

Abstract
Regulation of skeletal muscle fatty acid oxidation (FAO) and adaptation to exercise training have long been thought to depend on delivery of fatty acids (FAs) to muscle, their diffusion into muscle, and muscle mitochondrial content and biochemical machinery. However, FA entry into muscle occurs via a regulatable, protein-mediated mechanism, involving several transport proteins. Among these CD36 is key. Muscle contraction and pharmacological agents induce CD36 to translocate to the cell surface, a response that regulates FA transport, and hence FAO. In exercising CD36 KO mice, exercise duration (−44%), and FA transport (−41%) and oxidation (−37%) are comparably impaired, while carbohydrate metabolism is augmented. In trained CD36 KO mice, training-induced upregulation of FAO is not observed, despite normal training-induced increases in mitochondrial density and enzymes. Transfecting CD36 into sedentary WT muscle (+41%), comparable to training-induced CD36 increases (+44%) in WT muscle, markedly upregulates FAO to rates observed in trained WT mice, but without any changes in mitochondrial density and enzymes. Evidently, in vivo CD36-mediated FA transport is key for muscle fuel selection and training-induced FAO upregulation, independent of mitochondrial adaptations. This CD36 molecular mechanism challenges the view that skeletal muscle FAO is solely regulated by muscle mitochondrial content and machinery.
 |
Arend Bonen is professor emeritus in the Department of Human Health and Nutritional Sciences at the University of Guelph, Guelph, Ontario, Canada. His work has focused for many years on skeletal muscle substrate metabolism, with particular emphases in recent years on the regulation of skeletal and cardiac muscle protein-mediated fatty acid transport and oxidation. He was the Canada Research Chair in Metabolism and Health and is a Fellow of the Royal Society of Canada.
In skeletal muscle, carbohydrates and lipids are oxidized to provide energy (ATP) at rest and during exercise. The biochemical foundation for the understandings of muscle carbohydrate metabolism began with a series of studies beginning in the mid-1960s, when Eric Hultman and colleagues (Bergstrom & Hultman, 1966, 1967; Bergstrom et al. 1967; Hermansen et al. 1967; Nilsson & Hultman, 1973) established that muscle and hepatic glycogen (to provide glucose) are key carbohydrates for use by skeletal muscle. On the other hand, lipolysis of adipose tissue triacylglycerol was shown to provide fatty acids to exercising skeletal muscle via the circulation (cf. Hagenfeldt, 1979; Kiens, 2006). Intramuscular triacylglycerol hydrolysis can also provide exercising muscle with fatty acids, but quantification of muscle triacylglycerol hydrolysis has been fraught with some methodological difficulties, as discussed elsewhere (cf. Watt et al. 2002; Kiens, 2006).
The proportion of energy provided by carbohydrates and lipids can vary considerably in working muscle, as their relative usage is related to the intensity of exercise (Romijn et al. 1993; van Loon et al. 2001) (Fig. 1). However, the myriad signals involved in regulating fuel utilization and mobilization coordinately from three different fuel depots (liver, adipose tissue and muscle) during exercise are largely unknown. Nevertheless, a reciprocal relationship between carbohydrate and lipid utilization by skeletal muscle is a well-established phenomenon (Fig. 1). However, skeletal muscle is metabolically promiscuous, and the relative utilization of carbohydrates and fatty acids by muscle is not immutably fixed for given exercise intensities, particularly at intensities ≤65%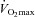 .
.
Figure 1. Relative and absolute quantities of fuel selection during exercise by humans at selected exercise intensities.
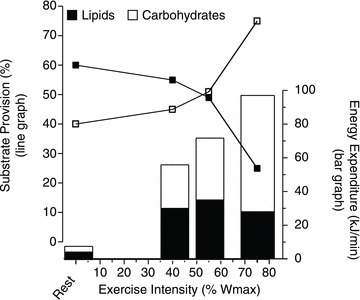
Bars show the energy expenditure at rest and at different exercise intensities (40, 55 and 75%Wmax) (scale on the right). The line graph shows the relative contributions (%) of lipid and carbohydrate use at rest and during each exercise condition (scale on the left). Redrawn fromvan Loon et al. (2001).
The regulation of skeletal muscle fatty acid oxidation has for many years been thought to depend on the blood borne delivery of fatty acids to muscle, their simple diffusion into muscle, the muscle mitochondrial content and the mitochondrial biochemical regulatory mechanisms (Holloszy & Booth, 1976; Hagenfeldt, 1979; Koves et al. 2005b; Bruce et al. 2006). The well-known, exercise training-induced upregulation of skeletal muscle fatty acid oxidation (cf. Holloszy & Booth, 1976; Kiens, 2006) and its induction by molecular mechanisms (e.g. PGC-1α, Koves et al. 2005a; PPARδ, Wang et al. 2004; ERRγ, Rangwala et al. 2010) have been attributed to mitochondrial biogenesis and the resulting increase in fatty acid oxidation enzymes.
The mitochondria-centric view of the acute and adaptive upregulation of fatty acid oxidation was developed prior to recent recognition that fatty acid entry into muscle occurs via a regulatable, protein-mediated mechanism (cf. Bonen et al. 2007a; Glatz et al. 2010). This fatty acid transport process may well be central to (a) allowing the upregulation of skeletal muscle fatty acid oxidation, particularly during acute exercise, and (b) regulating the adaptation of muscle fatty acid oxidation in response to exercise training. In this brief review we examine recent evidence that the fatty acid transporter CD36 is a key molecular component that controls the entry of fatty acids into muscle, and thereby (a) regulates skeletal muscle fatty acid oxidation at rest and during exercise, (b) influences muscle fuel selection and endurance exercise performance, and (c) is central to the well-known training-induced adaptation of skeletal muscle fatty acid oxidation in a mitochondria-independent manner.
Common assumptions in studies of fatty acid metabolism
In the many studies that have examined skeletal muscle fatty acid utilization (cf. Holloszy & Booth, 1976; Kiens, 2006), there are several underlying assumptions. The first assumption is that the blood borne delivery of fatty acids to muscle for their oxidation is more than adequate at rest and during exercise. With the possible exception of highly trained, endurance cyclists exercising at a high intensity (85%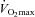 , Romijn et al. 1995), it appears that this general assumption is correct in view of both the older data and recent molecular data showing that exercise performance is impaired in animal models in which fatty acid delivery to muscle is altered experimentally (Hickson et al. 1977) or in genetic animal models in which fatty acid mobilization from adipose tissue and their delivery to muscle are reduced (i.e. hormone sensitive lipase (HSL) KO (Fernandez et al. 2008) and adipose triacylglycerol lipase (ATGL) KO mice (Huijsman et al. 2009; Schoiswohl et al. 2010)).
, Romijn et al. 1995), it appears that this general assumption is correct in view of both the older data and recent molecular data showing that exercise performance is impaired in animal models in which fatty acid delivery to muscle is altered experimentally (Hickson et al. 1977) or in genetic animal models in which fatty acid mobilization from adipose tissue and their delivery to muscle are reduced (i.e. hormone sensitive lipase (HSL) KO (Fernandez et al. 2008) and adipose triacylglycerol lipase (ATGL) KO mice (Huijsman et al. 2009; Schoiswohl et al. 2010)).
The second underlying assumption is that fatty acid entry into skeletal muscle occurs in an unregulated manner by simple diffusion. In light of recent work by our group and others showing that fatty acid entry into muscle involves a highly regulated, protein-mediated mechanism (cf. Bonen et al. 2007a; Glatz et al. 2010), it appears that this second assumption is likely to be incorrect.
Fatty acid transport, transporters and oxidation in skeletal muscle
In the late 1980s and through the 1990s evidence was obtained showing that there were a number of cell surface proteins (CD36, FABPpm, and FATP1–6) that appeared to be involved with transporting fatty acids into metabolically important tissues, including adipose tissue, liver, heart and skeletal muscle (cf. Bonen et al. 2007a; Glatz et al. 2010). At the same time, studies in our group provided key evidence that fatty acids were transported into skeletal muscle (cf. Bonen et al. 2007a; Glatz et al. 2010). These results challenged the dogma of the day, which held that such proteins were unnecessary (cf. Hamilton et al. 2002). While the debate over this matter continued for a number of years (cf. Bonen et al. 2007a; Kampf & Kleinfeld, 2007; Kampf et al. 2007), there is now a wide acceptance that membrane proteins are involved in facilitating the uptake of fatty acids across the plasma membrane (cf. Glatz et al. 2010). However, why there are a number of differing fatty acid transporters remains unknown, as in skeletal muscle they stimulate fatty acid transport independently and to differing extents (CD36 = FATP4 > FABPpm + FATP1) (Nickerson et al. 2009). It has been speculated that some of these proteins may interact with each other (cf. Bonen et al. 2007a; Glatz et al. 2010), but this remains to be demonstrated.
To date, the effects of CD36 have been extensively studied in skeletal muscle, as this was one of the first fatty acid transporters to be identified (Abumrad et al. 1993). In addition, a high quality antibody became available early (Matsuno et al. 1996; Bonen et al. 2000) and CD36 transgenic and KO models have existed for more than a decade (Febbraio et al. 1999; Ibrahimi et al. 1999; Coburn et al. 2000; Hajri et al. 2002; Bonen et al. 2007b). Comparable experimental tools have not been available for other fatty acid transporters. Thus, based on the currently existing evidence, it appears that CD36 is likely to be the key fatty acid transporter in skeletal muscle (cf. Glatz et al. 2010). However, this view may yet change when more experimental tools are developed for the other fatty acid transporters.
Work in our group has demonstrated that the rate of fatty acid transport into giant vesicles, derived from red and white skeletal muscle and the heart, (i) scaled with the well-known differences in fatty acid oxidation among these tissues and (ii) was highly correlated with the plasmalemmal content of CD36 and FABPpm, but not with plasmalemmal FATP1 (Luiken et al. 1999). More recent work has also shown high inter-correlations between CPT-1 and selected fatty acid transporters (CD36, FABPpm, and FATP4, but not FATP1) in metabolically heterogeneous rodent skeletal muscles (Nickerson et al. 2009) as well as between whole body fatty acid oxidation and FATP4 in trained muscle (Jeppesen et al. 2012). Collectively, these data suggest that protein-mediated fatty acid transport is likely to support fatty acid oxidation by skeletal muscle.
Acute and chronic regulation of fatty acid transport
Given that fatty acids are important for many cellular functions, it seemed from a physiological/metabolic perspective that it would be highly desirable to regulate the protein-mediated fatty acid transport into metabolically important tissues, none more so than skeletal muscle in which the rate of fatty acid oxidation can be rapidly altered. Indeed, the GLUT4 translocation paradigm in response to metabolic stimuli (muscle contraction, insulin) seemed particularly relevant in this regard. This working hypothesis stimulated us, and others, to examine whether fatty acid transport into skeletal muscle was (a) acutely regulatable (i.e. within minutes), (b) whether this was attributable to the translocation of a fatty acid transporter and (c) whether the fatty acid transport system was adaptable to exercise training.
Acute regulation of fatty acid transport into muscle
We (Bonen et al. 2000) used a muscle contraction model (electrical nerve stimulation) to alter the metabolic demand by rodent skeletal muscle. This rapidly induced an increase in fatty acid transport within 1 min, with further increases thereafter, and declining once contractile activity had ceased (Fig. 2A). When the demand for fuel use by muscle was upregulated, by increasing the rate of muscle contraction, there was a linear increase in the rate of fatty acid transport (Fig. 2B). A contraction-induced translocation of CD36 from an intracellular depot to the plasma membrane accounted for the increased rate of fatty acid transport (Fig. 2C), as blocking plasma membrane CD36 with a specific inhibitor (sulfo-N-succinimidyl oleate (SSO)) impaired the contraction-induced fatty acid transport (Fig. 2D) (Bonen et al. 2000). This work represented a paradigm shift, as it was the first evidence to show that fatty acid transport into muscle was acutely upregulated (i.e. within minutes), and involved the translocation of a fatty acid transporter, namely CD36, from an intracellular depot to the cell surface. Subsequently, this observation has now been confirmed by others (cf. Glatz et al. 2010).
Figure 2. Effects of electrically stimulated muscle contraction on fatty acid transport, and plasma membrane CD36.
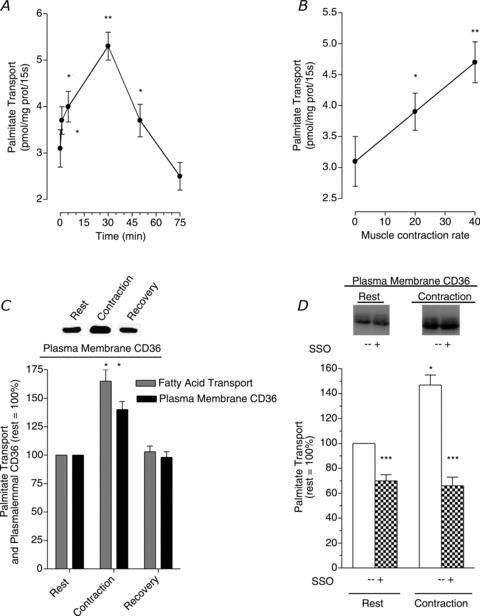
Data are means ± SEM. Fatty acid transport was examined in giant sarcolemmal vesicles obtained from rat skeletal muscles and plasma membrane CD36 was measured in these vesicles. A, the effect of muscle contraction and recovery from muscle contraction on the rate of fatty acid transport. B, the relationship between the rate of muscle contraction and the rate of fatty acid transport. C, the relative changes in the rate of fatty acid transport and plasma membrane CD36 at the end of 30 min muscle contraction and at the end of 55 min recovery (rest = 100%). D, the effect of the CD36 inhibitor (SSO) on fatty acid transport in resting and in 30 min contracted muscle. Note that control (–SSO) and the inhibition studies were conducted on vesicles from the same pools of resting and contracting muscles as shown by the similar plasma membrane CD36 in each of these two conditions. *P < 0.05, contraction vs. rest (A–D), and contraction vs. recovery (C). **P < 0.05, 40 Hz vs. 20 Hz (B). ***P < 0.05, +SSO treatment vs.–SSO treatment in resting and contracting muscle (D). From Bonen et al. (2000); © American Society for Biochemistry and Molecular Biology.
More recently it has also been shown that, except for FATP6, muscle fatty acid transporters can potentially be induced to translocate to the plasma membrane during electrically stimulated muscle contraction or exercise (Turcotte et al. 2005; Jain et al. 2009; Jeppesen et al. 2011; Bradley et al. 2012; McFarlan et al. 2012), as well as to the t-tubules during muscle contraction (Stefanyk et al. 2012). It appears that ERK1/2 and AMPK are the predominant signalling molecules regulating fatty acid uptake during low- to moderate-intensity muscle contraction and during moderate- to high-intensity muscle contraction, respectively (Turcotte et al. 2005; Raney & Turcotte, 2006). These signals induce the translocation of CD36 to the sarcolemma, but possibly not FABPpm (Turcotte et al. 2005; Raney & Turcotte, 2006; Jeppesen et al. 2009). This latter group (Jeppesen et al. 2011) has, however, also reported that AMPK is not necessarily essential in the regulation of CD36 translocation and FA uptake in skeletal muscle during muscle contraction (Jeppesen et al. 2011). Undoubtedly myriad signals, some of which may be redundant, are likely to be activated during muscle contraction. Which among these are critical for targeting CD36 and other fatty acid transporters remains to be established.
Adaptation of fatty acid transport with chronic muscle stimulation and exercise training
With chronic muscle stimulation and endurance exercise training a number of fatty acid transporters in muscle are increased, including CD36, FABPpm (Koonen et al. 2004; Perry et al. 2008; Talanian et al. 2010), and FATP4, while FATP1 was reduced (Jeppesen et al. 2012). These adaptive responses of fatty acid transporters may well be needed to support the increased need for fatty acids to support the training-induced increase in skeletal muscle fatty acid oxidation. This is also suggested by findings in our studies and others that PGC-1α not only induces muscle mitochondrial biogenesis and stimulates muscle fatty acid oxidation, but this nuclearly encoded transcriptional co-activator also induces the expression of CD36 (Benton et al. 2008, 2010).
Fatty acid oxidation and CD36-mediated fatty acid transport
We have shown that the skeletal muscle fatty acid transporter CD36 influences fatty acid oxidation, in mice overexpressing CD36 and in CD36 KO mice. Specifically, in CD36 transgenic mice, contraction of isolated soleus muscle increased fatty acid oxidation markedly more than in contracting WT soleus muscles (Ibrahimi et al. 1999) (Fig. 3A). Conversely, in CD36 KO mice, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR)-, (Bonen et al. 2007b) (Fig. 3B) or caffeine-stimulated fatty acid transport and oxidation (Lally et al. 2012) were both markedly impaired, despite the fact that FABPpm, and FATP1 and 4 were still induced to translocate to the plasma membrane in CD36 KO mice (Lally et al. 2012). It was calculated that CD36 accounted for ∼70% of the AICAR- or caffeine-stimulated increase in both muscle fatty acid transport and oxidation (Bonen et al. 2007b; Lally et al. 2012). Clearly, these studies demonstrate a very tight association between CD36-mediated fatty acid transport and fatty acid oxidation, when muscle was metabolically stimulated with pharmaceutical agents or with electrically induced muscle contraction.
Figure 3. Effects of CD36 overexpression and CD36 ablation on muscle fatty acid oxidation.
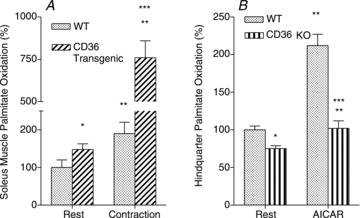
Data are means ± SEM. A, the effects of muscle contraction in isolated soleus muscle on fatty acid oxidation in WT and CD36 transgenic mice (redrawn from Ibrahimi et al. (1999)) with permission from the American Physiological Society for Biochemistry and Molecular Biology. B, the effects of AICAR stimulation on fatty acid oxidation by perfused hindquarter muscles of WT and CD36 KO mice (redrawn from Bonen et al. (2007b) with permission from the American Physiological Society). *P < 0.05, in resting muscle CD36 transgenic or CD36 KO vs. WT. **P < 0.05, contracting or AICAR-stimulated muscle vs. respective resting muscle. ***P < 0.05, contracting CD36 transgenic muscle vs. contracting WT muscle, and AICAR-stimulated CD36 KO muscle vs. AICAR-stimulated WT muscle.
CD36 critically regulates exercise- and training-induced upregulation of fatty acid oxidation in a mitochondria-independent manner
Recently we examined the role of CD36, in vivo, with respect to muscle fuel selection under basal conditions, during a metabolic challenge (exercise), and after exercise training. We also investigated whether CD36 overexpression mimicked the exercise training-induced upregulation of fatty acid oxidation (full details of these studies are found in McFarlan et al. (2012)).
Metabolic phenotype of CD36 KO mice
In WT and CD36 KO mice, the 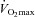 and muscle mitochondrial content and enzymes do not differ (Fig. 4). Under resting conditions CD36 KO mice display reduced rates of skeletal muscle fatty acid transport (−21%) and oxidation (−25%), and lower concentrations of intramuscular triacylglycerol (−31%), diacylglycerol (−18%) and ceramide (−15%), despite higher concentrations of circulating fatty acids (+43%) (Fig. 4). With respect to carbohydrate metabolism, hepatic (−20%) but not muscle glycogen was reduced, and the CD36 KO animals exhibited greater muscle insulin sensitivity (Fig. 4). This CD36 KO metabolic phenotype concurs with prior observations (Febbraio et al. 1999; Coburn et al. 2000; Goudriaan et al. 2003; Bonen et al. 2007b). Taken altogether it appears that ablation of CD36 increased the reliance on carbohydrate metabolism in skeletal muscle under resting conditions.
and muscle mitochondrial content and enzymes do not differ (Fig. 4). Under resting conditions CD36 KO mice display reduced rates of skeletal muscle fatty acid transport (−21%) and oxidation (−25%), and lower concentrations of intramuscular triacylglycerol (−31%), diacylglycerol (−18%) and ceramide (−15%), despite higher concentrations of circulating fatty acids (+43%) (Fig. 4). With respect to carbohydrate metabolism, hepatic (−20%) but not muscle glycogen was reduced, and the CD36 KO animals exhibited greater muscle insulin sensitivity (Fig. 4). This CD36 KO metabolic phenotype concurs with prior observations (Febbraio et al. 1999; Coburn et al. 2000; Goudriaan et al. 2003; Bonen et al. 2007b). Taken altogether it appears that ablation of CD36 increased the reliance on carbohydrate metabolism in skeletal muscle under resting conditions.
Figure 4. Phenotype of CD36 KO mice compared to WT mice (100%, see dashed line).
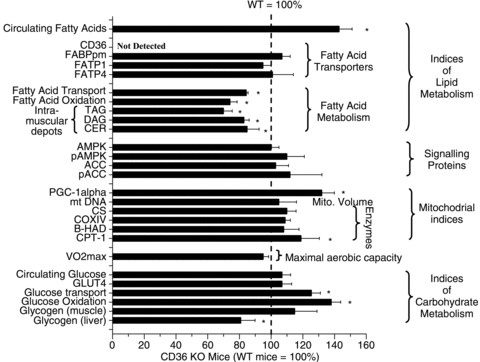
Data are means ± SEM. *P < 0.05, CD36 KO vs. WT. From McFarlan et al. (2012). © American Society for Biochemistry and Molecular Biology.
Acute exercise
In CD36 KO and WT mice exercising at 78%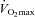 , there were profound differences in fuel selection and exercise performance (Fig. 5). In the exercising CD36 KO mice the reductions in fatty acid transport into muscle (−41%) and whole body fatty acid oxidation (−37%) and exercise performance (−44%) were all impaired similarly (Fig. 5A), despite comparable provision of circulating fatty acids to muscle in CD36 KO and WT mice (Fig. 5A). Despite similar, absolute reductions in hepatic (93–97%) and muscle glycogen (32–37%) in exercised WT and CD36 KO mice (McFarlan et al. 2012), carbohydrate metabolism during exercise, as determined from respiratory exchange ratio, was considerably greater in the CD36 KO mice (+89%, Fig. 5B). This was attributable to the enhanced rates of muscle and hepatic glycogen utilization in CD36 KO mice relative to WT mice (Fig. 5B). A similar phenomenon has been observed previously in other animal models in which fatty acid utilization by exercising muscle is diminished (Hickson et al. 1977; Fernandez et al. 2008; Huijsman et al. 2009). Since the depletion of hepatic glycogen has long been linked with an impaired exercise performance, it is likely that the accelerated and almost complete hepatic glycogen depletion in CD36 KO mice accounted for their premature fatigue (McFarlan et al. 2012).
, there were profound differences in fuel selection and exercise performance (Fig. 5). In the exercising CD36 KO mice the reductions in fatty acid transport into muscle (−41%) and whole body fatty acid oxidation (−37%) and exercise performance (−44%) were all impaired similarly (Fig. 5A), despite comparable provision of circulating fatty acids to muscle in CD36 KO and WT mice (Fig. 5A). Despite similar, absolute reductions in hepatic (93–97%) and muscle glycogen (32–37%) in exercised WT and CD36 KO mice (McFarlan et al. 2012), carbohydrate metabolism during exercise, as determined from respiratory exchange ratio, was considerably greater in the CD36 KO mice (+89%, Fig. 5B). This was attributable to the enhanced rates of muscle and hepatic glycogen utilization in CD36 KO mice relative to WT mice (Fig. 5B). A similar phenomenon has been observed previously in other animal models in which fatty acid utilization by exercising muscle is diminished (Hickson et al. 1977; Fernandez et al. 2008; Huijsman et al. 2009). Since the depletion of hepatic glycogen has long been linked with an impaired exercise performance, it is likely that the accelerated and almost complete hepatic glycogen depletion in CD36 KO mice accounted for their premature fatigue (McFarlan et al. 2012).
Figure 5. Effects of acute exercise on selected parameters in WT and CD36 KO mice, including run time to fatigue and fatty acid metabolism (A) and carbohydrate metabolism (B).
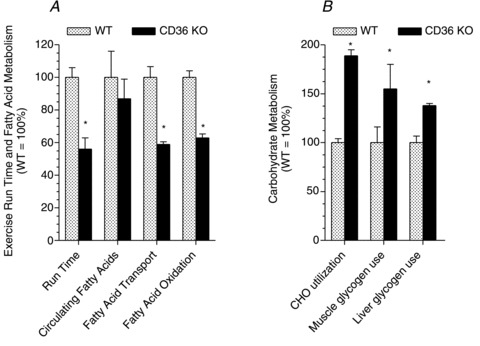
Data are means ± SEM. The exercise response in WT mice was set to 100% and the data in CD36 KO mice were expressed relative to the WT mice. Fatty acid oxidation (A) and carbohydrate (CHO) utilization (B) were determined from the respiratory exchange ratio during exercise. Circulating fatty acids increased during exercise in both WT and CD36 KO mice. The relative (%) comparisons in Fig. 5A of the circulating fatty acids are those at the end of exhaustive exercise. The rate of muscle and hepatic glycogen use (Fig. 5B) was based on their absolute reduction during exercise to exhaustion divided by the exercise time. It is recognized that these depletion rates are not necessarily linear as assumed by this calculation. WT muscle and hepatic glycogen depletion were set to 100%, and CD36 KO muscle and hepatic glycogen depletion were expressed relative to the respective WT tissues. Note that exercise-induced hepatic glycogen depletion is 5- to 10-fold greater than in skeletal muscle (see McFarlan et al. 2012). *P < 0.05, CD36 KO vs. WT. From McFarlan et al. (2012). © American Society for Biochemistry and Molecular Biology.
During exercise there were comparable differences between WT and CD36 KO mice with respect to whole body fatty acid oxidation (ratio of WT:CD36 KO = 1.6:1) and muscle fatty acid transport (ratio of WT:CD36 KO = 1.7:1), suggesting that fatty acid oxidation during exercise was highly dependent on CD36-mediated fatty acid transport into muscle. We have previously shown quantitatively comparable impairments in muscle fatty acid transport (−70%) and oxidation (−70%) in muscles of CD36 KO mice treated with AICAR (Fig. 3B) (Bonen et al. 2007b) or caffeine (Lally et al. 2012). In each instance, whether with exercise (McFarlan et al. 2012), with AICAR (Bonen et al. 2007b) or with caffeine (Lally et al. 2012), the underlying cause of the impairment in fatty acid oxidation in CD36 KO mice is their inability to upregulate fatty acid transport into muscle, a process that is dependent on the translocation of CD36 to the plasma membrane induced by metabolic stimuli (Bonen et al. 2000; Jain et al. 2009), including acute exercise (McFarlan et al. 2012).
Exercise training
A 6 week treadmill training programme induced the expected adaptive responses in skeletal muscle, namely comparable increases in muscle mitochondrial content of WT and CD36 KO mice, as shown by their upregulation of mtDNA (21–27%), citrate synthase (10–14%) and COXIV (22–30%) (Fig. 6). Training also induced an increase in CD36 (+44%, WT only) and a small increase in FABPpm in both WT and CD36 KO mice (9–13%). In contrast, skeletal muscle FATP1 and FATP4 content was not altered by 6 weeks of training in either group of mice (Fig. 6).
Figure 6. Exercise training induced changes in WT and CD36 KO mice relative to their respective sedentary control groups (100%).
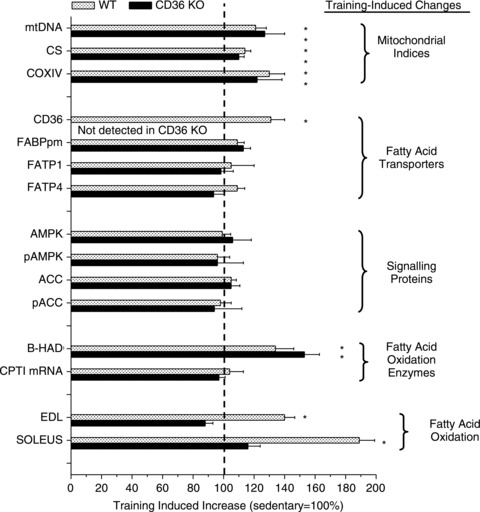
Data are means ± SEM. Fatty acid oxidation was examined when muscles were stimulated with caffeine (3 mm). From McFarlan et al. (2012). © American Society for Biochemistry and Molecular Biology.
Despite all these normal, and comparable, training-induced changes in skeletal muscle mitochondria and biochemical machinery in WT and CD36 KO mice, the training-induced changes in fatty acid oxidation in these animals were markedly different. Specifically, as expected a training-induced increase in muscle fatty acid oxidation occurred in the trained WT mice. In contrast, in the CD36 KO mice, 6 weeks of training failed to upregulate fatty acid oxidation, despite the normal training-induced increases in muscle mitochondrial content and enzymes (Fig. 6). This indicates that CD36 is an important molecular regulator of the training-induced increase in skeletal muscle fatty acid oxidation.
CD36 transfected soleus muscle
Although muscle CD36, mitochondrial content and fatty acid oxidation enzymes, and fatty acid oxidation were all concomitantly increased in trained WT animals, it appeared that the training-induced increase in fatty acid oxidation in WT skeletal muscle was dependent on CD36. Therefore, we transfected CD36 into sedentary WT muscles to perform a gain of function study. This permitted us to examine the independent role of CD36 on fatty acid oxidation in WT muscle, in the absence of concurrent changes in muscle mitochondrial content and fatty acid oxidation enzymes.
Since exercise training increased muscle CD36 by 44%, we matched the genetic overexpression in WT sedentary muscle to the same level (+41%). CD36 transfection of muscle did not alter muscle mitochondrial enzymes or enzymes involved in fatty acid oxidation. However, in CD36-transfected muscle, fatty acid oxidation was increased markedly in the absence of any changes at the level of the mitochondria. This CD36-specific increase in fatty acid oxidation (net change: +84%) paralleled the increase in fatty acid oxidation in trained muscle (net change: +90%) (Fig. 7) (McFarlan et al. 2012).
Figure 7. Comparison of exercise training-induced changes and CD36-transfection in WT soleus muscles on muscle CD36, mitochondrial content and enzymes, and caffeine-stimulated (3 mm) fatty acid oxidation.

Data are means ± SEM. Note that CD36 transfection increased muscle CD36 protein comparably to exercise training, and this increase in CD36 upregulated fatty acid oxidation independent of any changes in muscle mitochondrial content or enzymes. *P < 0.05, exercise-trained vs. sedentary, and CD36 transfected vs. transfection with empty vector in the same animal. **P < 0.05, exercise-trained vs. CD36 transfected. From McFarlan et al. (2012). © American Society for Biochemistry and Molecular Biology.
Influence of sarcolemmal CD36 and mitochondrial CD36 on fatty acid oxidation
We (Campbell et al. 2004; Bezaire et al. 2006; Holloway et al. 2009; Smith et al. 2011) and a number of other groups (Schenk & Horowitz, 2006; King et al. 2007; Aoi et al. 2008; Sebastian et al. 2009) have shown that CD36 is present on mitochondria, in skeletal muscle and other metabolically important tissues. At this location CD36 appears to contribute to regulating fatty acid oxidation (Campbell et al. 2004; Bezaire et al. 2006; Holloway et al. 2009; Smith et al. 2011). However, it seems that in skeletal muscle the quantitative contribution of mitochondrial CD36 to fatty acid oxidation is less than that of the sarcolemmal CD36. Specifically, in permeabilized muscles exposed to pyruvate, pyruvate + succinate, or palmitate, caffeine did not alter either mitochondrial state IV or state III respiration in either WT or CD36 KO muscles (McFarlan et al. 2012). In contrast, caffeine did stimulate (a) whole muscle fatty acid oxidation (+60%), (b) plasma membrane CD36 (+37%), and (c) fatty acid transport into muscle (+70%) (McFarlan et al. 2012). Thus, caffeine-induced stimulation of whole muscle fatty acid oxidation, and its further increase in trained WT muscle would seem to be largely attributable to the increased rate of CD36-mediated, sarcolemmal fatty acid transport into skeletal muscle.
Summary
Recent observations challenge the long-held belief that skeletal muscle fatty acid oxidation and its training-induced adaptation are regulated solely by the blood borne fatty acid delivery to muscle, and by the mitochondrial density and selected enzymes involved in fatty acid oxidation. In vivo, the fatty acid transporter CD36, independent of muscle mitochondrial content and enzymes, is a key component of the molecular machinery required for (a) regulating skeletal muscle fuel selection at rest and during exercise, and (b) the well-known, training-induced upregulation of fatty acid oxidation (Fig. 8).
Figure 8. Summary of experimental treatments showing schematically the dissociation between skeletal muscle fatty acid oxidation and mitochondrial biogenesis and enzymes, and the coordinate regulation between the fatty acid transporter CD36 and skeletal muscle fatty acid oxidation.
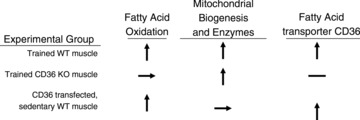
↑= increase; →= no change; –= not present
Acknowledgments
This research was supported by the Canadian Institutes of Health Research, the Heart and Stroke Foundation, and the Natural Sciences and Engineering Research Council of Canada and the Canada Research chair programme.
Author contributions
Y.Y., S.S.J., J.T.M., L.A.S. and A.C. contributed to the analyses and interpretation of the data, the drafting and revising of the manuscript, and the final approval of the published version of the manuscript. A.B. contributed to the conception and design of the work presented, as well as the analyses and interpretation of the data, the drafting and revising of the manuscript, and the final approval of the published version of the manuscript.
References
- Abumrad NA, El-Maghrabi MR, Amri E-Z, Lopez E, Grimaldi P. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268:17665–17668. [PubMed] [Google Scholar]
- Aoi W, Naito Y, Takanami Y, Ishii T, Kawai Y, Akagiri S, Kato Y, Osawa T, Yoshikawa T. Astaxanthin improves muscle lipid metabolism in exercise via inhibitory effect of oxidative CPT I modification. Biochem Biophys Res Commun. 2008;366:892–897. doi: 10.1016/j.bbrc.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Benton CR, Holloway GP, Han XX, Yoshida Y, Snook LA, Lally J, Glatz JF, Luiken JJ, Chabowski A, Bonen A. Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia. 2010;53:2008–2019. doi: 10.1007/s00125-010-1773-1. [DOI] [PubMed] [Google Scholar]
- Benton CR, Nickerson J, Lally J, Han X-X, Holloway GP, Glatz JFC, Luiken JJFP, Graham TE, Heikkila JJ, Bonen A. Modest PGC-1α overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in SS, not IMF, mitochondria. J Biol Chem. 2008;283:4228–4240. doi: 10.1074/jbc.M704332200. [DOI] [PubMed] [Google Scholar]
- Bergstrom J, Hermansen L, Hultman E, Saltin B. Diet, muscle glycogen and physical performance. Acta Physiol Scand. 1967;71:140–150. doi: 10.1111/j.1748-1716.1967.tb03720.x. [DOI] [PubMed] [Google Scholar]
- Bergstrom J, Hultman E. Muscle glycogen synthesis after exercise: an enhancing factor localized to the muscle cells in man. Nature. 1966;210:309–310. doi: 10.1038/210309a0. [DOI] [PubMed] [Google Scholar]
- Bergstrom J, Hultman E. A study of the glycogen metabolism during exercise in man. Scand J Clin Lab Invest. 1967;19:218–228. doi: 10.3109/00365516709090629. [DOI] [PubMed] [Google Scholar]
- Bezaire V, Bruce CR, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Identification of fatty acid translocase on human skeletal muscle mitochondrial membranes: essential role in fatty acid oxidation. Am J Physiol Endocrinol Metab. 2006;290:E509–E515. doi: 10.1152/ajpendo.00312.2005. [DOI] [PubMed] [Google Scholar]
- Bonen A, Chabowski A, Luiken JJFP, Glatz JFC. Is membrane transport of FFA mediated by lipid, protein, or both? Mechanisms and regulation of protein-mediated cellular fatty acid uptake: molecular, biochemical, and physiological evidence. Physiology (Bethesda) 2007a;22:15–29. doi: 10.1152/physiologyonline.2007.22.1.15. [DOI] [PubMed] [Google Scholar]
- Bonen A, Han X-X, Habets DDJ, Febbraio M, Glatz JFC, Luiken JJFP. A null mutation in skeletal muscle FAT/CD36 reveals its essential role in insulin-, and AICAR-stimulated fatty acid metabolism. Am J Physiol Endocrinol Metab. 2007b;292:E1740–E1749. doi: 10.1152/ajpendo.00579.2006. [DOI] [PubMed] [Google Scholar]
- Bonen A, Luiken JJFP, Arumugam Y, Glatz JFC, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275:14501–14508. doi: 10.1074/jbc.275.19.14501. [DOI] [PubMed] [Google Scholar]
- Bradley NS, Snook LA, Jain SS, Heigenhauser GJ, Bonen A, Spriet LL. Acute endurance exercise increases plasma membrane fatty acid transport proteins in rat and human skeletal muscle. Am J Physiol Endocrinol Metab. 2012;302:E183–189. doi: 10.1152/ajpendo.00254.2011. [DOI] [PubMed] [Google Scholar]
- Bruce CR, Thrush AB, Mertz VA, Bezaire V, Chabowski A, Heigenhauser GJ, Dyck DJ. Endurance training in obese humans improves glucose tolerance, mitochondrial fatty acid oxidation and alters muscle lipid content. Am J Physiol Endocrinol Metab. 2006;291:E99–E107. doi: 10.1152/ajpendo.00587.2005. [DOI] [PubMed] [Google Scholar]
- Campbell SE, Tandon NN, Woldegiorgis G, Luiken JJFP, Glatz JFC, Bonen A. A novel function for FAT/CD36: involvement in long chain fatty acid transfer into the mitochondria. J Biol Chem. 2004;279:36335–36341. doi: 10.1074/jbc.M400566200. [DOI] [PubMed] [Google Scholar]
- Coburn CT, Knapp FF, Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissue of CD36 knockout mice. J Biol Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Frieda S, Pearce A, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- Fernandez C, Hansson O, Nevsten P, Holm C, Klint C. Hormone-sensitive lipase is necessary for normal mobilization of lipids during submaximal exercise. Am J Physiol Endocrinol Metab. 2008;295:E179–186. doi: 10.1152/ajpendo.00282.2007. [DOI] [PubMed] [Google Scholar]
- Glatz JFC, Luiken JJFP, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- Goudriaan JR, Dahlmans VE, Teusink B, Ouwens DM, Febbraio M, Maassen JA, Romijn JA, Havekes LM, Voshol PJ. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J Lipid Res. 2003;44:2270–2277. doi: 10.1194/jlr.M300143-JLR200. [DOI] [PubMed] [Google Scholar]
- Hagenfeldt L. Metabolism of free fatty acids and ketone bodies during exercise in normal and diabetic man. Diabetes. 1979;28(Suppl 1):66–70. doi: 10.2337/diab.28.1.s66. [DOI] [PubMed] [Google Scholar]
- Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest. 2002;109:1381–1389. doi: 10.1172/JCI14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J, Guo W, Kamp F. Mechanisms of cellular uptake of long-chain fatty acids: Do we need cellular proteins. Mol Cell Biochem. 2002;239:17–23. [PubMed] [Google Scholar]
- Hermansen L, Hultman E, Saltin B. Muscle glycogen during prolonged severe exercise. Acta Physiol Scand. 1967;71:129–139. doi: 10.1111/j.1748-1716.1967.tb03719.x. [DOI] [PubMed] [Google Scholar]
- Hickson RC, Rennie MJ, Conlee RK, Winder WW, Holloszy JO. Effects of increased plasma fatty acids on glycogen utilization and endurance. J Appl Physiol. 1977;43:829–833. doi: 10.1152/jappl.1977.43.5.829. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- Holloway GP, Jain SS, Han X-X, Glatz JFC, Luiken JJFP, Bonen A. FAT/CD36 null mice reveal that mitochondrial FAT/CD36 is required to up-regulate mitochondrial fatty acid oxidation in contracting muscle. Am J Physiol Regul Integr Comp Physiol. 2009;297:R960–R967. doi: 10.1152/ajpregu.91021.2008. [DOI] [PubMed] [Google Scholar]
- Huijsman E, van de Par C, Economou C, van der Poel C, Lynch GS, Schoiswohl G, Haemmerle G, Zechner R, Watt MJ. Adipose triacylglycerol lipase deletion alters whole body energy metabolism and impairs exercise performance in mice. Am J Physiol Endocrinol Metab. 2009;297:E505–513. doi: 10.1152/ajpendo.00190.2009. [DOI] [PubMed] [Google Scholar]
- Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R, Abumrad NA. Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscles, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem. 1999;274:26761–26766. doi: 10.1074/jbc.274.38.26761. [DOI] [PubMed] [Google Scholar]
- Jain SS, Chabowski A, Snook LA, Schwenk RW, Glatz JFC, Luiken JJFP, Bonen A. Additive effects of insulin and muscle contraction on fatty acid transport and fatty acid transporters, FAT/CD36, FABPpm, FATP1, 4 and 6. FEBS Lett. 2009;583:2294–2300. doi: 10.1016/j.febslet.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Jeppesen J, Albers P, Luiken JJ, Glatz JF, Kiens B. Contractions but not AICAR increase FABPpm content in rat muscle sarcolemma. Mol Cell Biochem. 2009;326:45–53. doi: 10.1007/s11010-008-0006-0. [DOI] [PubMed] [Google Scholar]
- Jeppesen J, Albers PH, Rose AJ, Birk JB, Schjerling P, Dzamko N, Steinberg GR, Kiens B. Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J Lipid Res. 2011;52:699–711. doi: 10.1194/jlr.M007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen J, Jordy AB, Sjoberg KA, Fullekrug J, Stahl A, Nybo L, Kiens B. Enhanced fatty acid oxidation and FATP4 protein expression after endurance exercise training in human skeletal muscle. PLoS One. 2012;7:e29391. doi: 10.1371/journal.pone.0029391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampf JP, Kleinfeld AM. Is membrane transport of FFA mediated by lipid, protein, or both? An unknown protein mediates free fatty acid transport across the adipocyte plasma membrane. Physiology (Bethesda) 2007;22:7–14. doi: 10.1152/physiol.00011.2006. [DOI] [PubMed] [Google Scholar]
- Kampf JP, Kleinfeld AM, Bonen A, Chabowski A, Luiken JJFP, Glatz JFC. Is membrane transport of FFA mediated by lipid, protein, or both?: Points of agreement. Physiology (Bethesda) 2007;22:29. [Google Scholar]
- Kiens B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev. 2006;86:205–243. doi: 10.1152/physrev.00023.2004. [DOI] [PubMed] [Google Scholar]
- King KL, Stanley WC, Rosca M, Kerner J, Hoppel CL, Febbraio M. Fatty acid oxidation in cardiac and skeletal muscle mitochondria is unaffected by deletion of CD36. Arch Biochem Biophys. 2007;467:234–238. doi: 10.1016/j.abb.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonen DPY, Benton CR, Arumugam Y, Tandon NN, Calles-Escandon J, Glatz JFC, Luiken JJFP, Bonen A. Different mechanism can alter fatty acid transport when muscle contractile activity is chronically altered. Am J Physiol Endocrinol Metab. 2004;286:1042–1049. doi: 10.1152/ajpendo.00531.2003. [DOI] [PubMed] [Google Scholar]
- Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. Peroxisome proliferator-activated receptor-γ co-activator 1α-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005a;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- Koves TR, Noland RC, Bates AL, Henes ST, Muoio DM, Cortright RN. Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am J Physiol Cell Physiol. 2005b;288:C1074–1082. doi: 10.1152/ajpcell.00391.2004. [DOI] [PubMed] [Google Scholar]
- Lally JS, Jain SS, Han XX, Snook LA, Glatz JF, Luiken JJ, McFarlan J, Holloway GP, Bonen A. Caffeine-stimulated fatty acid oxidation is blunted in CD36 null mice. Acta Physiol (Oxf) 2012;205:71–81. doi: 10.1111/j.1748-1716.2012.02396.x. [DOI] [PubMed] [Google Scholar]
- Luiken JJFP, Turcotte LP, Bonen A. Protein-mediated palmitate uptake and expression of fatty acid transport proteins in heart giant vesicles. J Lipid Res. 1999;40:1007–1016. [PubMed] [Google Scholar]
- Matsuno K, Diaz-Ricard M, Montgomery RR, Aster T, Jamieson GA, Tandon NN. Inhibition of platelet adhesion to collagen by monoclonal anti-CD36 antibodies. Br J Haematol. 1996;92:960–967. doi: 10.1046/j.1365-2141.1996.422962.x. [DOI] [PubMed] [Google Scholar]
- McFarlan JT, Yuko Yoshida Y, Jain SS, Han X-X, Snook LA, Lally J, Smith BK, Glatz JFC, Luiken JJFP, Sayer RA, Tupling AR, Chabowski A, Holloway GP, Bonen A. In vivo, fatty acid translocase (CD36) critically regulates skeletal muscle fuel selection, exercise performance and training-induced adaptation of fatty acid oxidation. J Biol Chem. 2012;287:23502–23516. doi: 10.1074/jbc.M111.315358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson JG, Alkhateeb H, Benton CR, Lally J, Nickerson J, Han X-X, Wilson MH, Jain SS, Snook LA, Glatz JFC, Chabowski A, Luiken JJFP, Bonen A. Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1, and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J Biol Chem. 2009;284:16522–16530. doi: 10.1074/jbc.M109.004788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson LH, Hultman E. Liver glycogen in man – the effect of total starvation or a carbohydrate-poor diet followed by carbohydrate refeeding. Scand J Clin Lab Invest. 1973;32:325–330. doi: 10.3109/00365517309084355. [DOI] [PubMed] [Google Scholar]
- Perry CG, Heigenhauser GJ, Bonen A, Spriet LL. High-intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl Physiol Nutr Metab. 2008;33:1112–1123. doi: 10.1139/H08-097. [DOI] [PubMed] [Google Scholar]
- Raney MA, Turcotte LP. Regulation of contraction-induced FA uptake and oxidation by AMPK and ERK1/2 is intensity dependent in rodent muscle. Am J Physiol Endocrinol Metab. 2006;291:E1220–1227. doi: 10.1152/ajpendo.00155.2006. [DOI] [PubMed] [Google Scholar]
- Rangwala SM, Wang X, Calvo JA, Lindsley L, Zhang Y, Deyneko G, Beaulieu V, Gao J, Turner G, Markovits J. Estrogen-related receptor gamma is a key regulator of muscle mitochondrial activity and oxidative capacity. J Biol Chem. 2010;285:22619–22629. doi: 10.1074/jbc.M110.125401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, Wolfe RR. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol Endocrinol Metab. 1993;265:E380–E391. doi: 10.1152/ajpendo.1993.265.3.E380. [DOI] [PubMed] [Google Scholar]
- Romijn JA, Coyle EF, Sidossis LS, Zhang X-J, Wolfe RR. Relationship between fatty acid delivery and fatty acid oxidation during strenuous exercise. J Appl Physiol. 1995;79:1939–1945. doi: 10.1152/jappl.1995.79.6.1939. [DOI] [PubMed] [Google Scholar]
- Schenk S, Horowitz JF. Coimmunoprecipitation of FAT/CD36 and CPTI in skeletal muscle increases proportionally with fat oxidation after endurance exercise training. Am J Physiol Endocrinol Metab. 2006;291:E254–260. doi: 10.1152/ajpendo.00051.2006. [DOI] [PubMed] [Google Scholar]
- Schoiswohl G, Schweiger M, Schreiber R, Gorkiewicz G, Preiss-Landl K, Taschler U, Zierler KA, Radner FP, Eichmann TO, Kienesberger PC, Eder S, Lass A, Haemmerle G, Alsted TJ, Kiens B, Hoefler G, Zechner R, Zimmermann R. Adipose triglyceride lipase plays a key role in the supply of the working muscle with fatty acids. J Lipid Res. 2010;51:490–499. doi: 10.1194/jlr.M001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian D, Guitart M, Garcia-Martinez C, Mauvezin C, Orellana-Gavalda JM, Serra D, Gomez-Foix AM, Hegardt FG, Asins G. Novel role of FATP1 in mitochondrial fatty acid oxidation in skeletal muscle cells. J Lipid Res. 2009;50:1789–1799. doi: 10.1194/jlr.M800535-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BK, Jain S, Rimbaud S, Dam A, Quadrilatero J, Ventura-Clapier R, Bonen A, Holloway GP. FAT/CD36 is located on the outer mitochondrial membrane, upstream of long chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem J. 2011;437:125–134. doi: 10.1042/BJ20101861. [DOI] [PubMed] [Google Scholar]
- Stefanyk LE, Bonen A, Dyck DJ. Insulin and contraction-induced movement of fatty acid transport proteins to skeletal muscle transverse-tubules is distinctly different than to the sarcolemma. Metabolism. 2012 doi: 10.1016/j.metabol.2012.04.002. (in press) [DOI] [PubMed] [Google Scholar]
- Talanian JL, Holloway GP, Snook LA, Heigenhauser GJ, Bonen A, Spriet LL. Exercise training Increases sarcolemmal and mitochondrial fatty acid transport proteins in human skeletal muscle. Am J Physiol Endocrinol Metab. 2010;299:E180–E188. doi: 10.1152/ajpendo.00073.2010. [DOI] [PubMed] [Google Scholar]
- Turcotte LP, Raney MA, Todd MK. ERK1/2 inhibition prevents contraction-induced increase in plasma membrane FAT/CD36 content and FA uptake in rodent muscle. Acta Physiol Scand. 2005;184:131–139. doi: 10.1111/j.1365-201X.2005.01445.x. [DOI] [PubMed] [Google Scholar]
- van Loon LJ, Greenhaff PL, Constantin-Teodosiu D, Saris WH, Wagenmakers AJ. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol. 2001;536:295–304. doi: 10.1111/j.1469-7793.2001.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt MJ, Heigenhauser GJ, Spriet LL. Intramuscular triacylglycerol utilization in human skeletal muscle during exercise: is there a controversy. J Appl Physiol. 2002;93:1185–1195. doi: 10.1152/japplphysiol.00197.2002. [DOI] [PubMed] [Google Scholar]