCII) expression on the tumours in both DLBCL subtypes directly correlates with significant decreases in patient survival. One common mechanism accounting for MHCII down-regulation in DLBCL is reduced expression of the MHC class II transactivator (CIITA), the master regulator of MHCII transcription. Furthermore, reduced CIITA expression in ABC DLBCL correlates with the presence of the transcriptional repressor positive regulatory domain-I-binding factor-1 (PRDI-BF1). However, the mechanisms underlying down-regulation of CIITA in GCB DLBCL are currently unclear. In this study, we demonstrate that neither PRDI-BF1 nor CpG hypermethylation at the CIITA promoters are responsible for decreased CIITA in GCB DLBCL. In contrast, histone modifications associated with an open chromatin conformation and active transcription were significantly lower at the CIITA promoters in CIITA− GCB cells compared with CIITA+ B cells, which suggests that epigenetic mechanisms contribute to repression of CIITA transcription. Treatment of CIITA− or CIITAlow GCB cells with several different histone deacetylase inhibitors (HDACi) activated modest CIITA and MHCII expression. However, CIITA and MHCII levels were significantly higher in these cells after exposure to the HDAC-1-specific inhibitor MS-275. These results suggest that CIITA transcription is repressed in GCB DLBCL cells through epigenetic mechanisms involving HDACs, and that HDACi treatment can alleviate repression. These observations may have important implications for patient therapy.
Keywords: B cells, histone deacetylase inhibitor, human, lymphoma, MHC
Introduction
Cancers derived from B cells (B-cell lymphomas) account for 6% of all cancers in the USA, and are responsible for 70 000 newly diagnosed cases each year. Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin's lymphoma (NHL), accounting for approximately 30% of all NHL and 40% of B-lineage NHL. Response rates of DLBCL patients to standard chemotherapy regimens are highly variable, and only 50% of patients are cured. Gene expression profiling, immunophenotyping, genetic and morphological studies have identified distinct molecular subtypes of DLBCL. Based on these studies, DLBCL has been proposed to originate from B cells from at least two distinct stages of differentiation: peripheral B cells from the germinal centre (GCB), and post-germinal centre, activated B cells (ABC). GCB DLBCL has a better overall survival rate following chemotherapy compared with ABC DLBCL. Subsequent studies revealed that the levels of MHCII expression on DLBCL tumour cells directly correlated with patient survival in every cohort of patients examined.1–4 Furthermore, the levels of MHCII on DLBCL tumours were on average higher on GCB compared with ABC.5 MHCII expression is directly correlated with the numbers of tumour-infiltrating T lymphocytes in DLBCL.6–8 These collective observations suggest that decreases in MHCII antigen expression on DLBCL leads to loss of tumour immunosurveillance and ultimately to worse patient outcome. Therefore, defining the mechanisms responsible for down-regulating MHCII expression in DLBCL is critical to develop therapies for enhancing MHCII expression, tumour immunosurveillance and patient survival.
MHCII molecules are heterodimeric, cell surface proteins that present antigenic peptides to CD4+ T cells to initiate antigen-specific immune responses. MHCII gene expression is co-ordinately regulated by cell-type-specific and developmental mechanisms. Constitutive MHCII expression is restricted to antigen-presenting cells such as B cells, dendritic cells, macrophages and activated T cells, but fibroblasts, epithelial and endothelial cells express these molecules after exposure to interferon-γ (IFN-γ).9 During B-cell development, MHCII expression is differentially regulated: expression is absent in human pro-B cells, activated during early B-cell maturation, maximized in mature B cells, and silenced in plasma cells.9 Regulation of both constitutive and IFN-γ-inducible MHCII transcription is mediated by three highly conserved cis-acting DNA sequences termed the SXY module, and the trans-acting factors RFX, NF-Y and CREB.9 The class II transactivator (CIITA), a non-DNA binding co-factor that interacts with RFX, NF-Y, CREB, RNA polymerase and histone acetyltransferases, is also essential for MHCII transcription.10,11 CIITA enhances MHCII transcription by functioning as a scaffold between the DNA-bound transcription factors, chromatin modifiers and the transcriptional machinery.10,12
Differential expression of CIITA accounts for the cell-type specific and developmental regulation of MHCII transcription.10,11 Transcription of CIITA is constitutive in mature B cells and dendritic cells, and is activated in fibroblasts, epithelial and endothelial cells by IFN-γ.10,11 Silencing of CIITA expression mediates the down-regulation of MHCII transcription during plasma cell differentiation.13 Ectopic CIITA expression can restore MHCII expression in CIITA-deficient cells, including plasma cells.10,11 Hence, CIITA has been termed the ‘master regulator’ of MHCII transcription. The regulation of CIITA transcription is mediated by three distinct promoters that function in a cell-type-specific manner: promoter I (pI) is active in dendritic cells and macrophages, promoter III (pIII) is active in B cells, activated T cells and plasmacytoid dendritic cells, and pIV is activated by IFN-γ in a wide variety of cell types, including cells of the B-cell lineage.10,14,15
Several distinct mechanisms have been described that account for the silencing of CIITA expression. Down-regulation of CIITA transcription during plasma cell differentiation is mediated by a transacting repressor termed the positive regulatory domain-I-binding factor-1 (PRDI-BF1).16,17 PRDI-BF1, or B-lymphocyte-induced maturation protein 1 (Blimp-1) in mice, represses CIITA pIII and pIV activity in cells of B-cell ontogeny.16–19 Epigenetic mechanisms, including DNA hypermethylation and post-translational histone modifications, are also involved in controlling CIITA transcription.20–22 Hypermethylation of the CpG dinucleotides within CIITA pIV has been associated with the inability of haematopoietic, developmental, gastric, and colorectal tumour cells to induce CIITA transcription following exposure to IFN-γ.21,23–25 In contrast, repression of CIITA transcription in uveal melanoma cells does not correlate with DNA hypermethylation at CIITA pIV, but rather with trimethylation of histone 3 lysine 27 (H3-K27).21 In addition, H3 acetylation and H3-K4 methylation correlated with active CIITA transcription in mature B cells and IFN-γ-treated epithelial cells.20,26
Histone acetylation contributes to an accessible chromatin structure that permits gene transcription.27 Conversely, histone deacetylation and DNA hypermethylation are correlated with closed chromatin that mediates gene silencing.27 The acetylated state of histones at specific promoters is determined by the equilibrium between the enzymatic activities of histone acetyltransferases and histone deacetylases (HDACs).27 Histone acetyltransferases and HDACs are over-expressed or exhibit altered activity in some cancers, including DLBCL, which results in the dysregulation of gene transcription.27–30 Unlike genetic mutations, epigenetic dysregulation can potentially be reversed or modified therapeutically. For example, in vitro treatment with HDAC inhibitors (HDACi) can alter the acetylated state of chromatin and trigger the transcription of silenced genes, including CIITA and MHCII.31,32
The HDACi can directly induce the differentiation, growth arrest and apoptosis of multiple haematological malignant cell lines, by both induction and repression of critical genes that regulate these processes.28 HDACi have complex effects on immunity, altering both innate and adaptive immune responses.33–37 HDACi are currently being tested in clinical trials to treat a variety of cancers, including DLBCL.38–41 Two HDACi are currently approved by the US Food and Drug Administration: vorinostat for relapsed cutaneous T-cell lymphoma, and romidepsin for relapsed cutaneous T-cell lymphoma and peripheral T-cell lymphoma; however, the mechanism of action is unknown. Clinical trials in DLBCL show some single-agent efficacy.39–41 To date, the majority of recent trials have focused on combinations of HDACi with novel chemotherapeutic agents, radiotherapy and radioimmunotherapy.
Our previous studies in primary DLBCL tumours and established DLBCL cell lines demonstrated that the most common mechanism accounting for down-regulation of MHCII expression was decreased CIITA expression.42–45 In DLBCL with a more terminally differentiated phenotype (ABC), expression of PRDI-BF1 was inversely correlated with CIITA and MHCII.5 However, the mechanisms underlying decreased CIITA expression in GCB DLBCL have not been well defined. Therefore, in our current study we investigated the molecular basis for the down-regulation of CIITA transcription in GCB DLBCL cell lines. We demonstrate that the absence of CIITA transcription correlates with epigenetic silencing of the CIITA promoters in DB, a GCB, CIITA/MHCII-negative DLBCL cell line. Importantly, CIITA and MHCII expression were restored in DB cells treated with HDACi, suggesting that HDACs play an important role in repressing CIITA transcription in DLBCL.
Materials and methods
Cell culture
The DLBCL, Raji Burkitt's lymphoma and Jar choriocarcinoma cell lines were cultured as previously described.43,46 The molecular phenotype and CIITA/MHCII expression status of the DLBCL cell lines used in this study are shown in Table 1. NCI-H929 and U266 human plasma cell lines (kindly provided by Dr Martin Zand) were cultured in RPMI-1640 (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (Invitrogen), 50 U/ml penicillin/streptomycin (Invitrogen), 50 μm 2-mercaptoethanol and 1 mm sodium pyruvate (Invitrogen). Plasma cell lines are derived from a late stage of B-cell differentiation and therefore are known to have down-regulated CIITA, and therefore MHCII expression. Plasma cells also lack many of the usual B-cell and germinal centre-associated markers. Plasma cell lines were therefore used as controls for the typical physiological down-regulation of MHCII in benign B-cell development.
Table 1.
Phenotypes of the diffuse large B-cell lymphoma (DLBCL) cell lines used in this study.
| Cell line | DLBCL type | CIITA/HLA-DR levels |
|---|---|---|
| DB | GCB | Negative/negative |
| OCI-Ly3 | ABC | Intermediate/intermediate |
| OCI-Ly7 | GCB | Intermediate/intermediate |
| OCI-Ly10 | ABC | Intermediate/negative |
| OCI-Ly19 | GCB | High/high |
| SUDHL-4 | GCB | Low/low |
| SUDHL-6 | GCB | Low/low |
| Farage | GCB | High/high |
| Toledo | GCB | Intermediate/intermediate |
Cell lines were categorized as germinal centre B cells (GCB) or activated B cells (ABC) based on gene expression profiling studies. CIITA and HLA-DR expression levels were based on the results of quantitative RT-PCR and flow cytometric analyses43
Treatments
Interferon-γ was purchased from PBL Biomedical Laboratories (Piskataway, NJ). Trichostatin A (TSA) was purchased from Wako (Richmond, VA) and diluted in 100% ethanol prior to use. Apicidin, sodium butyrate, valproic acid, and MS-275 were purchased from CalBioChem (San Diego, CA) and reconstituted before use as follows: apicidin and valproic acid, 100% ethanol; sodium butyrate, molecular grade water; and MS-275, DMSO. Cells (2·5 × 106/5 ml) were treated with various concentrations of IFN-γ or HDACi for 24 or 48 hr and subsequently harvested for flow cytometry or RT-PCR.
Flow cytometry
Flow cytometry was performed as previously described.43 Flow cytometric analysis was performed using an LSRII FACS instrument (BD, Franklin Lakes, NJ) and FACSDiVa software. Histograms and the geometric mean fluorescence intensity values were obtained using WinMDI software.
RNA isolation and RT-PCR
RNA was isolated using Trizol (Invitrogen), and reverse transcriptase (RT) reactions were performed as described previously.43,47 The primer sequences, cycle numbers and annealing temperatures used in standard RT-PCR were: PRDI-BF1-F, 5′-ACACACGGGAGAAAAGCCAC-3′ and PRDI-BF1-R, 5′-CTTGTGGCACTGGGAGCAC-3′, (28 cycles at 55°); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) -F, 5′-CCATGGGGAAGGTGAAGGTCGGAGTC-3′ and GAPDH-R, 5′-GGT GGTGCAGGAGGCATTGCTGATG-3′ (20 cycles at 55°). Quantitative RT-PCR was performed as previously described using an iCycler (Bio-Rad, Hercules, CA) instrument and Sybr-Green master mix (Bio-Rad) as directed by the manufacturer.43 Primers used for quantitative RT-PCR were described previously.43,48 Standard curves were generated using plasmids containing the respective cDNAs for each gene examined. Melt peaks were measured for each reaction to confirm the specificity of the primers. Expression of the MHCII, CIITA and IRF-1 genes was normalized to GAPDH gene expression, and data are represented as copy number per 20 ng RNA.
Bisulphite sequencing
The CpG methylation status at the CIITA promoters III and IV was assessed in select DLBCL lines by bisulfite sequencing as previously described.49 Briefly, genomic DNA isolated with DNeasy Tissue kits (Qiagen, Valencia, CA) was subjected to bisulphite modification using the Epitect Bisulfite Kit (Qiagen). Modified DNA was subjected to PCR for 35 cycles at 55° using the following primer sequences: pIII-F: 5′-TTAAGGGAGTGTGGTAAAATTAGAGGGTG-3′, pIII-R-nest 5′-AAACACAAACTCCTATTCCCATCCTCAC-3′, pIII-R-out 5′-AAACAACTCTTTCACATCTTCCAATAA CCTAC-3′, and pIV-F-out 5′-GGTTGGATTGAGTTGGAGAGAAATAGAGAT-3′, pIV-F-nest 5′-TGGGGA TAAGTTTTTTGTAATTTAGGA-3′, pIV-R 5′-CTACTAATAACCTCTCCCTCCAGCCAA-3′. PCR products were purified with the Qiaquick PCR Purification Kit (Qiagen, Valencia, CA), ligated into the pGEM-T Easy plasmid (Promega, Madison, WI) and transformed into DH5α competent Escherichia coli cells (Invitrogen). Plasmid DNA was isolated from 10 individual colonies, purified with the Wizard Plus SV Miniprep DNA Purification system (Promega), and subsequently sequenced by the University of Arizona DNA core sequencing service with the primer PCM13R 5′-TCACAC AGGAAACAGCTATGAC-3′.
Transient transfection assays
DB cells were transiently transfected with pCIITApIII(322)luc (which contains 322 bp of the human CIITA type III promoter)47 or the empty vector control pGL3-basic as a negative control by electroporation as previously described.43 Twenty-four hours after transfection, aliquots of the transfected cells were transferred to fresh medium +/– HDACi. Cell extracts were prepared 24 hr later for measurement of luciferase activity using a Dual luciferase kit (Promega) as specified by the manufacturer. Luciferase values are represented as relative luciferase activity per μg protein, minus the background fluorescence.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed essentially as described by Choi et al.50 Genomic DNA was purified from immunoprecipitates using PCR Purification Kits (Invitrogen), and subsequently subjected to quantitative PCR using the primers previously described by Green et al.20 Data are represented as the ratio of histone modifications at the CIITA promoters versus GAPDH promoter in each of the respective cell lines.
Statistical analysis
Prism 5.0 software (GraphPad, La Jolla, CA) and the paired Student's t-test with Microsoft Office Excel 2003 and 2010 were used for statistical analysis of the data.
Results
IFN-γ treatment does not up-regulate MHCII expression in DLBCL cell lines
Previous studies demonstrated that patients whose DLBCL tumours expressed the lowest levels of MHCII had the worst survival rates. Although silencing of CIITA was found to be the most common mechanism accounting for MHCII loss in both GCB and ABC DLBCL, the underlying basis for down-regulation of CIITA in GCB DLBCL is currently not clear. Hence, we sought to both define the mechanisms responsible for silencing CIITA expression in GCB DLBCL and identify potential approaches for up-regulating CIITA and MHCII expression in these tumours. Since IFN-γ has been reported to enhance CIITA promoter III (pIII) and pIV activity in multiple different cell types, including B cells,14,15,51 the effects of IFN-γ on CIITA and MHCII expression were examined in GCB DLBCL cell lines. CIITA/MHCII− DB and CIITA/MHCIIlow SUDHL-4 and SUDHL-6 GCB cells were exposed to 100 or 500 U/ml IFN-γ for 24 and 48 hr, and subjected to flow cytometry and quantitative RT-PCR. MHCI (HLA-A/-B/-C) expression was examined as a positive control for a cell surface molecule up-regulated by IFN-γ. Treatment with IFN-γ had no effect on HLA-DR surface expression on DB, SUDHL-4 or SUDHL-6 cells (Fig. 1a), or any other MHCII+ DLBCL line tested (data not shown). However, HLA-ABC expression was up-regulated on all of the DLBCL lines following exposure to IFN-γ, which suggests that the IFN-γ signalling pathway is intact in these cells (Fig. 1b). Interferon-γ had no effect on HLA-DRα or HLA-DQα mRNA expression in any of the DLBCL lines examined (Fig. 1c; data not shown). Furthermore, CIITA mRNA expression was not activated by IFN-γ in DB cells (Fig. 1c), despite the fact that CIITA pIV luciferase-reporter activity (not shown) was increased more than 25-fold by IFN-γ in transient transfection assays. Moreover, only minor increases in CIITA mRNA expression were observed in IFN-γ-treated SUDHL-4 and SUDHL-6 cells (Fig. 1c). Together these results suggest that the minor increases in CIITA mRNA expression observed were not sufficient to mediate increases in MHCII expression in these two DLBCL lines. IFN-γ significantly increased IRF-1 mRNA expression in every DLBCL cell line analysed, confirming that the IFN-γ signalling pathway is intact (Fig. 1c; data not shown). These collective results demonstrate that IFN-γ does not up-regulate MHCII gene expression in these DLBCL cell lines. Furthermore, these results indicate that the endogenous CIITA pIII and pIV are both silenced in DB cells.
Figure 1.
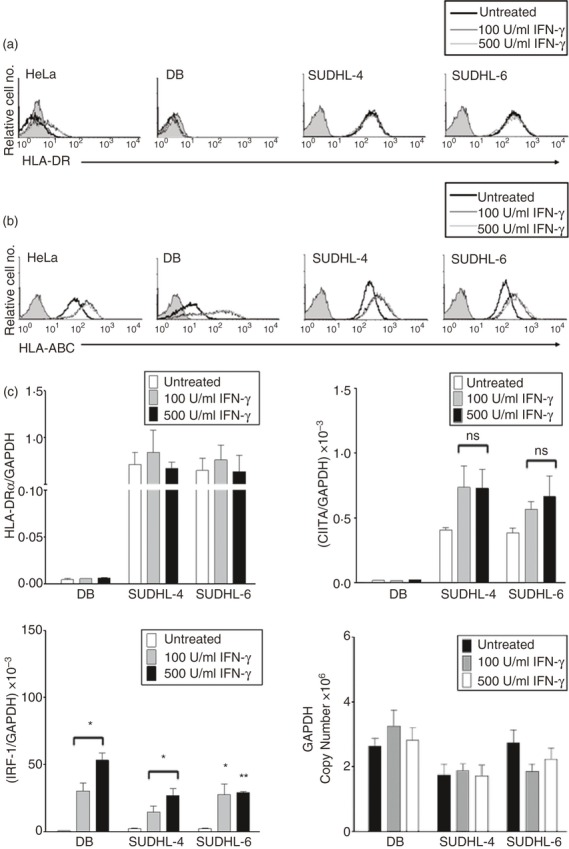
Interferon-γ (IFN-γ) treatment does not enhance MHCII expression on diffuse large B-cell lymphoma (DLBCL) cell lines. DB, SUDHL-4, and SUDHL-6 DLBCL germinal centre B (GCB) cells were treated with 0, 100 or 500 U/ml interferon-γ (IFN-γ) for 24 hr. Flow cytometry was performed using antibodies to (a) HLA-DR and (b) HLA-A/-B/-C/-G. Shaded histograms represent isotype control staining and open histograms represent specific staining. These experiments were repeated at least four times for each cell line with similar results. The histograms shown are representative. (c): RNA was isolated from each line following exposure to IFN-γ for 24 hr and subjected to quantitative RT-PCR using primers specific to HLA-DRα, CIITA, IRF-1, and GAPDH. Data are represented as the ratio of mRNA expression for each gene to GAPDH mRNA expression, and are the average of three independent experiments. Standard errors are shown. Treatment with IFN-γ resulted in a statistically significant enhancement of IRF-1, but not HLA-DRα or CIITA mRNA expression in all three cell lines examined (*P < 0·05; **P < 0·001; ns, not significant).
PRDI-BF1 does not correlate with decreased CIITA expression in GCB DLBCL cell lines
PRDI-BF1 was reported to repress transcription from both CIITA pIII and pIV in cells of B-cell ontogeny.16–19 In addition, studies of primary DLBCL biopsies showed that samples exhibiting loss of CIITA expression were more differentiated towards plasma cells and expressed PRDI-BF1.5,52,53 In contrast, the panel of cell lines we are studying are mostly GCB cells, and the mechanisms underlying loss of CIITA in these DLBCL cells are not well defined.54,55 To determine whether there is an inverse correlation between PRDI-BF1 and CIITA expression in GCB DLBCL cell lines, standard RT-PCR was performed. The human plasma cell lines U266 and NCI-H929 were included as positive controls for PRDI-BF1 expression, while Raji (Burkitt lymphoma) and HeLa (cervical carcinoma) cells were included as negative controls. As expected, PRDI-BF1 mRNA expression was observed in the U266 and NCI-H929 plasma cell lines, but not in Raji or HeLa cells (Fig. 2). In contrast, PRDI-BF1 mRNA expression was not detected in CIITA/MHCII− DB GCB cells, CIITA/MHCIIlow SUDHL-4 or SUDHL-6 GCB cells, or CIITA/MHCII+ OCI-Ly19, Toledo, or Farage DLBCL GCB cells (Fig. 2). Furthermore, relatively minimal levels of PRDI-BF1 mRNA expression were detected in OCI-Ly7 GCB cells and OCI-Ly3 and OCI-Ly10 ABC cells (Fig. 2), all of which express CIITA and MHCII as described in our previous work (for reference, levels of CIITA and HLA-DRα mRNA are also shown for DB, Raji and NCI-H929 in Fig. 6).43 These results demonstrate that PRDI-BF1 expression does not correlate with decreased CIITA or MHCII expression in DLBCL cell lines exhibiting a GCB phenotype, and furthermore, strongly suggest that the molecular mechanism(s) that these DLBCL cells use to silence CIITA transcription is distinct from plasma cells and ABC DLBCL.
Figure 2.
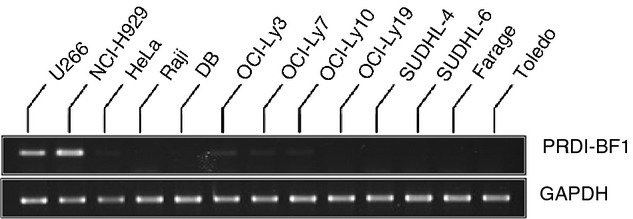
Positive regulatory domain-I-binding factor-1 (PRDI-BF1) mRNA expression in diffuse large B-cell lymphoma (DLBCL) cells. Standard RT-PCR was performed for PRDI-BF1 mRNA expression in the DLBCL cell lines. U266 and NCI-H929 plasma cell lines were included as positive controls and HeLa and Raji cells as negative controls for PRDI-BF1. GAPDH mRNA expression was examined as a control for mRNA integrity and quality of the cDNA reactions. The data are representative of three experiments using independent preparations of RNA from each cell line.
Figure 6.

Effects of histone deacetylase inhibitors (HDACi) on CIITA and MHCII mRNA expression in DB diffuse large B-cell lymphoma (DLBCL) cells. (a) RNA was isolated from DB cells exposed for 0 and 24 hr to increasing doses of trichostatin A (TSA) and subjected to quantitative RT-PCR using primers for HLA-DRα, CIITA and GAPDH. Data are represented as the ratio of HLA-DRα or CIITA mRNA expression to GAPDH mRNA expression and are the average of at least three independent experiments. Standard errors are shown. (b) DB cells were transiently transfected with a luciferase gene vector containing 322 bp of the human CIITA pIII, cultured for 24 hr +/– 100 nm TSA, and subjected to luciferase assays. Data are represented as relative luciferase units (RLU) and are the average of four independent experiments. RNA was isolated from (c) DB and (d) NCI-H929 cells cultured for 24 hr with various concentrations of MS-275 and subjected to quantitative RT-PCR as described for Fig. 6(a). Cells treated with 100 nm TSA were included as a positive control for the effects of HDACi on CIITA and HLA-DRα mRNA expression. Data are the average of three independent experiments. Statistical significance is shown for each of the treatments (*P < 0·05; **P < 0·001; ***P < 0·0001; ns; not significant).
DNA hypermethylation does not mediate the down-regulation of CIITA expression in DLBCL cell lines
Hypermethylation of CpG dinucleotides within the CIITA pIII and pIV regions correlated with the silencing of CIITA transcription in a variety of cancers exhibiting down-regulated MHCII expression.23–25 To define the relationship between DNA methylation at the CIITA promoters and CIITA transcription in DLBCL cell lines, bisulphite sequencing was performed. A 455-bp region of CIITA pIII and a 362-bp region of pIV containing 9 and 12 CpG dinucleotides (MeC), respectively, were analysed in 10 independent transformants for each cell line. Jar (choriocarcinoma) and Jurkat (T-cell lymphoma) cell DNAs were included as positive controls,56,57 and extensive CpG methylation was detected at both CIITA pIII and pIV in these cell lines (Fig. 3). In contrast, little or no CpG methylation was observed at the CIITA promoters in MHCII+ Raji control cells (Fig. 3). Importantly, little CpG methylation was observed at the CIITA promoters in CIITA− DB, or CIITAlow SUDHL-4 or SUDHL-6 DLBCL cells (Fig. 3). Consistent with these observations, similar results were observed in PCR studies of genomic DNA digested with methylation-sensitive restriction enzymes, and furthermore, treatment of DB cells with the demethylating agent 5-azacytidine had no effect on CIITA or MHCII expression (data not shown). Collectively, these results indicate that CpG hypermethylation within the CIITA promoter regions does not correlate with down-regulation of CIITA transcription in these DLBCL GCB cell lines.
Figure 3.
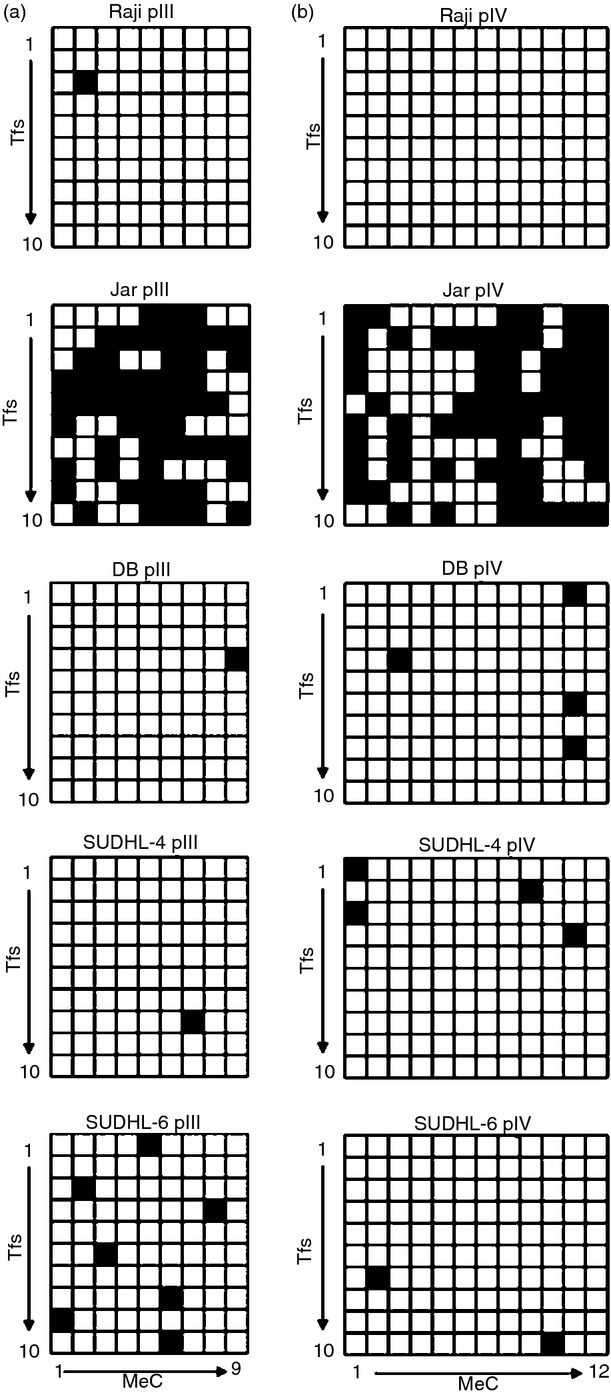
DNA methylation analysis at CIITA promoters III and IV (pIII, pIV) in diffuse large B-cell lymphoma (DLBCL) cells. Genomic DNA was isolated from DB, SUDHL-4, and SUDHL-6 cells and subjected to bisulphite sequencing analysis for methylated CpG dinucleotides (MeC) within (a) CIITA pIII (455 bp region) and (b) pIV (362 bp region). These CIITA pIII and pIV regions contain 9 and 12 CpG dinucleotides, respectively (x-axis). Ten transformants (y-axis) were sequenced for each CIITA promoter in each cell line. Black and white squares represent methylated and unmethylated CpG dinucleotides, respectively. Raji and Jar cells (shown) and Jurkat cells were included as negative and positive controls, respectively, for CpG methylation at the CIITA promoters.
Histone modifications at the CIITA promoters in DB DLBCL cells
Green et al.20 previously demonstrated that distinct changes in post-translational histone modifications at the CIITA promoters are associated with the down-regulation of CIITA transcription that occurs upon differentiation of B cells to plasma cells. To assess the histone modifications at CIITA pIII and pIV in DB DLBCL cells, ChIP assays were performed using antibodies specific for H3 acetylation, H3-K4 dimethylation and trimethylation and H3-K9 dimethylation and trimethylation. Raji and NCI-H929 cells were included as positive and negative controls, respectively, for these histone modifications at the CIITA promoters. Furthermore, the GAPDH promoter was also examined as a positive control for H3 acetylation and H3-K4 methylation, and the levels of these histone modifications at CIITA pIII and pIV in each cell line were normalized to the levels at the GAPDH promoter (Fig. 4). The levels of H3 acetylation at CIITA pIII were substantially lower in DB and NCI-H929 cells relative to Raji cells (Fig. 4a). Similarly, significantly lower levels of H3 acetylation were observed at CIITA pIV in DB and NCI-H929 cells compared with Raji cells (Fig. 4b). While DB cells exhibited significantly lower levels of H3-K4 dimethylation at both CIITA pIII and pIV compared with Raji cells, the levels of this histone modification were higher at both CIITA promoters in NCI-H929 cells (Fig. 4). However, H3-K4 trimethylation, which is associated with actively transcribed loci, was significantly lower at both CIITA pIII and pIV in DB and NCI-H929 cells versus Raji cells (Fig. 4). Neither H3-K9 dimethylation nor trimethylation were detected at CIITA pIII or pIV in DB, NCI-H929, or Raji cells (data not shown). Taken together, the ChIP assays demonstrate that the levels of both H3 acetylation and H3-K4 methylation are reduced at CIITA pIII and pIV in DB cells compared with Raji cells. Hence, diminished levels of the histone modifications associated with active transcription correlate with silencing of CIITA transcription in DB DLBCL cells.
Figure 4.
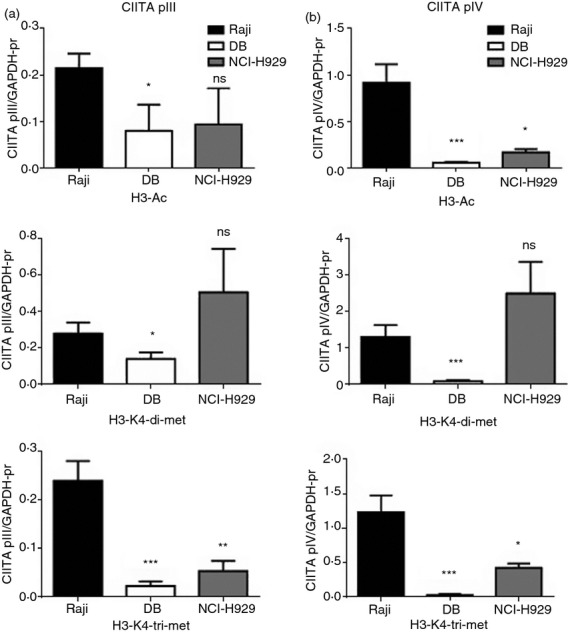
Histone modifications at CIITA promoter III and IV (pIII, pIV) in DB cells. Chromatin immunoprecipitation assays were performed on Raji, DB and NCI-H929 cells for CIITA pIII, CIITA pIV and the GAPDH promoter using antibodies specific for acetylated histone 3 (H3) and dimethylated and trimethylated H3-K4, and quantitative PCR was performed. Data are represented as the ratio of each histone modification at (a) CIITA pIII and (b) pIV versus the GAPDH promoter, and are the average of four independent experiments. Standard errors and statistical significance (*P < 0·05; **P < 0·001; ***P < 0·0001; ns, not significant) are shown.
Effects of HDAC inhibitor treatments on CIITA and MHCII gene expression in DLBCL cells
The results of the ChIP assays suggest that the relative activities of the histone acetyltransferases and HDACs that control histone acetylation at the CIITA locus are skewed towards HDACs in DB GCB cells. Treatment with HDACi is known to up-regulate MHCII expression in a number of different cell types, including human neuroblastoma and murine plasma and trophoblast cell lines, through CIITA-dependent and CIITA-independent mechanisms.32,47 However, the role of these molecules in regulating CIITA and MHCII expression in DLBCL is not known. Furthermore, HDACi are currently being used in clinical trials to treat a variety of cancers, including DLBCL, but the mechanism of action is currently unclear and is thought to be primarily related to increased apoptosis.28,38,40 To investigate the potential role of HDACs in repressing CIITA and/or MHCII expression in DLBCL, flow cytometry and quantitative RT-PCR analyses were performed on DLBCL cell lines exposed to various HDACi. As treatment with HDACi has been shown to up-regulate CIITA and MHCII in murine plasma cells,32 NCI-H929 human plasma cells were included in these experiments. Treatment of DB cells with the pan-HDACi trichostatin A (TSA) for 24 hr resulted in a modest dose-dependent increase in cell surface expression of the representative MHCII marker, HLA-DR (Fig. 5a). At the optimal dose of TSA (100 nm), the mean fluorescence intensity of HLA-DR surface expression was ∼ fourfold above the MFI of untreated DB cells and only minor effects on DB cell viability were observed. In contrast, TSA treatment did not activate HLA-DR expression on NCI-H929 plasma cells (Fig. 5c and Table 2). The HDACi treatments enhanced MHCI (HLA-ABC) expression on both DB and NCI-H929 cells in a dose-dependent manner, demonstrating that HDACi exposure can modulate gene expression in both of these cell lines (Fig. 5a,c, Table 2). Consistent with the flow cytometry results, quantitative RT-PCR demonstrated that HLA-DRα mRNA expression was increased in TSA-treated DB cells (Fig. 6a). Importantly, enhanced HLA-DRα mRNA expression correlated with increased CIITA expression in these cells (Fig. 6a). The maximal levels of CIITA and HLA-DRα mRNA expression detected in TSA-treated DB cells were only ∼ 5% of the levels observed in Raji cells (Fig. 6a). TSA also modestly enhanced CIITA and HLA-DR expression in SUDHL-6 cells, but had no effect on OCI-Ly3 and OCI-Ly7 or NCI-H929 plasma cells (Table 2, data not shown). Treatment of DB cells with other chemically distinct HDACi, including the pan-HDACi sodium butyrate, or class I-specific HDACi apicidin and valproic acid, also modestly increased HLA-DR expression on DB cells but not NCI-H929 cells (Table 2).
Figure 5.
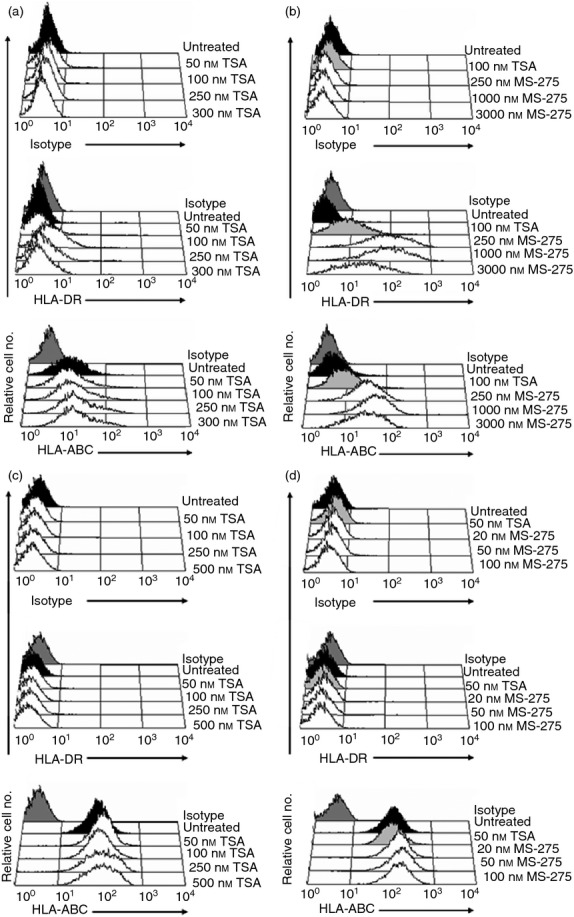
Histone deacetylase inhibitors (HDACi) activate surface MHCII expression on DB diffuse large B-cell lymphoma (DLBCL) cells. MHCII− DLBCL DB cells were exposed for 0 or 24 hr to: (a) the pan-HDACi trichostatin A (TSA) or (b) the HDAC-1-specific inhibitor MS-275, and NCI-H929 plasma cells to (c) TSA and (d) MS-275. Cells were subsequently subjected to flow cytometry using antibodies to the MHCII molecule HLA-DR and the MHCI pan antibody (HLA-ABC). Experiments were repeated at least three times with similar results. The histograms shown are representative.
Table 2.
Effects of histone deacetylase inhibitor (HDACi) treatment on cell surface MHC expression
| Cells/Treatment | Isotype | HLA-DR | HLA-ABC |
|---|---|---|---|
| Raji | 3 | 487 | 601 |
| RL | 2 | 329 | 434 |
| Daudi | 4 | 452 | 6 |
| Ramos | 3 | 296 | 474 |
| DB | 3 | 2 | 6 |
| 100 nm trichostatin A | 3 | 9 | 15 |
| 2 mm sodium butyrate | 2 | 19 | 21 |
| 3 μm apicidin | 4 | 8 | 12 |
| 5 mm valproic acid | 2 | 8 | n.d. |
| 250 nm MS-275 | 3 | 71 | 28 |
| SUDHL-6 | 4 | 108 | 97 |
| 100 nm trichostatin A | 4 | 151 | 147 |
| NCI-H929 | 3 | 2 | 81 |
| 50 nm trichostatin SA | 4 | 2 | 110 |
| 2 mm sodium butyrate | 3 | 2 | 203 |
| 100 nm MS-275 | 3 | 3 | 133 |
n.d., not done.
Cells were exposed for 24 hr to the HDACi shown and subsequently subjected to flow cytometry using antibodies to HLA-DR or HLA-A/-B/-C as described for Fig. 5. Values indicate the geometric mean fluorescence intensities and are representative of at least three independent experiments for each condition
MS-275 is an HDAC-1-specific inhibitor currently being tested in clinical trials that has shown promising activity in both rituximab-chemotherapy-refractory and rituximab-chemotherapy-sensitive DLBCL cell lines and primary lymphoma cells isolated from patients.58 Therefore, the effects of MS-275 treatment on CIITA and MHCII expression in DLBCL cells were examined, and TSA (100 nm) treatments were included as controls in these experiments. Treatment of DB cells with MS-275 also resulted in dose-dependent increases of HLA-DR expression (Fig. 5b). Strikingly, HLA-DR surface expression on DB cells exposed to the optimal dose of MS-275 (250 nm) was eightfold higher compared with TSA-treated cells (Table 2 and Fig. 5b). Moreover, MS-275 also dose-dependently activated CIITA and HLA-DRα mRNA expression in DB cells, and maximal levels were higher than those observed with TSA (Fig. 6c). Indeed, the levels of CIITA and HLA-DRα mRNA expression in DB cells treated with 250 nm MS-275 corresponded to 63% and 35%, respectively, of the levels observed in Raji cells, which were significantly higher levels than those observed in response to TSA treatment. RT-PCR analyses demonstrated that exposure of DB cells to MS-275 and other HDACi selectively activated expression of CIITA type III transcripts (data not shown). Consistent with these results, transient luciferase-reporter assays demonstrated that HDACi treatment resulted in a marked increase in CIITA pIII activity in DB cells (Fig. 6b). In addition, MS-275 dose-dependently enhanced CIITA and HLA-DRα mRNA expression in CIITA/MHCIIlow SUDHL-6 GCB cells (see Supplementary material, Fig. S1). Although MS-275 treatment modestly enhanced CIITA mRNA expression in NCI-H929 plasma cells, neither HLA-DRα surface protein expression nor mRNA expression were enhanced (Figs 5d and 6d, Table 2). Increased MHCI surface expression was observed on DB and NCI-H929 cells treated with MS-275, confirming that this HDACi has the capacity to alter gene expression in both of these cell lines (Fig. 5b,d). Collectively, these results demonstrate that treatment with HDACi up-regulate CIITA and MHCII expression in CIITA/MHCII− and CIITA/MHCIIlow GCB DLBCL cells.
Discussion
Understanding the mechanisms responsible for decreased MHCII expression in DLBCL tumours is essential to determine whether therapies can be designed that can up-regulate MHCII expression, and so potentially improve patient outcome. We previously demonstrated that decreased CIITA expression is the most common mechanism responsible for MHCII down-regulation in DLBCL cell lines.43 As sequencing analyses failed to detect any loss-of-function mutations in the cell lines or in the majority of primary DLBCL samples, we hypothesized that epigenetic mechanisms mediate the silencing of CIITA transcription in DLBCL.43,44 We therefore examined the regulation of CIITA expression in these DLBCL cell lines. We found that the levels of histone modifications associated with an accessible chromatin conformation and active transcription were significantly lower at the CIITA promoters in MHCII− DLBCL GCB cells versus MHCII+ B cells, suggesting that epigenetic mechanisms play a role in silencing CIITA expression in DLBCL cells. Importantly, we demonstrate for the first time that pan-acting and class I HDAC-specific inhibitors currently being tested in clinical trials up-regulate CIITA and MHCII expression, but the HDAC-1-specific inhibitor MS-275 had the greatest effect. Hence, our results strongly suggest that CIITA transcription is silenced in CIITA− DLBCL GCB cells by mechanisms involving HDAC-mediated repression of histone acetylation at the CIITA promoters, and that HDACi therapy may be a viable strategy to reverse this phenomenon.
PRDI-BF1 is a transacting repressor that triggers physiological differentiation of B cells into plasma cells, and extinguishes CIITA transcription by directly binding to CIITA pIII and pIV.15–18,55 PRDI-BF1 was previously detected in 43% of 235 primary DLBCL samples.52 Interestingly, DLBCL patients with PRDI-BF1+ tumours exhibited significantly shorter failure-free survival (P < 0·05), and decreased overall and disease-free survival,52 which is similar to MHCII-deficient DLBCL patients. In subsequent studies of primary DLBCL biopsies, a correlation was identified between absence of CIITA expression and differentiation towards a plasma cell phenotype with corresponding expression of PRDI-BF1.5,53 We therefore examined PRDI-BF1 expression in our panel of DLBCL GCB cell lines. While relatively high levels of PRDI-BF1 mRNA expression were detected in the plasma cell lines, PRDI-BF1 expression was detected in only one GCB and two ABC DLBCL lines, and the relative levels were low. Moreover, PRDI-BF1 is reportedly mutated and non-functional in the ABC cell line OCI-Ly3, and we previously reported that this DLBCL cell line exhibited relatively high levels of CIITA expression.43,59 Importantly, PRDI-BF1 mRNA expression was not observed in DB, SUDHL-4 or SUDHL-6 cells, which is consistent with gene expression profiling studies demonstrating that these cells are of a GCB cell phenotype.54,55 Hence, although PRDI-BF1 is associated with repression of CIITA expression in ABC DLBCL, our results demonstrate that PRDI-BF1 expression does not correlate with the down-regulation of CIITA expression in our panel of GCB lines.
We previously hypothesized that the silencing of CIITA transcription in DLBCL was mediated by epigenetic mechanisms.43 This hypothesis was based on two key observations: (i) sequencing analysis failed to detect any loss-of-function mutations in the CIITA gene in DLBCL cells, and (ii) CIITA is epigenetically silenced in other cancers.21–25 One epigenetic mechanism that has been correlated with the silencing of CIITA transcription in various cancer types is DNA methylation.21–25,47 The silencing of IFN-γ-inducible CIITA transcription in colorectal and gastric cancers is associated with hypermethylation of CpG dinucleotides at CIITA pIV.24 Similarly, CpG hypermethylation and histone hypoacetylation correlate with the repression of CIITA pIII and pIV transcription in T-cell leukaemia and developmental tumours.21,25 In contrast, we previously detected methylation of the CIITA promoters in a fibrosarcoma cell line that expresses CIITA and MHCII following exposure to IFN-γ.47 Moreover, no relationship between silencing of CIITA transcription and DNA methylation at the CIITA promoters was detected in uveal melanoma.21 Hence, the precise role that CpG methylation at the CIITA locus plays in regulating transcription, if any, is currently unclear. DNA hypermethylation has been shown to correlate with the down-regulation of other subsets of genes in DLBCL.60 However, in our study, CpG hypermethylation was not observed at either CIITA pIII or pIV in DLBCL GCB cell lines, which agrees with our previously published data in patient samples.49 Taken together, these results strongly suggest that DNA methylation does not repress CIITA transcription in DLBCL.
The levels of histone 3 (H3) acetylation were significantly lower at both CIITA pIII and pIV in CIITA− DLBCL GCB cells relative to CIITA+ B cells. In addition, H3-K4 trimethylation was also significantly lower at both CIITA pIII and pIV in MHCII− DB DLBCL and NCI-H929 plasma cells versus Raji cells, which is consistent with the fact that transcription is silenced at this locus. In contrast, while H3-K4 dimethylation, which is associated with open and permissive chromatin, was reduced at CIITA pIII and pIV in DB cells, the levels were reproducibly higher in NCI-H929 cells. These observations are similar to those reported by Green et al.,20 who demonstrated that the H3 acetylation and H3-K4 dimethylation and trimethylation modifications were collectively lost at CIITA pIII as primary B cells differentiated into plasma cells. Although we detected higher levels of H3-K4 dimethylation at the CIITA promoters than Green et al., the differences in our studies are likely due to the fact that we normalized the levels of these modifications to those detected at the GAPDH promoter, whereas in the other study the data were represented as fold difference over antibody control. Down-regulation of CIITA transcription during maturation of dendritic cells is also accompanied by significant decreases in histone acetylation at the CIITA locus.61 The H3 and H4 acetylation and H3-K4 trimethylation marks also increase at CIITA pIV in epithelial and fibroblast cells following exposure to IFN-γ.26 Conversely, H3-K9 dimethylation was associated with the silencing of CIITA transcription in plasma cell lines and unstimulated epithelial cells.20,26 However, in contrast to these studies, we did not detect H3-K9 dimethylation or trimethylation at the CIITA promoters in either plasma cells or MHCII− DLBCL GCB cells, despite using several different commercially available antibodies. It is currently unclear why our results differ, but the levels of H3-K9 methylation observed previously at CIITA pIII in NCI-H929 cells were only three-fold higher than background, and could not be detected in primary plasma cells.20 Taken together, these results demonstrate that silencing of CIITA transcription in DB cells is associated with a paucity of H3 acetylation and H3-K4 methylation at pIII and pIV.
The hypoacetylation of histone-3 at CIITA pIII and pIV in DB cells is consistent with a role for HDACs in silencing of CIITA transcription in these cells. Indeed, treatment of DB cells with several chemically distinct HDACi, including the pan-acting HDACi TSA and sodium butyrate, and the class I-specific HDACi apicidin and valproic acid, modestly activated CIITA and HLA-DR expression. Interestingly, treatment with the HDAC-1-specific inhibitor MS-275 induced significantly higher levels of CIITA mRNA and HLA-DR expression in DB cells compared with the other HDACi tested. HDAC-1 over-expression has been observed in some types of cancers, including DLBCL, and HDAC-1 has been reported to specifically inhibit CIITA function.29,62,63 To the best of our knowledge, this is the first study reporting that MS-275 treatment efficiently activates CIITA transcription. Whether this dramatic effect is mediated directly by HDAC-1 inhibition, or by some other consequence of MS-275 exposure requires further investigation. At higher concentrations, MS-275 can also inhibit HDAC-3 function, but the doses that maximally activated CIITA and MHCII expression in DLBCL in our study have been reported to affect only HDAC-1.64 Knockdown of HDAC-1 and/or HDAC-3 expression by siRNA would clearly provide important information on the relative role(s) of these factors in silencing CIITA transcription in DB cells, but unfortunately these experiments were hampered by the extremely low transfection efficiency of these cells.
The precise mechanism(s) by which HDACs inhibit CIITA transcription are also unclear. The transcription factors Sp1 and PU.1, which are required for activation of transcription from CIITA pIII in B cells, have been shown to interact directly with HDAC-1 and repress transcription of other target genes.65,66 Hence, Sp1 and PU.1 may differentially recruit HDAC-1 to the CIITA promoters in DB cells versus MHCII+ B cells, thereby facilitating silencing of CIITA transcription. Alternatively, HDACs may inhibit CIITA transcription in DB cells by a mechanism that blocks the ability of the requisite transacting factors such as Sp1 and PU.1 to bind the CIITA promoters. A third possibility is that HDACs interact with novel factor(s) to block CIITA transcription. The precise mechanism, whatever it may be, is sufficient to repress transcription from both CIITA pIII and pIV.
The mechanisms responsible for repressing CIITA and MHCII expression appear to be similar but distinct in DLBCL GCB cells versus plasma cells, which may have implications for therapy of B-cell versus plasma cell malignancies. Specifically, the key transcriptional repressor PRDI-BF1, which is bound in vivo to both CIITA pIII and pIV in plasma cells,19 is not expressed in the CIITA− DLBCL GCB cell lines in this study. We detected lower levels of H3 acetylation and H3-K4 trimethylation at CIITA pIII and pIV in plasma cells and MHCII− DLBCL compared with MHCII+ Raji cells, suggesting that the chromatin structure at the CIITA locus differs in these cell types. However, although the necessary transcription factors are not bound to CIITA pIII in plasma cells,67 it is not yet known whether this is the case in CIITA− DLBCL cells. Moreover, in our study the pan-HDACi TSA and sodium butyrate activated CIITA transcription in DB DLBCL cells, but not NCI-H929 plasma cells. This is consistent with the observations that HDACi enhanced CIITA pIII reporter activity in DB cells (this study), but not plasma cells.16 Furthermore, in contrast to DB cells, MS-275 induced only low levels of CIITA expression in human plasma cells, but MHCII expression was not activated. The induction of CIITA expression in MS-275-treated NCI-H929 cells was actually higher than the levels induced in TSA-treated DB cells, suggesting that additional silencing mechanism(s) or levels of repression may inhibit MHCII expression in this plasma cell line. In addition, as HDACi activate CIITA transcription and MHCII expression in murine plasma cells,32 distinctions may also exist in the mechanisms mediating the silencing of CIITA transcription in mouse versus human plasma cells.
We previously reported that in the mouse L1210 B-cell lymphoma, the expression of MHCI, MHCII, and the co-stimulatory molecules B7-1, B7-2 and CD40 on immunogenic clones conferred the capacity to act as antigen-presenting cells and activate naive antigen-specific T cells ex vivo.68 Similarly, tumour cells that ectopically express MHC and co-stimulatory molecules can also function as antigen-presenting cells for tumour-specific antigens, and promote tumour rejection in mouse model systems.69–71 HDACi up-regulate endogenous MHC and/or co-stimulatory molecule expression on certain tumour cell-types, by both CIITA-dependent and -independent mechanisms.32,63,71 Importantly, these epigenetically modified tumour cells also have the capacity to function as antigen-presenting cells to stimulate antigen-specific T cells, and can serve as effective tumour vaccines.71,72 Survival curves based on MHCII expression on DLBCL tumours suggest that incremental increases in MHCII expression are paralleled by incremental increases in patient survival.2 Since our in vitro studies demonstrate that MS-275 activated levels of MHCII expression on DB cells that are comparable to those observed on other mature B-cell lines, the ability of these agents to function in a similar manner in vivo may lead to enhanced immunosurveillance, and therefore increased patient survival. It will therefore be important to discern whether clinical HDACi therapies up-regulate CIITA and MHCII expression in the DLBCL tumours of patients. Clinical trials addressing this question are currently open for enrolment.
Acknowledgments
This work was supported in part by National Institutes of Health grant RO1-HD056183 (to SPM) and American Cancer Society Grant ACS# RSG0605501LIB (to LMR).
Disclosure
The authors have no financial conflicts of interest to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Activation of CIITA and HLA-DRα mRNA expression in SUDHL-6 germinal centre B cells by MS-275. RNA was isolated from SUDHL-6 cells cultured for 24 hr with various concentrations of MS-275 and subjected to quantitative RT-PCR as described for Fig. 6(c). Data are the average of two independent experiments.
References
- 1.Rimsza LM, Farinha P, Fuchs DA, Masoudi H, Connors JM, Gascoyne RD. HLA-DR protein status predicts survival in patients with diffuse large B-cell lymphoma treated on the MACOP-B chemotherapy regimen. Leuk Lymphoma. 2007;48:542–6. doi: 10.1080/10428190601078605. [DOI] [PubMed] [Google Scholar]
- 2.Rimsza LM, Roberts RA, Miller TP, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251–8. doi: 10.1182/blood-2003-07-2365. [DOI] [PubMed] [Google Scholar]
- 3.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 4.Veelken H, Vik Dannheim S, Schulte Moenting J, Martens UM, Finke J, Schmitt-Graeff A. Immunophenotype as prognostic factor for diffuse large B-cell lymphoma in patients undergoing clinical risk-adapted therapy. Ann Oncol. 2007;18:931–9. doi: 10.1093/annonc/mdm012. [DOI] [PubMed] [Google Scholar]
- 5.Wilkinson ST, Vanpatten KA, Fernandez DR, et al. Partial plasma cell differentiation as a mechanism of lost major histocompatibility complex class II expression in diffuse large B-cell lymphoma. Blood. 2012;119:1459–67. doi: 10.1182/blood-2011-07-363820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lippman SM, Spier CM, Miller TP, Slymen DJ, Rybski JA, Grogan TM. Tumor-infiltrating T-lymphocytes in B-cell diffuse large cell lymphoma related to disease course. Mod Pathol. 1990;3:361–7. [PubMed] [Google Scholar]
- 7.List AF, Spier CM, Miller TP, Grogan TM. Deficient tumor-infiltrating T-lymphocyte response in malignant lymphoma: relationship to HLA expression and host immunocompetence. Leukemia. 1993;7:398–403. [PubMed] [Google Scholar]
- 8.Stopeck AT, Gessner A, Miller TP, Hersh EM, Johnson CS, Cui H, Frutiger Y, Grogan TM. Loss of B7.2 (CD86) and intracellular adhesion molecule 1 (CD54) expression is associated with decreased tumor-infiltrating T lymphocytes in diffuse B-cell large-cell lymphoma. Clin Cancer Res. 2000;6:3904–9. [PubMed] [Google Scholar]
- 9.Glimcher LH, Kara CJ. Sequences and factors: a guide to MHC class-II transcription. Annu Rev Immunol. 1992;10:13–49. doi: 10.1146/annurev.iy.10.040192.000305. [DOI] [PubMed] [Google Scholar]
- 10.Harton JA, Ting JP. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol. 2000;20:6185–94. doi: 10.1128/mcb.20.17.6185-6194.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mach B, Steimle V, Martinez-Soria E, Reith W. Regulation of MHC class II genes: lessons from a disease. Annu Rev Immunol. 1996;14:301–31. doi: 10.1146/annurev.immunol.14.1.301. [DOI] [PubMed] [Google Scholar]
- 12.Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl):S21–33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 13.Silacci P, Mottet A, Steimle V, Reith W, Mach B. Developmental extinction of major histocompatibility complex class II gene expression in plasmocytes is mediated by silencing of the transactivator gene CIITA. J Exp Med. 1994;180:1329–36. doi: 10.1084/jem.180.4.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piskurich JF, Gilbert CA, Ashley BD, Zhao M, Chen H, Wu J, Bolick SC, Wright KL. Expression of the MHC class II transactivator (CIITA) type IV promoter in B lymphocytes and regulation by IFN-γ. Mol Immunol. 2006;43:519–28. doi: 10.1016/j.molimm.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao M, Flynt FL, Hong M, et al. MHC class II transactivator (CIITA) expression is upregulated in multiple myeloma cells by IFN-γ. Mol Immunol. 2007;44:2923–32. doi: 10.1016/j.molimm.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh N, Gyory I, Wright G, Wood J, Wright KL. Positive regulatory domain I binding factor 1 silences class II transactivator expression in multiple myeloma cells. J Biol Chem. 2001;276:15264–8. doi: 10.1074/jbc.M100862200. [DOI] [PubMed] [Google Scholar]
- 17.Piskurich JF, Lin KI, Lin Y, Wang Y, Ting JP, Calame K. BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat Immunol. 2000;1:526–32. doi: 10.1038/82788. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Gilbert CA, Hudson JA, Bolick SC, Wright KL, Piskurich JF. Positive regulatory domain I-binding factor 1 mediates repression of the MHC class II transactivator (CIITA) type IV promoter. Mol Immunol. 2007;44:1461–70. doi: 10.1016/j.molimm.2006.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tooze RM, Stephenson S, Doody GM. Repression of IFN-γ induction of class II transactivator: a role for PRDM1/Blimp-1 in regulation of cytokine signaling. J Immunol. 2006;177:4584–93. doi: 10.4049/jimmunol.177.7.4584. [DOI] [PubMed] [Google Scholar]
- 20.Green MR, Yoon H, Boss JM. Epigenetic regulation during B cell differentiation controls CIITA promoter accessibility. J Immunol. 2006;177:3865–73. doi: 10.4049/jimmunol.177.6.3865. [DOI] [PubMed] [Google Scholar]
- 21.Holling TM, van Eggermond MC, Jager MJ, van den Elsen PJ. Epigenetic silencing of MHC2TA transcription in cancer. Biochem Pharmacol. 2006;72:1570–6. doi: 10.1016/j.bcp.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 22.Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006;27:405–12. doi: 10.1016/j.it.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Morimoto Y, Toyota M, Satoh A, et al. Inactivation of class II transactivator by DNA methylation and histone deacetylation associated with absence of HLA-DR induction by interferon-γ in haematopoietic tumour cells. Br J Cancer. 2004;90:844–52. doi: 10.1038/sj.bjc.6601602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satoh A, Toyota M, Ikeda H, et al. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-γ-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene. 2004;23:8876–86. doi: 10.1038/sj.onc.1208144. [DOI] [PubMed] [Google Scholar]
- 25.van den Elsen PJ, Holling TM, van der Stoep N, Boss JM. DNA methylation and expression of major histocompatibility complex class I and class II transactivator genes in human developmental tumor cells and in T cell malignancies. Clin Immunol. 2003;109:46–52. doi: 10.1016/s1521-6616(03)00200-6. [DOI] [PubMed] [Google Scholar]
- 26.Ni Z, Karaskov E, Yu T, et al. Apical role for BRG1 in cytokine-induced promoter assembly. Proc Natl Acad Sci U S A. 2005;102:14611–6. doi: 10.1073/pnas.0503070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 28.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107:600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Min SK, Koh YH, Park Y, Kim HJ, Seo J, Park HR, Cho SJ, Kim IS. Expression of HAT1 and HDAC1, 2, 3 in Diffuse Large B-Cell Lymphomas, Peripheral T-Cell Lymphomas, and NK/T-Cell Lymphomas. Korean J Pathol. 2012;46:142–50. doi: 10.4132/KoreanJPathol.2012.46.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta M, Han JJ, Stenson M, Wellik L, Witzig TE. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: implications for therapy. Leukemia. 2012;26:1356–64. doi: 10.1038/leu.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou SD, Khan AN, Magner WJ, Tomasi TB. Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int Immunol. 2005;17:1483–94. doi: 10.1093/intimm/dxh326. [DOI] [PubMed] [Google Scholar]
- 32.Magner WJ, Kazim AL, Stewart C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–24. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 33.Mombelli M, Lugrin J, Rubino I, Chanson AL, Giddey M, Calandra T, Roger T. Histone deacetylase inhibitors impair antibacterial defenses of macrophages. J Infect Dis. 2011;204:1367–74. doi: 10.1093/infdis/jir553. [DOI] [PubMed] [Google Scholar]
- 34.Roger T, Lugrin J, Le Roy D, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011;117:1205–17. doi: 10.1182/blood-2010-05-284711. [DOI] [PubMed] [Google Scholar]
- 35.Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, Ferrara JL. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004;101:3921–6. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson J, Pahuja A, Graham M, Hering B, Hancock WW, Bansal-Pakala P. Effects of histone deacetylase inhibitor SAHA on effector and FOXP3+ regulatory T cells in rhesus macaques. Transplant Proc. 2008;40:459–61. doi: 10.1016/j.transproceed.2008.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 38.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 39.Crump M, Coiffier B, Jacobsen ED, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol. 2008;19:964–9. doi: 10.1093/annonc/mdn031. [DOI] [PubMed] [Google Scholar]
- 40.O'Connor OA, Heaney ML, Schwartz L, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24:166–73. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- 41.Persky DO, Bernstein SH, Goldman B, Rimsza LM, Fisher RI, Miller TP. A phase II study of PXD101 (belinostat) in relapsed and refractory aggressive B-cell lymphomas (rel/ref ABCL): SWOG S0520. J Clin Oncol. 2012;30:e18536. [Google Scholar]
- 42.Bushway M, Cycon KA, Mulvaney K, Murphy SP. Coordinate loss of MHC class II expression in the diffuse large B cell lymphoma cell line OCI-Ly2 is due to a novel mutation in RFX-AP. Immunogenetics. 2010;62:109–16. doi: 10.1007/s00251-009-0418-3. [DOI] [PubMed] [Google Scholar]
- 43.Cycon KA, Rimsza LM, Murphy SP. Alterations in CIITA constitute a common mechanism accounting for downregulation of MHC class II expression in diffuse large B-cell lymphoma (DLBCL) Exp Hematol. 2009;37:184–94. doi: 10.1016/j.exphem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Rimsza LM, Chan WC, Gascoyne RD, et al. CIITA or RFX coding region loss of function mutations occur rarely in diffuse large B-cell lymphoma cases and cell lines with low levels of major histocompatibility complex class II expression. Haematologica. 2009;94:596–8. doi: 10.3324/haematol.2008.000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rimsza LM, Roberts RA, Campo E, et al. Loss of major histocompatibility class II expression in non-immune-privileged site diffuse large B-cell lymphoma is highly coordinated and not due to chromosomal deletions. Blood. 2006;107:1101–7. doi: 10.1182/blood-2005-04-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi JC, Holtz R, Petroff MG, Alfaidy N, Murphy SP. Dampening of IFN-γ-inducible gene expression in human choriocarcinoma cells is due to phosphatase-mediated inhibition of the JAK/STAT-1 pathway. J Immunol. 2007;178:1598–607. doi: 10.4049/jimmunol.178.3.1598. [DOI] [PubMed] [Google Scholar]
- 47.Holtz R, Choi JC, Petroff MG, Piskurich JF, Murphy SP. Class II transactivator (CIITA) promoter methylation does not correlate with silencing of CIITA transcription in trophoblasts. Biol Reprod. 2003;69:915–24. doi: 10.1095/biolreprod.103.017103. [DOI] [PubMed] [Google Scholar]
- 48.Morris AC, Riley JL, Fleming WH, Boss JM. MHC class II gene silencing in trophoblast cells is caused by inhibition of CIITA expression. Am J Reprod Immunol. 1998;40:385–94. doi: 10.1111/j.1600-0897.1998.tb00423.x. [DOI] [PubMed] [Google Scholar]
- 49.Wilkinson ST, Fernandez DR, Murphy SP, et al. Decreased major histocompatibility complex class II expression in diffuse large B-cell lymphoma does not correlate with CpG methylation of class II transactivator promoters III and IV. Leuk Lymphoma. 2009;50:1875–8. doi: 10.3109/10428190903297531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi JC, Holtz R, Murphy SP. Histone deacetylases inhibit IFN-γ-inducible gene expression in mouse trophoblast cells. J Immunol. 2009;182:6307–15. doi: 10.4049/jimmunol.0802454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piskurich JF, Linhoff MW, Wang Y, Ting JP. Two distinct γ interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor β. Mol Cell Biol. 1999;19:431–40. doi: 10.1128/mcb.19.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia JF, Roncador G, Garcia JF, et al. PRDM1/BLIMP-1 expression in multiple B and T-cell lymphoma. Haematologica. 2006;91:467–74. [PubMed] [Google Scholar]
- 53.Montes-Moreno S, Gonzalez-Medina AR, Rodriguez-Pinilla SM, et al. Aggressive large B-cell lymphoma with plasma cell differentiation: immunohistochemical characterization of plasmablastic lymphoma and diffuse large B-cell lymphoma with partial plasmablastic phenotype. Haematologica. 2010;95:1342–9. doi: 10.3324/haematol.2009.016113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 55.Sarosiek KA, Nechushtan H, Lu X, Rosenblatt JD, Lossos IS. Interleukin-4 distinctively modifies responses of germinal centre-like and activated B-cell-like diffuse large B-cell lymphomas to immuno-chemotherapy. Br J Haematol. 2009;147:308–18. doi: 10.1111/j.1365-2141.2009.07851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holling TM, Van der Stoep N, Van den Elsen PJ. Epigenetic control of CIITA expression in leukemic T cells. Biochem Pharmacol. 2004;68:1209–13. doi: 10.1016/j.bcp.2004.03.046. [DOI] [PubMed] [Google Scholar]
- 57.Morris AC, Beresford GW, Mooney MR, Boss JM. Kinetics of a γ interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Mol Cell Biol. 2002;22:4781–91. doi: 10.1128/MCB.22.13.4781-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashraf U, Czuczman MS, Marvis C, Gibbs J, Hernandez-Ilizaliturri FJ. Entinostat (SNDX-275), a novel DAC inhibitor, is highly effective in rituximab-[chemotherapy]-sensitive or rituximab-[chemotherapy]-resistant lymphomas and has synergistic anti-tumor activity when combined with bortezomib. Blood. 2009;114:1435–6. [Google Scholar]
- 59.Tam W, Gomez M, Chadburn A, Lee JW, Chan WC, Knowles DM. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood. 2006;107:4090–100. doi: 10.1182/blood-2005-09-3778. [DOI] [PubMed] [Google Scholar]
- 60.Pike BL, Greiner TC, Wang X, et al. DNA methylation profiles in diffuse large B-cell lymphoma and their relationship to gene expression status. Leukemia. 2008;22:1035–43. doi: 10.1038/leu.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Landmann S, Muhlethaler-Mottet A, Bernasconi L, et al. Maturation of dendritic cells is accompanied by rapid transcriptional silencing of class II transactivator (CIITA) expression. J Exp Med. 2001;194:379–91. doi: 10.1084/jem.194.4.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Conley BA, Wright JJ, Kummar S. Targeting epigenetic abnormalities with histone deacetylase inhibitors. Cancer. 2006;107:832–40. doi: 10.1002/cncr.22064. [DOI] [PubMed] [Google Scholar]
- 63.Zika E, Greer SF, Zhu XS, Ting JP. Histone deacetylase 1/mSin3A disrupts γ-interferon-induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol Cell Biol. 2003;23:3091–102. doi: 10.1128/MCB.23.9.3091-3102.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khan N, Jeffers M, Kumar S, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 65.Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, Wintersberger E, Seiser C. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol. 1999;19:5504–11. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzuki M, Yamada T, Kihara-Negishi F, Sakurai T, Oikawa T. Direct association between PU.1 and MeCP2 that recruits mSin3A-HDAC complex for PU.1-mediated transcriptional repression. Oncogene. 2003;22:8688–98. doi: 10.1038/sj.onc.1207182. [DOI] [PubMed] [Google Scholar]
- 67.Ghosh N, Piskurich JF, Wright G, Hassani K, Ting JP, Wright KL. A novel element and a TEF-2-like element activate the major histocompatibility complex class II transactivator in B-lymphocytes. J Biol Chem. 1999;274:32342–50. doi: 10.1074/jbc.274.45.32342. [DOI] [PubMed] [Google Scholar]
- 68.Cycon KA, Clements JL, Holtz R, Fuji H, Murphy SP. The immunogenicity of L1210 lymphoma clones correlates with their ability to function as antigen-presenting cells. Immunology. 2009;128:e641–51. doi: 10.1111/j.1365-2567.2009.03052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baskar S, Glimcher L, Nabavi N, Jones RT, Ostrand-Rosenberg S. Major histocompatibility complex class II+B7-1+ tumor cells are potent vaccines for stimulating tumor rejection in tumor-bearing mice. J Exp Med. 1995;181:619–29. doi: 10.1084/jem.181.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dissanayake SK, Thompson JA, Bosch JJ, Clements VK, Chen PW, Ksander BR, Ostrand-Rosenberg S. Activation of tumor-specific CD4+ T lymphocytes by major histocompatibility complex class II tumor cell vaccines: a novel cell-based immunotherapy. Cancer Res. 2004;64:1867–74. doi: 10.1158/0008-5472.can-03-2634. [DOI] [PubMed] [Google Scholar]
- 71.Khan AN, Magner WJ, Tomasi TB. An epigenetically altered tumor cell vaccine. Cancer Immunol Immunother. 2004;53:748–54. doi: 10.1007/s00262-004-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maeda T, Towatari M, Kosugi H, Saito H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood. 2000;96:3847–56. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.