

Abstract
Understanding immunoregulatory mechanisms is essential for the development of novel interventions to improve long-term allograft survival. Programmed death 1 (PD-1) and its ligands, PD-L1 and PD-L2, have emerged as critical inhibitory signaling pathways that regulate T cell response and maintain peripheral tolerance. PD-1 signaling inhibits alloreactive T cell activation, and can promote induced regulatory T cell development. Furthermore, the upregulation of PD-L1 on nonhematopoietic cells of the allograft may actively participate in the inhibition of immune responses and provide tissue-specific protection. In murine transplant models, this pathway has been shown to be critical for the induction and maintenance of graft tolerance. In this review, we discuss the current knowledge of the immunoregulatory functions of PD-1 and its ligands and their therapeutic potential in transplantation.
Keywords: costimulation, transplantation, alloimmunity, PD-1, PD-L1, PD-L2, regulatory T cells, tolerance
Introduction
T cells play a major role in coordinating the immune response against allo-antigens during organ transplantation. While T cell activation depends on the initial antigen-specific signal provided to T cell receptors via the antigen-loaded MHC complex, additional signals provided by costimulatory molecules fine-tune this response, determining its strength, nature and duration. Some costimulatory interactions potentiate the activation and proliferation of naïve T cells, while others inhibit T cell activation and promote regulation (1, 2). The founding members of the B7:CD28 costimulatory family are the CD28 and CTLA-4 co-receptors that both bind to the B7-1 (CD80) and B7-2 (CD86) molecules. CD28 acts as a strong positive costimulatory receptor and CTLA-4 as a potent coinhibitory receptor. The programmed death 1 (PD-1) receptor: PD-Ligand (PD-L) pathway is another major receptor-ligand network that functions primarily to provide a coinhibitory signal. PD1: PD-L interactions maintain peripheral tolerance and are exploited by tumors and viruses that cause chronic infection to evade immune eradication. As such, this pathway has emerged as a potential therapeutic target for either enhancing or dampening the immune response. This review will summarize our current understanding of the immunoregulatory functions of the PD-1: PD-L pathway and its therapeutic potential, focusing on its relevance to the field of transplantation.
Structure and Expression of PD-1 and its Ligands
The inhibitory receptor programmed death 1 (PD-1; CD279) is a cell surface molecule with a single immunoglobulin (Ig) superfamily domain and a cytoplasmic domain containing two tyrosine-based signaling motifs: a tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) (Figure 1) (3). PD-1 has two ligands, PD-L1 (B7-H1; CD274) (4) and PD-L2 (B7-DC; CD273) (5, 6), both of which have Ig-V-like and IgC-like extracellular domains and a short intracellular domain. PD-1 is inducibly expressed by T cells and B cells after activation (7) as well as natural killer T (NKT) cells, NK cells, activated monocytes and some subsets of dendritic cells (DCs) [reviewed in (8)]. PD-1 is upregulated after TCR or BCR engagement on naïve lymphocytes and persistent antigen stimulation maintains high PD-1 expression (9). The common γ-chain cytokines (IL-2, IL-7, IL-15 and IL-21), TLRs and interferons also can potentiate PD-1 expression on T cells (10). Expression of PD-1 is in part mediated by the recruitment of the nuclear factor of activated T cells c1 (NFATc1) to the nucleus (11). NFATc1 together with AP-1 and NF-κB constitute the most critical transcription factors activated upon antigen recognition by T cells. Interestingly, the calcineurin inhibitor Cyclosporine A markedly reduces PD-1 expression through its effect on NFATc1 (11). While PD-1 upregulation on naïve T cells peaks at 48 hours after anti-CD3 or anti-CD3/anti-CD28 stimulation in vitro (12), PD-1 on allogeneic CD4+ T cells progressively increases over time following skin transplantation in vivo, reaching the highest levels on day 10 post-transplant (13). Determination of the level of PD-1 expression in different immune cell subsets at various time points in transplantation remains to be explored. Lastly, certain subsets of T cells express high levels of PD-1, including CD4+Foxp3+ regulatory T cells (Tregs) (14), T follicular helper cells (TFH) (15), memory T cells (16) and “exhausted” CD8 cells (17).
Figure 1. Effect of PD-1 signaling in T cells.

PD-1 signaling dephosphorylates proximal signaling molecules and augments PTEN expression, inhibiting PI3K and AKT activation. The consequences include decreased T cell proliferation, cytokine production and cell survival. PD-1 inhibition can be overcome by strong TCR signaling, CD28 signaling or IL-2 signaling (not shown). SHP2, protein tyrosine phosphatase; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homolog; AKT, serine/threonine protein kinase; PIP3, Phosphatidylinositol (3,4,5)-triphosphate.
Of the PD-1 ligands, PD-L1 has very broad expression, whereas PD-L2 is inducibly expressed in a more restricted fashion. PD-L1 is expressed constitutively on murine T and B cells, DCs, macrophages, mesenchymal stem cells and bone marrow-derived mast cells (12, 18), and induced to higher levels by inflammation. In addition, it can be upregulated on non-hematopoietic cells, including vascular endothelial cells, epithelial cells, muscle cells, hepatocytes, placental cells and pancreatic islet cells (19, 20). In humans, PD-L1 is mainly an inducible molecule. PD-L2 is upregulated on DCs, macrophages, bone marrow-derived mast cells and on a subset of peritoneal B1 B cells as well as on germinal center B cells (21). PD-L2 is also expressed on human, but not mouse, vascular endothelial cells and T cells (6, 22). Interferon-γ (IFN-γ) potently upregulates PD-L1, and to a lesser extent PD-L2. IL-4 and GM CSF are the strongest known stimuli for inducing PD-L2 expression (5, 12, 19, 23, 24). In addition to binding PD-1, PD-L1 can also bind the B7-1 (CD80) molecule, thus connecting the PD-1:PD-L1 pathway with the B7-1:CD28/CTLA-4 pathway (25). PD-L2, however, does not bind to B7-1. There are data to suggest that PD-L2 may have another receptor, the identity of which is still unknown (26).
Function of PD-1: PD-L Pathway
A major role of the PD-1: PD-L pathway is the inhibition of T cell function by engagement of the PD-1 receptor on T cells by PD-L1 or PD-L2 on antigen presenting cells (APCs). APCs transfected with PD-L1 or PD-L2 inhibit T cell responses, while blockade or genetic ablation of PD-L1 or PD-L2 on DCs or other APCs enhances their capacity to stimulate T cell responses in vitro as compared to wild-type (WT) APCs (27, 28). Conversely, T cells that lack PD-1 are hyper-responsive relative to WT T cells (5, 27, 29–31). These inhibitory interactions not only suppress T cells during the priming phase of an immune response in secondary lymphoid tissues, but also modulate effector T cell responses, either during migration to the site of inflammation or in the target tissue itself (8, 32).
PD-1 transduces an inhibitory signal when it is bound by its ligands in the presence of TCR or BCR activation (5, 33, 34). Phosphorylation of a tyrosine residue in the immunoreceptor tyrosine-based switch motif (ITSM) of PD-1 appears to have a key functional role in mediating PD-1 immunoinhibition. Phosphorylation of the ITSM motif leads to the recruitment of SH2-domain containing tyrosine phosphatase 2 (SHP-2), and possibly SHP-1, to the cytoplasmic domain of PD-1, which then down-regulates CD28-mediated PI3K activity and consequently, leads to less activation of Akt (Figure 1) (35). The exact mechanism of PD-1-mediated antagonism of the PI3K pathway is not yet clear (35). PD-1 ligation also inhibits the phosphorylation of other signaling molecules including CD3, ZAP70 and PCK (35). Thus, a major function of PD-1 signaling is to directly inhibit antigen receptor signaling.
Signaling through PD-1 exerts major effects on cytokine production by T cells, inhibiting production of IFN-γ, tumor necrosis factor-α and interleukin-2 (IL-2). PD-1 can also inhibit T cell proliferation (5, 36), and inhibit the upregulation of Bcl-xL, an anti-apoptotic protein (33). Lastly, PD1 signaling decreases the expression of the transcription factors GATA-3, Tbet and Eomes, which are associated with T cell effector function (37). However, a strong positive signaling through CD28 and/or IL-2 receptor can overcome PD-1 inhibitory effects on T cell proliferation, differentiation and survival (5, 18, 37, 38). PD-1 signaling has also been implicated in reversal of the “stop signal” that is mediated by TCR signaling (39). This means that in the presence of PD-1, T cells have a shortened dwell time in their interactions with APCs, which can lead to decreased T cell activation and may also favor the induction of Tregs.
PD-1 can also inhibit signaling through B cell receptor. The role of PD-1 in controlling antibody production may be directly related to PD-1 on the B cells or secondary to effects of PD-1 on T cells. T cell interactions with B cells involve recognition of antigen by helper T cells, which then stimulate B cell expansion, isotype switching and affinity maturation. Among T cells, follicular helper cells (TFH) have emerged as key supporters of the B cell response (40). TFH express high levels of PD-1 (15, 41), and PD-L1 and PD-L2 are upregulated on germinal center B cells (42). PD-1 has been shown to be important for the regulation of the germinal center B cell response; PD-1−/− BALB/c mice have a reduced number of long-lived plasma cells after immunization with (4-hydroxy-3-nitrophenyl) acetyl-chicken-γ-globulin (42). In contrast, in two immunization models with either keyhole limpet hemocyanin or extract of S. mansoni eggs in B6 background mice, PD-L1 deficiency led to a significant expansion of TFH cells and enhanced Ag-specific antibody responses (43). PD-1 deficiency can lead to generation of increased numbers of TFH cells with aberrant phenotypes that lead to dysregulated selection of B cells and antibody diversity in germinal centers (44). Further studies are needed to delineate the functions of this pathway in regulating TFH cell function and B cell responses in the germinal center.
Recently described roles for PD-1 expression on DCs and monocytes highlight the possibility that PD-1 signaling may also occur independently of T cell or B cell antigen receptor signaling, possibly by impinging on other receptor signaling pathways (45, 46). For example, PD-1 ligation in monocytes has been shown to stimulate the production of IL-10 during HIV infection, which in turn contributes to reducing T cell function (45). These findings demonstrate that PD-1 expression on a non-lymphocyte population also may influence T cell immune function in HIV infection and this finding may extend to other settings.
In addition to PD-1 mediated signaling, there are data to suggest that signals may be transduced by PD-1 ligands. However, the cytoplasmic tail of PD-L1 has no known function. The cytoplasmic domains of human and mouse PD-L2 differ, with the mouse version being only 4 amino acids long and the human bearing 30 amino acids. While this longer form has no known signaling motifs, it is conserved in a number of mammals (but not rodents), which suggests functional significance. Data supporting PD-L1 and/or PD-L2 signaling are primarily based on experiments using soluble PD-1 reagents that engage PD-L1/2 and lead to upregulation of IL-10 production and reduction in DC function (4, 47). The physiological roles of signaling through PD-L1 or PD-L2 remain to be explored.
PD-1: PD-L Pathway in Peripheral Tolerance
The immune system has the daunting task of responding to foreign antigens, while avoiding self-reactivity. Central tolerance mechanisms prevent the emergence of most self-reactive T lymphocytes from the thymus, but some self-antigen-specific T cells escape into the periphery. To prevent the development of autoimmunity, multiple mechanisms of peripheral tolerance have evolved, including T cell deletion, anergy and the suppressive function of Tregs. Failure of any of these mechanisms might result in autoimmunity. The PD-1:PD-L1 pathway plays a critical role in regulating the delicate balance between protective immunity and tolerance.
Initial evidence suggesting an important role for PD-1 in tolerance came from studies using blocking antibodies and knockout mice (27, 48, 49). In contrast to the CTLA-4 knockout mouse, which dies of an aggressive autoimmune lymphoproliferative disease within three to four weeks of birth (50, 51), the PD-1 knockout mouse is viable on a number of different backgrounds. However, susceptibility to autoimmune disease is exacerbated in PD-1−/− mice on an autoimmune-prone background (52, 53). PD-1−/− and PD-L1−/− NOD mice develop accelerated spontaneous diabetes, which manifests in 100% of male and female mice by 10 weeks (36, 54). Similarly, PD-L1 deficiency on the MRL-Faslpr background results in development of myocarditis and pneumonitis (55). These studies indicate that the PD-1 pathway is critical for self-tolerance.
PD-1 regulates both thymic selection and peripheral tolerance. PD-1 plays a role in inhibiting positive selection of thymocytes during the transition from the `double negative' to CD4+CD8+ 'double positive' stage (56, 57). The role of PD-1 in negative selection is less clear. Two studies using the alloreactive 2C TCR transgenic model showed increased negative selection in the absence of PD-1 (53, 58), while analysis of HY-specific CD4 and CD8 TCR transgenic mice revealed no role for PD-1 in negative selection (59). In addition, PD-1 is critical for peripheral tolerance, restraining the development of self-reactive CD4 and CD8 T cells. PD-1 has an important role in controlling the outcome of initial encounters between naive self-reactive T cells and DCs. PD-1:PD-L1 interactions are required for both the induction and maintenance of CD4 T cell tolerance. For example, the PD-1 pathway is a critical mediator of tolerance induced by administration of antigen-coupled fixed splenocytes, which can reverse diabetes in NOD mice (60). PD-1 deficiency on self antigen-specific T cells increases CD8 T cell responses to antigen-bearing resting DCs, and abrogates CD8 T cell tolerance to peripheral self-antigen expression in vivo (30, 31, 61). PD-L2 is required for oral tolerance, as shown by failure of oral tolerance in PD-L2−/− mice (62).
The use of T-cell depletion therapy is expanding in solid organ transplantation. Upon lymphodepletion, regulatory mechanisms, including PD-1 signaling, prevent the emergence of autoreactive T cells during homeostatic proliferation. In a lymphopenic environment, a subpopulation of PD-1high T cells develop that have skewed TCR repertoires and appear to be pre-apoptotic (63). It has been speculated that PD-1 may inhibit expansion of potentially pathogenic self-reactive CD8 T cells during homeostatic reconstitution of lymphopenic environments (63). Using an adoptive cell transfer model, Thangavelu et al. demonstrated that the reconstitution of Rag−/− recipients with PD1−/− hematopoietic stem cell (HSC) precursors, but not WT HSC, causes severe autoimmunity, which does not develop when mature WT or PD-1−/− T cells are used as donor cells (59). These findings support a critical role for PD-1 in regulating homeostatic proliferation, in particular of recent thymic emigrants. Therefore, preservation of an intact PD-1 pathway might be important for the prevention of autoimmunity in patients undergoing reconstitution of their immune system after lymphoablation in solid organ and hematopoietic stem cell transplantation.
Regulatory T cells
Regulatory T cell populations are critical for the maintenance of peripheral tolerance, are potent inhibitors of many immune responses, and play an important role in the prevention of graft rejection (64, 65). CD4+Foxp3+ Tregs, the most widely studied suppressive T cell population, are critical for peripheral tolerance as illustrated by the fatal autoimmune condition of scurfy mice and the human immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), both caused by mutations in the foxp3 gene (66, 67). Foxp3+ Tregs can be divided into `natural' Treg (nTreg) that arise as committed regulatory cells from the thymus, and `induced' Treg (iTreg) that are generated in the periphery by polarization of naïve CD4+ T cells in a TGFβ- and IL-2-dependent fashion (68–76).
Foxp3+ Tregs highly express PD-1 and PD-L1 (1) and a role for the PD1:PD-L pathway in the generation of Tregs has been supported by a number of studies (77–81). PD-L1−/− DCs were shown to be defective at supporting the TGFβ-induced conversion of naïve T cells to Tregs in vitro, and conversion and maintenance of iTreg function in vivo was also dependent on PD-L1 (79). PD-L1 engagement of its receptors (PD-1, and possibly B7-1) on naïve T cells leads to iTreg development, at least in part, by inhibiting mTOR/Akt signaling, as shown by experiments using microbeads coated with anti-CD3, anti-CD28 and PD-L1 fusion protein as artificial APCs (77). There are multiple studies that have identified AKT as a strong repressor of Tregs (82–84). It is proposed that AKT diminishes TGF-β-induced Foxp3 expression in a kinase-dependent manner and via a rapamycin-sensitive pathway (82). There is also evidence that weak TCR signals during T cell differentiation, and limited costimulation, can favor Foxp3 induction (85–88). Using human Th1-polarized T cells in a human-into-mouse graft-versus-host disease model (xGVHD), Amarnath et al. showed that PD-L1: PD-1 signaling was critical for the conversion of human Th1 cells into Tregs in vivo, preventing the development of xGVHD (81). The role of PD-1 in Treg conversion was partially mediated by the activation of intracellular SHP1/2 and consequent reduction of STAT1 phosphorylation, thereby abrogating IFN-γ-mediated maintenance of T-bet and favoring Foxp3 expression (81). Thus, the PD-1 pathway may promote tolerance induction in vivo by inducing and sustaining iTreg.
The role of PD-L1 in natural or existing Tregs is less well understood. Antibody blockade studies suggest that this pathway inhibits expansion of established Tregs, as well as effector T cells (89, 90). In the setting of hepatitis C virus infection, it has been suggested that PD-L1 negatively regulates Tregs by blocking STAT5 phosphorylation (89). However, PD-L1 maintains Foxp3 expression and enhances the suppressive capacity of induced Tregs in vitro (77). It may be that the PD-1 pathway exerts distinct effects on induced Tregs and natural Tregs. Further work is needed to clarify this issue as well as the question of whether PD-1 plays a role in regulating the dichotomy between iTreg and Th17 differentiation. In addition, a subpopulation of regulatory allo-specific CD8+ T cells expressing PD-1, which is induced by ICOS: B7h blockade, was found to be important for immune regulation (91). Thus, it appears that the PD-1 pathway may regulate the generation and/or functions of multiple types of regulatory T cells.
PD-1:PD-L Pathway in Transplantation
Understanding immunoregulatory mechanisms is essential for the development of novel interventions that might help improve long-term graft survival. Coinhibitory signals play a key role in the regulation of the alloimmune response against the transplanted organ. In particular, a number of studies have demonstrated that an intact PD-1: PD-L interaction is important for the induction and maintenance of graft tolerance (13, 32, 92–96).
PD-1, PD-L1 and PD-L2 mRNA expression are significantly increased in cardiac allografts during rejection, while expression is minimal in syngeneic grafts or naïve hearts, indicating that upregulation is related to the host alloresponse, rather than a consequence of ischemia/reperfusion injury (97). A similar observation was made in mouse liver transplantation, where allogeneic livers had a significantly higher expression of PD-L1, both in the sinusoidal and parenchymal areas (92). One of the unique characteristics of PD-L1, compared to other costimulatory molecules, is its pattern of broad expression, not only on hematopoietic cells, but also on non-hematopoietic cells such as endothelium, placental trophoblasts and islet cells (1). After transplantation, there is significant upregulation of PD-L1 on endothelial cells in heart allografts (32). The potential function of PD-L1 in the endothelium is particularly intriguing, since the vasculature is the first interface between the immune cells and the target graft, residing in an optimal location for controlling the alloimmune response.
Manipulation of the PD-1:PD-L1 pathway in Alloimmunity
Animal models of solid organ transplantation
Functions of the PD-1:PD-L1 pathway have been investigated in multiple different murine solid organ transplant models (Table 1), as well as in bone marrow transplantation. Blockade of PD-1 and PD-L1, but not PD-L2, using an antibody approach, significantly accelerated cardiac graft rejection in a fully MHC mismatched allogeneic model (BALB/c into B6), particularly in the absence of CD28 costimulation (98). In less allogeneic models, such as the MHC class II mismatched model (bm12 into B6), blockade of PD-L1 also precipitated rejection, whereas PD-1 and PD-L2 blockade had no effect on graft survival (96, 99). The absence of a significant effect of PD-L2 blockade in transplantation suggests that PD-L1 and PD-L2 may have different roles in tolerance induction, possibly related, in part, to diverse expression of these ligands. For example, PD-L1 expression is high in tolerogenic subsets of DCs, Tregs and mesenchymal stem cells (32, 100, 101), whereas the expression of PD-L2 is mostly restricted to professional APCs.
Table 1.
PD-1: PD-L1 function in various transplant models.
| Transplanted Organ | Model | Intervention | Effect | Potential Mechanisms | Ref. |
|---|---|---|---|---|---|
| Heart | BALB/c → B6 | PD-L1.Ig or PD-L2.Ig +/− cyclosporine or rapamycin | PD-L1.Ig prolongs graft survival in combination with limited immunosuppression | ↓ IFN-γ ↓RANTES, MIP-1, IL 10, CXCR3, CCR5 |
(97) |
| BALB/c → B6 | 1) CTLA-4-Ig+ anti-PD-L1 or 2 mAb 2) CTLA-4-Ig in PD-L1KO or PD-L2KO recipients |
PD-L1 blockade or PD-L1KO recipient prevents tolerance development (early and late) | ↑CD8 eff mem ↑IFN-γ, IL4 ↓Tregs in grafts |
(94) | |
| BALB/c → B6 | 1) Anti-PD-1 mAb + CD154mAb/DST 2) PD-1KO recipients + CD154mAb/DST |
PD-1 blockade or PD-1KO recipient prevents tolerance development | ↑IFN-γ, IL2, IP10, RANTES, CXCR3, CCR5 | (95) | |
| BALB/c → B6 CD28KO or B7DKO | Anti-PD-1 or anti-PD-L1 mAb | PD-L1 blockade accelerates rejection at a faster tempo than PD-1 blockade | ↑CD4 and CD8eff mem ↑IFN-γ |
(98) | |
| BALB/c PD-L1 chimera → B6 | PD-L1 chimeric donor | Graft PD-L1 expression on both hematopoietic and nonhematopoietic cells are essential for tolerance | ↑CD8 eff mem ↑IFN-γ, GrB |
(32) | |
| Bm12→ B6 B6→ bm12 B6 → bm1 |
1) Anti-PD-L1 or Anti-PD-L2 mAb 2)PD-L1KO or PD-L2KO recipients 3)PD-L1KO or PD-L2KO donor hearts |
On bm12 model: PD-L1 blockade accelerates rejection; PD-L1KO donor accelerates rejection | ↑CD4 eff mem ↑IFN-γ, IL4 |
(96) | |
| Bm12→ B6 Bm12→ B6 B7-1KO or B7-2KO or PD-1KO |
Selective blockade of PD-L1: B7-1 pathway via antibody approach (2H11) or dual blockade with anti-PD-L1 mAb (MIH-6) | PD-L1:B7-1 blockade exacerbates chronic injury; dual blockade of PD-L1 has a greater deleterious effect. | ↓Tregs ↑IFN-γ, IL4, IL6 |
(99) | |
| F344→ Lewis (rat model) | Overexpression of PD-L1 on donor heart via adenovirus +/− cyclosporine | Slight delay in rejection in combination with cyclosporine | ↓CD4 graft infiltration | (119) | |
| Skin | Bm12→B6 and bm12→ABM transgenic mice | 1) Anti-PD-1, anti-PD-L1 oranti-PD-L2 mAb | Only PD-L1 blockade accelerated rejection; CD25-dependent effect | ↑allogeneic T cell proliferation ↑IFN-γ ↓ apoptosis of alloreactive CD4 cells |
(13) |
| mOva skin→B6 | Anti-PD-1 and anti-PD-L1 mAb +/− anti-CD28 and anti-CD154 mAb | PD-1 and PD-L1 blockade accelerated skin rejection | ↑ allogeneic T cell proliferation ↑IFN-γ |
(102) | |
| Islet cells | Syngeneic islets from WT or PD-L1KO→ NOD mice | PD-L1 and/or PD-L2 deficiency on islets | PD-L1 deficiency accelerates diabetes development PD-L1 expressed on islets protects against infiltration of autoreactive T cells | PD-L1KO islets: ↑ CD4+ T cells infiltrating islets ↑IFN-γ, TNF-α | (36) |
| Allogeneic islets from DBA/2→ B6 mice | PD-L1.Ig and anti-CD154mAb | PD-L1.Ig prolonged islet allograft survival | ↓ T cell activation | (118) | |
| Liver | B6 → C3H (85% tolerance) | 1) Anti-PD-L1 mAb 2) Anti-PD-1 mAb 3) PD-L1 deficiency in liver |
PD1 and PDL1 blockade precipitates rejection; PDL1KO liver prevents tolerance development | ↑cytotoxic T cell infiltration ↑GrB, FasL and perforin in grafts |
(92) |
Abbreviations: DST, donor-specific transfusion; ABM, anti-bm12 (ABM) transgenic (tg) model; NOD, non-obese diabetic mice
PD-L1 plays a critical role in the induction and maintenance of peripheral transplant tolerance. In a fully MHC mismatched cardiac transplant model in which tolerance was induced by multiple doses of CTLA-4-Ig, early administration of an anti-PD-L1 mAb prevented tolerance induction, while delayed administration abrogated graft survival (94). Accelerated rejection was associated with a significant increase in the frequency of CD8+ effector memory T cells and IFN-γ-producing alloreactive T cells in the periphery, while Foxp3+-graft infiltrating Tregs were decreased (94). This finding has been confirmed in PD-L1-deficient recipients. Furthermore, blockade or elimination of PD-L1 also abrogates other tolerogenic strategies, such as anti-CD154 mAb combined with donor-specific transfusion (95). In summary, the PD-1:PD-L1 pathway is key for tolerance development, however, strong TCR signaling might overcome PD-1 mediated inhibition, which may occur in non-immunosuppressed recipients of fully MHC mismatched transplanted organs.
In order to more finely assess the effects of PD-1 and PD-L1 in allo-specific T cells, our group has used the anti-bm12 (ABM) transgenic (tg) model, in which CD4+ TCR tg cells are specifically reactive to I-Abm12 (13). Following bm12 skin transplantation, both PD-1 and PD-L1 blockade enhanced alloreactive T cell proliferation and Th1 cell differentiation, although PD-L1 blockade led to a greater effect compared to PD-1 and was unique in its capacity to inhibit alloantigen-specific T cell apoptosis. The effects of PD-L1 blockade were dependent on the presence of CD4+CD25+ Tregs in vivo (13). Another group used a single-antigen mismatched transplant model where allo-specific CD8+ T cells can recognize mOVA skin grafts and be tracked over time (102). Similarly, blockade of PD-1 resulted in rapid expansion of donor-specific T cells and accelerated skin graft loss, even in the presence of combined CD28/CD40L blockade (102), reinforcing the pivotal role of PD-1:PD-L1 interactions in the regulation of alloreactive T cells.
Mouse models of liver transplantation (103) are very interesting to investigate, since the majority of liver grafts are accepted across MHC barriers without the requirement for immunosuppressive therapy. When B6 WT livers are transplanted into allogeneic C3H recipients, more than 85% of transplanted livers survive long-term (>100 days). PD-L1 deficiency in the liver graft (B6) or administration of anti-PD-1 or anti-PD-L1 blocking antibody prevented tolerance development in all C3H recipient mice (92), and was associated with significantly higher infiltration of cytotoxic T lymphocytes and increased levels of inflammatory cytokines (92). These findings suggest that PD-1: PD-L1 interactions may be sufficient for the development of spontaneous liver allograft tolerance in mice.
In light of the increased awareness of donor-specific alloantibodies as contributors to chronic rejection, interest in follicular helper cells (TFH) in alloimmune responses has expanded. TFH cells provide B cell help necessary for the generation of an effective antibody-mediated response (40) and express high amounts of PD-1 (CD44+CXCR5highPD-1high) (104). However, as discussed earlier in this review, the precise role of the PD-1 pathway in regulating T cell-dependent antibody responses is still unclear (42, 43). Similarly, the importance of this pathway in TFH generation in alloimmunity remains to be determined.
Murine models of bone marrow transplantation
Graft-versus-host disease (GVHD) is one of the most feared complications of bone marrow transplantation, and PD-1+ infiltrating cells are found at increased frequency in multiple GVHD target organs (spleen, colon and liver). Blazar et al. have shown that deficiency of the PD-1: PD-L1 pathway increases the lethality of GVHD in a model where B6 bone marrow cells and splenocytes are transferred into lethally irradiated B10.BR recipients (105). The accelerated GVHD was dependent on IFN-γ, and additional blockade of CTLA-4 further hastened the disease, supporting a non-redundant role of CTLA-4 and PD-1 in GVHD (105). Furthermore, in a model of acute myeloid leukemia, adoptively transferred AML-reactive cytotoxic T cells were more effective at eradicating the tumor after PD-L1 blockade (106). In sum, although blockade of the PD-1 inhibitory pathway may enhance anti-tumor responses, blocking this pathway might be deleterious in bone marrow transplantation due to worsening GVHD.
Novel Role of PD-L1:B7-1 in Alloimmunity
Our studies revealed a significantly greater effect of a blocking anti-PD-L1 mAb as compared to a blocking anti-PD-1 mAb in alloimmunity (98, 99). This finding was initially hypothesized to be related to the differences in expression of PD-1 and PD-L1, or half-lives/affinity of the antibodies. Subsequently, the discovery that PD-L1 could bind to B7-1 in addition to PD-1 suggested a different reason for this difference: PD-L1 interactions with B7-1 also might be important in regulating alloimmune responses (Figure 2) (25). Our group dissected the importance of PD-L1: B7-1 interactions in transplantation with the use of knockout animals (B7-1−/−, B7-2−/−, PD-L1−/−, PD-1−/−) and a newly characterized anti-PD-L1 antibody that selectively blocks the PD-L1: B7-1 interaction (10F.2H11) (99). Selective blockade of the PD-L1: B7-1 pathway led to significantly worse vasculopathy in bm12 allografts in B6 recipients and was associated with an increase in alloreactivity and a decrease in Tregs (99). However, the sole blockade of this pathway (PD-L1: B7-1) was not as powerful as dual blockade (PD-L1:PD-1/PD-L1:B7-1) with the anti-PD-L1 MIH-6 mAb in enhancing the alloimmune response, suggesting non-redundant roles of PD-L1: B7-1 and PD-L1: PD-1 interactions.
Figure 2. Costimulatory signaling network.
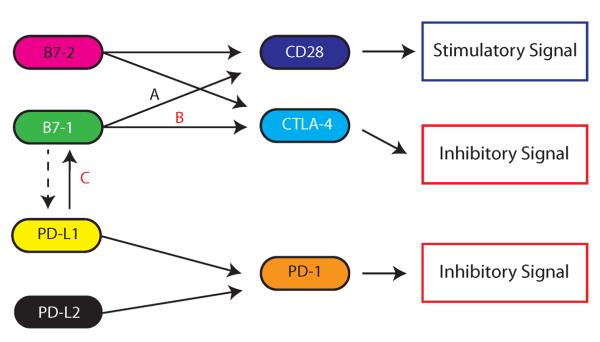
Integration of both positive and negative costimulatory signals during and after initial T cell activation will determine the fate and intensity of the alloimmune response. The complexity of the costimulatory network is exemplified by the potential effects of B7-1 targeting (such as accomplished by CTLA-4-Ig) – while blockade of B7-1 decreases positive costimulatory signaling through CD28 (A), it may affect two inhibitory signaling pathways, preventing B7-1 engagement of CTLA-4 (B) and PD-L1 (C). Dashed line represent the still controversial issue of reverse signaling through PD-L1 or PD-L2.
Since the affinity of the interaction between PD-L1 and B7-1 is higher than the affinity of B7-1 for CD28, it is possible that this interaction achieves sequestration of B7-1 from CD28 (25). However, in vitro studies suggest that the PD-L1: B7-1 interaction may function bidirectionally to inhibit T cells responses, by signaling through PD-L1 and/or B7-1 (25). Since both PD-L1 and B7-1 can be expressed on T cells, we further analyzed PD-L1: B7-1 interactions in an allo-setting using a combination of sensitized T cells and allo-DCs from WT and PD-L1−/− mice in an in vitro culture assay in the presence of either IgG control or 2H11 Ab (selective B7-1: PD-L1 blocking Ab) (99). In this setting, a potential dominant direction of effect was seen by the ligation of B7-1 on T cells by PD-L1 on DCs compared to PD-L1 on T cells and B7-1 on DCs, suggesting active, inhibitory signaling of B7-1 in T cells (99). A similar predominant direction of signaling was observed in a model of tolerance, in which OVA-reactive OT-I T cells undergo activation in response to injection of high-dose OVA257–264 peptide (107). In a minor mismatched GVHD model, Yi et al. showed that IFN-γ leads to upregulation of PD-L1 on host APCs, and PD-L1 on these cells interacts with B7-1, and not PD-1, to expand donor-derived Tregs (90). A function for B7-1 on T cells has been suggested previously (108). However, there are some studies indicating that B7-1 may signal into DCs and lead to the upregulation of IDO (109, 110). In sum, the PD-L1: B7-1 interaction plays a non-redundant inhibitory role compared to PD-L1: PD-1 interactions. How CTLA-4-Ig, which blocks B7-1 and B7-2 ligands, affects the balance among B7-1 interactions with its possible binding partners (CD28, CTLA-4 and PD-L1) remains to be determined (Figure 2), but the affinity of CTLA-4 for B7-1 is the highest of these interactions (25), so presumably CTLA-4-Ig has a significant ability to prevent B7-1:CD28 and B7-1:PD-L1 interactions.
Fetal maternal tolerance
Pregnancy is the most physiological model of tolerance to alloantigens, in which the mother accepts the fetus expressing allogeneic paternal antigens. The placenta is the central organ involved in immune regulation during pregnancy, since it contains both maternal and fetal cells in close proximity (111). However, some maternal cells have been shown to cross the placenta and reside in fetal lymph nodes, inducing the development of Tregs that suppress anti-maternal immunity in the fetus (112). PD-L1 is highly expressed on Tregs and in trophoblasts lining the decidua layer of the placenta (111). Interestingly, PD-L1 blockade or deficiency in B6 females mated with male allogeneic CBA mice results in decreased allogeneic fetal survival rates (113). The decrease in litter size and number upon PD-L1 blockade was shown to be dependent on CD25+ Tregs. Using an adoptive transfer model with alloantigen-specific TCR transgenic T cells in a bm12 × B6 mating, PD-L1 blockade was capable of breaking fetomaternal tolerance through a decrease in Treg induction, an increase in Treg apoptosis and a shift towards Th17 cells (114). Therefore, PD-L1 seems to play a role, not only in the regulation of effector T cells, but also in the promotion of Ag-specific Tregs in the context of fetomaternal tolerance. Further dissection of the importance of PD-L1 in different subpopulation of cells, including trophoblasts, is required to fully understand the role of PD-L1 in the fetomaternal immune interaction.
Importance of Donor Tissue PD-L1 Expression
One of the most intriguing aspects of the PD-1: PD-L1 pathway is related to the expression of PD-L1 by a variety of parenchymal cells, including heart, lung, kidney, pancreas, endothelium and placenta (1). This expression pattern raises the possibility that non-hematopoietic cells actively participate in the regulation of immune responses and can protect tissues against excessive inflammation and/or alloimmune responses. Indeed, non-hematopoietic expression of PD-L1 contributes to prevention of diabetes in the NOD mouse model (36) and regulates self-reactive CD8+ T cell responses in other models of self-tolerance (30, 115).
To address the question of whether donor PD-L1 on APCs and/or nonhematopoietic cells are important for tolerance development in transplantation, we generated PD-L1 chimeric mice on the B6 background and transplanted their hearts into BALB/c recipients treated with CTLA-4-Ig for tolerance induction (32). Both donor expression of PD-L1 on hematopoietic and nonhematopoietic cells (mainly endothelium) was essential for tolerance induction in this fully MHC mismatched model. However, PD-L1 deficiency on nonhematopoietic cells led to an earlier and more aggressive rejection with higher frequency of IFN-γ-producing alloreactive cells and CD8+ effector T cells (32).
Similarly, in a single MHC class II mismatched model of chronic rejection (B6 into bm12), PD-L1 deficiency in the donor heart also accelerated rejection (MST=16 vs >56 days on WT control hearts) and enhanced alloreactivity in recipients (96). In the setting of IFN-γ and/or allostimulation, the endothelium is known to upregulate PD-L1 (19, 32, 116). Endothelial PD-L1 is capable of downregulating CD8+ T cell responses and promoting the generation of allogeneic Tregs (19, 78, 116). Furthermore, in human kidney transplantation, tubular epithelial cells were found to upregulate PD-L1 during acute allograft rejection and PD-L1 on human tubular epithelial cell cultures was capable of inhibiting proliferation and cytokine production of CD4+ and CD8+ T cells (117). These findings illustrate how donor PD-L1 expression on nonhematopoietic cells can play a functional role in alloimmune regulation. PD-L1 on nonhematopoietic cells may be important for the induction of allograft tolerance and prevention of rejection by inhibiting T cells previously activated in secondary lymphoid organs within the allograft (Figure 3).
Figure 3. PD-1: PD-L1 signaling in allograft tolerance.

This figure illustrates some of the potential interactions of this pathway in lymphoid organs and in a cardiac allograft. (A), Tolerogenic DCs interact with naïve T cells and may induce either alloantigen-specific Tregs (iTregs) or decreased activation of effector T cell (T eff), depending on the presence (or absence) of TGF-β. (B), After activation by DCs in secondary lymphoid organs, alloantigen-specific effector T cells migrate to the graft where initial interaction with PD-L1 in the endothelium might inhibit T effector responses. Additional T eff cells that infiltrate the graft may be suppressed by iTregs or local cells expressing PD-L1 (APCs or non-hematopoieitic cells). Whether PD1/PDL1 on iTregs play a significant role in the direct suppression of T eff remains to be determined. Other potential costimulatory interactions are not depicted in this figure for simplicity.
Modulation of PD-1:PD-L1 to Improve Allograft Survival
All of the above experiments dissected the role of PD-1:PD-L1 pathway in transplantation with the use of loss-of-function approaches, with either blocking antibodies or knockout mice. Another way to investigate PD-1:PD-L1 function in transplantation is by potentiating its interaction. Several studies have used a PD-L1-Ig fusion protein that apparently signals through PD-1. This PD-L1-Ig was able to prolong cardiac allograft survival in a fully MHC-mismatched transplant model (BALB/c into B6) treated with concomitant low dose immunosuppression (cyclosporine A or rapamycin), however it did not affect graft survival when PD-L1-Ig was used alone (97). PD-L1-Ig in combination with low dose rapamycin for 2 weeks was able to induce long-term graft survival in all B6 recipients of BALB/c hearts (MST>100 days vs. 14 days on rapamycin alone group, p<0.0001) (97). Similarly, PD-L1-Ig prevented islet allograft rejection and facilitated tolerance induction when combined with anti-CD154 or suboptimal doses of rapamycin (118). Moreover, PD-L1-Ig was also capable of minimizing transplant arteriosclerosis in combination with anti-CD154 in another cardiac transplant model (97). These observations confirm that solely targeting the PD-1 pathway is probably not sufficient to inhibit the alloimmune response, and that additional control of T cell activation is required. Nonetheless, enhancing PD-1 signaling might be an important adjuvant approach for the protection of the graft against chronic injury.
Recent studies suggest another therapeutic strategy to improve graft survival could be enhancing the expression of PD-1 or PD-L1. Dudler et al. constructed an adenoviral vector expressing a PD-L1-Ig fusion protein and transfected F344 rat heart donor hearts, which were then transplanted into Lewis recipients (119). Cardiomyocytes were the predominant cell type transduced by the adenovirus, while only rare endothelial cells were transduced. Even though PD-L1 expression was only transient, graft rejection was slightly delayed, especially in combination with subtherapeutic cyclosporine (MST=25 vs 15, p<0.05) (119). Moreover, DCs transfected with PD-L1 recombinant adenovirus and injected into kidney recipients also minimized proteinuria and delayed renal allograft loss in a rat transplant model, in which Fisher 344 kidneys were transplanted orthotopically into a Lewis host after native kidneys were removed (120). While all control rats died prior to 12 days after transplant, PD-L1-DC-treated rats had greater than 60% survival at 3 weeks post-transplant (120). These observations suggest that PD-1: PD-L1 interactions are important in tissue-specific tolerance and methods to enhance PD-1/PD-L1 expression could be a promising approach in the induction of long-term tolerance.
Clinical Translation
While the therapeutic potential of promoting the PD-1 pathway has not yet been realized, the PD-1: PDL-1 pathway has become an attractive therapeutic target in the setting of chronic infection and cancer. The critical role for the PD-1 pathway in mediating T cell exhaustion was first revealed by blocking studies in models of chronic viral infection (121). PD-1 is highly expressed on “exhausted” T cells that develop during chronic viral infections in animals and humans (122). These T cells develop in the setting of chronic antigenic stimulation (17) and progressively lose effector functions, such as cytokine production and proliferation, leading to a significant deficit in viral clearance (123). Blockade of PD-L1 or PD-1 can reinvigorate the function of “exhausted” T cells and reduce viral load (123). Though PD-1 is highly expressed on “exhausted” T cells, other T cell subsets, including effector memory T cells present in peripheral blood of healthy humans, might also express high PD-1 levels (16). Therefore, additional markers such as CD223 (LAG-3), CD244 (2B4) and TIM-3 might be required to correctly identify this subset of T cells (121). Whether “exhausted” T cells develop in the setting of alloimmunity is not known, and is a subject for future investigation (124).
In the oncology field, there are a number of clinical trials in progress examining antibody-mediated blockade of PD-1 (anti-PD-1 mAb; Bristol-Myers Squibb, CureTech/Teva and Merck) (125) and PD-L1 (anti-PD-L1 mAb; Bristol-Myers Squibb, Genentech) in the treatment of refractory solid tumors, including melanoma, renal cell carcinoma, colorectal cancer and non-small cell lung cancer as well as in hematologic malignancies [reviewed in (126)]. Based on the pre-clinical data, blockade of PD-1 has the potential to lead to immunopathology and activation of self-reactive T cells. Initial safety reports from a phase I clinical trial with anti-PD-1 mAb in refractory malignancies (BMS-936558) revealed that therapy was well tolerated and the most common side effects reported were fatigue and diarrhea (127).
At present, there are no PD-1 agonist reagents in clinical trials. Several approaches are being investigated, including development of agonist antibodies, PD-L1 or PD-L2 fusion proteins, or enhanced local induction of tissue PD-1 ligand expression. How PD-1 engagement would fit in the currently available immunosuppressive armamentarium and what additional benefits it might promote are still undetermined. However, preclinical data suggest that PD-1 agonists alone may not be sufficient to prevent graft rejection, and that combination therapy with CTLA-4Ig, reagents targeting other coinhibitory receptors or administration of antigen-specific therapies may be needed. Preliminary work in mice from our group suggests that enhanced signaling of PD-1 is able to protect the allograft against chronic rejection and induce long-term graft survival in combination with single-dose CTLA-4-Ig in a fully allogeneic HLA mismatched murine cardiac transplant model (Riella et al. unpublished data). Therefore, the simultaneous blockade of B7:CD28 costimulation with enhancement of the coinhibitory PD-1:PD-L1 might translate into an effective immunomodulatory strategy.
Concluding Remarks
PD-1 and its ligands play critical and diverse regulatory roles in the immune system. There are multiple potential mechanisms by which PD-1: PD-L1 interactions might participate in the induction of allograft tolerance. PD-L1 can limit effector T cell function and expansion, as well as induce regulatory T cells, providing several means by which this pathway can tip the balance away from immunity, toward tolerance. The upregulation of PD-1 on T cells and PD-L1 on hematopoietic and nonhematopoietic cells might serve as an important negative feedback mechanism for controlling the alloimmune response and limiting allo-specific T cell activation and proliferation against the allograft. An agonist agent would have the potential to simultaneously inhibit function of effector T cells and promote de novo Treg generation. Such a PD-1 agonist not only could be beneficial in the prevention of allograft rejection, but also has the potential to ameliorate autoimmunity. The fundamental and therapeutic importance of the PD-1: PD-L1 pathway in immune regulation gives impetus to further investigation in transplantation.
Acknowledgments
This work was supported by the following grants: research grant from the American Heart Association to L.V.R., National Institute of Health (NIH) Grant P01 AI56299 to AHS and RO1 AI51559 to M.H.S.;
Abbreviations
- BCR
B cell receptor
- CTLA-4
cytotoxic T lymphocyte-associated antigen 4
- DC
dendritic cell
- EAE
experimental autoimmune encephalomyelitis
- MHC
major histocompatibility complex
- mOVA
membrane-bound chicken OVA
- NOD
non-obese diabetic
- PD-1
Programmed cell death-1
- PD-L1
Programmed cell death-1 ligand
- TCR
T cell receptor
- TLR
Toll-like receptor
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
REFERENCES
- 1.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010 Jul;236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li XC, Rothstein DM, Sayegh MH. Costimulatory pathways in transplantation: challenges and new developments. Immunol Rev. 2009 May;229(1):271–93. doi: 10.1111/j.1600-065X.2009.00781.x. [DOI] [PubMed] [Google Scholar]
- 3.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO journal. 1992;11(11):3887–982. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nature medicine. 1999;5(12):1365–74. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 5.Latchman Y, Wood C, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nature immunology. 2001;2(3):261–9. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 6.Tseng S, Otsuji M, Gorski K, Huang X, Slansky J, Pai S, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. The Journal of experimental medicine. 2001;193(7):839–85. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. International immunology. 1996;8(5):765–837. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 8.Keir M, Butte M, Freeman G, Sharpe A. PD-1 and its ligands in tolerance and immunity. Annual review of immunology. 2008;26:677–1381. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med. 2006 Oct 2;203(10):2223–7. doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O'Shea MA, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008 Nov 15;181(10):6738–46. doi: 10.4049/jimmunol.181.10.6738. [DOI] [PubMed] [Google Scholar]
- 11.Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008 Oct 1;181(7):4832–9. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002 Nov 15;169(10):5538–45. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 13.Sandner SE, Clarkson MR, Salama AD, Sanchez-Fueyo A, Domenig C, Habicht A, et al. Role of the programmed death-1 pathway in regulation of alloimmune responses in vivo. J Immunol. 2005 Mar 15;174(6):3408–15. doi: 10.4049/jimmunol.174.6.3408. [DOI] [PubMed] [Google Scholar]
- 14.Baecher-Allan C, Brown J, Freeman G, Hafler D. CD4+CD25+ regulatory cells from human peripheral blood express very high levels of CD25 ex vivo. Novartis Foundation symposium. 2003;252:67. doi: 10.1002/0470871628.ch6. [DOI] [PubMed] [Google Scholar]
- 15.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007 Oct 15;179(8):5099–108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 16.Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol. 2011 Apr 1;186(7):4200–12. doi: 10.4049/jimmunol.1001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007 Oct;27(4):670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Freeman G, Long A, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of experimental medicine. 2000;192(7):1027–61. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, et al. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 2002 Apr;9(2):133–45. doi: 10.1038/sj/mn/7800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang S, Latchman Y, Buhlmann J, Tomczak M, Horwitz B, Freeman G, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. European journal of immunology. 2003;33(10):2706–22. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- 21.Zhong X, Tumang J, Gao W, Bai C, Rothstein T. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. European journal of immunology. 2007;37(9):2405–15. doi: 10.1002/eji.200737461. [DOI] [PubMed] [Google Scholar]
- 22.Messal N, Serriari N-E, Pastor S, NunÃs̈ J, Olive D. PD-L2 is expressed on activated human T cells and regulates their function. Molecular immunology. 2011;48(15–16):2214–23. doi: 10.1016/j.molimm.2011.06.436. [DOI] [PubMed] [Google Scholar]
- 23.Loke Pn, Allison J. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5336–77. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schreiner B, Mitsdoerffer M, Kieseier B, Chen L, Hartung H-P, Weller M, et al. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. Journal of neuroimmunology. 2004;155(1–2):172–254. doi: 10.1016/j.jneuroim.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Butte M, Keir M, Phamduy T, Sharpe A, Freeman G. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–33. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S, Bajorath Jr, Flies D, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. The Journal of experimental medicine. 2003;197(9):1083–174. doi: 10.1084/jem.20021752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004 Jul 20;101(29):10691–6. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W, Carper K, Malone F, Latchman Y, Perkins J, Fu Y, et al. PD-L1/PD-1 signal deficiency promotes allogeneic immune responses and accelerates heart allograft rejection. Transplantation. 2008 Sep 27;86(6):836–44. doi: 10.1097/TP.0b013e3181861932. [DOI] [PubMed] [Google Scholar]
- 29.Brown J, Dorfman D, Ma F-R, Sullivan E, Munoz O, Wood C, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. Journal of immunology (Baltimore, Md : 1950) 2003;170(3):1257–323. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 30.Keir M, Freeman G, Sharpe A. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. Journal of immunology (Baltimore, Md : 1950) 2007;179(8):5064–134. doi: 10.4049/jimmunol.179.8.5064. [DOI] [PubMed] [Google Scholar]
- 31.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005 Mar;6(3):280–6. doi: 10.1038/ni1165. [DOI] [PubMed] [Google Scholar]
- 32.Riella LV, Watanabe T, Sage PT, Yang J, Yeung M, Azzi J, et al. Essential role of PDL1 expression on nonhematopoietic donor cells in acquired tolerance to vascularized cardiac allografts. Am J Transplant. 2011 Apr;11(4):832–40. doi: 10.1111/j.1600-6143.2011.03451.x. [DOI] [PubMed] [Google Scholar]
- 33.Chemnitz J, Parry R, Nichols K, June C, Riley J. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. Journal of immunology (Baltimore, Md : 1950) 2004;173(2):945–99. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 34.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(24):13866–937. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parry R, Chemnitz J, Frauwirth K, Lanfranco A, Braunstein I, Kobayashi S, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and cellular biology. 2005;25(21):9543–96. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006 Apr 17;203(4):883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, Kaja M-K, et al. T-cell tolerance or function is determined by combinatorial costimulatory signals. The EMBO journal. 2006;25(11):2623–56. doi: 10.1038/sj.emboj.7601146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carter L, Fouser L, Jussif J, Fitz L, Deng B, Wood C, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. European journal of immunology. 2002;32(3):634–77. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 39.Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009 Nov;10(11):1185–92. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005 Nov;5(11):853–65. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- 41.Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med. 2011 Jul 4;208(7):1377–88. doi: 10.1084/jem.20102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010 Jun;11(6):535–42. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hams E, McCarron MJ, Amu S, Yagita H, Azuma M, Chen L, et al. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. 2011 May 15;186(10):5648–55. doi: 10.4049/jimmunol.1003161. [DOI] [PubMed] [Google Scholar]
- 44.Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012 Apr 27;336(6080):485–9. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- 45.Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. 2010 Apr;16(4):452–9. doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009 Jun 4;113(23):5811–8. doi: 10.1182/blood-2009-02-203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuipers H, Muskens F, Willart M, Hijdra D, van Assema FB, Coyle AJ, et al. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. Eur J Immunol. 2006 Sep;36(9):2472–82. doi: 10.1002/eji.200635978. [DOI] [PubMed] [Google Scholar]
- 48.Ansari M, Salama A, Chitnis T, Smith R, Yagita H, Akiba H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. The Journal of experimental medicine. 2003;198(1):63–72. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004 Mar;20(3):327–36. doi: 10.1016/s1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- 50.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995 Nov;3(5):541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 51.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995 Nov 10;270(5238):985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A. 2005 Aug 16;102(33):11823–8. doi: 10.1073/pnas.0505497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999 Aug;11(2):141–51. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 54.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001 Jan 12;291(5502):319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 55.Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J Immunol. 2008 Aug 15;181(4):2513–21. doi: 10.4049/jimmunol.181.4.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishimura H, Honjo T, Minato N. Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med. 2000 Mar 6;191(5):891–8. doi: 10.1084/jem.191.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keir M, Latchman Y, Freeman G, Sharpe A. Programmed death-1 (PD-1):PD-ligand 1 interactions inhibit TCR-mediated positive selection of thymocytes. Journal of immunology (Baltimore, Md : 1950) 2005;175(11):7372–81. doi: 10.4049/jimmunol.175.11.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blank C, Brown I, Marks R, Nishimura H, Honjo T, Gajewski TF. Absence of programmed death receptor 1 alters thymic development and enhances generation of CD4/CD8 double-negative TCR-transgenic T cells. J Immunol. 2003 Nov 1;171(9):4574–81. doi: 10.4049/jimmunol.171.9.4574. [DOI] [PubMed] [Google Scholar]
- 59.Thangavelu G, Parkman JC, Ewen CL, Uwiera RR, Baldwin TA, Anderson CC. Programmed death-1 is required for systemic self-tolerance in newly generated T cells during the establishment of immune homeostasis. J Autoimmun. 2011 May;36(3–4):301–12. doi: 10.1016/j.jaut.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 60.Fife B, Guleria I, Gubbels Bupp M, Eagar T, Tang Q, Bour-Jordan H, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. The Journal of experimental medicine. 2006;203(12):2737–84. doi: 10.1084/jem.20061577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarrio ML, Grabie N, Bu DX, Sharpe AH, Lichtman AH. PD-1 Protects against Inflammation and Myocyte Damage in T Cell-Mediated Myocarditis. J Immunol. 2012 May 15;188(10):4876–84. doi: 10.4049/jimmunol.1200389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Chung Y, Bishop C, Daugherty B, Chute H, Holst P, et al. Regulation of T cell activation and tolerance by PDL2. Proc Natl Acad Sci U S A. 2006 Aug 1;103(31):11695–700. doi: 10.1073/pnas.0601347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin SJ, Peacock CD, Bahl K, Welsh RM. Programmed death-1 (PD-1) defines a transient and dysfunctional oligoclonal T cell population in acute homeostatic proliferation. J Exp Med. 2007 Oct 1;204(10):2321–33. doi: 10.1084/jem.20062150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joffre O, Santolaria T, Calise D, Al Saati T, Hudrisier D, Romagnoli P, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med. 2008 Jan;14(1):88–92. doi: 10.1038/nm1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008 May 30;133(5):775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 66.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001 Jan;27(1):68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 67.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001 Jan;27(1):20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 68.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003 Apr;4(4):330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 69.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003 Feb 14;299(5609):1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 70.Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004 Oct;21(4):491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 71.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005 Apr 4;201(7):1061–7. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pyzik M, Piccirillo CA. TGF-beta1 modulates Foxp3 expression and regulatory activity in distinct CD4+ T cell subsets. J Leukoc Biol. 2007 Aug;82(2):335–46. doi: 10.1189/jlb.1006644. [DOI] [PubMed] [Google Scholar]
- 73.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007 Aug 6;204(8):1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007 Jun;7(6):443–53. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- 75.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007 Apr 1;178(7):4022–6. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 76.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004 May 17;199(10):1401–8. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PDL1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009 Dec 21;206(13):3015–29. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krupnick AS, Gelman AE, Barchet W, Richardson S, Kreisel FH, Turka LA, et al. Murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T cells. J Immunol. 2005 Nov 15;175(10):6265–70. doi: 10.4049/jimmunol.175.10.6265. [DOI] [PubMed] [Google Scholar]
- 79.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008 Jul 8;105(27):9331–6. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang C, Li Y, Proctor TM, Vandenbark AA, Offner H. Down-modulation of programmed death 1 alters regulatory T cells and promotes experimental autoimmune encephalomyelitis. J Neurosci Res. 2010 Jan;88(1):7–15. doi: 10.1002/jnr.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011 Nov 30;3(111):111ra20. doi: 10.1126/scitranslmed.3003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008 Mar 17;205(3):565–74. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qu Y, Zhang B, Zhao L, Liu G, Ma H, Rao E, et al. The effect of immunosuppressive drug rapamycin on regulatory CD4+CD25+Foxp3+T cells in mice. Transpl Immunol. 2007 Apr;17(3):153–61. doi: 10.1016/j.trim.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 84.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007 Jan 1;178(1):320–9. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 85.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007 Aug 6;204(8):1765–74. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005 Dec;6(12):1219–27. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 87.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008 Jun 3;105(22):7797–802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007 Aug 1;179(3):1427–30. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 89.Franceschini D, Paroli M, Francavilla V, Videtta M, Morrone S, Labbadia G, et al. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. The Journal of clinical investigation. 2009;119(3):551–615. doi: 10.1172/JCI36604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yi T, Li X, Yao S, Wang L, Chen Y, Zhao D, et al. Host APCs augment in vivo expansion of donor natural regulatory T cells via B7H1/B7.1 in allogeneic recipients. J Immunol. 2011 Mar 1;186(5):2739–49. doi: 10.4049/jimmunol.1002939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Izawa A, Yamaura K, Albin MJ, Jurewicz M, Tanaka K, Clarkson MR, et al. A novel alloantigen-specific CD8+PD1+ regulatory T cell induced by ICOS-B7h blockade in vivo. J Immunol. 2007 Jul 15;179(2):786–96. doi: 10.4049/jimmunol.179.2.786. [DOI] [PubMed] [Google Scholar]
- 92.Morita M, Fujino M, Jiang G, Kitazawa Y, Xie L, Azuma M, et al. PD-1/B7-H1 interaction contribute to the spontaneous acceptance of mouse liver allograft. Am J Transplant. 2010 Jan;10(1):40–6. doi: 10.1111/j.1600-6143.2009.02859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Salama A, Chitnis T, Imitola J, Ansari M, Akiba H, Tushima F, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. The Journal of experimental medicine. 2003;198(1):71–9. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tanaka K, Albin MJ, Yuan X, Yamaura K, Habicht A, Murayama T, et al. PDL1 is required for peripheral transplantation tolerance and protection from chronic allograft rejection. J Immunol. 2007 Oct 15;179(8):5204–10. doi: 10.4049/jimmunol.179.8.5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang L, Han R, Hancock WW. Programmed cell death 1 (PD-1) and its ligand PD-L1 are required for allograft tolerance. Eur J Immunol. 2007 Oct;37(10):2983–90. doi: 10.1002/eji.200737583. [DOI] [PubMed] [Google Scholar]
- 96.Yang J, Popoola J, Khandwala S, Vadivel N, Vanguri V, Yuan X, et al. Critical role of donor tissue expression of programmed death ligand-1 in regulating cardiac allograft rejection and vasculopathy. Circulation. 2008 Feb 5;117(5):660–9. doi: 10.1161/CIRCULATIONAHA.107.741025. [DOI] [PubMed] [Google Scholar]
- 97.Ozkaynak E, Wang L, Goodearl A, McDonald K, Qin S, O'Keefe T, et al. Programmed death-1 targeting can promote allograft survival. J Immunol. 2002 Dec 1;169(11):6546–53. doi: 10.4049/jimmunol.169.11.6546. [DOI] [PubMed] [Google Scholar]
- 98.Ito T, Ueno T, Clarkson MR, Yuan X, Jurewicz MM, Yagita H, et al. Analysis of the role of negative T cell costimulatory pathways in CD4 and CD8 T cell-mediated alloimmune responses in vivo. J Immunol. 2005 Jun 1;174(11):6648–56. doi: 10.4049/jimmunol.174.11.6648. [DOI] [PubMed] [Google Scholar]
- 99.Yang J, Riella LV, Chock S, Liu T, Zhao X, Yuan X, et al. The novel costimulatory programmed death ligand 1/B7.1 pathway is functional in inhibiting alloimmune responses in vivo. J Immunol. 2011 Aug 1;187(3):1113–9. doi: 10.4049/jimmunol.1100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abe M, Wang Z, de Creus A, Thomson AW. Plasmacytoid dendritic cell precursors induce allogeneic T-cell hyporesponsiveness and prolong heart graft survival. Am J Transplant. 2005 Aug;5(8):1808–19. doi: 10.1111/j.1600-6143.2005.00954.x. [DOI] [PubMed] [Google Scholar]
- 101.Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, Korthauer U, et al. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. 2003 Apr 1;170(7):3637–44. doi: 10.4049/jimmunol.170.7.3637. [DOI] [PubMed] [Google Scholar]
- 102.Koehn BH, Ford ML, Ferrer IR, Borom K, Gangappa S, Kirk AD, et al. PD-1-dependent mechanisms maintain peripheral tolerance of donor-reactive CD8+ T cells to transplanted tissue. J Immunol. 2008 Oct 15;181(8):5313–22. doi: 10.4049/jimmunol.181.8.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chadha R, Heidt S, Jones ND, Wood KJ. Th17: contributors to allograft rejection and a barrier to the induction of transplantation tolerance? Transplantation. 2011 May 15;91(9):939–45. doi: 10.1097/TP.0b013e3182126eeb. [DOI] [PubMed] [Google Scholar]
- 104.Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. 2010 Aug 27;33(2):241–53. doi: 10.1016/j.immuni.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blazar BR, Carreno BM, Panoskaltsis-Mortari A, Carter L, Iwai Y, Yagita H, et al. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-gamma-dependent mechanism. J Immunol. 2003 Aug 1;171(3):1272–7. doi: 10.4049/jimmunol.171.3.1272. [DOI] [PubMed] [Google Scholar]
- 106.Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010 Oct 7;116(14):2484–93. doi: 10.1182/blood-2010-03-275446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010 Aug 26;116(8):1291–8. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Paust S, Lu L, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(28):10398–801. doi: 10.1073/pnas.0403342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dejean A, Beisner D, Ch'en I, Kerdiles Y, Babour A, Arden K, et al. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nature immunology. 2009;10(5):504–17. doi: 10.1038/ni.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nature immunology. 2002;3(11):1097–198. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 111.Guleria I, Sayegh MH. Maternal acceptance of the fetus: true human tolerance. J Immunol. 2007 Mar 15;178(6):3345–51. doi: 10.4049/jimmunol.178.6.3345. [DOI] [PubMed] [Google Scholar]
- 112.Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. 2008 Dec 5;322(5907):1562–5. doi: 10.1126/science.1164511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guleria I, Khosroshahi A, Ansari MJ, Habicht A, Azuma M, Yagita H, et al. A critical role for the programmed death ligand 1 in fetomaternal tolerance. J Exp Med. 2005 Jul 18;202(2):231–7. doi: 10.1084/jem.20050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.D'Addio F, Riella LV, Mfarrej BG, Chabtini L, Adams LT, Yeung M, et al. The link between the PDL1 costimulatory pathway and Th17 in fetomaternal tolerance. J Immunol. 2011 Nov 1;187(9):4530–41. doi: 10.4049/jimmunol.1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation. 2007 Oct 30;116(18):2062–71. doi: 10.1161/CIRCULATIONAHA.107.709360. [DOI] [PubMed] [Google Scholar]
- 116.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003 Nov;33(11):3117–26. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 117.Starke A, Lindenmeyer MT, Segerer S, Neusser MA, Rusi B, Schmid DM, et al. Renal tubular PD-L1 (CD274) suppresses alloreactive human T-cell responses. Kidney Int. 2010 Jul;78(1):38–47. doi: 10.1038/ki.2010.97. [DOI] [PubMed] [Google Scholar]
- 118.Gao W, Demirci G, Strom TB, Li XC. Stimulating PD-1-negative signals concurrent with blocking CD154 co-stimulation induces long-term islet allograft survival. Transplantation. 2003 Sep 27;76(6):994–9. doi: 10.1097/01.TP.0000085010.39567.FB. [DOI] [PubMed] [Google Scholar]
- 119.Dudler J, Li J, Pagnotta M, Pascual M, von Segesser LK, Vassalli G. Gene transfer of programmed death ligand-1.Ig prolongs cardiac allograft survival. Transplantation. 2006 Dec 27;82(12):1733–7. doi: 10.1097/01.tp.0000250757.69384.79. [DOI] [PubMed] [Google Scholar]
- 120.Peng W, Ran B, Ma Y, Huang X, Chang Q, Wang X. Dendritic cells transfected with PDL1 recombinant adenovirus induces T cell suppression and long-term acceptance of allograft transplantation. Cell Immunol. 2011;271(1):73–7. doi: 10.1016/j.cellimm.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 121.Wherry EJ. T cell exhaustion. Nat Immunol. 2011 Jun;12(6):492–9. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 122.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009 Jul 10;138(1):30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 123.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009 Mar 12;458(7235):206–10. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Valujskikh A, Li XC. Memory T cells and their exhaustive differentiation in allograft tolerance and rejection. Curr Opin Organ Transplant. 2012 Feb;17(1):15–9. doi: 10.1097/MOT.0b013e32834ee443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010 Jul 1;28(19):3167–75. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012 Jan 9; doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mc Dermott DF DC, Sznol M, Sosman JA, Smith DC, Powderly JD, Fetquate DM, Kollia G, Gupta K, Wigginton J. A phase I study to evaluate safety and antitumor activity of biweekly BMS-936558 in patients with RCC and other advanced refractory malignancies. J Clin Oncol. 2011;29(Suppl 7) abstr 331. [Google Scholar]