

Abstract
Balanced immunoregulatory networks are essential for maintenance of systemic tolerance. Disturbances in the homeostatic equilibrium between inflammatory mediators, immune regulators and immune effector cells are implicated directly in the pathogenesis of autoimmune diseases, including rheumatoid arthritis (RA). In this study we characterize the peripheral blood CD8+CD28− regulatory T cells (Treg) contribution to the immunoregulatory network in health and in RA. In health, CD8+CD28− Treg are suppressive but, unlike CD4+Treg, they function predominantly through the action of soluble mediators such as interleukin (IL)-10 and transforming growth factor (TGF)-β. Neutralization of TGF-β consistently reduced CD8+CD28− Treg suppressor function in vitro. RA, CD8+CD28− Treg are increased numerically, but have reduced expression of inducible co-stimulator (ICOS) and programmed death 1 (PD-1) compared to healthy or disease controls. They produce more IL-10 but autologous T cells express less IL-10R. This expression was found to be restored following in-vitro addition of a tumour necrosis factor inhibitor (TNFi). Deficiencies in both the CD8+CD28− Treg population and reduced sensitivity of the T responder cells impact upon their regulatory function in RA. TNFi therapy partially restores CD8+CD28− Treg ability in vivo and in vitro, despite the defects in expression of functionally relevant molecules by RA CD8+CD28− Treg compared to healthy controls. This study places CD8+CD28− Treg cells in the scheme of immune regulation alongside CD4+ Treg cells, and highlights the importance of understanding impaired responsiveness to regulation that is common to these suppressor subsets and their restored function in response to TNFi therapy.
Keywords: anti-TNF therapy, CD8+ T cells, immunotherapy, regulatory T cells, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease [1] driven ultimately by the overwhelming production of proinflammatory cytokines that hinder the return to immunological homeostasis. T cell defects resulting in imbalance of the critical network of cellular and soluble immune effectors, and their regulators that maintain self-tolerance, are implicated in the pathogenesis of RA. Research over several decades indicate that RA T cells are dysfunctional and show reduced responsiveness to recall antigens [2].
Perhaps the most compelling evidence for the importance of cytokine imbalance in RA is the success of tumour necrosis factor (TNF) inhibitor based-therapies (TNFi) in generating disease remission. Several studies have since proposed that CD4+CD25hiforkhead box protein 3 (FoxP3)+ regulatory T cells (Treg) are functionally deficient in RA patients and regain some function in patients who were responsive to TNF inhibitor therapy [3].
In 2005, Davila et al. showed that CD8+CD28−CD56+ cells could suppress memory T cell responses. CD8+CD28−CD56+ clones were generated from RA synovial membrane (SM), which inhibited inflammatory cytokine production by RASM engrafted into severe combined immunodeficiency–non-obese diabetic (SCID–NOD) mice [4]. Increasing evidence now supports the case for a regulatory role for CD8+CD28− T cells in immune suppression in cancer [5], transplantation [6] and autoimmune disease, such as systemic lupus erythematosus (SLE) [7]. As an alternative regulatory link in the immune network, these cells may prove as important as CD4+CD25hiFoxP3+ Treg in controlling immune homeostasis in a disease where accelerated immune ageing enhances the loss of CD28 [8].
This study investigated the ex vivo phenotypic and functional characteristics of the CD8+CD28− Treg in RA. CD8+CD28− Treg were more abundant in RA patients treated with methotrexate [RA(MTX)], although fewer cells expressed inducible co-stimulator (ICOS) and programmed death (PD)-1 when compared with healthy controls. CD8+CD28− Treg from RA(MTX) failed to mediate suppression in the presence of a blocking transforming growth factor (TGF)-β antibody and produced high levels of interleukin (IL)-10. Concomitantly, RA T cell cultures expressed fewer cell surface IL-10 receptors (IL-10R) which may account, in part, for the relative insensitivity of the RA responder cells. CD8+CD28− Treg function, but not the reduced expression of ICOS and PD-1, was improved following TNF inhibitor therapy. This study identifies CD8+ Treg as a potential immunosuppressive force that is compromised in RA.
Materials and methods
Patient information
Donors provided informed written consent in the Academic Department of Rheumatology out-patient clinic at Guy's Hospital and King's College Hospital London UK. Ethical approval for the study was obtained from Bromley Hospital and Guy's and St Thomas's Hospital Local Research Ethical Committees. Heparinized peripheral blood (PB) samples were collected from healthy controls (HC), osteoarthritis (OA) patients used as disease controls, RA patients treated with MTX only, RA(MTX) and RA patients treated with TNF-α inhibitors (adalimumab, infliximab or etanercept in combination with MTX only) RA(TNFi). Paired PB and synovial fluid (SF) samples were obtained from RA(MTX) and RA(TNFi). All donors were age- and sex-matched. No patients on steroids or alternative disease modifying anti-rheumatic drugs were used. Patient demographics are shown in Table 1.
Table 1.
Patient characteristics of rheumatoid arthritis (RA) patients.†
| HC (n = 24) | OA (n = 17) | RA(MTX) (n = 60) | RA(TNFi) (n = 57) | |
|---|---|---|---|---|
| Age (years) | 55 ± 9 | 58 ± 11 | 61 ± 17 | 41 ± 15 |
| Females : males | 10:14 | 10:7 | 42:18 | 46:11 |
| Responder : non-responders | n.a. | n.d. | n.d. | 50:7 |
Demographic information of healthy controls (HC), disease controls: osteoarthritis, RA patients treated with methotrexate(MTX); RA(MTX) or MTX in combination with a tumour necrosis factor (TNF) inhibitor; RA(TNFi). Not applicable (n.a.) and values are shown as mean ± standard deviation; n.d.: not done.
Antibodies
Antibodies conjugated directly to fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinium chlorophyll cyanin 5·5 (PerCP.Cy5·5) or allophycocyanin (APC) were used for flow cytometric analysis: CD3, CD8, CD28, CD56, CD94, CD137/4-1BB, CD152/cytotoxic T lymphocyte antigen-4 (CTLA-4), CD210/IL-10R, CD278/ICOS, CD279/PD-1, isotype mouse immunoglobulin (Ig)G or rat IgG controls [Becton Dickinson (BD), Oxford, UK] were used as required.
Cell isolation
Mononuclear cell isolation
Heparinized peripheral blood mononuclear cells (PBMC) and synovial fluid mononuclear cells (SFMC) were isolated using Lymphoprep (Axis Shield, Oslo, Norway) by density gradient centrifugation and resuspended in tissue culture medium (TCM) [RPMI-1640 medium supplemented with 10% inactivated fetal calf serum (FCS); Invitrogen, Paisley, UK].
Negative isolation of CD8+CD28− Treg
Carbonyl iron was added to PBMC at 37°C for 60 min to remove phagocytic cells (Invitrogen). The B and CD4+ T cells were removed by positive selection with immunomagnetic beads: CD19 pan B cell and CD4 beads (Dynal; Invitrogen) at 4°C for 30 min. The remaining cells were incubated with 0·4 μg/ml purified anti-CD28 antibody (BD Biosciences, Oxford, UK) (4°C for 20 min) followed by anti-mouse IgG beads (Dynal; Invitrogen) at 4°C for 30 min. The purity of the negatively isolated CD8+CD28− Treg expressing CD3 was >95%, as determined by flow cytometric analysis. For T cell and monocyte isolation the T cell-negative (Dynal; Invitrogen) and CD14-positive isolation kits (Dynal; Invitrogen) were used, respectively, according to the manufacturer's instructions.
Immunofluorescent staining and flow cytometry
Whole blood staining
Heparinized PB (100 μl) was incubated with antibodies for 20 min at 4°C, then with 2 ml fluorescence activated cell sorter (FACS) lysing solution (BD Biosciences) for 10 min at room temperature and washed twice in immunofluorescence buffer (IFB) (phosphate-buffered saline with 0·05% sodium azide and 0·1% bovine serum albumin) for 5 min and fixed in 1% paraformaldehyde in IFB (Sigma, Poole, UK).
PBMC surface staining
PBMC in IFB were surface-stained with required antibodies for 20 min on ice, washed twice in IFB and fixed for analysis. Analysis for all samples was carried out with a FACSCalibur flow cytometer (BD Biosciences) using CellQuest software (BD Biosciences).
Co-cultures and suppression assays
Suppression assays
CD8+CD28− Treg were placed in co-culture with autologous responder PBMC at ratios of 1:1, 0·2:1 and 0·1:1 (PBMC 105 cells/well).
Cultures were stimulated with anti-CD3 antibody (1/1000 dilution) [muromonab-CD3 (OKT3)] [American Type Cell Collection (ATCC), Rockville, MD, USA] in 96-well flat-bottomed plates (Corning Costar, Sunderland, UK) and incubated in a 5% CO2 humidified atmosphere at 37°C for 72 h.
Cross-over co-culture assays
CD8+CD28− Treg were co-cultured with either allogeneic responder T cells from HC or RA(MTX). Each HC or RA(MTX) CD8+CD28− Treg sample was co-cultured with autologous T cells or allogeneic T cells isolated from two HC and two RA(MTX). Cultures were stimulated with CD3/CD28 beads (Dynal, Invitrogen) and incubated for 72 h at 37°C.
Blocking experiments
TNF inhibitor [infliximab (IFX), 10 ng/ml; Remicade®, Centocor, the Netherlands], anti-TGF-β1 antibody (5 μg/ml, clone 1D11, mIgG1; R&D Systems, Abingdon, UK) and LEAF™ purified mouse IgG1, k isotype control (clone MG1-45; Biolegend, Cambridge, UK), were added at the start of culture in the functional assays. All reagents were added to either the 1:1 co-culture or PBMC alone.
Transwell (TW) experiments
CD8+CD28− Treg were co-cultured with autologous responder PBMC and CD14+ cells at a ratio of 1:1:1 in the presence or absence of a semi-permeable membrane held in a TW (0·4 μm pore size) (Corning Costar). In 24-well plates, PBMC were placed into the well and CD14+ monocytes and CD8+CD28− Treg into the upper chamber of the TW. Cells were stimulated with anti-CD3 antibody (1:1000 dilution; ATCC) for 72 h.
All cultures were pulsed with 20 μCi tritiated [3H]-thymidine (GE Healthcare, Little Chalfont, UK) for the last 18 h and the uptake measured on a Topcount scintillation counter (Perkin Elmer, Cambridge, UK). Proliferation was determined as counts per minute (cpm) ± standard error of the mean (s.e.m.).
Cytokine determination
Supernatants were harvested and stored in aliquots at −80°C until required. IL-2, IL-17, IL-10, TNF-α and interferon (IFN)-γ concentrations were determined using the human FlowCytomix Simplex kits (Bender MedSystems GmbH, Vienns, Austria), according to the manufacturer's instructions.
Statistical analysis
Statistical analysis was performed with GraphPad Prism version 5·00 (GraphPad, San Diego, CA, USA) using the appropriate statistical tests, as stated in the figure legends.
Results
Detection of CD8+CD28− Treg and subsets
To ensure the correct population of cells was accessed for whole blood analysis, total CD3+CD8+ cells were gated and used in subsequent analysis for the absence of CD28 and any additional marker (Fig. 1a). The relative frequency of CD8+CD28− Treg in RA(MTX) was significantly higher when compared with HC, OA and RA(TNFi) (Fig. 1b). The OA disease control group also showed raised levels of CD8+CD28− Treg when compared with HC. Similarly, subsets expressing CD56 (Fig. 1c) and CD94 (Fig. 1d) were found to be significantly higher in RA(MTX) in comparison with HC, OA and RA(TNFi). No significant correlation was found with the disease activity score or erythrocyte sedimentation rate. A significant positive correlation was found between CD8+CD28− Treg and age in RA(MTX) (r = 0·26; P = 0·042) and RA(TNFi) (r = 0·27; P = 0·042).
Fig. 1.
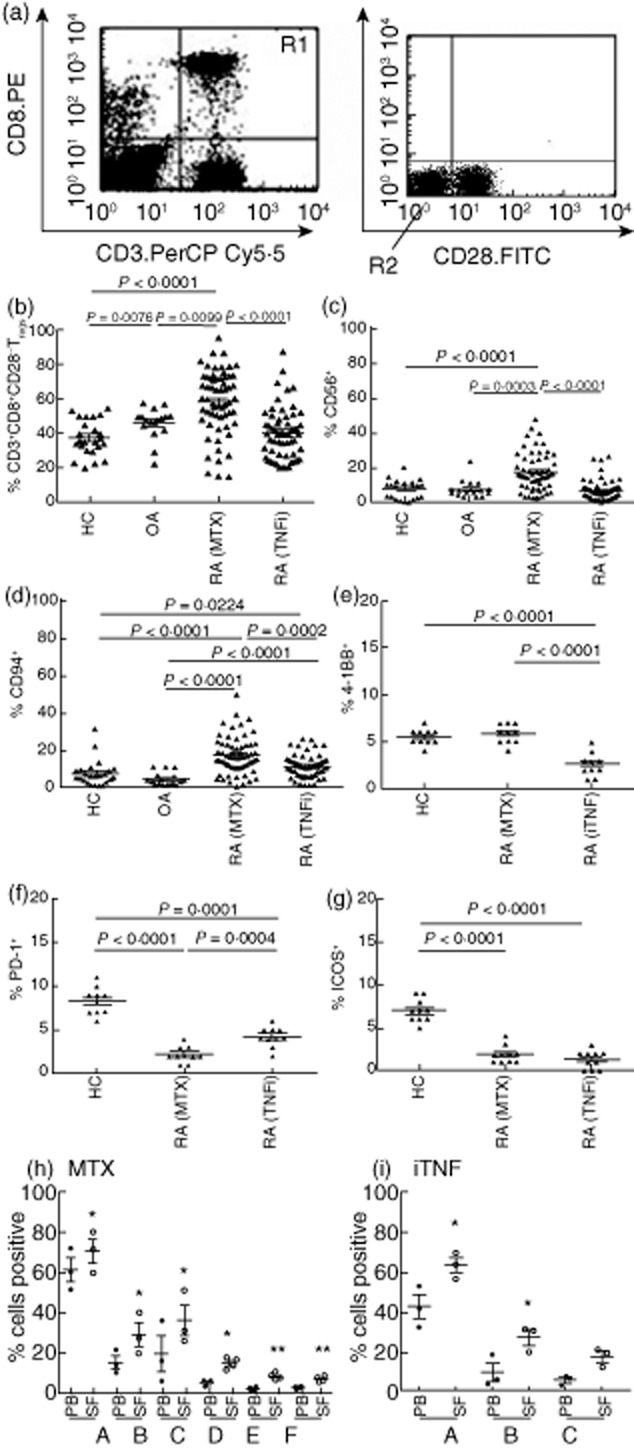
CD8+CD28− regulatory T cells (Treg) in healthy control (HC) and diseased groups. Peripheral blood (PB) samples were stained with antibodies. (a) CD3.peridinin chlorophyll cyanin 5·5 (PerCPCy5·5) and CD8. phycoerythrin (PE)-positive cells were live gated (R1) and analysed for CD28.fluorescein isothiocyanate (FITC)-negative cells (R2). (b) CD8+CD28− Tregs from HC (n = 24), osteoarthritis (OA) patients (n = 17), rheumatoid arthritis (RA) patients treated with methotrexate(MTX); RA(MTX) (n = 60) or tumour necrosis factor (TNF)-α inhibitors RA(TNFi) (n = 57) and subsets co-expressing CD56 (c) or CD94 (d) was determined. Next, peripheral blood mononuclear cells (PBMC) were stimulated with anti-CD3antibodies (1:1000) for 48 h and 4-1BB (e), programmed death 1 (PD-1) (f) or inducible co-stimulator (ICOS) (g) on CD8+CD28− Tregs was examined. In paired PB and synovial fluid (SF) samples, RA (MTX) (n = 3) CD8+CD28− Tregs (h, A), subsets co-expressing CD56 (h, B), CD94 (h, C), 4-1BB (h, D), PD-1 (h, E) or ICOS (h, F) and RA (iTNF) (n = 3) CD8+CD28− Tregs (iA), subsets co-expressing CD56 (iB) or CD94 (iC) were examined. Significant differences were determined using Student's unpaired and paired t-test. *P < 0·04; **P < 0·005.
In parallel with the measurement of CD8+CD28− Treg ex vivo, the ability of these cells to up-regulate expression of the alternative co-stimulatory molecules, 4-1BB, PD-1 and ICOS, was investigated. No expression of these molecules was observed prior to stimulation. Following anti-CD3 antibody stimulation 4-1BB expression was up-regulated on CD8+CD28− Treg at a similar frequency in HC and RA(MTX) groups but expression was reduced significantly in RA(TNFi) (Fig. 1e). In contrast, the up-regulation of PD-1 expression on CD8+CD28− Treg varied between groups, but RA(MTX) expression was reduced significantly compared with both HC and RA(TNFi) (Fig. 1f). The expression of ICOS by CD8+CD28− Treg was found to be significantly lower in both RA(MTX) and RA(TNFi) when compared with HC (Fig. 1g). In addition, although CTLA-4 was detectable in CD4+ cells, there was no expression, intracellular or surface, by the CD8+CD28− Treg subset (data not shown).
Subsequently, the phenotype of CD8+CD28− Treg was examined in paired PBMC and SFMC. The relative frequency of CD8+CD28− Treg was increased significantly in the SF of RA(MTX) (Fig. 1hA) and RA(TNFi) (Fig. 1iA). The co-expression of CD56 (Fig. 1hB) and CD94 (Fig. 1hC) by CD8+CD28− Treg in paired RA(MTX) PBMC and SFMC samples was significantly higher in the SF. In contrast, RA(TNFi) showed significantly increased co-expression of CD56 (Fig. 1iB), but not CD94 expression (Fig. 1iC), by SF CD8+CD28− Treg.
Neither RA(MTX) PB nor SF CD8+CD28− Treg expressed alternative co-stimulatory molecules, 4-1BB, PD-1 or ICOS ex vivo. However, following anti-CD3 stimulation, 4-1BB (Fig. 1hD), PD-1 (Fig. 1hE) and ICOS (Fig. 1hF) were up-regulated on CD8+CD28− Treg and a significantly higher expression by SF Treg was observed.
Deficient CD8+CD28− T function in RA(MTX)
Functional studies of CD8+CD28− Treg showed that HC CD8+CD28− Treg could suppress autologous PBMC proliferation significantly at a 1:1 ratio of CD8+CD28− Treg : PBMC (Fig. 2a). Suppression was dose-dependent, as determined by initial assays. Responder PBMC proliferation was suppressed significantly at 1:1 (PBMC responder, 13 347 ± 2417 cpm versus 1:1, 7164 ± 3535 cpm, P = 0·04) and 0·2:1 (10 759 ± 1496 cpm, P = 0·03). No suppression was observed at the ratio of: 0·1:1 [13 606 ± 1905 cpm, P = not significant (n.s.)]. In contrast, RA(MTX) CD8+CD28− Treg were unable to suppress autologous responder PBMC proliferation (Fig. 2b), although RA(TNFi) CD8+CD28− Treg showed limited but significant suppressor function (Fig. 2c). To ensure that suppression at 1:1 was not due to competition for nutrients or space, two PBMC controls were included: PBMC 1 (2·105 cells/well) and PBMC 2 (1·105 cells/well). To determine if natural killer (NK) cell activity was part of the suppressor mechanism we compared purified subpopulations of CD8+CD28− Treg, free of NK cells and CD8+CD28−CD56+ Treg compared with CD8+CD28− Treg. No significant differences were found between the groups. For example, responder PBMC proliferation (13 347 ± 1209 cpm) was suppressed significantly at a ratio of 1:1 by CD8+CD28−CD16− Treg (9017 ± 854 cpm P = 0·04) and CD8+CD28−CD56+Treg (7164 ± 3535 cpm, P = 0·04). In addition, total cell counts and viability were investigated and no reduction was observed.
Fig. 2.

CD8+CD28− regulatory T cells (Treg) from rheumatoid arthritis (RA) patients treated with methotrexate (RA(MTX) are functionally defective. CD8+CD28– Tregs were isolated by immunomagnetic bead separation from the peripheral blood (PB) of (a) healthy controls (HC) (n = 11), (b) RA(MTX) (n = 6); and (c) RA patients on tumour necrosis factor (TNF)-α inhibitor therapy; RA(TNFi) (n = 12). Cells were co-cultured with autologous responder peripheral blood mononuclear cells (PBMC) in the presence of anti-CD3 antibody. CD8+CD28− Tregs from (d) HC (n = 3), (e) RA(MTX) (n = 3) or (f) RA(TNFi) (n = 3) were co-cultured with autologous CD14+ monocytes in the upper chamber of a transwell with autologous responder PBMC in the lower chamber, or in direct contact at 1:1 and stimulated with anti-CD3 antibody. (g) To determine the effect of a TNF inhibitor in vitro, infliximab (IFX) was added. RA(MTX) CD8+CD28− Tregs were co-cultured with autologous responder peripheral blood mononuclear cells (PBMC) (ratio 1:1) and stimulated with anti-CD3 antibody in the absence or presence of infliximab (IFX) (a representative figure from five experiments is shown). (h) To determine if transforming growth factor (TGF)-β is critical for suppressor function, CD8+CD28− Tregs were co-cultured with autologous responder PBMC (ratio 1:1) stimulated with anti-CD3 antibody in the absence or presence of an anti-TGF-β antibody. Cell proliferation was determined by the uptake of [3H]-thymidine for the final 18 h of culture (mean cpm ± standard error of the mean). Statistical significance in functional assays and transwell experiments was determined by the Student's paired t-test (P < 0·05) and one-way analysis of variance (ANOVA) (*P < 0·04), respectively.
Regulatory function is mainly dependent on soluble mediators
The relative importance of soluble mediators and/or direct cell-contact as a mechanism for the suppressive function of CD8+CD28− Treg was investigated by co-culture of CD8+CD28− Treg separated from the autologous responder PBMC by a semi-permeable membrane TW. The TW contained autologous CD14+ monocytes (MO) to ensured full stimulation of CD8+CD28− T cells by anti-CD3. Parallel cultures contained cells in direct contact. HC (Fig. 2d) and RA(TNFi) (Fig. 2f) CD8+CD28− Treg suppressed responder PBMC proliferation in the presence and absence of a TW; however, no suppression was seen in experiments with RA(MTX) CD8+CD28− Treg (Fig. 2e). It was noted that the degree of suppression in HC and RA(TNFi) cultures tended to be greater in the presence of TW, suggesting that direct cell contact did not enhance the suppressive function of these cells and that soluble mediators were involved.
To establish whether the improved regulatory function of RA(TNFi) CD8+CD28− Treg ex vivo was a result of TNF-α blockade, anti-TNF antibody was added to RA(MTX) cultures. The addition of TNFi significantly improved the RA(MTX) CD8+CD28− Treg suppressive function (Fig. 2g).
To investigate the importance of IL-10 for CD8+CD28− Treg function, neutralizing antibodies were added to the HC functional assays. In the presence of a neutralizing IL-10 antibody, inhibition of the suppressor function was observed in some HC, but this was not consistent. In contrast, in the presence of neutralizing anti-TGF-β antibody, CD8+CD28− T cell suppressor function was reduced significantly (Fig. 2h).
Stimulated RA(MTX) CD8+CD28− Treg secrete IL-10 but fail to up-regulate IL-10R
Because the CD8+CD28− Treg effector mechanism involved soluble mediators, the cytokine production of the cells was examined. IL-2, IL-17 and TNF-α were detected at low levels but showed no detectable difference in concentration between the cultures (data not shown). In contrast, high concentrations of IFN-γ (Fig. 3a) were produced by stimulated CD8+CD28− Treg from all three subject groups, although there appeared to be no additive effect in the 1:1 co-cultures.
Fig. 3.
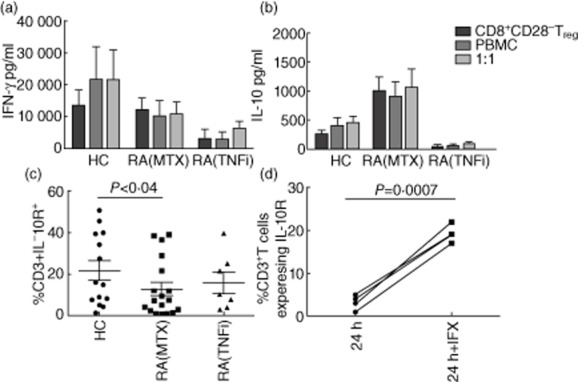
Cytokine profiles of stimulated CD8+CD28− regulatory T cells (Treg), peripheral blood mononuclear cells (PBMC) and co-cultures. CD8+CD28− Tregs were isolated by immunomagnetic bead separation from the PB of healthy controls (HC) (n = 9), RA patients treated with methotrexate, RA(MTX) (n = 9) or tumour necrosis factor (TNF)-α inhibitors, RA(TNFi) (n = 3) and cultured alone or in 1:1 co-culture with autologous PBMC and stimulated with anti-CD3 antibody. Supernatants from cultures were collected at 24 h. (a) Interferon (IFN)-γ and (b) interleukin (IL)-10 production was detected using FlowCytomix beads. IL-10 receptor (IL-10R) expression by CD3+ T cells (c) of PBMC was determined over 48 h culture with anti-CD3. (d) To determine the effect of in vitro addition, of blocking TNF in RA(MTX) cultures, infliximab (IFX) was added in vitro and IL-10R expression on RA(MTX) T cells was determined 24 h after stimulation.
Significantly different concentrations of IL-10 were produced by RA(MTX) CD8+CD28− Treg (1013 ± 231 pg/ml) compared with HC (271 ± 69 pg/ml, P = 0·0072) or RA(TNFi) [RA(TNFi) (49 ± 27 pg/ml, P = 0·041)] (Fig. 3b). As the concentration of cytokine detected in in-vitro cultures is dependent upon the balance between production and use of the cytokine, high concentrations of IL-10, in the dysfunctional RA(MTX) CD8+CD28− Treg cultures following stimulation may be due to abnormal uptake and, thus, lead to deficient downstream signalling by IL-10. On investigation over 48 h, IL-10R expression on RA(MTX) CD3+ T cells was significantly lower than HC T cells (Fig. 3c) and reduced on CD8+CD28− Treg. In-vitro addition of TNFi to RA(MTX) cultures showed a significant increase in IL-10R expression on responder CD3+ T cells from RA(MTX) (Fig. 3d). However, the RA(TNFi) IL-10R expression was only marginally improved and remained lower that that of the HC (Fig. 3c).
RA(MTX) responder cells are less sensitive to CD8+CD28− Treg suppression
To address the question of whether the deficient regulatory function of RA(MTX) CD8+CD28− Treg was due to an intrinsic defect or reduced sensitivity of the responder cells, cross-over co-culture experiments were performed using highly purified T cells from HC and RA(MTX). HC CD8+CD28− Treg suppressed proliferative responses significantly by autologous responder T cell (Tresp) to CD3/CD28 stimulation (Fig. 4a). However, in co-culture with each of two different allogeneic Tresp from RA(MTX) or HC, HC CD8+CD28− Treg failed to suppress proliferation by RA Tresp (RA1 and RA2) while significantly suppressing allogeneic Tresp from two HC (HC1 and HC2) (Fig. 4a).
Fig. 4.
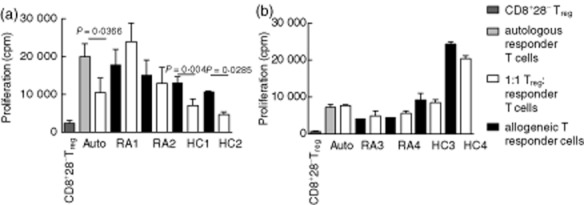
Responder T cells from rheumatoid arthritis (RA) patients treated with methotrexate(MTX) patients are resistant to suppression by CD8+CD28− regulatory T cells (Treg). CD8+CD28− Tregs isolated by immunomagnetic bead separation from peripheral blood (PB) samples from (a) healthy controls (HC) (n = 5) or (b) RA(MTX) (n = 3) were co-cultured with autologous responder T cells, two allogeneic RA(MTX) responder T cells or two allogeneic HC responder T cells. All cultures were stimulated with CD3/CD28 beads. Cell proliferation was determined by the uptake of [3H]-thymidine for the last 18 h of culture (mean ± counts per minute). Statistical significance was determined using the Student's paired t-test.
The reverse experiments showed that RA(MTX) CD8+CD28− Treg failed to suppress proliferation by autologous Tresp, two allogeneic RA Tresp (RA3 and RA4) and two allogeneic HC Tresp (HC3 and HC4) (Fig. 4b).
Discussion
This study has revealed for the first time that despite an in-vivo abundance of CD8+CD28− Treg in RA patients they are functionally deficient. Our studies suggest that their function can be improved by TNFi therapy in vivo, although a longitudinal study is needed for confirmation.
The expansion of the CD8+CD28− Treg population in both the PB or SF of RA(MTX) patients was similar to the reported increase in CD4+ Tregs in RA patients [9]. We confirmed the findings from a previous report [8] that CD8+CD28− Treg numbers correlate with age. Indeed, the expansion may simply highlight the accelerated immune ageing in RA patients resulting in terminally differentiated T cells lacking CD28 expression [10]. In the synovial fluid this growth may be accelerated further by the local cytokine milieu, where high local concentrations of IL-7 and IL-15 promote CD8+CD28− growth [11], while high TNF-α concentrations abrogate CD28 transcription [12].
The inability of ex-vivo RA(MTX) CD8+CD28− Treg to suppress activation of autologous responder cells raised three questions: (i) what is the mechanism of action of this particular Treg; (ii) are RA(MTX) CD8+CD28− capable of suppressing healthy allogeneic responder cells; and (iii) would the addition of TNFi in vitro or in vivo restore their function? TW cultures established that HC and RA(TNFi) CD8+CD28− Treg, in contrast to CD4+CD25+ Treg [13], required little or no direct responder cell contact, suggesting that soluble mediators were the dominant mode of action. IL-10 is a critical mediator for CD8+ Tregs [14]. IL-10 was detected at significantly higher levels in RA(MTX) compared with HC CD8+CD28− Treg cultures, therefore we hypothesized that this may be due partially to defective uptake and signalling by IL-10 in the RA(MTX) cells. Indeed, we show evidence that IL-10R is not up-regulated to the same extent by activated RA(MTX) as it is on HC T cells. This may be exacerbated by the concomitant low expression of ICOS CD8+CD28− Treg in RA(MTX), which stabilizes IL-10R [15]. In contrast, IL-10 levels were reduced compared with HC in anti-CD3 antibody stimulated RA(TNFi) CD3+CD8+CD28−Treg cultures. An explanation for this finding may be the counter-regulation between IL-10 and TNF-α. IL-10 production requires the initial presence of TNF-α but IL-10 regulates the stability of TNF-α mRNA [16]. Inconsistent inhibition of suppression, using neutralizing anti-IL-10, may be due to IL-10 gene polmorphisms that relate to high/low IL-10 production [17]. Less variable results may be obtained by blocking the IL-10 receptor. In addition to IL-10, it has been reported that TGF-β is critical for both CD4+ and CD8+ Treg suppressor function; we show that blocking TGF-β in vitro reduces suppression of responder PBMC proliferation by CD8+ CD28− Treg. Further analysis of this mechanism will be explored in future studies.
In addition, all activated CD8+CD28− Treg cultures produced high levels of IFN-γ similar to that produced by CD4+CD28− T cells [18]. Far from being inflammatory this may be associated with suppressor function, particularly as CD8+ T cells mediating suppression in systemic lupus erythematosus (SLE) [19] and in murine collagen-induced arthritis are dependent upon IFN-γ [20].
While autoimmune diseases have been linked with genetic polymorphisms of co-stimulatory markers [21,22], the functional implications have not yet been fully deciphered. Genetic polymorphism, of course, may compromise not only the function of these molecules but their detection by antibodies. The lack of cell surface CD28 prompted the investigation of the possible expression of alternative co-stimulatory molecules, PD-1, ICOS and 4-1BB, by CD8+CD28− Treg. The expression of all these molecules was higher on RA SF CD8+CD28− cells compared with paired PB Treg, perhaps reflecting the higher activation status of the SF cells. The SF cytokine milieu also contains high local concentrations of IL-15 and IL-12 which down-regulate CD28 but enhance 4-1BB, ICOS and PD-1 expression by CD8+ T cells and increase CD8+ cell survival [23]. CD4+CD25+ Treg display attenuated regulatory function following 4-1BB expression [24]. As 4-1BB expression was reduced in RA(TNFi), this raises the question as to whether or not it might be a component of the improved suppressor function by CD8+CD28− Treg following therapy in RA(TNFi) patients. The ability to suppress T cell responses may therefore be a balance between the pro-proliferative drive of 4-1BB and the inhibitory effect of other mediators, such as PD-1.
Overall, a relatively low expression of PD-1 and ICOS was shown by all CD8+CD28− Treg samples. Nevertheless, PD-1 has been linked positively to CD8+CD28− Treg with suppressor function in lupus-prone mice [25]. Therefore, it was notable that PD-1 expression by RA(TNFi) was increased compared with RA(MTX), although still below healthy control levels.
For further insight into the defective CD8+CD28− Treg in RA, cells were used in cross-over co-culture experiments between the RA(MTX) and HC subjects. RA(MTX) CD8+CD28− Treg remained unable to suppress allogeneic healthy or RA responder cells, whereas HC CD8+CD28− cells suppressed allogeneic HC responder cells but not RA(MTX) responder T cells. This finding complements the fact that responder T cells had reduced sensitivity to CD4+CD25hi Tregs in active SLE [26] and type 1 diabetes patients [27], suggesting that in autoimmune diseases Treg activity is hampered by both defective Treg function and the relative insensitivity of the responder cells. The effect of TNF inhibitor on the ex-vivo phenotype and function of CD8+CD28− cells, such as the increase in IL-10R expression on RA(MTX) T cells, suggests strongly that these cells are only temporarily incapacitated by TNF-α and when this is removed from the environment the activity appears to return to normal. However, RA(TNFi) expression of IL-10R remained lower than normal HC expression and suggests that other mediators are involved. Continuing these studies, the role of IL-10 and TGF-β is under further investigation. Longitudinal studies will be performed to address the effect of therapy on CD8+CD28− Treg. In addition, signalling defects in HC, RA(MTX) and RA(TNFi) CD8+CD28− Treg will be investigated.
In conclusion, this study places CD8+CD28− Treg alongside CD4+CD25hi Treg in the network of immune regulation in RA and highlights the importance of understanding impaired responsiveness to regulation and the beneficial effect of TNF inhibitor therapy at the cellular level.
Acknowledgments
The authors would like to thank all the clinical staff at Guy's Hospital and King's College Hospital, London and all patients who donated blood. They would also like to acknowledge the help of Dr L. Taams for a critical review of the manuscript. This work was supported by a Medical Research Council (UK) PhD studentship for S. Ceeraz.
Disclosure
The authors have no financial or commercial conflicts of interest.
References
- 1.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 2.Ponchel F, Morgan AW, Bingham SJ, et al. Dysregulated lymphocyte proliferation and differentiation in patients with rheumatoid arthritis. Blood. 2002;100:4550–4556. doi: 10.1182/blood-2002-03-0671. [DOI] [PubMed] [Google Scholar]
- 3.Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davila E, Kang YM, Park YW, et al. Cell-based immunotherapy with suppressor CD8+ T cells in rheumatoid arthritis. J Immunol. 2005;174:7292–7301. doi: 10.4049/jimmunol.174.11.7292. [DOI] [PubMed] [Google Scholar]
- 5.Filaci G, Fenoglio D, Fravega M, et al. CD8+ CD28- T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J Immunol. 2007;179:4323–4334. doi: 10.4049/jimmunol.179.7.4323. [DOI] [PubMed] [Google Scholar]
- 6.Colovai AI, Mirza M, Vlad G, et al. Regulatory CD8+CD28– T cells in heart transplant recipients. Hum Immunol. 2003;64:31–37. doi: 10.1016/s0198-8859(02)00742-5. [DOI] [PubMed] [Google Scholar]
- 7.Tulunay A, Yavuz S, Direskeneli H, Eksioglu-Demiralp E. CD8+CD28–, suppressive T cells in systemic lupus erythematosus. Lupus. 2008;17:630–637. doi: 10.1177/0961203308089400. [DOI] [PubMed] [Google Scholar]
- 8.Fagnoni FF, Vescovini R, Mazzola M, et al. Expansion of cytotoxic CD8+ CD28– T cells in healthy ageing people, including centenarians. Immunology. 1996;88:501–507. doi: 10.1046/j.1365-2567.1996.d01-689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004;50:2775–2785. doi: 10.1002/art.20499. [DOI] [PubMed] [Google Scholar]
- 10.Goronzy JJ, Shao L, Weyand CM. Immune aging and rheumatoid arthritis. Rheum Dis Clin North Am. 2010;36:297–310. doi: 10.1016/j.rdc.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Churchman SM, Ponchel F. Interleukin-7 in rheumatoid arthritis. Rheumatology. 2008;47:753–759. doi: 10.1093/rheumatology/ken053. [DOI] [PubMed] [Google Scholar]
- 12.Bryl E, Vallejo AN, Weyand CM, Goronzy JJ. Down-regulation of CD28 expression by TNF-alpha. J Immunol. 2001;167:3231–3238. doi: 10.4049/jimmunol.167.6.3231. [DOI] [PubMed] [Google Scholar]
- 13.Leipe J, Skapenko A, Lipsky PE, Schulze-Koops H. Regulatory T cells in rheumatoid arthritis. Arthritis Res Ther. 2005;7:S4–14. doi: 10.1186/ar1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Endharti AT, Rifa IM, Shi Z, et al. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J Immunol. 2005;175:7093–7097. doi: 10.4049/jimmunol.175.11.7093. [DOI] [PubMed] [Google Scholar]
- 15.Tuettenberg A, Huter E, Hubo M, et al. The role of ICOS in directing T cell responses: ICOS-dependent induction of T cell anergy by tolerogenic dendritic cells. J Immunol. 2009;182:3349–3356. doi: 10.4049/jimmunol.0802733. [DOI] [PubMed] [Google Scholar]
- 16.Rajasingh J, Bord E, Luedemann C, et al. IL-10-induced TNF-alpha mRNA destabilization is mediated via IL-10 suppression of p38 MAP kinase activation and inhibition of HuR expression. FASEB J. 2006;20:2112–2114. doi: 10.1096/fj.06-6084fje. [DOI] [PubMed] [Google Scholar]
- 17.Huizinga TW, Keijsers V, Yanni G, et al. Are differences in interleukin 10 production associated with joint damage? Rheumatology. 2000;39:1180–1188. doi: 10.1093/rheumatology/39.11.1180. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7– CD28– T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996;97:2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filaci G, Bacilieri S, Fravega M, et al. Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J Immunol. 2001;166:6452–6457. doi: 10.4049/jimmunol.166.10.6452. [DOI] [PubMed] [Google Scholar]
- 20.Seo SK, Choi JH, Kim YH, et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088–1094. doi: 10.1038/nm1107. [DOI] [PubMed] [Google Scholar]
- 21.Kim YO, Kim HJ, Kim SK, Chung JH, Hong SJ. Association of the CD28/CTLA4/ICOS polymorphisms with susceptibility to rheumatoid arthritis. Clin Chem Lab Med. 2010;48:345–353. doi: 10.1515/CCLM.2010.074. [DOI] [PubMed] [Google Scholar]
- 22.Kong EK, Prokunina-Olsson L, Wong WH, et al. A new haplotype of PDCD1 is associated with rheumatoid arthritis in Hong Kong Chinese. Arthritis Rheum. 2005;52:1058–1062. doi: 10.1002/art.20966. [DOI] [PubMed] [Google Scholar]
- 23.Pulle G, Vidric M, Watts TH. IL-15-dependent induction of 4-1BB promotes antigen-independent CD8 memory T cell survival. J Immunol. 2006;176:2739–2748. doi: 10.4049/jimmunol.176.5.2739. [DOI] [PubMed] [Google Scholar]
- 24.Choi BK, Bae JS, Choi EM, et al. 4-1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. J Leukoc Biol. 2004;75:785–791. doi: 10.1189/jlb.1003491. [DOI] [PubMed] [Google Scholar]
- 25.Singh RP, La Cava A, Hahn BH. pConsensus peptide induces tolerogenic CD8+ T cells in lupus-prone (NZB × NZW)F1 mice by differentially regulating Foxp3 and PD1 molecules. J Immunol. 2008;180:2069–2080. doi: 10.4049/jimmunol.180.4.2069. [DOI] [PubMed] [Google Scholar]
- 26.Venigalla RK, Tretter T, Krienke S, et al. Reduced CD4+,CD25– T cell sensitivity to the suppressive function of CD4+,CD25high,CD127–/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum. 2008;58:2120–2130. doi: 10.1002/art.23556. [DOI] [PubMed] [Google Scholar]
- 27.Lawson JM, Tremble J, Dayan C, et al. Increased resistance to CD4+CD25hi regulatory T cell-mediated suppression in patients with type 1 diabetes. Clin Exp Immunol. 2008;154:353–359. doi: 10.1111/j.1365-2249.2008.03810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]