

Abstract
Human monocyte-derived dendritic cells (DC) (MoDC) are utilized for immunotherapy. However, in-vitro immunological effects are often not mirrored in vivo. We studied the tissue-homing potential of MoDC. Circulating monocytes and DC expressed different tissue-homing markers and, during in-vitro development of MoDC, homing marker expression was lost resulting in a ‘homeless’ phenotype. Retinoic acid (RA) induced gut-homing markers (β7 and CCR9) and a regulatory phenotype and function [decreased human leucocyte antigen D-related (HLA-DR) and increased ILT3 and fluorescein isothiocyanate (FITC-dextran uptake) in MoDC]. RA-MoDC were less stimulatory and primed conditioned T cells with a gut-homing profile (β7+CLA−). Unlike the normal intestinal microenvironment, that from inflamed colon of ulcerative colitis (UC) patients did not induce regulatory properties in MoDC. However, RA-MoDC maintained their regulatory gut-specific properties even in the presence of UC microenvironment. Therefore, MoDC may be ineffectual for immunotherapy because they lack tissue-homing and tissue-imprinting specificity. However, MoDC rehabilitation with gut-homing potential by RA could be useful in promoting immunotherapy in pathologies such as UC.
Keywords: dendritic cell, homing markers, immunotherapy, retinoic acid, ulcerative colitis
Introduction
Dendritic cells (DC) are unique in their ability to generate primary immune responses, determining both the nature and type of immune responses [1,2]. Manipulation of DC phenotype and function are promising strategies to perform immunotherapy. However, due their low numbers in human tissues, ‘naturally occurring’ human DC are not the usual source of DC for human studies. Given their easy accessibility and large numbers, monocyte-derived DC (MoDC) generated from fresh circulating monocytes during 5–7-day in-vitro culture are the standard human DC model [3]. They may be reinjected in vivo to elicit their function. This strategy has been proposed for individualized immunotherapy in several pathologies [4–7]. Generation of tolerogenic DC may help to restore homeostasis lost in the gastrointestinal tract in patients with inflammatory bowel disease (IBD) [7–10]. However, despite their in-vitro efficacy, DC immunotherapy is far from ideal when translated to the clinics. Factors affecting their success include preparation, source and dose of DC, route of administration, antigen-pulsing strategy, maturation status and even the selected antigen itself [5]. However, we consider that there is an essential missing factor which may explain the lack of efficacy in vivo: the compartmentalization of immune responses through DC homing marker expression and migration.
Immune responses are elicited in a tissue-/organ-specific way. Immune compartmentalization is controlled by DC which imprint tissue specific homing markers on stimulated T cells, restricting their migration to target tissues [11–14]. The requirement for tissue specificity for inducing inflammation in the human gut has been demonstrated in patients with Crohn's disease where, despite systemic lymphocyte sensitization to antigens encountered through the gut, inflammation was induced only by challenge with these antigens in the gut, and not by skin challenge [15]. Therefore, appropriate tissue specificity is likely to be required for immunotherapy. However, information about the expression of homing markers on human DC themselves is scarce.
We demonstrated recently that human peripheral circulating DC display a multi-homing profile which may be necessary for them to populate different tissues. However, within skin and gut, DC express tissue-specific markers due to the effect of the surrounding microenvironment [16]. Thus, multi-homing circulating human DC can acquire a phenotype characteristic of gut tissue-specific DC when cultured in the normal tissue microenvironment in vitro [17–20]. Retinoic acid (RA), the active form of vitamin A, mediates several tolerogenic properties of intestinal DC [14,21–23], including the acquisition of gut-homing properties on human DC and their capacity to imprint gut-homing properties on stimulated T cells [16]. However, when human blood DC are cultured in vitro in the absence of tissue factors and/or RA, DC lost expression of homing markers [16].
We hypothesized that MoDC may also lose homing-marker expression upon in-vitro culture during development. Therefore, in this study we have (i) characterized the homing profile of human circulating monocytes and subsequent MoDC in comparison with those of fresh blood DC; and (ii) studied whether we could generate stable gut-specific regulatory MoDC. Our findings revealed that human MoDC displayed a homeless phenotype, but RA rehabilitated them with a regulatory gut-homing profile which was maintained even when exposed to an inflamed microenvironment from ulcerative colitis (UC) patients. Our findings may open new doors for performing targeted tissue-specific immunotherapy.
Materials and methods
Blood samples and MoDC
Human peripheral blood was collected from healthy volunteers with no known autoimmune or inflammatory diseases, allergies or malignancies, following informed consent. Peripheral blood mononuclear cells (PBMC) were obtained by centrifugation over Ficoll-Paque Plus (Amersham Biosciences, Chalfont St Giles, UK). Monocytes were obtained by positive selection of CD14+ cells from PBMC [suspended in miniMACS buffer: phosphate-buffered saline (PBS) containing 5% bovine serum albumin (BSA) and 2 mM ethylenediamine tetraacetic acid (EDTA)] with immunomagnetic beads (Miltenyi Biotech, Bisley, UK), following the manufacturer's instructions. Human MoDC were generated following 5-day culture (37°C, 5% CO2, high humidity) of monocytes (0·5 million/ml/well) in 24-well microtitre plates (Becton Dickinson, Oxford, UK) in complete medium [Dutch modified RPMI-1640 (Sigma-Aldrich, Dorset, UK) containing 100 u/ml penicillin/streptomycin, 2 mM L-glutamine and 10% fetal calf serum (FCS) (TCS Cellworks, Buckingham, UK)] supplemented with interleukin (IL)-4 (50 U/ml; Promega, Southampton, UK) and granulocyte–macrophage colony-stimulating factor (GM-CSF) (0·1 μg/ml; Promega) for 5 days. On day 5, MoDC were pooled, washed and cultured for 24 h (0·5 million/ml) in complete medium in the presence of different doses (10−6 M, 10−7 M, 10−8 M) of all-trans-retinoic acid (RA) (Sigma-Aldrich), lipopolysaccharide (LPS) (0·1 μg/ml; Sigma-Aldrich) or preconditioned with 10−6 M RA for 2 h before being cultured subsequently for 22 h with LPS after gentle RA-washing.
Colonic samples and UC biopsy conditioning
Colonic biopsies from inflamed areas of three UC patients were collected in ice-chilled complete medium and cultured within 1 h in complete medium in 24-well culture dishes (one biopsy/well/ml) for 24 h, as explained previously [16]. Complete medium (1 ml) was added to empty wells and incubated for use as a basal condition. Media were centrifuged and either medium-only or cell-free UC colonic biopsy supernatants (UC-SN) were used to condition MoDC for 24 h. All experiments used freshly obtained cultured supernatants or medium. MoDC were also pulsed (t = 2 h) with and without RA (10−6 M) and washed twice before being conditioned.
Antibody labelling
Monoclonal antibodies with the following specificities, conjugations and clones were used: β7 [either phycoerythin (PE) or PE-cyanin 5 (Cy5)] (FIB504), CCR5-fluorescein isothiocyanate (FITC) (2D7/CCR5), CD3-PE-Cy5 (UCHT1), cutaneous lymphocyte-associated antigen (CLA) (either FITC or biotin) (HECA-452), human leucocyte antigen D-related (HLA-DR) (either FITC or PE-Cy5 (G46-6), interferon (IFN)-γ-allophycocyanin (APC) (XMG1·2) and streptavidin-APC were purchased from BD Biosciences (Oxford, UK); CCR4-APC (205410), CCR7-PE (150503), CCR8-APC (191704), CCR9-PE (112509), CCR10-APC (314315) and immunoglobulin-like transcript (ILT)-3 (PE) (293623) were purchased from R&D Systems (Abingdon, UK); CD14 (PE-Cy5) (TÜK4) and IL-10-PE (JES3-9D7) were purchased from AbD Serotec (Oxford, UK). Appropriate isotype-matched control antibodies were purchased from the same manufacturers. Cells were labelled in PBS containing 1 mM EDTA and 0·02% sodium azide [fluorescence activated cell sorter (FACS) buffer]. Labelling was performed on ice and in the dark for 20 min. Cells were washed twice in FACS buffer, fixed with 1% paraformaldehyde in 0·85% saline and stored at 4°C prior to acquisition on the flow cytometer (within 48 h). For intracellular staining, cells were fixed with Leucoperm A following surface staining, and permeabilized with Leucoperm B before adding antibodies for intracellular labelling. After incubation cells were washed in FACS buffer, fixed and acquired on the flow cytometer.
Phagocytic activity of DC
Phagocytic activity of MoDC was determined by uptake of FITC-dextran (molecular weight 40 kDa) (Sigma-Aldrich), following the manufacturer's instructions. Briefly, after conditioning of MoDC with different doses of RA, MoDC were incubated with 1 mg/ml of FITC-dextran for 2 h either at 37°C or on ice (internal control), and then washed twice in PBS. Flow cytometry acquisition was performed subsequently.
Flow cytometry and data analysis
Data were acquired on a FACSCalibur cytometer (BD Biosciences) and analysed using WinList version 5·0 software (Verity, ME, USA). The proportion of cells positive for a given marker was determined by reference to staining with an isotype-matched control antibody. For single-parameter analysis, WinList was used to subtract the normal cumulative histogram for isotype control staining from a similar histogram of staining with the test antibody using the superenhanced Dmax (SED) normalized subtraction [24] to obtain the percentage of positive cells for a given marker and the intensity ratio (IR) of the staining of cells identified as positive by the subtraction compared with that of the isotype.
Proliferation assays
T cells (>95% pure) were obtained from freshly isolated PBMC after depletion of CD14-, CD19- and HLA-DR-positive cells with immunomagnetic beads (Miltenyi Biotech, Bisley, UK). Alternatively, T cells were also depleted for β7 expression using β7 APC-labelled antibody (BD Biosciences) and anti-APC microbeads (Miltenyi Biotech) (>95% CD3+β7−). In all cases, T cells were labelled with 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen Ltd, Paisley, UK), following the manufacturer's instructions. CFSE-labelled T cells (400 000) were incubated for 5 days in round-bottomed 96-well microtitre plates (Becton Dickinson) with different concentrations of allogeneic MoDC (0, 1, 2 or 3% of T cells). Cells were harvested on day 5, and the proportion and phenotype of stimulated T cells (CFSElow) were quantified by flow cytometry within total CD3+ cells in the lymphocyte gate.
Statistical analyses
One- or two-way paired analysis of variance (anova) corrected for multiple comparisons, or paired t-test, were applied as stated in the figure legends. The level of significance was fixed at P < 0·05.
Results
CCR9 and CCR7 were the predominant homing markers on circulating monocytes
Human circulating monocytes were identified as CD14+ cells by flow cytometry (Fig. 1a). Homing-marker expression was determined by subtraction of isotype control staining from the corresponding test staining (Fig. 1b) to avoid any subjectivity in interpretation of the total percentage of cells which were positive for a given marker. No gender differences in any of the assayed homing markers (12 females and eight males) were detected (data not shown).
Fig. 1.
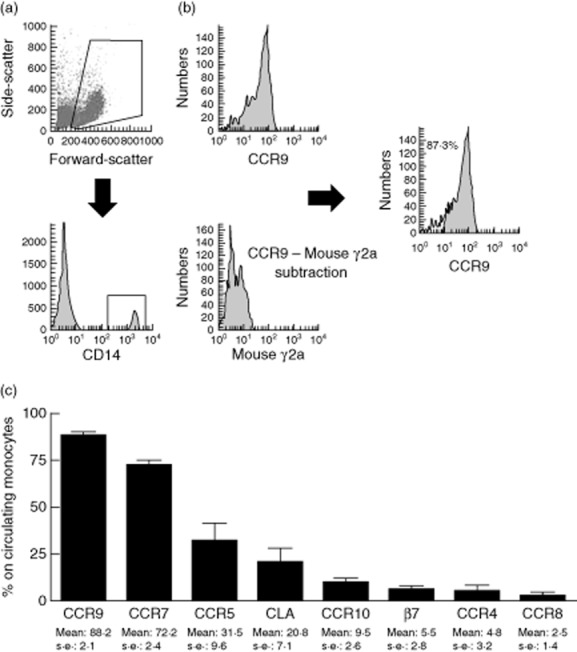
Homing profile of circulating monocytes. (a) Identification of monocytes from peripheral blood mononuclear cells according to the forward- and side-scatter and subsequent CD14 expression. (b) Isotype control staining histogram was subtracted from the respective test staining for homing markers on monocytes (CCR9 in this example). Closed histogram after subtraction represents the proportion of positive cells assessed by superenhanced Dmax (SED) normalized subtraction from the isotype histrogram. Homing profile of circulating monocytes in healthy controls (mean ± standard error of the mean; n = 20) is displayed in panel (c).
Human circulating DC displayed a multi-homing profile characterized mainly by co-expression of gut-homing β7 integrin and skin-homing CLA molecule, as described previously [16]. Conversely, gut- and lymph node-homing markers [chemokine receptors (CCR)9 and CCR7, respectively] were the predominant homing markers expressed by circulating monocytes from healthy controls (Fig. 1c). CCR5 (involved in migration to sites of inflammation) and skin-homing molecule CLA were also expressed on some circulating monocytes. CCR10 (associated with migration to skin and mucosal sites), β7 integrin (gut-homing), CCR4 and CCR8 (both skin-homing) were virtually absent on circulating monocytes (Fig. 1c).
Monocytes lost homing marker expression upon differentiation into MoDC
Human monocytes were cultured for 5 days in a classic in-vitro culture in the presence of IL-4 and GM-CSF to generate MoDC as assessed by up-regulated HLA-DR and loss of expression of the monocyte marker CD14 with time (Fig. 2). During in-vitro differentiation, monocytes lost expression of all assayed homing markers, leaving day 5 MoDC with a ‘homeless’ profile (Fig. 2).
Fig. 2.

Monocyte-derived dendritic cells do not express homing markers. Human monocytes differentiated into monocyte-derived dendritic cells (MoDC) in the presence of interleukin (IL)-4 and granulocyte–macrophage colony-stimulating factor (GM-CSF), as assessed by up-regulation of human leucocyte antigen D-related (HLA-DR) and loss of CD14 expression. Results are displayed in each histogram as percentage of expression or as intensity ratio (IR). Expression of homing markers was lost during culture as monocytes differentiated into MoDC, rendering them devoid of homing marker expression. Monocytes were obtained by positive selection of CD14+ cells from peripheral blood mononuclear cells (PBMC) (>99% purity) and their homing marker expression assessed on all cells in the lymphogate. Results are representative of two independent experiments performed in triplicate with similar results.
RA induced an immature, gut-homing phenotype on MoDC
We next studied whether RA could induce a gut-homing profile on MoDC. None of the assayed RA concentrations had any effect on cell survival, as determined by total cell numbers (data not shown).
When cultured with different doses of RA, MoDC acquired expression of both β7 and CCR9 gut-homing markers in a dose-dependent manner (Fig. 3a). We also assessed the maturation status of RA-conditioned cells. Because all MoDC expressed HLA-DR and the inhibitory receptor ILT3 (data not shown), these markers were studied by IR compared to the isotype control. Expression of both markers increased in a dose-dependent fashion following RA treatment (Fig. 3b), while RA also increased the phagocytic capacity of MoDC, a characteristic of immature DC for sampling antigen (Fig. 3b).
Fig. 3.
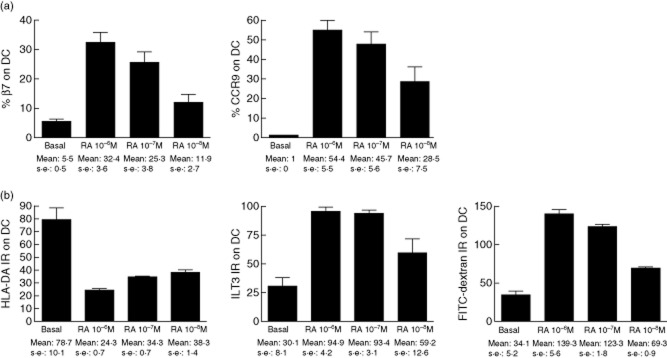
Retinoic acid induces an immature gut-homing profile on monocyte-derived dendritic cells. Monocyte-derived dendritic cells (DC) were cultured for 24 h in the presence of different doses of retinoic acid (RA). Expression of (a) gut-homing markers β7 and CCR9 and (b) intensity ratio (IR) human leucocyte antigen D-related (HLA-DR) and immunoglobulin-like transcript (ILT)-3 (as all DC expressed those markers) and update of fluorescein isothiocyanate (FITC)-dextran were assessed by flow cytometry. Results are displayed as mean ± standard error of the mean of one experiment (assayed in triplicate) representative of three independent experiments.
RA-conditioned MoDC had decreased T cell stimulatory capacity and primed T cells with a gut-homing profile
The stimulatory capacity of MoDC for allogeneic CFSE-labelled T cells and the homing profile of stimulated T cells were also studied. Consistent with the induced immature phenotype (Fig. 3b), RA decreased the stimulatory capacity of MoDC in a dose-dependent manner (Fig. 4a). We also studied the homing-imprinting property of MoDC on stimulated T cells (CFSElow). MoDC-stimulated T cells (CFSElow) expressed CLA only, β7 only or were double-positive or double-negative for both homing markers. However, RA-conditioned MoDC, which had acquired a gut-homing phenotype (Fig. 3a), also induced a gut-homing phenotype on stimulated T cells via decreased expression of CLA, rendering most of the stimulated T cells potentially gut-homing with a β7+CLA− phenotype (Fig. 4b). T cells stimulated by RA-conditioned MoDC were mainly β7+CLA−, so we also studied the intensity of β7 expression via IR, proving that β7 expression was also increased upon stimulation with RA-treated MoDC (Fig. 4c).
Fig. 4.
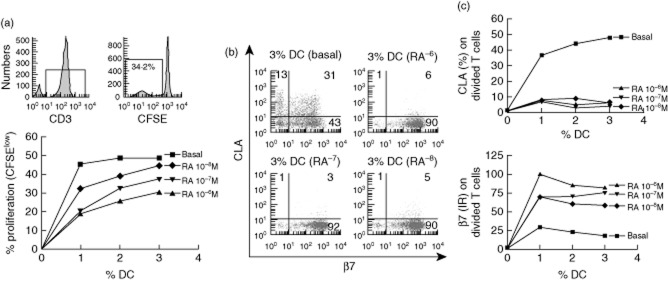
Retinoic acid reduces dendritic cell stimulatory capacity and increases their gut-homing imprinting capacity on stimulated T cells. (a) T cell proliferation following mixed lymphocyte reaction (MLR) of 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE)-labelled T cells cultured for 5 days in the presence of different doses (1, 2 and 3%) of monocyte-derived dendritic cells (DC), conditioned previously with different doses of retinoic acid (RA) or in complete medium only (basal). T cells were identified as CD3+ lymphocytes and the percentage of dividing T cells referred to the total number of cultured cells determined as CFSElow T cells. (b) β7/CLA dot-plots of dividing T cells cultured with 3% allogeneic DC and (c) β7/CLA expression on stimulated T cells against the percentage of conditioned DC.
RA-MoDC maintain their decreased stimulatory and β7 T cell imprinting capacity in the presence of LPS
We next studied if the decreased stimulatory capacity of RA-MoDC (Fig. 4a) was maintained in the presence of a proinflammatory stimulus such as LPS. Also, as β7 was expressed on both resting T cells and most dividing T cells (irrespective of MoDC conditioning), we determined whether β7 expression was imprinted by MoDC or due to the selective survival of β7+ T cells.
β7− T cells were enriched from total PBMC (Fig. 5a) and stimulated with MoDC that had been conditioned previously for 24 h with/without RA. Monocyte-derived DC had also been conditioned with LPS (0·1μg/ml) for 24 h with/without previous pulsing with 10−6 M RA for 2 h. Pretreatment of MoDC with RA abrogated the increased stimulatory capacity induced by LPS (Fig. 5b). RA-conditioned DC also abrogated CLA expression on β7− T cells and induced β7 expression, even following MoDC LPS conditioning after RA treatment (Fig. 5c,d). Thus, RA specifically confers the ability of DC to prime gut-homing T cells.
Fig. 5.
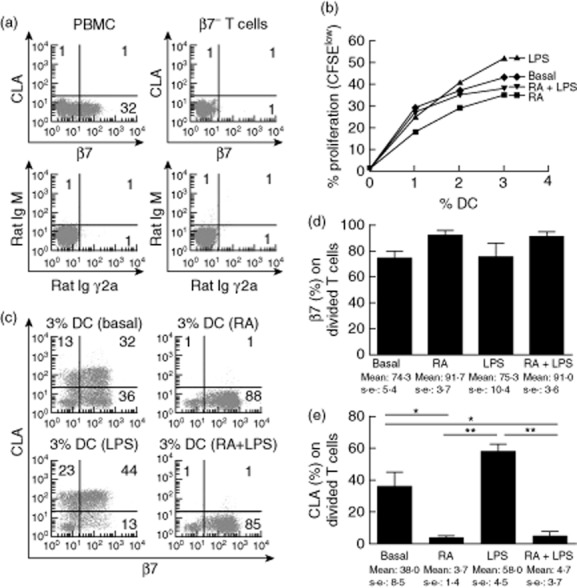
Retinoic acid (RA) conditioned dendritic cells (DC) maintain their decreased stimulatory and gut-homing imprinting capacity on β7− T cells in the presence of lipopolysaccharide (LPS). (a) β7-negative T cells were enriched from total peripheral blood mononuclear cells (PBMC), as confirmed when compared with the isotypes. (b) β7-negative T cells were used to assess the stimulatory capacity and (c) homing imprinting capacity of DC conditioned previously with RA (24 h, 10−6 M), LPS (24 h, 0·1 μg/ml), RA (2 h, 10−6 M) and subsequent LPS (22 h 0·1 μg/ml) or in complete medium (basal). β7 and cutaneous lymphocyte-associated antigen (CLA) expression were determined on stimulated [5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE)low] T cells. (d) One-way repeated-measures analysis of variance (anova) corrected for multiple comparisons was applied on pooled data from three independent experiments. P-value below 0·05 was considered statistically significant; *P < 0·05; **P < 0·01.
RA-MoDC maintained their gut-homing, immature phenotype in the presence of colonic biopsy supernatants from UC patients
The ability of RA to induce gut-homing and gut-imprinting properties on MoDC may enable their use in immunotherapy strategies to treat inflammatory pathologies of the gastrointestinal tract, such as UC. However, DC phenotype and function can be altered by the tissue microenvironment [25]. Thus, although RA-MoDC maintained their regulatory capacity even in the presence of LPS (Fig. 5b), once DC enter the inflamed mucosal sites in vivo they could potentially acquire proinflammatory properties and even contribute to the ongoing inflammation [26,27]. Supporting this effect of the inflamed microenvironment on DC, when human blood-enriched DC are exposed colonic microenvironments from healthy controls [16] or non-inflamed areas from UC patients [28] they acquire an immature phenotype. However, when conditioned with inflamed areas from UC patients, these properties are not acquired. In this case, DC do not decrease their stimulatory capacity and, furthermore, DC acquire an increased capacity to prime responding T cells with a skin-homing profile [28].
To address the effect of the UC microenvironment on MoDC, cells – previously pulsed with and without 10−6 M of RA – were conditioned with supernatants from colonic biopsies of UC patients (inflamed areas; UC-SN). Similar to blood-enriched DC [28], MoDC conditioned with UC-SN did not differ in their stimulatory capacity from basal MoDC (Fig. 6a). However, when MoDC had been pulsed previously with 10−6 M RA, their stimulatory capacity remained decreased compared with both basal (P < 0·05 at 3%) and no-RA supplemented with UC-SN MoDC (P < 0·05 at 2% and P < 0·01 at 3% (Fig. 6a). RA-treated MoDC retained their capacity to induce a gut-homing profile on conditioned T cells following UC-SN conditioning with virtually none of the stimulated T cells expressing CLA, unlike those MoDC conditioned with UC-SN without previous RA pulsing (Fig. 6b,c).
Fig. 6.

Dendritic cells (DC) conditioned with retinoic acid (RA) maintain their homeostatic, gut-specific properties in the presence of a proinflammatory microenvironment. (a) Monocyte-derived DC were conditioned with culture supernatants from inflamed intestinal biopsies of ulcerative colitis patients [(UC)-supernatants (SN)] with and without previous conditioning with RA and compared to untreated (basal) DC. T cell proliferation following mixed lymphocyte reaction (MLR) of carboxyfluorescein diacetate succinimidyl ester (CFSE)-labelled T cells with allogeneic DC was determined as explained in Fig. 4a. (b) Gut-homing (β7) and skin-homing [cutaneous lymphocyte-associated antigen (CLA)] markers were assessed on resting T cells (unstimulated) and on T cells (CFSElow) stimulated with DC preconditioned with medium only, UC-SN or RA and UC-SN. (c) Pooled data of tissue-homing profile of stimulated T cells. (d) Intracellular production of interleukin (IL)-10 and interferon (IFN)-γ by stimulated T cells following MLR and (e) pooled data of cytokine production by stimulated T cells. Graphs show the mean ± standard error of the mean of three independent experiments. Two-way anova (a) and paired t-test (c,e) were applied. P-value below 0·05 was considered statistically significant. *P < 0·05; **P < 0·01.
Finally, we studied the cytokine profile of stimulated T cells. Pulsing MoDC with RA prior to UC-SN conditioning enabled MoDC to stimulate T cells with no proinflammatory cytokine profile; the T cells had reduced IFN-γ production and increased IL-10 production compared with T cells stimulated by MoDC conditioned with UC-SN only (Fig. 6d,e). Therefore, RA-conditioned MoDC exposed to an inflamed microenvironment showed no evidence of reversion towards a proinflammatory phenotype.
Discussion
Human circulating DC display a multi-homing β7+CLA+ profile [16] which was not shared by monocytes where CCR9 and CCR7 were the main homing markers. The differences between homing properties of DC and monocytes may be determinants of their different tissue distributions. Monocytes differentiated in vitro in the presence of IL-4 and GM-CSF lost expression of their homing markers and failed to express those found on normal circulating DC, rendering MoDC with a homeless phenotype. The complete loss of homing markers studied in MoDC contrasts with the multi-homing potential of circulating DC [16]. The homeless phenotype of MoDC is likely to indicate incapacity to home appropriately to skin or mucosal surfaces by these in-vitro-generated DC. MoDC also failed to induce any tissue-specific homing profile of stimulated T cells as opposed to tissue-resident DC [16]. This blanket loss in homing potential may limit the in-vivo usefulness of these cells for immunotherapy.
Human circulating monocytes and DC have different immunological properties but also different homing marker expression. Although both populations have ‘mixed’ or multi-homing profiles, the homing markers that they express are different. Circulating DC are mainly β7+CLA+ [16] (gut- and skin-homing). In contrast, both homing markers are hardly expressed by circulating monocytes, which are mainly CCR7+CCR9+ (lymph node- and small bowel-homing markers) (Fig. 1). These differences might account for different tissue distributions in vivo. During in-vitro culture, in the absence of external factors monocytes lose their homing marker expression (Fig. 2), rendering MoDC with a homeless phenotype different from that of circulating DC. We cannot exclude expression of other homing markers by MoDC. However, it is significant that all studied homing markers were lost in time following in-vitro differentiation to MoDC and that similar loss of homing markers has been described in cultured blood DC [16]. Therefore, the loss of homing markers following in-vitro culture in the absence of tissue and/or homing-inducing factors may be a general mechanism in antigen-presenting cells. Utilizing DC as therapeutic agents is effective in autoimmune and inflammatory diseases, and DC are now even being used to enhance survival rates of solid organ transplants [5,7]. Practical issues of the small numbers of DC obtainable from tissues predicate the use of MoDC for immunotherapy in humans. However, we demonstrate here that, unlike human blood and/or tissue DC, MoDC are devoid of homing marker expression. Thus, when reinjected in vivo into patients the lack of homing marker expression may abrogate their trafficking into effector sites, providing an explanation for the limited success of DC immunotherapy strategies in vivo [3].
DC control the migration of antigen-specific T cells to target tissues by imprinting their own homing markers on T cells that they stimulate [16], so antigen-specific T cells are directed back to the target tissue where the antigen had been sampled. The last aim of immunotherapy approaches utilizing MoDC is to trigger T cell specific responses. Subcutaneous immunization is the most clinically relevant administration route. However, MoDC must enter lymph nodes and present antigen, so MoDC used in immunotherapy are usually matured with cytokine cocktails [29] to induce CCR7 expression so that they acquire the capacity to migrate to the lymph nodes and are able to stimulate a T cell response. This approach is likely to abrogate the immature phenotype of MoDC. In addition, it is not only a matter of triggering immune responses, but also of directing them to the target tissues; although these DC may enter lymph nodes directly and stimulate T cells, they are unlikely to give tissue-homing potential to those stimulated cells, as it is the source of DC and not the lymph node cells or stroma that determines the homing property of the stimulated T cells [11]. Thus, even if MoDC enter the lymph and generate antigen-specific T cells, the lack of homing markers of MoDC may cause the failure to induce a tissue-specific homing profile on T cells.
An alternative approach would be to induce homing markers directly on MoDC, so that they would migrate to the target tissue and once there, following conditioning by the tissue microenvironment [16], enter the respective lymph nodes (mesenteric and/or Peyer's patches in the gut) to perform antigen presentation and imprint their own homing profile on responding T cells. However, intestinal tissue microenvironment not only induces CCR7 expression on DC [16], it also affects their phenotype and function; when blood DC are conditioned with inflamed colonic microenvironment from UC patients, DC lose their homeostatic properties and imprint a skin-homing profile on T cells in the absence of RA [28]. Therefore, a major concern regarding use of DC for immunotherapeutic purposes in IBD is that RA-MoDC could potentially adopt a proinflammatory phenotype and function in vivo in the inflamed colonic microenvironment, contributing to the ongoing inflammation [26,27]. However, RA-MoDC maintained their regulatory phenotype even in the presence or an inflamed microenvironment derived from UC patients (Fig. 6); a gut-specific immunomodulation might thus be achieved with such cells, negating the necessity for systemic immunosuppression produced with current treatments. However, a recent study demonstrated that RA-conditioned DC adopt proinflammatory properties in the presence of IL-15, abrogating oral tolerance to dietary antigens [30]. Several differences between these studies may account for the discrepancies. The major difference is the use of human material for this study compared with murine models; data from animal models may not be transferrable directly into human mechanisms of disease [31]. Also, the MoDC in this study were exposed to an inflamed colonic microenvironment derived directly from human UC colonic biopsies rather than IL-15; our previous studies suggest that IL-6 is the dominant cytokine in UC pathogenesis [28]. Finally, higher RA concentrations were used to pulse MoDC in this study. These factors may explain why RA-pulsed DC were not recruited towards a proinflammatory phenotype when exposed to an intestinal microenvironment from UC patients.
RA is a key player of immune homeostasis in the gut controlling immunoglobulin (Ig)A switching of B cells, the development of T cells with a regulatory phenotype/function and induction of gut-homing properties of stimulated B and T cells [14,21–23,32]. RA also mediates acquisition of gut-homing properties on human DC and regulates their ability to imprinting homing properties onto stimulated T cells [16,33], while decreasing their T cell stimulatory capacity [34], presumably through secondary transforming growth factor (TGF)-β induction [35]. We are aware that we did not study the capacity of RA-MoDC to induce regulatory T cells. That is because our recent evidence has shown that forkhead box protein 3 (FoxP3) expression may not be a valid marker to identify T cells with a regulatory phenotype; because its expression is dynamic, dependent upon T cell densities, it is subsequently lost in time following removal of the stimulant, and FoxP3+ T cells developed in vitro do not perform a suppressive activity [36]. However, we have shown that RA-MoDC displayed a decreased stimulatory capacity in a dose-dependent fashion (Fig. 4), even in the presence of LPS (Fig. 5) or an inflamed microenvironment from UC patients (Fig. 6), and T cells that they stimulated decreased their IFN-γ production and increased their IL-10 content (Fig. 6). Together, these results suggest that RA had the capacity to rehabilitate homeless MoDC with an immature and gut-homing phenotype and the capacity to generate gut-homing T cells that were not proinflammatory.
In summary, in addition to factors usually considered for utilizing DC in immunotherapy, the homing profile of DC themselves and, more importantly, their capacity to imprint homing profiles on the T cells they stimulate will influence a DC immunomodulatory approach. Our data suggest that RA conditioning may increase the efficacy of MoDC as therapeutic agents, providing tissue specificity and sustained homeostatic effects for DC-specific therapy targeted at mucosal inflammatory diseases with tissue compartmentalization. Therapeutic approaches for IBD would, ideally, involve gut-specific prevention of dysregulated inflammation without compromising immunogenicity towards harmful or invading pathogens; our data provide a potential method of doing so while minimizing systemic immunosuppression, which is a side effect of almost all current IBD therapies. Our findings could also be extended to other gastrointestinal autoimmune disease such as Crohn's disease and/or coeliac disease. Future studies may identify ways of inducing other tissue-specific homing markers in order to develop tissue-homing specific MoDC to perform tissue-specific immunotherapy.
Acknowledgments
This work was supported by Marie Curie Intra European Fellowship (FP7-people-IEF-2008-235993), St Mark's Hospital Foundation the Brigid Balfour Fund and the BBSRC (WMNI P33458). The authors also thank Isabel Comino for her helpful technical support.
Disclosures
No authors had financial conflicts of interest that might be construed to influence the results or interpretation of their manuscript.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Niess JH, Reinecker HC. Dendritic cells: the commanders-in-chief of mucosal immune defenses. Curr Opin Gastroenterol. 2006;22:354–360. doi: 10.1097/01.mog.0000231807.03149.54. [DOI] [PubMed] [Google Scholar]
- 3.Collin M, Bigley V, Haniffa M, Hambleton S. Human dendritic cell deficiency: the missing ID? Nat Rev Immunol. 2011;11:575–583. doi: 10.1038/nri3046. [DOI] [PubMed] [Google Scholar]
- 4.Figdor CG, de Vries IJ, Lesterhuis WJ, et al. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;5:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 5.García F, Routy JP. Challenges in dendritic cells-based therapeutic vaccination in HIV-1 infection Workshop in dendritic cell-based vaccine clinical trials in HIV-1. Vaccine. 2011;29:6454–6463. doi: 10.1016/j.vaccine.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM. Dendritic cells in vivo: a key target for a new vaccine science. Immunity. 2008;29:319–324. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7:610–621. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- 8.Ricart E, Panés J, Benítez-Ribas D. Dendritic cells: a new horizon in cell therapy for inflammatory bowel disease? Gastroenterol Hepatol. 2011;34:100–106. doi: 10.1016/j.gastrohep.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Yamanishi H, Murakami H, Ikeda Y, et al. Regulatory dendritic cells pulsed with carbonic anhydrase I protect mice from colitis induced by CD4(+)CD25(–) T cells. J Immunol. 2012;188:2164–2172. doi: 10.4049/jimmunol.1100559. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Rey E, Delgado M. Therapeutic treatment of experimental colitis with regulatory dendritic cells generated with vasoactive intestinal peptide. Gastroenterology. 2006;131:1799–1811. doi: 10.1053/j.gastro.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Stagg AJ, Kamm MA, Knight SC. Intestinal dendritic cells increase T cell expression of alpha4beta7 integrin. Eur J Immunol. 2002;32:1445–1454. doi: 10.1002/1521-4141(200205)32:5<1445::AID-IMMU1445>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 12.Mora JR, Bono MR, Manjunath N, et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 13.Johansson-Lindbom B, Svensson M, Wurbel MA, et al. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med. 2003;198:963–969. doi: 10.1084/jem.20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mora JR, Iwata M, Eksteen B, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 15.van den Bogaerde J, Kamm MA, Knight SC. Immune sensitization to food, yeast and bacteria in Crohn's disease. Aliment Pharmacol Ther. 2001;15:1647–1653. doi: 10.1046/j.1365-2036.2001.01032.x. [DOI] [PubMed] [Google Scholar]
- 16.Mann ER, Bernardo D, Al-Hassi HO, et al. In humans, gut-specific homeostatic dendritic cells are generated from blood precursors by the gut microenvironment. Inflamm Bowel Dis. 2012;18:1275–1286. doi: 10.1002/ibd.21893. [DOI] [PubMed] [Google Scholar]
- 17.Rimoldi M, Chieppa M, Salucci V, et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol. 2005;6:507–514. doi: 10.1038/ni1192. [DOI] [PubMed] [Google Scholar]
- 18.Butler M, Ng CY, van Heel DA, et al. Modulation of dendritic cell phenotype and function in an in vitro model of the intestinal epithelium. Eur J Immunol. 2006;36:864–874. doi: 10.1002/eji.200535497. [DOI] [PubMed] [Google Scholar]
- 19.Iliev ID, Mileti E, Matteoli G, et al. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol. 2009;2:340–350. doi: 10.1038/mi.2009.13. [DOI] [PubMed] [Google Scholar]
- 20.Iliev ID, Spadoni I, Mileti E, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. 2009;58:1481–1489. doi: 10.1136/gut.2008.175166. [DOI] [PubMed] [Google Scholar]
- 21.Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mora JR, Iwata M, Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwata M, Hirakiyama A, Eshima Y, et al. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Bagwell CB. Hyperlog – a flexible log-like transform for negative, zero, and positive valued data. Cytometry A. 2005;64:34–42. doi: 10.1002/cyto.a.20114. [DOI] [PubMed] [Google Scholar]
- 25.Dudda JC, Lembo A, Bachtanian E, et al. Dendritic cells govern induction and reprogramming of polarized tissue-selective homing receptor patterns of T cells: important roles for soluble factors and tissue microenvironments. Eur J Immunol. 2005;35:1056–1065. doi: 10.1002/eji.200425817. [DOI] [PubMed] [Google Scholar]
- 26.Laffont S, Siddiqui KR, Powrie F. Intestinal inflammation abrogates the tolerogenic properties of MLN CD103+ dendritic cells. Eur J Immunol. 2010;40:1877–1883. doi: 10.1002/eji.200939957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aymeric R, Jianping H, Abhisake K, et al. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernardo D, Vallejo-Diez S, Mann ER, et al. IL-6 controls the intestinal inflammation in human ulcerative colitis and mediates the conditioning of dendritic cells towards a pro-inflammatory phenotype with increased skin-homing phenotype and skin-imprinting capacity on T-cells. Eur J Immunol. 2012;42:1337–1353. doi: 10.1002/eji.201142327. [DOI] [PubMed] [Google Scholar]
- 29.Karlsen M, Hovden AO, Vogelsang P, et al. Bromelain treatment leads to maturation of monocyte-derived dendritic cells but cannot replace PGE2 in a cocktail of IL-1β, IL-6, TNF-α and PGE2. Scand J Immunol. 2011;74:135–143. doi: 10.1111/j.1365-3083.2011.02562.x. [DOI] [PubMed] [Google Scholar]
- 30.DePaolo RW, Abadie V, Tang F, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471:220–224. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbons DL, Spencer J. Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal Immunol. 2011;4:148–157. doi: 10.1038/mi.2010.85. [DOI] [PubMed] [Google Scholar]
- 32.von Boehmer H. Oral tolerance: is it all retinoic acid? J Exp Med. 2007;204:1737–1739. doi: 10.1084/jem.20071251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammerschmidt SI, Friedrichsen M, Boelter J, et al. Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J Clin Invest. 2011;121:3051–3061. doi: 10.1172/JCI44262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bedford PA, Knight SC. The effect of retinoids on dendritic cell function. Clin Exp Immunol. 1989;75:481–486. [PMC free article] [PubMed] [Google Scholar]
- 35.Feng T, Cong Y, Qin H, et al. Generation of mucosal dendritic cells from bone marrow reveals a critical role of retinoic acid. J Immunol. 2010;185:5915–5925. doi: 10.4049/jimmunol.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernardo D, Al-Hassi HO, Mann ER, et al. T-cell proliferation and forkhead box P3 expression in human T cells are dependent on T-cell density: physics of a confined space? Hum Immunol. 2012;73:223–231. doi: 10.1016/j.humimm.2011.12.017. [DOI] [PubMed] [Google Scholar]