

Abstract
CD8+ T cells are essential for controlling Trypanosoma cruzi infection. During Brazil strain infection, C57BL/6 mice expand parasite-specific CD8+ T cells recognizing the dominant TSKB20 (ANYKFTLV) and sub-dominant TSKB74 (VNYDFTLV) trans-sialidase gene (TS)-encoded epitopes with up to 40% of all CD8+ T cells specific for these epitopes. Though this is one of the largest immunodominant T cell responses described for any infection, most mice fail to clear T. cruzi and subsequently develop chronic disease. To determine if immunodominant TS-specific CD8+ T cells are necessary for resistance to infection, we epitope-tolerized mice by high-dose intravenous injections of TSKB20 or TSKB74 peptides. Tolerance induction led to deletion of TS-specific CD8+ T cells but did not prevent the expansion of other effector CD8+ T cell populations. Mice tolerized against either TSKB20 or TSKB74, or both epitopes simultaneously, exhibited transient increases in parasite loads, though ultimately they controlled the acute infection. Furthermore, BALB/c mice tolerized against the TSKD14 peptide effectively controlled acute T. cruzi infection. These data are consistent with the hypothesis that development of high frequency CD8+ T cell populations focused on TS-derived epitopes contributes to optimal control of acute infection, but is not required for the development of immune resistance.
Introduction
CD8+ T cells are critical for adaptive immune control of intracellular pathogens by virtue of their ability to produce a variety of cytokines and to directly target infected host cells for destruction. Pathogen-specific CD8+ T cells recognize foreign peptide epitopes presented in the context of surface bound class I major histocompatibility complexes (MHC I) using clonally diverse T cell receptors (TCR). During the course of infection with viral, bacterial, and protozoan pathogens, clones of pathogen-specific CD8+ T cells expand in number, providing the host with the effector cells capable of controlling pathogen load (1–2). A focused, reproducible hierarchy of epitope-specific CD8+ T cells often occurs in which certain clones are represented at higher numbers (dominant) than other pathogen-specific T cells (sub-dominant), a phenomenon termed immunodominance (3).
Much of our knowledge concerning the role of dominant and sub-dominant CD8+ T cells in control of infection has been derived from mouse infections with model bacterial (e.g. Listeria monocytogenes) (4) and viral (Lymphocytic Choriomeningitis virus, Influenza virus, and Vaccinia virus) (5) pathogens. For these less complex viral and bacterial pathogens, dominant CD8+ T cells recognizing a small subset of pathogen-derived peptides are sufficient for adaptive immune control of infection. In comparison, protozoan parasites are more complex antigenically due to their larger genomes and proteomes as well as lifecycles involving distinct extracellular and intracellular stages occurring within a host. Though dominant CD8+ T cells have recently been described for several intracellular parasites (6–10), the role that these populations play in immune resistance to infection is not fully understood.
In addition to being larger, many parasite genomes also contain greatly expanded sets of variant gene families encoding surface expressed and secreted proteins (11–12). Our group has recently identified immunodominant CD8+ T cells responding to acute and chronic Trypanosoma cruzi infection that are specific for the (H-2Kb-binding) TSKB20 (ANYKFTLV) and TSKB18 (ANYDFTLV) epitopes encoded by trans-sialidase (TS)4 gene family members (8). The dominant TSKB20-specific response (which also recognizes the cross-reactive TSKB21 peptide) represents approximately 20–30% of the total CD8+ T cell compartment and the sub-dominant TSKB18-specific population (which recognizes the cross-reactive TSKB74 peptide) represents 4–10% of effector CD8+ T cells at the peak of acute infection with Brazil strain T. cruzi in C57BL/6 (B6) mice. This degree of immunodominance is remarkable considering there are >1,400 annotated TS family gene members in the CL Brenner reference genome (of >12,000 annotated genes) (12). Moreover, distinct strains of T. cruzi appear to have unique sets of TS genes (8, 13), suggesting that this gene family has evolved at a population level under considerable immune pressure. Unlike persistent viral infections (14), the CD8+ T cells recognizing TS-derived epitopes remain highly competent throughout this chronic infection, despite persistent antigen exposure (8, 15–18). However there is as yet no evidence that these sustained effector responses select for TS epitope-loss mutants of the parasite (17).
Although some TS gene products have been experimentally confirmed as capable of performing the critical enzymatic function of transferring sialic acid residues from host glycoproteins to molecules on the parasite’s surface, the vast majority of TS gene family members lack evidence of this activity (19). Thus, it is unclear what the selective advantage for expansion of the TS gene family is if they provide numerous targets for adaptive immunity (20–21). Some have proposed that TS genes participate in immune evasion, promoting the chronic nature of T. cruzi infection (12, 17, 19, 22–26). The strong immunodomination by TS-derived epitopes predictably results in the out-competition of other epitope-specific CD8+ T cell populations. However, the significance of the tight focusing of the CD8+ T cell response on only a few of the vast array of possible parasite-derived epitopes is not known. Herein, we explore the role of immunodominant CD8+ T cells in immune resistance to T. cruzi infection by inducing immunological tolerance to TS-derived epitopes during the course of acute infection. Though infection with this parasite elicits one of the strongest immunodominant CD8+ T cells responses documented, we find the focus of the adaptive immune response to be remarkably plastic.
Materials and Methods
Mice and parasites
C57BL/6 and BALB/c mice were obtained from the National Cancer Institute at Frederick (Frederick, MD) and kept under specific pathogen-free conditions at the Coverdell Center animal facility (University of Georgia, Athens, GA). For T. cruzi infections, 8 week-old female mice were infected intraperitoneally (ip) with 1 × 103 trypomastigotes of the Brazil strain. Trypomastigotes were maintained in tissue culture by serial passage through Vero cells. Mice were euthanized by CO2 inhalation. The University of Georgia Institutional Animal Care and Use Committee approved all animal use protocols.
Peptide treatments
Peptides were synthesized by SigmaGenosys (Saint Louis, Missouri) or GenScript (Piscataway, New Jersey). Peptides used were the H-2Kb-restricted TSKB20 (ANYKFTLV), TSKB74 (VNYDFTLV), and OVA257-264 (SIINFEKL) peptides, or the H-2Kd-restricted peptides TSKD14 (IYNVGQVSI) and LL091-99 (GYKDGNEYI). Lyophilized peptide was suspended in dimethyl sulfoxide (DMSO) at a concentration of 100 mg/ml and stored at −20°C. Stock peptide was diluted to the desired concentration in sterile saline (PBS) for intravenous (iv) injection (each mouse received 100 μl per injection). Peptide-treated mice initially received 300 μg peptide on day -7 and 100 μg on days -4 and -1. Mice were infected on day 0 and injected with 100 μg peptide weekly until the end of the experiment. An equal quantity of peptide was injected whether mice received one or two peptides simultaneously. Tolerized mice were sacrificed 7 days after final peptide treatment.
T cell phenotyping
For ex vivo lymphocyte phenotyping, spleens were removed and dissociated by rubbing between two glass slides in a medium of hypotonic ammonium chloride to lyse red blood cells. Cell numbers were determined on a Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter, Fullerton, CA). 5 × 106 washed splenocytes were suspended for staining in PBS with 1% bovine serum albumin and 0.05% sodium azide (PAB) (both from Sigma). TSKB20/Kb, TSKB74/Kb, and TSKD14/Kd tetramers were synthesized at the Tetramer Core Facility (Emory University, Atlanta, Georgia) and were labeled with PE (Molecular Probes, Carlsbad, CA). Antibodies used were CD8 Pacific Blue, CD4 PE-Cy5, CD127 PE-Cy7, FoxP3 PE (ebioscience, San Diego, CA), CD11b PE-Cy5, B220 PE-Cy5, CD25 APC (CALTAG), CD44 APC, CD11a FITC, and CD62L FITC (BD bioscience, San Jose, CA). Cells were stained at 4°C for 30 minutes, washed with PAB and fixed in 2% formaldehyde. The ebioscience intracellular staining kit was used for FoxP3 staining. At least 500,000 cells were collected for each sample on a Cyan ADP using Summit version 4.3 (Beckman Coulter). FlowJo Flow Cytometry Analysis Software Version 7 (Tree Star, Ashland, OR) was used for analyses.
T cell stimulation and intracellular cytokine staining
1.5 × 106 splenocytes were stimulated in 96-well round-bottom tissue culture plates (Costar, Corning, NY) at 37°C for 5 hr in the presence 1 μM peptide and brefeldin A (Golgi Plug, BD biosciences). For polyclonal activation, wells were pulsed with 30 μg anti-mouse CD3ε (ebioscience) for 1 hr at 37°C and excess antibody was removed prior to the addition of cells. Cells were stained with CD8 Pacific Blue and CD4 FITC (ebioscience) followed by intracellular staining with IFNγ APC (BD biosciences) and IL-10 PE (ebioscience) according to the cytofix/cytoperm kit (BD biosciences). At least 150,000 cells were collected for analysis.
In vivo cytotoxicity assay
Spleen cells from naïve mice were incubated for 1 hour at 37°C with 10 μM peptide or media alone, and then labeled with different concentrations of carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes) as described (8) to produce CFSE high, medium, and low populations. Equal numbers of CFSE-labeled cells were transferred ip into recipients, and after 16 hours, splenocytes were isolated and CFSE-labeled cells were detected by flow cytometry. Percentage of specific killing was determined using the formula 1−[(% CFSElo naïve/% CFSEmed/hi naïve)/(% CFSElo infected/CFSEmed/hi infected)] × 100%.
Real-time PCR
Mouse hind leg muscles were collected and popliteal lymph nodes were removed as well as extraneous adipose tissue prior to DNA extraction as described (27). Extracted DNA was analyzed by real-time PCR essentially as described (27). PCR reactions consisted of iQ SYBR Green Supermix (BioRad) and primers specific for T. cruzi or mouse genomic DNA (27). An iQ5 Multi-Color Real-Time PCR Detection System was used with iQ5 Standard Edition Optical System Software Version 2 (both BioRad). T. cruzi equivalents were calculated as the quantity of T. cruzi satellite DNA divided by the quantity of mouse TNFα DNA in each sample.
Statistical analysis
Statistical significance was calculated using a two-tailed students t test.
Results
Repetitive intravenous administration of peptide results in depletion of epitope-specific CD8+ T cells during acute T. cruzi infection
In order to determine the importance of immunodominant TS-peptide-specific CD8+ T cell responses in T. cruzi infection, we first developed a system in which immunodominant TS-specific CD8+ T cell responses were ablated. Because the TSKB20 and TSKB74 epitopes (and cross-reactive epitopes) are encoded by numerous TS genes (more than 200 in the case of TSKB20/21; (8)), it was not feasible to generate gene knock-out parasites which do not express either antigen. We instead induced immune tolerance to these epitopes by administration of high doses of soluble peptide intravenously (iv) (Fig. 1A), a method previously used to induce and maintain epitope-specific tolerance in models of viral infection (28–32).
Figure 1. Repetitive intravenous administration of peptide depletes epitope-specific CD8+ T cells during acute T. cruzi infection.

(A) Protocol used to induce epitope-specific tolerance using high-dose peptide administration. Mice were injected with 100 μl peptide on indicated days before and after infection with 1,000 Brazil trypomastigotes. The primary injection was with 300 μg peptide and 100 μg was administered in the following treatments. Tolerized mice were allowed to rest for 7 days after final treatment before the experimental endpoint. Spleens of peptide-treated mice were assayed for the presence of epitope-specific CD8+ T cells at 21 days post-infection with Brazil strain T. cruzi. (B) Splenocytes from OVA257-264-, TSKB20-, and TSKB74-treated B6 mice were stained for CD44 and TSKB20/Kb or TSKB74/Kb tetramers. Histograms are gated on CD8+ cells that were CD4− CD11b− B220−. Numbers indicate percentage of tetramer+ cells of total CD8+ T cells. Data are from individual mice and are representative of five experiments. The total numbers of TSKB20/Kb (C) or TSKB74/Kb (D) CD8+ T cells per spleen were calculated. Data are mean ± SEM and are cumulative from 3 separate experiments (n=4–11 per group). *, p<0.05 compared to OVA Tx group.
At the peak of the T cell response (~3 weeks post-infection; (8) and data not shown), TSKB20- and TSKB74-specific CD8+ T cells in the spleens of mice injected with the respective peptides were nearly undetectable (Fig. 1B). Infected control mice injected with the irrelevant OVA257-264 peptide had normal proportions of TSKB20 and TSKB74 tetramer+ CD8+ T cells compared with infected mice treated with PBS + DMSO or non-treated mice (data not shown). Importantly, TSKB20-tolerized mice had a normal complement of TSKB74 tetramer+ CD8+ T cells and TSKB74-injected mice had TSKB20 tetramer+ CD8+ T cells (Fig. 1B), demonstrating that depletion via TS-derived peptides was epitope-specific and did not prevent priming of other CD8+ T cells specific for homologous TS epitopes. Peptide treatments effectively prevented expansion of peptide-specific T cells throughout the course of acute infection (Fig. 1C–D). Interestingly, spleens of TSKB20-treated mice had significantly more TSKB74 tetramer+ CD8+ T cells at the peak of expansion compared to OVA257-264-treated mice (Fig. 1D). A similar compensation in immunodominance hierarchies has been noted in virus infection with epitope-loss variants and deletion mutants (33–36), and suggests that T. cruzi-specific CD8+ T cells with alternative specificities expand in the absence of competition by the normally dominant TSKB20-specific CD8+ T cell population.
Peptide tolerized mice lack epitope-specific CD8+ T cell effector functions
Depletion of tetramer+ CD8+ T cells by repetitive peptide treatment also resulted in epitope-specific immune tolerance as assessed by the failure to produce IFNγ in response to stimulation with peptide ex vivo (Fig. 2A–C). Additionally, both TSKB20 and TSKB74 tolerized mice were deficient in peptide-specific cytotoxicity in vivo at 28 days post-infection (Fig. 2D–E). Compared to OVA257-264-treated mice, TSKB20-treated mice killed the majority of TSKB74-pulsed target cells but not TSKB20-pulsed targets, and TSKB74-treated mice efficiently killed most TSKB20-loaded targets but not TSKB74-loaded targets (Fig. 2D–E). Though peptide-treated mice exhibited essentially background levels of tolerizing epitope-specific CD8+ T cells (Fig. 1 and 2A–C), low levels of tolerizing epitope-specific cytotoxicity were apparently maintained in vivo (Fig. 2D–E). Nevertheless, this residual killing was less than that previously observed for very sub-dominant T. cruzi-specific CD8+ T cells (8, 37). Notably, CD8+ T cells from OVA257-264-treated mice produced IFNγ after stimulation with TSKB20 or TSKB74 peptide but did not produce IFNγ after stimulation with OVA257-264 peptide (Fig. 2A), indicating that the peptide treatment does not stimulate T cell priming in the face of infection-induced inflammation. From these experiments we conclude that peptide treatments prevented the normally robust expansion of functional CD8+ T cells specific for the tolerizing peptide.
Figure 2. Peptide tolerized mice lack epitope-specific CD8+ T cell effector functions.

Peptide-treated mice were assayed for epitope-specific effector functions during acute T. cruzi infection. Splenocytes were incubated for 5 hours in the presence of 1 μM peptide and brefeldin A. (A) Representative intracellular IFNγ staining at 21 days post-infection. Histograms are gated on CD8+ CD4− lymphocytes and numbers indicate percentage of cytokine-producing CD8+ T cells. Data are from individual mice and are representative of five experiments. The percentage of CD8+ T cells producing IFNγ in response to (B) TSKB20- or (C) TSKB74-peptide stimulation over the course of acute infection. Data are mean ± SEM from 1 experiment (n=4–5 per group) and are representative of five experiments. *, p<0.05 compared to OVA Tx group. Naïve splenocytes were pulsed with 1 μM TSKB20, 1 μM TSKB74, or no peptide and then labeled with high, medium, or low concentrations of CFSE, respectfully. At 28 days post-infection, equal numbers of each population were co-transferred ip into mice then detected in the spleens after 16 hours. Numbers indicate the percentage of specific lysis measured for individual mice. (E) Data are mean ± SEM and are cumulative of two in vivo CTL experiments (n=6–7 per group). *, p<0.05 compared to OVA Tx group.
Peptide induced tolerance does not lead to enhanced regulatory T cell populations
Although the evidence above indicates that the decreased tetramer+ populations and functions were epitope-specific, we considered the possibility that the high-dose of peptide administered could induce enhanced regulatory T cell (Treg) populations capable of general immune suppression. Approximately 15% of splenic CD4+ T cells in naive mice expressed the FoxP3 transcription factor (two-thirds co-expressed the IL-2Rα chain (CD25)) (Fig. 3A), whereas the proportion of Tregs decreased in spleens after T. cruzi infection (Fig. 3A–B). Importantly, the proportion of Tregs in spleens of all peptide treated groups were similar over the course of acute infection (Fig. 3A–B), indicating that tolerization with T. cruzi-derived epitopes did not lead to an atypical expansion of Tregs.
Figure 3. Peptide induced tolerance does not enhance regulatory T cell populations.

Tolerized mice were monitored for regulatory T cell populations (A–B) as well as IL-10 production (C) during infection. (A) Representative intracellular staining for the FoxP3 transcription factor at 21 days post-infection. Histograms are gated on CD4+, and were co-stained with CD25. Numbers indicate the percentage of cells in each quadrant. (B) The percentage of FoxP3 expressing CD4+ T cells in spleens over the course of infection. Data are mean ± SEM from 2 experiments (n=3–12 per group). Splenocytes were incubated for 5 hours in media alone or with plate-bound mouse-CD3 monoclonal antibody in the presence of brefeldin A. Histograms are gated on CD4+ CD8− cells, and numbers indicate the percentage of IL-10 and/or IFNγ positive cells per quadrant. Data are from individual mice and are representative of three experiments.
Production of IL-10 by CD4+ T cells has also been shown to suppress CD8+ T cell function during infections (38). Polyclonally stimulated CD4+ T cells from infected peptide-treated mice exhibited robust IFNγ production whereas few IL-10-producing CD4+ T cells were detectable (Fig. 3C). Both OVA257-264 and TS peptide-treated mice maintained similar populations of cytokine producing CD4+ T cells during acute infection (Fig. 3C and data not shown). Furthermore, stimulation of splenocytes with TSKB20 or TSKB74 peptides in vitro failed to elicit IL-10 production irrespective of the in vivo peptide treatment (data not shown). Thus, we found no evidence that peptide-induced T cell tolerance was due to extrinsic T cell regulation, and conclude that it likely was the result of deletion of peptide-specific CD8+ T cells.
Mice tolerized against immunodominant T. cruzi epitopes control acute infection
T. cruzi Brazil strain-infected mice genetically deficient for, or depleted of, CD8+ T cells exhibit uncontrolled parasitemia and mortality by approximately one month post-infection (39–41). However, neither TSKB20- nor TSKB74-tolerized mice deficient in the respective immunodominant T cell population succumbed to acute infection of up to 35 days. Since skeletal muscle is a site of T. cruzi persistence in this model, we measured parasite load in muscle to determine the quality of immune control of T. cruzi infection in tolerized mice. The level of parasites, as measured by real-time PCR, was similar between peptide-treated groups of mice through-out infection (Fig. 4), with the exception that TSKB74-treated mice had an increased number of parasites at day 21 post-infection compared with control OVA257-264-treated mice (p=0.03) (Fig. 4). Although several TSKB20-tolerized mice had elevated numbers of parasites at 21 days post-infection, the group average was not statistically different (p=0.4) when compared to OVA257-264 -treated mice. We observed slightly greater cellular infiltration as well as parasitized host cells in muscle sections of individual tolerized mice exhibiting increased parasite loads (data not shown), further suggesting that depleting TSKB20- or TSKB74-specific CD8+ T cells can have a negative, though minor, impact on control of infection. Ultimately, both TS-peptide tolerized groups controlled parasite loads similar to OVA257-264-treated mice (Fig. 4), demonstrating that immune control of T. cruzi infection occurs despite the absence of the normal immunodominant CD8+ T cell population.
Figure 4. Mice tolerized against immunodominant T. cruzi epitopes control acute infection.
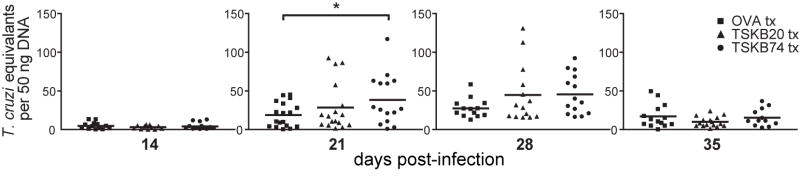
The quantity of T. cruzi DNA in skeletal muscle of peptide-treated mice was detected at indicated time points by real-time PCR. Data points are for individual mice and bars are the mean from five experiments (n=11–17 per group). Several individuals were removed from the analysis because they passed the Grubbs’ outlier test (GraphPad software). *, p<0.05.
Tolerized mice generate protective effector CD8+ T cell responses despite the absence of immunodominant CD8+ T cells
Since the TSKB20- and TSKB74-specific CD8+ T cells can represent as much as 40% of the total T. cruzi-specific CD8+ T cell population in infected mice (8), we next assessed the effect of depleting the immunodominant T cells on the overall size of the responding CD8+ T cell population bearing an activated phenotype. Most CD8+ T cells in a naïve spleen express the lymph node homing receptor, CD62L, and the IL-7Rα chain, CD127, but very few of these naïve T cells have an antigen-experienced phenotype (CD44hi CD11ahi) (Fig. 5A and C). However, after T. cruzi infection, a large proportion of CD8+ T cells from the control OVA257-264-treated mice had up-regulated surface expression of both CD44 and CD11a (Fig. 5A) and down-regulated CD62L and CD127 (Fig. 5C). Spleens of TSKB20- and TSKB74-tolerized mice contained similar proportions of antigen-experienced (CD44hi CD11ahi, CD62Llo CD127lo) CD8+ T cells (Fig. 5A–C) indicating that T. cruzi-specific T cells recognizing alternative parasite epitopes expand during T. cruzi infection when the normally immunodominant T cell populations are absent. The effector function of these T cells was confirmed by demonstrating their production of IFNγ in response to αCD3 stimulation (Fig. 5D).
Figure 5. Tolerized mice generate normal effector CD8+ T cell populations despite the absence of immunodominant CD8+ T cells.

Spleens of peptide-treated mice were assayed for the presence of antigen-experienced effector CD8+ T cells. (A) Representative staining for CD44 and CD11a expression at 21 days post-infection. Histograms are gated on CD8+ cells that were CD4− CD11b− B220−. Similar results were obtained in three separate experiments. (B) The total number of CD44hi CD11ahi CD8+ T cells per spleen was calculated. Data are mean ± SEM from one experiment (n=4–5 per group). *, p<0.05 compared to the OVA Tx group. (C) Representative staining for CD62L and CD127 at 21 days post-infection. Histograms are gated on CD8+ cells that were CD4− CD11b− B220−. Numbers indicate the percentage of CD8+ T cells that have lost (left) or retained (right) expression of each marker (bold lines). Shaded lines are a fluorescence-minus-one control for the indicated marker. Similar results were obtained in three separate experiments. (D) Splenocytes were incubated for five hours in media alone or with plate-bound mouse-CD3 monoclonal antibody in the presence of brefeldin A at 35 days post-infection. Data are the mean (± SEM) percentage of CD8+ T cells producing IFNγ for each condition (n=3–17 per group from three separate experiments). The percentage of CD8+ T cells capable of producing IFNγ was also similar between groups at 14, 21, and 28 days post-infection (data not shown).
B6 mice tolerized simultaneously against both TSKB20 and TSK74 (labeled as TS tx) showed a predictable decrease in both TSKB20- and TSKB74-specific CD8+ T cells (Fig. 6A and B) but interestingly had increased numbers of activated (CD44hi CD11ahi) CD8+ T cells (Fig. 6C) and a greater percentage of CD8+ T cells capable of producing IFNγ in response to αCD3 stimulation (Fig. 6B). Few activated CD8+ T cells had decreased expression of CD3 or TCRβ-chain in either control or TS-peptide tolerized mice (data not shown), excluding the possibility that the CD44hi CD11ahi CD8+ T cell population consisted of expanded TSKB20- or TSKB74-specific cells that were undetected by tetramer staining due to downregulation of the TCR. Furthermore, the TS-tolerized mice had an increased number of tissue parasites at the peak of infection (p=0.018 at 21 days post-infection), although they had effectively controlled their parasite load by 28 days post-infection (Fig. 6D). Thus, TSKB20- and TSKB74-specific CD8+ T cells are required for optimal control of T. cruzi at the peak of the infection, but other CD8+ T cells of unknown specificity can substitute to eventually contain the acute infection.
Figure 6. Mice tolerant to both TSKB20 and TSKB74 are ultimately resistant to T. cruzi infection.

B6 mice were injected with both TSKB20 and TSKB74 peptides (TS tx) similar to the description in Fig. 1A. TS Tx mice received the same total quantity of peptide as OVA Tx mice, i.e. half the effective dose of each individual peptide compared to experiments described in Fig. 1–5. (A) Representative tetramer staining at 21 days post-infection. Histograms are gated on CD8+ cells that were CD4− CD11b− B220−. Numbers indicate percentage of tetramer+ cells out of CD8+ T cells. Data are from individual mice and are representative of five experiments. (B) Representative intracellular staining for IFNγ produced in response to the indicated stimulus (see Materials and Methods). Histograms are gated on CD8+ CD4− cells. Numbers indicate percentage of IFNγ+ CD8+ T cells. (C) The total number of CD44hi CD11ahi CD8+ T cells per spleen was calculated. Data are mean ± SEM from one experiment (n=3–5 per group). *, p<0.05 compared to the OVA Tx group (same control individuals as in Fig 5B). (D) Quantity of T. cruzi DNA in skeletal muscle of peptide-treated mice detected by real-time PCR. Data points are individual mice and bars are the mean from two experiments (n=5–8 per group). One individual outlier was removed from the analysis. *, p<0.05.
BALB/c mice tolerized against a dominant TS-derived epitope do not exhibit enhanced susceptibility to T. cruzi infection
BALB/c mice infected with Brazil strain T. cruzi also generate immunodominant CD8+ T cells specific for a TS-derived epitope (13, 42), although the response to the H-2Kd-restricted TSKD14 (IYNVGQVSI) is at a substantially lower frequency than observed for TSKB20 and TSKB74 in B6 mice (Fig. 7A–D) (13, 42). However, similar to tolerized B6 mice, BALB/c mice tolerized with TSKD14 peptide showed the expected reduction in the TSKD14-specific response (Fig. 7A–E) and also effectively controlled acute T. cruzi infection with parasite loads similar to control LL091-99-tolerized mice throughout the acute infection (Fig. 7F). Furthermore, TSKD14-tolerization did not enhance regulatory T cell populations (data not shown) and a normal expansion of activated CD8+ T cells was observed (data not shown) in mice depleted of TSKD14-specific CD8+ T cells.
Figure 7. TSKD14 tolerized BALB/c mice remain resistant to T. cruzi infection.

BALB/c mice were treated with H-2Kd-restricted TSKD14 or LLO91-99 peptide as described in Fig. 1A. (A) Splenocytes from TSKD14- or LL091-99-treated BALB/c mice were stained for CD44 and TSKD14/Kd. Histograms are gated on CD8+ cells that were CD4− CD11b− B220−. Numbers indicate percentage of tetramer+ cells of total CD8+ T cells. Data are from individual mice and are representative of three experiments. (B) The total number of TSKD14/Kd CD8+ T cells per spleen. Data are mean ± SEM and are cumulative from one experiment (n=4–5 per group). *, p<0.05 compared to LLO Tx group. (C) Representative intracellular staining for IFNγ produced in response to TSKD14 or LL091-99 peptide stimulation. Histograms are gated on CD8+ CD4− cells. (D) The percentage of IFNγ producing CD8+ T cells specific for TSKD14 during acute infection. Data are mean ± SEM from two experiments (n=4–11 per group). *, p<0.05 compared to LLO Tx group. (E) TSKD14-specific in vivo cytotoxicity at 28 days post-infection. (F) Quantity of T. cruzi DNA in skeletal muscle of peptide-treated mice detected by real-time PCR. Data points are individual mice and bars are the mean from two experiments (n=4–10 per group). Several individuals were removed from the analysis because they passed the Grubbs’ outlier test (GraphPad software).
Discussion
Adaptive immunity to intracellular pathogens depends on CD8+ T cell recognition of host cells presenting foreign antigen. The consequences of antigen-specific CD8+ T cell responses focusing on a restricted versus broader set of pathogen-derived epitopes is not fully understood. The issue of immunodominance is particularly complicated for understanding immune control of protozoan parasites potentially presenting an expansive set of antigenic determinates, especially in comparison to viral pathogens where immunodominance has been extensively investigated. In this study, we addressed the role that immunodominant CD8+ T cells play in host resistance to T. cruzi infection by ablating epitope-specific T cells via administrating high-doses of peptide. We achieved significant epitope-specific tolerance against the dominant TSKB20 and sub-dominant TSKB74 peptides in H-2Kb-restricted B6 mice, and the dominant TSKD14 peptide in H-2Kd-restricted BALB/c mice during acute T. cruzi Brazil strain infection. B6 mice tolerized to TSKB20, TSKB74, or both epitopes simultaneously, exhibited modest and transitory increases in parasite load, suggesting that these greatly expanded T cell populations contribute to control of T. cruzi. Although these immunodominant TS-specific CD8+ T cells represent a significant portion of the parasite-specific response, deleting them during infection ultimately had minor consequences for the outcome of infection; thus, they are not required for the acute resistance provided by the adaptive immune response to T. cruzi.
Immunodominant CD8+ T cells are implicated as important for control of intracellular pathogens since they represent a majority of the responding T cell pool in circulation and at sites of infection. Attempts to determine the necessity of immunodominant T cells have often relied upon experimental infections with natural mutants or engineered viruses and bacteria that lack the epitope of interest (34–36, 43–49). Tolerance induction has also been used as a means of depleting mice of epitope-specific CD8+ T cells during viral infection (30–31, 50–55). In some cases, loss of the immunodominant CD8+ T cell population impaired viral control (31, 34, 36, 44, 47, 50–54) or enhanced disease without affecting viral load (34), while in other situations, pathogen load was not affected (30, 43, 46, 49) or disease manifestations were ameliorated (30, 47, 52, 55). Compensation in the dominance hierarchy in the absence of immunodominant T cells occurred in many of these infection models (34–36, 43, 46–48), though often T cells recognizing minor epitopes emerge instead of enhanced sub-dominant responses (35–36, 46–47). Thus, elimination of immunodominant CD8+ T cell responses has variable results depending on the infection model employed and is not readily predictable.
The observation that TS-peptide tolerized mice are resistant to acute T. cruzi infection directed us to question which parasite-derived peptides protective CD8+ T cells respond to in the absence of the normally dominant responses. In our studies, both B6 and BALB/c mice depleted of the previously identified immunodominant TS-specific T cells expanded effector CD8+ T cell populations to a similar level as control mice. These responding effector CD8+ T cells had an antigen-experienced CD44hi CD11ahi phenotype and rapidly produced IFNγ in response to stimulation with anti-CD3 antibodies, whole T. cruzi lysate, or T. cruzi-infected dendritic cells (Rosenberg, unpublished results). However, we were unable to identify the antigen specificity of these compensating T cells in screens against previously predicted CD8+ T cell targets (8) in either mouse strain (Rosenberg, unpublished results). A broader screen for epitope-specific responses will help identify the focus of compensating CD8+ T cells in TS-peptide tolerized mice, though T. cruzi’s large proteome (>12,000 genes) may preclude a full description of all epitopes recognized in the mouse model.
Comparison of the reference genomes for the related trypanosomatids T. cruzi, Trypanosoma brucei, and Leishmania major revealed massive expansion in several gene families encoding surface proteins uniquely in T. cruzi (12, 56). Since surface expressed or secreted proteins are excellent sources of epitopes for both B cell and T cell recognition (57–58), it is hypothesized that these large gene families have expanded because of immune selective pressure (26) and likely are involved in immune evasion (19, 59–60). The TS gene-family has drastically expanded to represent upwards of 6% of the annotated T. cruzi CL Brener genome (12), and this may underestimate by half the true number of full and partial TS sequences (Weatherly, in preparation). Several hundred TS genes encode epitopes recognized by TSKB20-specific CD8+ T cells (8), and many of these gene products are represented in the proteome of the mammalian-dwelling stages of T. cruzi Brazil strain (22). Furthermore, distinct strains likely have distinct sets of TS genes (8), resulting in strain-variant immunodominance patterns (8, 13, 61). The benefit of carrying within an otherwise fairly compact genome a large number of genes encoding related but variable surface proteins is clear in cases where a pathogen expresses only one variant at a time, as with African trypanosomes (62). The relative benefit in terms of immune evasion of simultaneously expressing variants, some of which contain the same immunodominant epitope, is less evident. However, the fact that T. cruzi persists in hosts despite highly functional parasite-specific immune responses suggests that its strategy of immune evasion is successful – if not entirely obvious.
Rodrigues et al. (13, 23) have proposed that the strong immunodominance by TS-specific T cells restricts the generation of a broader, more protective immune response and allows T. cruzi to escape complete destruction. While immunodomination certainly restricts the focus of the immune response, it is probably not the primary reason why the majority of T. cruzi-infected hosts are unable to achieve sterile immunity. First, the documented immunodominant CD8+ T cells of known specificity do not account for all of the T cells responding during infection; it is possible that these T cells of as yet unknown specificity are reactive to a broader set of T. cruzi epitopes. Second, vaccination to boost TS-specific CD8+ T cells enhances protection in mice (17, 20, 63); if immunodominance prevented immune control then one would expect a stronger dominant response induced by prior vaccination to be deleterious for these hosts. Third, though humans and mice generate TS-specific CD8+ T cells, the strong immunodominance observed in B6 mice is somewhat anomalous compared to that observed for other mouse haplotypes (13, 42, 63) and humans (8, 64–65). Therefore, either there are highly immunodominant CD8+ T cell responses whose specificity has yet to be identified in these hosts, or immunodominance per se is not required for persistence. Finally, as shown in this study, tolerizing B6 mice against the immunodominant TS-epitopes had a transient negative impact on host control of T. cruzi replication, but little influence on the ultimate outcome of acute infection. It remains to be determined if diverting the focus of the CD8+ T cell response away from these particular TS-encoded epitopes allows for the recognition of a broader set of epitopes (encoded by TS and other large gene-families or perhaps a more conserved set of genes), or otherwise alters the development of chronic disease due to the long-term persistence of T. cruzi in its host.
Since intracellular protozoan parasites do not rely on host cell machinery for gene expression, the pool of proteins readily introduced into the MHC I presentation pathway is controlled at the level of the parasite. Dominant antigens from numerous parasites are surface expressed and secreted proteins (6–8, 10, 66), therefore it seems likely that these pathogens have evolved to balance the necessary function of these proteins with the possibility of them serving as targets for immune recognition. Intriguingly, protective epitopes encoded in several secreted antigens of other parasites display significant variation both within (10) and between strains (6), similar to TS genes in T. cruzi. This variation has significant outcomes in terms of immunodominance (10) and cross-protection (6, 67). The issue of if and how variant T cell epitopes influence the outcome of these human diseases remains to be determined.
Acknowledgments
The authors are grateful to Angel Padilla, Juan Bustamante, and Matthew Collins for technical assistance and Julie Nelson of the Center for Tropical and Emerging Global Diseases Flow Cytometry Facility at the University of Georgia. We thank members of the Tarleton Research Group for helpful discussion.
Footnotes
This work was supported by US National Institutes of Health grants R01 AI22070 and R01 AI33106 to R.L.T.
Abbreviations used in this paper: TS, trans-sialidase; Treg, regulatory T cell
References
- 1.Wong P, Pamer EG. CD8 T cell responses to infectious pathogens. Annu Rev Immunol. 2003;21:29–70. doi: 10.1146/annurev.immunol.21.120601.141114. [DOI] [PubMed] [Google Scholar]
- 2.Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol. 2000;18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- 3.Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- 4.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 5.Yewdell JW. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity. 2006;25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Blanchard N, Gonzalez F, Schaeffer M, Joncker NT, Cheng T, Shastri AJ, Robey EA, Shastri N. Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat Immunol. 2008;9:937–944. doi: 10.1038/ni.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar KA, Sano G, Boscardin S, Nussenzweig RS, Nussenzweig MC, Zavala F, Nussenzweig V. The circumsporozoite protein is an immunodominant protective antigen in irradiated sporozoites. Nature. 2006;444:937–940. doi: 10.1038/nature05361. [DOI] [PubMed] [Google Scholar]
- 8.Martin DL, Weatherly DB, Laucella SA, Cabinian MA, Crim MT, Sullivan S, Heiges M, Craven SH, Rosenberg CS, Collins MH, Sette A, Postan M, Tarleton RL. CD8+ T-Cell responses to Trypanosoma cruzi are highly focused on strain-variant trans-sialidase epitopes. PLoS Pathog. 2006;2:e77. doi: 10.1371/journal.ppat.0020077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doolan DL, Southwood S, Freilich DA, Sidney J, Graber NL, Shatney L, Bebris L, Florens L, Dobano C, Witney AA, Appella E, Hoffman SL, Yates JR, 3rd, Carucci DJ, Sette A. Identification of Plasmodium falciparum antigens by antigenic analysis of genomic and proteomic data. Proc Natl Acad Sci U S A. 2003;100:9952–9957. doi: 10.1073/pnas.1633254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frickel EM, Sahoo N, Hopp J, Gubbels MJ, Craver MP, Knoll LJ, Ploegh HL, Grotenbreg GM. Parasite stage-specific recognition of endogenous Toxoplasma gondii-derived CD8+ T cell epitopes. J Infect Dis. 2008;198:1625–1633. doi: 10.1086/593019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coppel RL, Black CG. Parasite genomes. Int J Parasitol. 2005;35:465–479. doi: 10.1016/j.ijpara.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 12.El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 13.Tzelepis F, de Alencar BC, Penido ML, Claser C, Machado AV, Bruna-Romero O, Gazzinelli RT, Rodrigues MM. Infection with Trypanosoma cruzi restricts the repertoire of parasite-specific CD8+ T cells leading to immunodominance. J Immunol. 2008;180:1737–1748. doi: 10.4049/jimmunol.180.3.1737. [DOI] [PubMed] [Google Scholar]
- 14.Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007;19:408–415. doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Martin DL, Tarleton RL. Antigen-specific T cells maintain an effector memory phenotype during persistent Trypanosoma cruzi infection. J Immunol. 2005;174:1594–1601. doi: 10.4049/jimmunol.174.3.1594. [DOI] [PubMed] [Google Scholar]
- 16.Bustamante JM, Bixby LM, Tarleton RL. Drug-induced cure drives conversion to a stable and protective CD8+ T central memory response in chronic Chagas disease. Nat Med. 2008;14:542–550. doi: 10.1038/nm1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoft DF, Eickhoff CS, Giddings OK, Vasconcelos JR, Rodrigues MM. Trans-sialidase recombinant protein mixed with CpG motif-containing oligodeoxynucleotide induces protective mucosal and systemic trypanosoma cruzi immunity involving CD8+ CTL and B cell-mediated cross-priming. J Immunol. 2007;179:6889–6900. doi: 10.4049/jimmunol.179.10.6889. [DOI] [PubMed] [Google Scholar]
- 18.Bixby LM, Tarleton RL. Stable CD8+ T cell memory during persistent Trypanosoma cruzi infection. J Immunol. 2008;181:2644–2650. doi: 10.4049/jimmunol.181.4.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frasch AC. Functional diversity in the trans-sialidase and mucin families in Trypanosoma cruzi. Parasitol Today. 2000;16:282–286. doi: 10.1016/s0169-4758(00)01698-7. [DOI] [PubMed] [Google Scholar]
- 20.Miyahira Y, Takashima Y, Kobayashi S, Matsumoto Y, Takeuchi T, Ohyanagi-Hara M, Yoshida A, Ohwada A, Akiba H, Yagita H, Okumura K, Ogawa H. Immune responses against a single CD8+-T-cell epitope induced by virus vector vaccination can successfully control Trypanosoma cruzi infection. Infect Immun. 2005;73:7356–7365. doi: 10.1128/IAI.73.11.7356-7365.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araujo AF, de Alencar BC, Vasconcelos JR, Hiyane MI, Marinho CR, Penido ML, Boscardin SB, Hoft DF, Gazzinelli RT, Rodrigues MM. CD8+-T-cell-dependent control of Trypanosoma cruzi infection in a highly susceptible mouse strain after immunization with recombinant proteins based on amastigote surface protein 2. Infect Immun. 2005;73:6017–6025. doi: 10.1128/IAI.73.9.6017-6025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atwood JA, 3rd, Weatherly DB, Minning TA, Bundy B, Cavola C, Opperdoes FR, Orlando R, Tarleton RL. The Trypanosoma cruzi proteome. Science. 2005;309:473–476. doi: 10.1126/science.1110289. [DOI] [PubMed] [Google Scholar]
- 23.Rodrigues MM, Alencar BC, Claser C, Tzelepis F. Immunodominance: a new hypothesis to explain parasite escape and host/parasite equilibrium leading to the chronic phase of Chagas’ disease? Braz J Med Biol Res. 2009;42:220–223. doi: 10.1590/s0100-879x2009000300001. [DOI] [PubMed] [Google Scholar]
- 24.Martin D, Tarleton R. Generation, specificity, and function of CD8+ T cells in Trypanosoma cruzi infection. Immunol Rev. 2004;201:304–317. doi: 10.1111/j.0105-2896.2004.00183.x. [DOI] [PubMed] [Google Scholar]
- 25.Millar AE, Wleklinski-Lee M, Kahn SJ. The surface protein superfamily of Trypanosoma cruzi stimulates a polarized Th1 response that becomes anergic. J Immunol. 1999;162:6092–6099. [PubMed] [Google Scholar]
- 26.Tarleton RL. Immune system recognition of Trypanosoma cruzi. Curr Opin Immunol. 2007;19:430–434. doi: 10.1016/j.coi.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Cummings KL, Tarleton RL. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Mol Biochem Parasitol. 2003;129:53–59. doi: 10.1016/s0166-6851(03)00093-8. [DOI] [PubMed] [Google Scholar]
- 28.Aichele P, Brduscha-Riem K, Oehen S, Odermatt B, Zinkernagel RM, Hengartner H, Pircher H. Peptide antigen treatment of naive and virus-immune mice: antigen-specific tolerance versus immunopathology. Immunity. 1997;6:519–529. doi: 10.1016/s1074-7613(00)80340-4. [DOI] [PubMed] [Google Scholar]
- 29.Redmond WL, Marincek BC, Sherman LA. Distinct requirements for deletion versus anergy during CD8 T cell peripheral tolerance in vivo. J Immunol. 2005;174:2046–2053. doi: 10.4049/jimmunol.174.4.2046. [DOI] [PubMed] [Google Scholar]
- 30.Johnson AJ, Upshaw J, Pavelko KD, Rodriguez M, Pease LR. Preservation of motor function by inhibition of CD8+ virus peptide-specific T cells in Theiler’s virus infection. FASEB J. 2001;15:2760–2762. doi: 10.1096/fj.01-0373fje. [DOI] [PubMed] [Google Scholar]
- 31.Mendez-Fernandez YV, Johnson AJ, Rodriguez M, Pease LR. Clearance of Theiler’s virus infection depends on the ability to generate a CD8+ T cell response against a single immunodominant viral peptide. Eur J Immunol. 2003;33:2501–2510. doi: 10.1002/eji.200324007. [DOI] [PubMed] [Google Scholar]
- 32.Howe CL, Ure D, Adelson JD, LaFrance-Corey R, Johnson A, Rodriguez M. CD8+ T cells directed against a viral peptide contribute to loss of motor function by disrupting axonal transport in a viral model of fulminant demyelination. J Neuroimmunol. 2007;188:13–21. doi: 10.1016/j.jneuroim.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Most RG, Murali-Krishna K, Lanier JG, Wherry EJ, Puglielli MT, Blattman JN, Sette A, Ahmed R. Changing immunodominance patterns in antiviral CD8 T-cell responses after loss of epitope presentation or chronic antigenic stimulation. Virology. 2003;315:93–102. doi: 10.1016/j.virol.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Webby RJ, Andreansky S, Stambas J, Rehg JE, Webster RG, Doherty PC, Turner SJ. Protection and compensation in the influenza virus-specific CD8+ T cell response. Proc Natl Acad Sci U S A. 2003;100:7235–7240. doi: 10.1073/pnas.1232449100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreansky SS, Stambas J, Thomas PG, Xie W, Webby RJ, Doherty PC. Consequences of immunodominant epitope deletion for minor influenza virus-specific CD8+-T-cell responses. J Virol. 2005;79:4329–4339. doi: 10.1128/JVI.79.7.4329-4339.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas PG, Brown SA, Keating R, Yue W, Morris MY, So J, Webby RJ, Doherty PC. Hidden epitopes emerge in secondary influenza virus-specific CD8+ T cell responses. J Immunol. 2007;178:3091–3098. doi: 10.4049/jimmunol.178.5.3091. [DOI] [PubMed] [Google Scholar]
- 37.Kotner J, Tarleton R. Endogenous CD4(+) CD25(+) regulatory T cells have a limited role in the control of Trypanosoma cruzi infection in mice. Infect Immun. 2007;75:861–869. doi: 10.1128/IAI.01500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 39.Tarleton RL. Depletion of CD8+ T cells increases susceptibility and reverses vaccine-induced immunity in mice infected with Trypanosoma cruzi. J Immunol. 1990;144:717–724. [PubMed] [Google Scholar]
- 40.Tarleton RL, Koller BH, Latour A, Postan M. Susceptibility of beta 2-microglobulin-deficient mice to Trypanosoma cruzi infection. Nature. 1992;356:338–340. doi: 10.1038/356338a0. [DOI] [PubMed] [Google Scholar]
- 41.Tarleton RL, Grusby MJ, Postan M, Glimcher LH. Trypanosoma cruzi infection in MHC-deficient mice: further evidence for the role of both class I- and class II-restricted T cells in immune resistance and disease. Int Immunol. 1996;8:13–22. doi: 10.1093/intimm/8.1.13. [DOI] [PubMed] [Google Scholar]
- 42.Tzelepis F, de Alencar BC, Penido ML, Gazzinelli RT, Persechini PM, Rodrigues MM. Distinct kinetics of effector CD8+ cytotoxic T cells after infection with Trypanosoma cruzi in naive or vaccinated mice. Infect Immun. 2006;74:2477–2481. doi: 10.1128/IAI.74.4.2477-2481.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gallimore A, Hombach J, Dumrese T, Rammensee HG, Zinkernagel RM, Hengartner H. A protective cytotoxic T cell response to a subdominant epitope is influenced by the stability of the MHC class I/peptide complex and the overall spectrum of viral peptides generated within infected cells. Eur J Immunol. 1998;28:3301–3311. doi: 10.1002/(SICI)1521-4141(199810)28:10<3301::AID-IMMU3301>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 44.Moskophidis D, Zinkernagel RM. Immunobiology of cytotoxic T-cell escape mutants of lymphocytic choriomeningitis virus. J Virol. 1995;69:2187–2193. doi: 10.1128/jvi.69.4.2187-2193.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature. 1990;346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 46.Stock AT, Jones CM, Heath WR, Carbone FR. CTL response compensation for the loss of an immunodominant class I-restricted HSV-1 determinant. Immunol Cell Biol. 2006;84:543–550. doi: 10.1111/j.1440-1711.2006.01469.x. [DOI] [PubMed] [Google Scholar]
- 47.Vallbracht S, Jessen B, Mrusek S, Enders A, Collins PL, Ehl S, Krempl CD. Influence of a single viral epitope on T cell response and disease after infection of mice with respiratory syncytial virus. J Immunol. 2007;179:8264–8273. doi: 10.4049/jimmunol.179.12.8264. [DOI] [PubMed] [Google Scholar]
- 48.Weidt G, Utermohlen O, Heukeshoven J, Lehmann-Grube F, Deppert W. Relationship among immunodominance of single CD8+ T cell epitopes, virus load, and kinetics of primary antiviral CTL response. J Immunol. 1998;160:2923–2931. [PubMed] [Google Scholar]
- 49.Vijh S, I, Pilip M, Pamer EG. Noncompetitive expansion of cytotoxic T lymphocytes specific for different antigens during bacterial infection. Infect Immun. 1999;67:1303–1309. doi: 10.1128/iai.67.3.1303-1309.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Getts MT, Kim BS, Miller SD. Differential outcome of tolerance induction in naive versus activated Theiler’s virus epitope-specific CD8+ cytotoxic T cells. J Virol. 2007;81:6584–6593. doi: 10.1128/JVI.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin X, Njenga MK, Johnson AJ, Pavelko KD, David CS, Pease LR, Rodriguez M. Transgenic expression of Theiler’s murine encephalomyelitis virus genes in H-2(b) mice inhibits resistance to virus-induced demyelination. J Virol. 2002;76:7799–7811. doi: 10.1128/JVI.76.15.7799-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Myoung J, Bahk YY, Kang HS, Dal Canto MC, Kim BS. Anticapsid immunity level, not viral persistence level, correlates with the progression of Theiler’s virus-induced demyelinating disease in viral P1-transgenic mice. J Virol. 2008;82:5606–5617. doi: 10.1128/JVI.02442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pavelko KD, Pease LR, David CS, Rodriguez M. Genetic deletion of a single immunodominant T-cell response confers susceptibility to virus-induced demyelination. Brain Pathol. 2007;17:184–196. doi: 10.1111/j.1750-3639.2007.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schildknecht A, Welti S, Geuking MB, Hangartner L, van den Broek M. Absence of CTL responses to early viral antigens facilitates viral persistence. J Immunol. 2008;180:3113–3121. doi: 10.4049/jimmunol.180.5.3113. [DOI] [PubMed] [Google Scholar]
- 55.von Herrath M, Coon B, Homann D, Wolfe T, Guidotti LG. Thymic tolerance to only one viral protein reduces lymphocytic choriomeningitis virus-induced immunopathology and increases survival in perforin-deficient mice. J Virol. 1999;73:5918–5925. doi: 10.1128/jvi.73.7.5918-5925.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, Ghedin E, Peacock C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark UC, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM, Hauser C, Ivens AC, Kummerfeld SK, Pereira-Leal JB, Nilsson D, Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall N. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- 57.Deitsch KW, Lukehart SA, Stringer JR. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat Rev Microbiol. 2009;7:493–503. doi: 10.1038/nrmicro2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garg N, Nunes MP, Tarleton RL. Delivery by Trypanosoma cruzi of proteins into the MHC class I antigen processing and presentation pathway. J Immunol. 1997;158:3293–3302. [PubMed] [Google Scholar]
- 59.Kahn SJ, Wleklinski M. The surface glycoproteins of Trypanosoma cruzi encode a superfamily of variant T cell epitopes. J Immunol. 1997;159:4444–4451. [PubMed] [Google Scholar]
- 60.Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haolla FA, Claser C, de Alencar BC, Tzelepis F, de Vasconcelos JR, de Oliveira G, Silverio JC, Machado AV, Lannes-Vieira J, Bruna-Romero O, Gazzinelli RT, Dos Santos RR, Soares MB, Rodrigues MM. Strain-specific protective immunity following vaccination against experimental Trypanosoma cruzi infection. Vaccine. 2009 doi: 10.1016/j.vaccine.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 62.Pays E, Vanhamme L, Perez-Morga D. Antigenic variation in Trypanosoma brucei: facts, challenges and mysteries. Curr Opin Microbiol. 2004;7:369–374. doi: 10.1016/j.mib.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 63.Miyahira Y. Trypanosoma cruzi infection from the view of CD8+ T cell immunity--an infection model for developing T cell vaccine. Parasitol Int. 2008;57:38–48. doi: 10.1016/j.parint.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 64.Alvarez MG, Postan M, Weatherly DB, Albareda MC, Sidney J, Sette A, Olivera C, Armenti AH, Tarleton RL, Laucella SA. HLA Class IT Cell Epitopes from trans-Sialidase Proteins Reveal Functionally Distinct Subsets of CD8 T Cells in Chronic Chagas Disease. PLoS Negl Trop Dis. 2008;2:e288. doi: 10.1371/journal.pntd.0000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wizel B, Palmieri M, Mendoza C, Arana B, Sidney J, Sette A, Tarleton R. Human infection with Trypanosoma cruzi induces parasite antigen-specific cytotoxic T lymphocyte responses. J Clin Invest. 1998;102:1062–1071. doi: 10.1172/JCI3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Millar AE, Kahn SJ. The SA85-1.1 protein of the Trypanosoma cruzi trans-sialidase superfamily is a dominant T-cell antigen. Infect Immun. 2000;68:3574–3580. doi: 10.1128/iai.68.6.3574-3580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Douradinha B, Mota MM, Luty AJ, Sauerwein RW. Cross-species immunity in malaria vaccine development: two, three, or even four for the price of one? Infect Immun. 2008;76:873–878. doi: 10.1128/IAI.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]