

Abstract
Merkel cell carcinoma (MCC) is a rare but devastating skin disease that is increasing in incidence within the United States. The poor prognosis of MCC patients and limited understanding of MCC pathogenesis warrants innovative treatments to control MCC. Several lines of evidence have pointed to Merkel cell polyomavirus (MCPyV) as the etiological agent of MCC. In particular, the amino terminus of MCPyV large T antigen (LT) (aa1-258) is expressed in all MCPyV-positive tumors and plays an important role in MCC oncogenesis, rendering it an ideal therapeutic target for vaccination. In the current study, we developed a DNA vaccine encoding MCPyV LT aa1-258 (pcDNA3-LT). Within our pcDNA3-LT DNA vaccine, we identified that MCPyV LT aa136-160 likely contains an LT-specific CD4+ T helper epitope. We have also created an LT-expressing B16/LT tumor model using B16, a murine melanoma cell line, to characterize the potency of our DNA vaccine. Using this tumorigenic B16/LT tumor model, we found that pcDNA3-LT DNA vaccine generates antitumor effects mainly mediated by CD4+ T cells against B16/LT tumors in vaccinated C57BL/6 mice. Thus, immunotherapy using pcDNA3-LT DNA vaccine may represent a promising approach for the control of MCPyV-associated lesions. The B16/LT tumor model further serves as a useful model for testing various vaccine strategies against MCC.
Keywords: DNA vaccine, Merkel cell polyomavirus, Merkel cell carcinoma
1. Introduction
Merkel cell carcinoma (MCC) is a rare but devastating disease of the skin that is increasing in incidence within the United States [1]. Current treatment options are limited to surgery alone or in combination with adjuvant radiation [2]. However, the 2-year mortality rate remains at 28%, with a median survival time of 6.8 months [3]. The limited understanding of the pathogenesis of MCC and the poor prognosis of MCC patients underscores the need for innovative treatments to control MCC.
Recently, a new human polyomavirus termed Merkel cell polyomavirus (MCPyV) was discovered to be associated with MCC [4]. The carcinogenicity of MCPyV is supported by several lines of evidence. It is well established that polyomaviruses can transform cells and induce tumors in experimental animal models. MCPyV has been detected in ∼80% of MCC tumors worldwide. In these tumors, the virus is present as a chromosomally integrated copy, a characteristic of some viruses that cause cancers in humans. The MCPyV integration is clonal and the site is unique for each patient, indicating that the virus was present in the cell before or during oncogenic transformation. Compared to MCPyV in normal tissue, MCPyV genomes in tumor tissue harbor stop codon mutations in the large T antigen (LT) open reading frame that are predicted to ablate viral replication capacity [5]. Specifically, the truncated LT protein loses the C terminal helicase domain required for viral DNA replication while the RB-interacting domain is retained. A lack of viral replication is characteristic of some oncogenic viruses, as viral replication would cause death of the host cell. A necessary role for large T antigen expression in maintenance of the proliferative capacity and survival of MCPyV-positive MCC cell lines harboring truncated forms of the large T antigen was recently demonstrated [6]. Thus, sufficient evidence has accumulated to provisionally consider MCPyV as the etiological agent of MCC and to explore therapies that target the virus.
The importance of MCPyV LT in MCC oncogenesis renders it a potentially ideal target for molecular intervention. A unique feature of LT in MCC is that the protein is always truncated by the introduction of stop codons. Of the mutations that have been described, the stop codons are predicted to truncate the protein at or beyond amino acid 258. Thus, the amino terminus of MCPyV LT (aa1-258) represents an ideal therapeutic target, as it is likely to be present and expressed in all MCPyV-positive MCC tumors and it plays an important role in MCC oncogenesis. In addition, MCPyV LT is a foreign antigen and therefore faces no issue of immune tolerance. Based on this potentially ideal vaccine target, we sought to develop a vaccine for the control of MCC.
DNA vaccines are an attractive form of vaccination due to their safety, simplicity, stability, capacity for repeated administration, and ability to generate potent antigen-specific CD8+ T cell immunity (for reviews, see [7–9]). It has been recently demonstrated that CD8 T cell infiltration of MCC tumors correlates with better prognosis, supporting the idea that T cells may play a role in the clearance of MCC. Thus, we created a DNA vaccine to generate potent antigen-specific CD8+ T cell immunity for the control of MCC. In the current study, we generate a DNA vaccine encoding MCPyV LT aa1-258 (pcDNA3-LT). We found that within our pcDNA3-LT DNA vaccine, MCPyV LT aa136-160 likely contains a LT-specific CD4+ T helper epitope. To characterize the potency of our DNA vaccine, we created a MCC tumor model in which B16 melanoma tumor cell line was transduced with a lentivirus vector expressing MCC LT amino acids 1-258 to generate a novel LT-expressing tumorigenic cell line, B16/LT. Since melanomas, like Merkel cell carcinoma, are of cutaneous origin, the B16 melanoma cell line is suitable for the development of a MCPyV LT-expressing murine tumor model. Using this tumorigenic B16/LT tumor model, we found that pcDNA3-LT DNA vaccine generates protective and therapeutic antitumor effects, mainly mediated by CD4+ T cells, against LT-expressing B16/LT tumors in mice. Our study shows that a DNA vaccine encoding MCPyV LT has the potential for future clinical applications in the control of MCC.
2. Materials and methods
2.1. Mice
C57BL/6 mice (6–8 weeks old) were purchased from National Cancer Institute (Frederick, MD). All animals were maintained under specific pathogen-free conditions, and all procedures were performed according to approved protocols and in accordance with the recommendations for the proper use and care of experimental animals.
2.2. Constructs
For the generation of a DNA vaccine (pcDNA3-LT) encoding truncated large T antigen (aa1-258) of Merkel cell polyomavirus (strain 350), large T antigen cDNA (1-774 nt) was codon-optimized and synthesized by GeneScript Corporation (Piscataway, NJ) and cloned into XbaI and NotI sites of pcDNA3 (Invitrogen, Carlsbad, CA). For the generation of a lentivirus encoding truncated large T antigen, the large T antigen DNA was cloned into XbaI/NotI sites of pCDH1-EF1-GFP vector (System Bioscience, Mountain View, CA) to generate pCDH1-LTop-EF1-GFP. The vector contains two promoters, CMV promoter for the large T antigen and EF1 promoter for GFP expression.
2.3. Peptides
A total of 49 overlapping 20-mer peptides (overlapped by 15 amino acids) spanning Merkel cell polyomavirus large T antigen aa1-258 were synthesized by GeneScript Corporation (Piscataway, NJ). The identities of the peptides were validated by mass spectrometric analysis and the purity of the peptides was confirmed by high-performance liquid chromatography.
2.4. Cell lines
Murine melanoma B16/F10 cells were purchased from American Type Culture Collection. A large T and GFP-expressing B16F10 (B16/LT) cell line was generated by transduction with a lentiviral vector encoding large T antigen and GFP. Lentiviral vector pCDH1-LT-EF1-GFP, pCMVΔR8.91, and pMDG were transfected into 293 T cell line using lipofectamine (Invitrogen, Carlsbad, CA, USA) and the virion-containing supernatant was collected 48 h after transfection. The supernatant was then filtered through a 0.45 mm cellulose acetate syringe filter (Nalgene, Rochester, NY, USA) and used to infect B16F10 cells in the presence of 8mg/ml polybrene (Sigma–Aldrich, St. Louis, MO, USA). Transduced cells were isolated using preparative flow cytometry by GFP signal.
2.5. Western Blot analysis
Proteins from both B16 and sorted B16/LT cells were extracted by M-PER reagent. Equal amounts of proteins (20 μg) were loaded and separated by 10% gradient ready SDS-PAGE. The proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA) using electrophroesis. Blots were blocked with phosphate-buffered saline (PBS)-Tween 20 (PBST) containing 5% non-fat milk for 1 h at room temperature. Membranes were probed with anti-beta actin (St. Louis, MO) at 1:1000 dilution or sera from mice vaccinated pcDNA3-LTat 1:100 dilution in Tris-buffered saline with Tween 20 (TTBS) for 2 h, washed four times with TTBS, and then incubated with goat anti-mouse IgG conjugated to alkaline phosphatase (Amersham, Piscataway, NJ) at 1:1000 dilution in TTBS containing 5% nonfat milk. Membranes were washed four times with TTBS and developed using Hyperfilm-enhanced chemiluminescence (Amersham, Piscataway, NJ).
2.6. Intramuscular DNA vaccination by electroporation
Electroporation-mediated DNA vaccination was performed with methods similar to those described by Jacob et al. [10]. C57BL/6 mice (5 per group) were injected in the tibialis muscle of the shaved hind leg with either pcDNA3-LT or pcDNA3 empty vector DNA vaccine at a dose of 10 μg/20 μl per mouse [11]. The DNA plasmid was diluted to the appropriate concentration in a total volume of 20 μl of PBS. DNA injection was immediately followed by square wave electroporation at the injection site using a BTX830 (BTX Harvard Apparatus, Holliston, MA). A tweezer electrode was used to deliver eight pulses at 100 V for 20 ms. The number of doses delivered varied according to the experiment, as described in Section 3.
2.7. In vivo tumor protection experiments
C57BL/6 mice (5 per group) were immunized intramuscularly followed by electroporation with either pcDNA3-LT or pcDNA3 empty vector DNA vaccine on the first day of Weeks 0, 1, and 2. On the first day of Week 4, vaccinated mice were challenged subcutaneously in the right flank with B16/LT tumor (2 × 106 cells per mouse). Mice were monitored for evidence of tumor growth by inspection and palpation.
2.8. In vivo tumor treatment experiments
C57BL/6 mice (5 per group) were subcutaneously inoculated with 1 × 105 B16/LT tumor cells per mouse in the right flank on Day 0. Mice were monitored for evidence of tumor growth by inspection and palpation. Tumor growth was measured twice a week starting from Day 8 after tumor challenge. B16/LT-tumor bearing mice were treated with pcDNA3-LT or pcDNA3 empty vector DNA vaccine intramuscularly followed by electroporation three times at a 1-week interval 3 days after tumor inoculation.
2.9. In vivo immune cell depletion experiments
C57BL/6 mice (5 per group) were vaccinated with pcDNA3-LT DNA or pcDNA3 empty vector DNA vaccine intramuscularly followed by electroporation on Day 0. Vaccinated mice were boosted at the same dose and regimen on Day 7. On Day 8, vaccinated mice were intraperitoneally injected with anti-CD4, anti-CD8 or anti-NK1.1 monoclonal antibody every other day until Day 14 of the experiment (4 doses). Immune cell-depleted mice were then challenged with 1 × 105 B16/LT tumor cells per mouse subcutaneously in the right flank on Day 18. Mice were monitored for tumor growth by inspection and palpation. Tumor size was measured twice a week.
2.10. Tumor size measurement
Tumor growth was monitored by visual inspection and palpation. In addition, tumor size was measured by Vernier caliper twice a week. Tumor volumes were evaluated using the formula
2.11. Flow cytometry
Detection of cell surface CD8a and intracellular IFNγ was performed using flow cytometry as described previously [12,13]. Splenocytes isolated from vaccinated mice were incubated overnight with 1 μg/ml of GolgiPlug (BD Pharmingen) in the presence of 1 μg/ml of the large T antigen overlapping peptides or individual peptide (Supplementary Table 1). After washing twice with FACScan buffer, the cells were stained with phycoerythrin-conjugated anti-mouse CD8a antibody. The cells were then incubated with BD cytofix/cytoperm solution (BD Pharmingen) followed by staining with FITC-conjugated anti-mouse IFNγ antibody. Flow cytometry analysis was performed on Becton-Dickinson FACSCalibur with CELLQuest software (BD Biosciences, Mountain View, CA).
2.12. Statistical analysis
All data (expressed as mean ± s.d.) are representative of at least two different experiments. Comparisons between individual data points were made using Student's t-test. Kaplan–Meier survival curve analysis was used to evaluate tumor treatment experiments. All p values were calculated using the log-rank test. Values of p<0.05 were considered significant.
3. Results
3.1. Creation of a murine tumor model that expresses Merkel Cell polyomavirus (MCPyV) large T (LT) antigen
B16 murine melanoma cells were transduced with a lentiviral vector containing a mammalian gene encoding a truncated form of MCPyV large T antigen (aa1-258) under the control of a cytolomegalovirus (CMV) promoter and a GFP reporter gene under that of a EF1 promoter to generate a tumorigenic LT-expressing cell line, B16/LT. Successful transduction of the lentivirus would lead to the expression of GFP and allow isolation of the transduced tumor cells. As shown in Fig. 1A, the LT retrovirus-transduced B16 tumor cells demonstrated greater GFP expression compared to non-transduced B16 tumor cells. The GFP-positive cells were further isolated by preparative flow cytometry by GFP signal for the characterization of the expression of LT antigen. In order to generate antibody against LT antigen, we constructed a pcDNA3 vaccine encoding MCPyV LT aa1-258 (pcDNA3-LT). Mice were vaccinated with pcDNA3-LT intramuscularly followed by electroporation three times at a 1-week interval and boosted using the same dose and regimen 2 months later, as outlined in Fig. 2A. One week after the last vaccination, sera from vaccinated mice were collected for Western Blot analysis. As shown in Fig. 1B, left panel, cell lysate from B16/LT cells demonstrated a specific band consistent in size with LT antigen. In comparison, the band is absent in cell lysate from B16 melanoma cells. To confirm that loading of cell lysate was comparable between samples, we used antibody specific for beta actin as a loading control (Fig. 1B, right panel). Thus, our data show that we have successfully created a murine LT-expressing B16 tumor cell line.
Fig. 1.
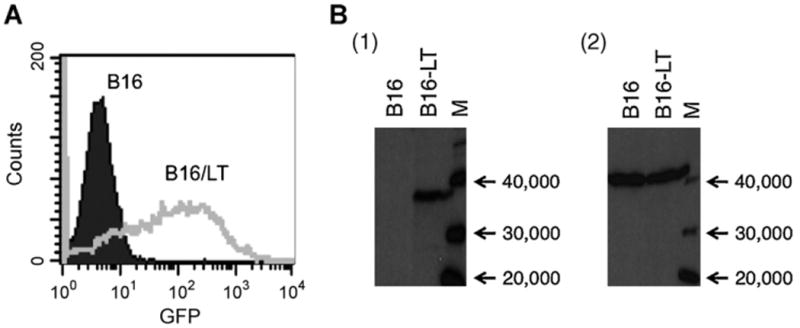
Generation and characterization of LT-expressing B16/LT tumor cell line. B16 mouse melanoma cells were transduced with a lentiviral vector containing a mammalian codon-optimized gene encoding the initial 258 amino acids of Merkel cell polyomavirus (strain 350) large T antigen (LT) under the control of a cytolomegalovirus promoter and a GFP reporter under the control of the EF1 promoter to generate tumorigenic B16/LT tumor cell line. (A) Characterization of transduction of B16/LT tumor cells by flow cytometry analysis after sorting. B16/LT tumor cells (Gray line, clear histogram) or control B16 melanoma cells (black line, filled black histogram) were sorted and characterized for GFP expression by flow cytometry analysis. (B) Characterization of LT expression by Western Blot analysis. C57BL/6 mice were vaccinated intramuscularly by electroporation with pcDNA3-LT (10 (μg/20 μl per mouse) three times at a 1-week interval. Two months later, the vaccinated mice were boosted with the same dose and regimen. Western Blot analysis of sera from vaccinated mice was performed 1 week after the last vaccination to characterize LT expression of B16/LT cells. Membranes were probed with either (1) sera from mice vaccinated with pcDNA3-LT or (2) loading control anti-beta actin antibody. Lane A, negative control B16. Lane B, B16/LT. Lane M, size marker.
Fig. 2.
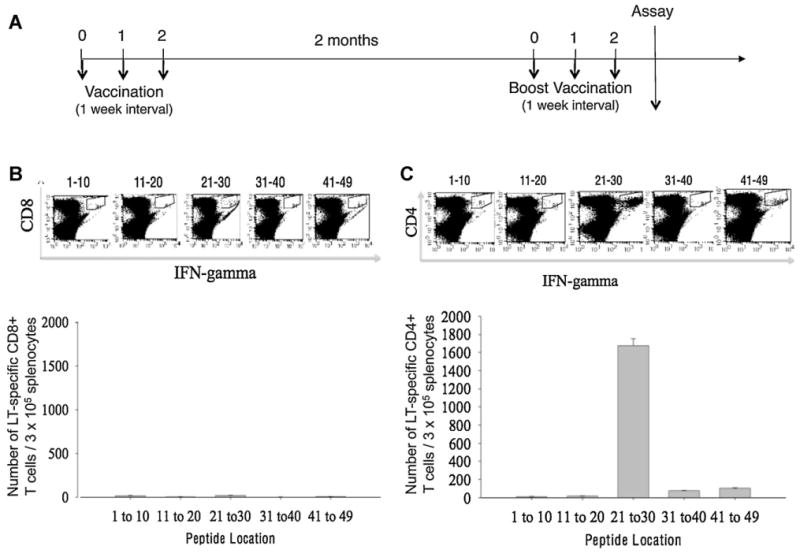
Characterization of LT-specific CD4+ and CD8+ T cell epitope(s) using LT overlapping peptides and splenocytes from mice vaccinated with pcDNA3-LT. (A) Schematic diagram of vaccination regimen. C57BL/6 mice (5 mice per group) were immunized with pcDNA3-LT (10 μg/20 μl per mouse) intramuscularly followed by electroporation on Weeks 0, 1, and 2. Vaccinated mice were boosted with the same dose and regimen 2 months later. Pooled splenocytes from vaccinated mice were collected, cultured in vitro with various overlapping LT peptides (see Supplementary Table 1) overnight, and then stained for intracellular IFN-γ and either CD8 or CD4 cell surface marker. (B) Intracellular cytokine staining followed by flow cytometry analysis to characterize LT-specific CD8+ T cell epitope using various LT overlapping peptides and splenocytes harvested from mice vaccinated with pcDNA3-LT. Note that no LT-specific CD8+ T cells were activated. (C) Intracellular cytokine staining followed by flow cytometry analysis to characterize LT-specific CD4+ T cell epitope using various LT overlapping peptides and splenocytes harvested from mice vaccinated with pcDNA3-LT. Data presented in this figure are from one experiment representative of two performed. Note that overlapping peptide pool #21–30 activated the most LT-specific CD4+ T cells.
3.2. MCPyV LT aa136-160 contains a LT-specific CD4 T helper epitope
We immunized mice with the above pcDNA3-LT vaccine in order to generate LT-specific T cell-mediated responses in vaccinated mice. An empty pcDNA3 vector was used as a control. To map the LT-specific CD8+ and/or CD4+ T cell epitope, we synthesized 49 overlapping 20-mer peptides, with an overlap of 15 amino acids spanning MCPyV LT aa1-258 (Supplementary Table 1). Five pools containing 10 individual peptides and one pool containing 9 peptides were prepared. The pools of peptides were each incubated with splenocytes from mice vaccinated with pcDNA3-LT using the regimen depicted in Fig. 2A. Mice vaccinated with pCDNA3-LT generated a significant LT-specific CD4+ T cell response when stimulated with the pool containing peptides #21–30 (Fig. 2C). In comparison, the vaccinated mice did not generate numbers of LT-specific CD8+ T cells above background levels when stimulated with any of the LT peptides (Fig. 2B). Mice vaccinated with the empty pcDNA3 vector also failed to generate significant LT-specific T cell-mediated responses (Supplementary Fig. 1). In order to further delineate the LT-specific CD4+ T cell epitope, we incubated the individual peptides from the pool containing peptides #21–30 with splenocytes from vaccinated mice. As shown in Fig. 3A, splenocytes from vaccinated mice stimulated with peptide #28 (aa136-155) and overlapping peptide #29 (aa141-160) demonstrated a significantly greater LT-specific CD4+ T cell immune responses compared to splenocytes stimulated with other candidate peptides. The amino acid sequence shared by peptide #28 and #29 is SSSSGYGS-FSASQASD (Fig. 3B) and this region likely contains the core region of the CD4+ T cell epitope.
Fig. 3.
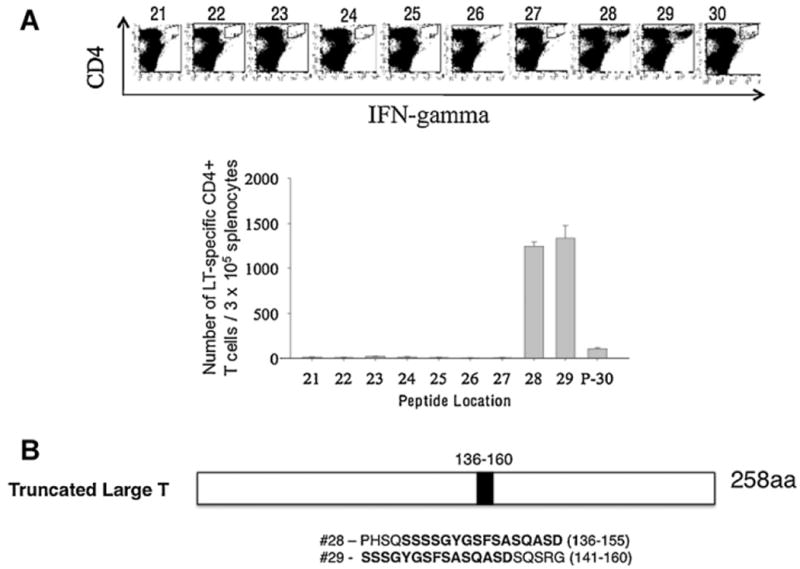
Identification of LT-specific CD4+ T cell epitope using splenocytes stimulated with various LT overlapping peptides. (A) Intracellular cytokine staining followed by flow cytometry analysis to characterize LT-specific CD4+T cell epitope using various LT overlapping peptides screened from the group composed of peptide pool 21–30 and splenocytes harvested from mice vaccinated with pcDNA3-LT using methods similar to those described in Fig. 2. (B) Schematic diagram of region of truncated form (258aa) of large T antigen where LT-specific CD4+ T cell epitope is located and the sequences of our two candidate LT-specific CD4+T cell epitopes. Note that overlapping peptides encoding LT-specific epitope #29 activated the most LT-specific CD4+ T cells.
3.3. Vaccination with pcDNA3-LT DNA vaccine provides protection against B16/LT tumor challenge
From the epitope mapping experiment, we observed that the pcDNA3-LT DNA vaccine-generated LT-specific CD4+T cell immune response had potential anti-tumor effect. However, a shorter 3-dose regimen eliciting the same level of immune response is more practical compared to the 6-dose regimen used in the mapping experiment as depicted in Fig. 2A In order to determine whether a more abbreviated regimen of 3 intramuscular administrations followed by electroporation, depicted in Fig. 4A, would also generate robust LT peptide (aa141-160)-specific CD4+ T cell immune responses, we immunized mice accordingly and performed intracellular cytokine staining for IFN-γ followed by flow cytometry. As shown in Fig. 4B, splenocytes from mice vaccinated with pcDNA3-LT and stimulated with candidate LT-specific CD4 epitope #29 (MCPyV LT aa141-160) generated significantly greater numbers of IFN-γ-secreting LT-specific CD4+ T cells compared to splenocytes from mice vaccinated with pcDNA3 empty vector and similarly stimulated. Thus, our data indicate that a vaccination regimen using three doses is able to generate significant numbers of LT-specific CD4+ T cells.
Fig. 4.
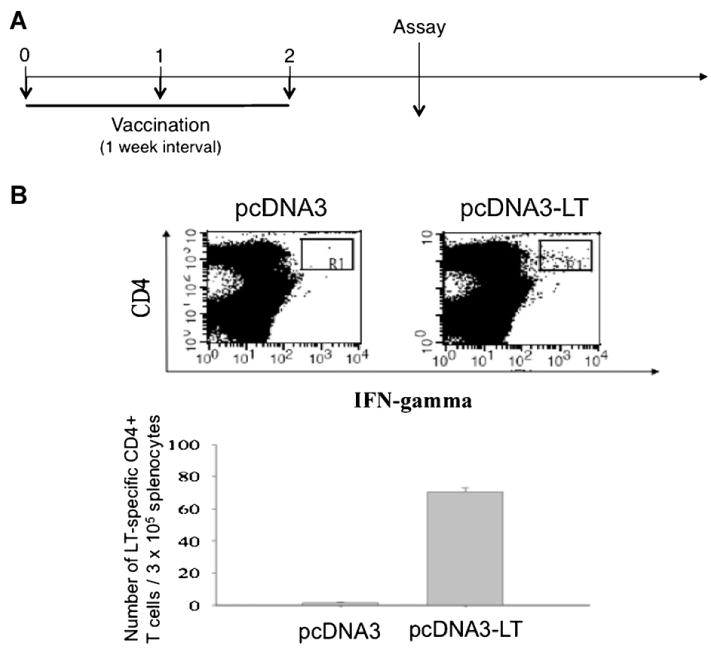
Characterization of LT-specific CD4+ T cells using splenocytes stimulated with LT peptide #29. (A) Schematic regimen of vaccination schedule used for in vivo tumor protection experiments. (B) Intracellular cytokine staining using phycoerythrin (CD4 labeling) and fluorescein isothiocyanate (IFN-γ labeling) as fluorescent tracers followed by flow cytometry analysis to characterize LT-specific CD4+ T cell responses using splenocytes stimulated with candidate peptide #29.
Using this vaccination regimen as depicted in Fig. 5A, we performed in vivo tumor protection experiments to determine the protective antitumor effects of our pcDNA3-LT DNA vaccine. C57BL/6 mice were vaccinated with pcDNA3-LT DNA vaccine or control empty pcDNA3 vector. Vaccinated mice were challenged with B16/LT tumor cells subcutaneously 1 week after the last vaccination. As shown in Fig. 5B, mice vaccinated with pcDNA3-LT demonstrated significantly smaller tumor volume than mice vaccinated with control empty pcDNA3 vector. As shown in Fig. 5C, mice vaccinated with pcDNA3-LT had significantly higher survival rate compared to mice vaccinated with pcDNA3. Thus, our data show that pcDNA3-LT DNA vaccine elicits a strong protective antitumor effect in vaccinated mice.
Fig. 5.
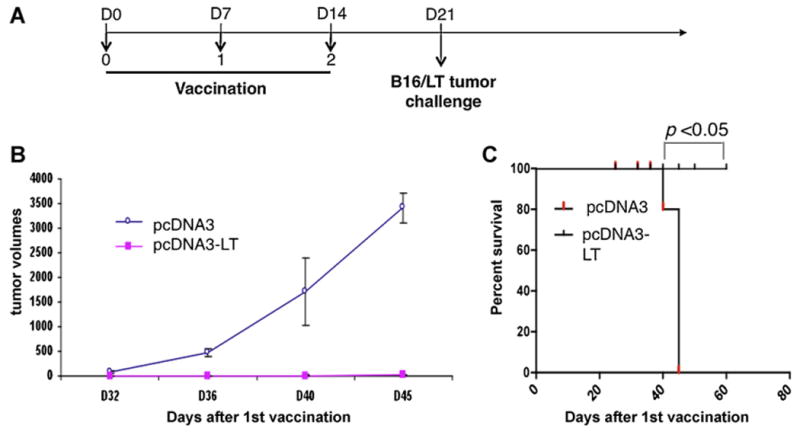
In vivo tumor protection experiments. (A) Schematic diagram of the vaccination regimen with either pcDNA3-LT DNA vaccine or control pcDNA3 empty vector. C57BL/6 mice (5 per group) were immunized with either pcDNA3-LT or pcDNA3 empty vector DNA vaccine (10 μg/20 μl per mouse) intramuscularly by electroporation on Days 0, 7, and 14. On Day 21, vaccinated mice were challenged subcutaneously in the right flank with 2 × 106 B16/LT tumor cells per mouse. (B) Line graph depicting the tumor volume in vaccinated mice. (C) Kaplan–Meier survival analysis of mice vaccinated with either pcDNA3-LT DNA vaccine or control pcDNA3 empty vector. Data shown are representative of two experiments performed.
3.4. Vaccination with pcDNA3-LT DNA vaccine generates potent therapeutic antitumor effects against LT-expressing tumors in mice
To determine whether pcDNA3-LT DNA vaccination is capable of generating a therapeutic antitumor effect, we first inoculated naïve C57BL/6 mice with B16/LT tumors and then treated the mice with pcDNA3-LT using the vaccination regimen depicted in Fig. 6A. As shown in Fig. 6B, B16/LT tumor-bearing mice vaccinated with pcDNA3-LT had significantly smaller tumor volume than mice vaccinated with control empty pcDNA3 vector. Furthermore, tumor-bearing mice vaccinated with pcDNA3-LT showed significantly longer survival compared to mice vaccinated with pcDNA3. Therefore, our data show that pcDNA3-LT DNA vaccine is capable of generating a therapeutic antitumor effect in B16/LT tumor-bearing mice.
Fig. 6. In vivo.
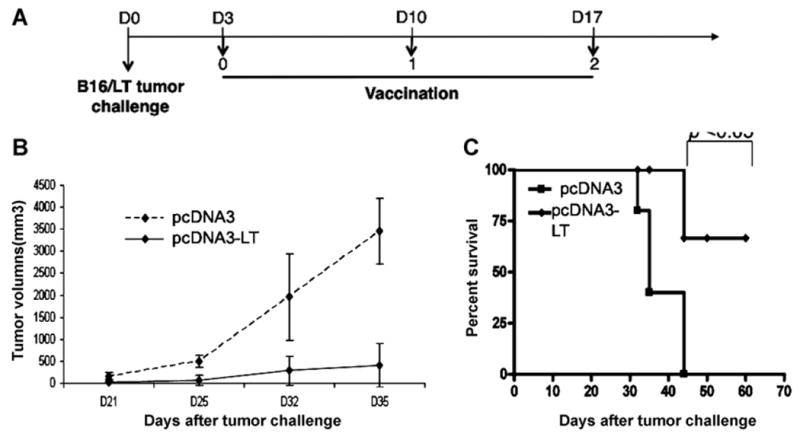
tumor treatment experiments. (A) Schematic diagram of the treatment regimen with either pcDNA3-LT DNA vaccine or control pcDNA3 empty vector. C57BL/6 mice (5 per group) were subcutaneously inoculated with 1 × 105 B16/LT tumor cells per mouse in the right flank on Day 0 (D0). The B16/LT-tumor bearing mice were treated with pcDNA3-LT or pcDNA3 intramuscularly followed by electroporation three times at a 1-week interval beginning on Day 3. Mice were monitored for evidence of tumor growth by inspection and palpation. Tumor growth was measured twice a week starting on Day 8. (B) Line graph depicting the tumor volume in B16/LT tumor-bearing mice treated with pcDNA3 or pcDNA3-LT DNA vaccine. (C) Kaplan–Meier survival analysis of B16/LT tumor-bearing mice treated with pcDNA3 or pcDNA3-LT DNA vaccine. Data shown are representative of two experiments performed.
3.5. CD4+ T cells play the most important role in the antitumor effects generated by pcDNA3-LT vaccination
To determine the subsets of lymphocytes that are important for antitumor effects generated by pcDNA3-LT DNA vaccine, we performed in vivo immune cell depletion experiments with anti-immune cell antibody using the regimen as specified in Fig. 7A. As shown in Fig. 7B, all of the mice depleted of CD4+ T cells grew tumors dramatically faster compared to non-depleted mice and mice depleted of CD8+ T cells or NK cells. Depletion of CD8+ T cells or NK cells also led to loss of protective antitumor effects, although the loss was not as significant as the depletion of CD4+ T cells. These results indicate that all subsets of lymphocytes are important in antitumor effects, with CD4+ T cells as the most important factor in the antitumor immunity generated by the pcDNA3-LT DNA vaccine.
Fig. 7.
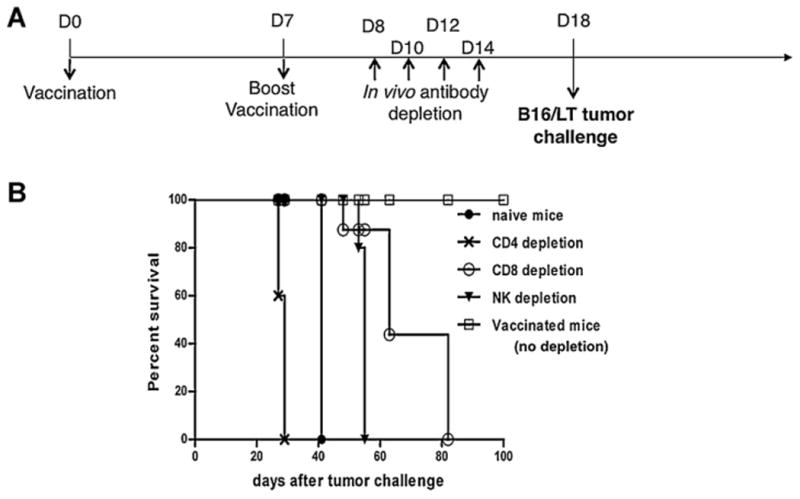
Characterization of the effects of lymphocyte subsets on the tumor protection of the pcDNA3-LT DNA vaccine. pcDNA3 and pcDNA3-LT DNA vaccinated mice were given intraperitoneal injection of the anti-CD4, anti-CD8 or anti-NK1.1 monoclonal antibody. The mice were then challenged with B16/LT tumor cells. (A) Schematic diagram of the vaccination regimen for in vivo immune cell depletion experiments. C57BL/6 mice (5 per group) were vaccinated intramuscularly by electroporation with pcDNA3 or pcDNA3-LT DNA vaccine (10 μg/20 μl per mouse) on Day 0. Vaccinated mice were boosted at the same dose and regimen on Day 7. On Days 8, 10, 12, and 14, the vaccinated mice were intraperitoneally injected with anti-CD4, anti-CD8 or anti-NK1.1 monoclonal antibody. Immune cell-depleted mice were then subcutaneously challenged with 1 × 105 B16/LT tumor cells per mouse in the right flank on Day 18. Mice were monitored for evidence of tumor growth by inspection, palpation and tumor size was measured twice a week. (B) Kaplan–Meier survival analysis of B16/LT tumor-bearing mice treated with pcDNA3 or pcDNA3-LT DNA vaccine. Data shown are representative of two experiments performed.
4. Discussion
In this study, we have successfully demonstrated that a pcDNA3-LT DNA vaccine, encoding MCPyV (aa1-258), can generate a LT-specific CD4+ T cell immune response in vaccinated mice. We have also identified a LT-specific CD4+ T helper epitope at MCPyV aa131-160. Furthermore, we created a preclinical murine MCC tumor model suitable for testing our DNA vaccine. Further characterization of our DNA vaccine demonstrated that the vaccine was capable of generating potent protective and therapeutic antitumor effects. We further determined that CD4+ T cells are the primary mediators for the observed antitumor effects in vaccinated mice.
Our B16/LT tumor model represents an innovative avenue for testing vaccines targeting MCPyV. To the best of our knowledge, there is currently no reported preclinical MCC tumor model for the testing of cancer immunotherapy. At present there is also no reported vaccine to treat MCPyV-induced MCC tumors in either preclinical or clinical settings. Thus, the B16/LT model serves as a potentially important tumor model for the testing of different vaccination strategies, including other forms of vaccines such as peptide-based, protein-based, dendritic cell-based, and vector-based vaccines targeting MCPyV.
In our study, we observed that CD4+ T cells are essential for the observed antitumor effects generated by vaccination with pcDNA3-LT DNA vaccine. It has been shown that CD4+ T cell immune responses can generate antitumor effects in cutaneous tumor models [14,15]. Furthermore, it was shown that naïve tumor-specific CD4+ T cells can differentiate into cytotoxic T cells in lymphopenic mice, as marked by expression of cytotoxic T cell-associated genes perforin, granzyme B, and LAMP-1 (CD107a), all of which are involved in degranulating cytotoxic T cells [15]. In this model, the activated naïve tumor-specific CD4+ T cells were independent of NK cells and were important for tumor clearance [15]. Thus, it will be important to further explore the role of tumor-specific CD4+ T cells in mediating antitumor immunity through direct cytotoxicity.
A mechanistic understanding of how a vaccine works can guide future efforts to further improve the vaccine. While in the current study we show that the antitumor effects of our pcDNA3-LT DNA vaccine is primarily mediated by CD4+ T cells, CD8+ T cells and NK cells also contributed to the efficacy of the vaccine in our tumor model (Fig. 7). Although the DNA vaccine in the current study does not generate appreciable LT-specific CD8+ T cell immune responses, different vaccination strategies may potentially lead to the enhancement of LT-specific CD8+ T cells. For example, we have previously found that DNA vaccine encoding calreticulin (CRT) [12,13,16–18] or heat shock protein 70 [19,20] linked to HPV-16 E7 can lead to the enhancement of HPV-16 E7-specific CD8+ T cell immune responses. It will be important in future investigations to determine whether these strategies can induce LT-specific CD8+ T cell immune responses.
For future clinical translation of a therapeutic MCPyV vaccine, it will be important to identify the most desirable vaccination strategy to generate optimal MCPyV LT-specific CD4+ and/or CD8+ T cell immune responses and antitumor effects against MCPyV LT-expressing tumor. We have previously generated a panel of therapeutic DNA vaccines employing various intracellular targeting strategies that can route model target antigen to desired subcellular compartments in order to enhance MHC class I and/or MHC class II antigen processing and presentation pathway(s) to enhance both antigen-specific CD4+ and CD8+ T cell immune responses. Strategies to enhance CD4+ T cell immune responses include the linkage of target antigen to the sorting signal of lysosome-associated membrane protein (LAMP-1) [21] to improve MHC class II antigen presentation to CD4+ T cells. Strategies to enhance CD8+ T cell immune responses include the linkage of target antigen to Mycobacterium tuberculosis heat-shock protein 70 (HSP70) [19], calreticulin (CRT) [16], gamma-tubulin [22], or the translocation domain (dII) of Pseudomonas aeruginosa exotoxin A (ETA) [23] to enhance MHC class I antigen presentation to CD8+ T cells. These DNA vaccination strategies have been proven to be effective in other antigenic systems (for review, see [8]). It will be of interest to further explore whether such DNA vaccination strategy can be applied to LT antigen to further enhance LT-specific CD4+ and/or CD8+ T cell responses as well as therapeutic antitumor effects for future clinical translation.
In summary, we have created a pcDNA3-LT DNA vaccine targeting MCPyV LT. We have also successfully developed a LT-expressing preclinical tumor model, B16/LT, for evaluating our LT-specific DNA vaccine strategy. Our study serves as an important platform for further development of immunotherapeutic strategies for the control of MCPyV-associated Merkel cell carcinoma.
Supplementary Material
Acknowledgments
This work was supported by the American Cancer Society, the National Cancer Institute SPORE P50 CA098252 and the 1 RO1 CA114425-01.
Abbreviations
- LT
large T antigen
- MCC
Merkel cell carcinoma
- MCPyV
Merkel cell polyomavirus
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.vaccine.2011.12.072.
References
- 1.Hodgson NC. Merkel cell carcinoma: changing incidence trends. J Surg Oncol. 2005 Jan;89(1):1–4. doi: 10.1002/jso.20167. [DOI] [PubMed] [Google Scholar]
- 2.Mojica P, Smith D, Ellenhorn JD. Adjuvant radiation therapy is associated with improved survival in Merkel cell carcinoma of the skin. J Clin Oncol. 2007 Mar;25(9):1043–7. doi: 10.1200/JCO.2006.07.9319. [DOI] [PubMed] [Google Scholar]
- 3.Allen PJ, Bowne WB, Jaques DP, Brennan MF, Busam K, Coit DG. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005 Apr;23(10):2300–9. doi: 10.1200/JCO.2005.02.329. [DOI] [PubMed] [Google Scholar]
- 4.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008 Feb;319(5866):1096–100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA. 2008 Oct;105(42):16272–7. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010 Jul;84(14):7064–72. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung CF, Monie A, Alvarez RD, Wu TC. DNA vaccines for cervical cancer: from bench to bedside. Exp Mol Med. 2007 Dec;39(6):679–89. doi: 10.1038/emm.2007.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsen SW, Paik AH, Hung CF, Wu TC. Enhancing DNA vaccine potency by modifying the properties of antigen-presenting cells. Expert Rev Vaccines. 2007 Apr;6(2):227–39. doi: 10.1586/14760584.6.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin K, Roosinovich E, Ma B, Hung CF, Wu TC. Therapeutic HPV DNA vaccines. Immunol Res. 2010 Jul;47(1–3):86–112. doi: 10.1007/s12026-009-8141-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacob J, Radkevich O, Forni G, Zielinski J, Shim D, Jones RF, et al. Activity of DNA vaccines encoding self or heterologous Her-2/neu in Her-2 or neu transgenic mice. Cell Immunol. 2006 Apr;240(2):96–106. doi: 10.1016/j.cellimm.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Best SR, Peng S, Juang CM, Hung CF, Hannaman D, Saunders JR, et al. Administration of HPV DNA vaccine via electroporation elicits the strongest CD8+ T cell immune responses compared to intramuscular injection and intradermal gene gun delivery. Vaccine. 2009 Sep;27(40):5450–9. doi: 10.1016/j.vaccine.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim D, Gambhira R, Karanam B, Monie A, Hung CF, Roden R, et al. Generation and characterization of a preventive and therapeutic HPV DNA vaccine. Vaccine. 2008 Jan;26(3):351–60. doi: 10.1016/j.vaccine.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng S, Ji H, Trimble C, He L, Tsai YC, Yeatermeyer J, et al. Development of a DNA vaccine targeting human papillomavirus type 16 oncoprotein E6. J Virol. 2004 Aug;78(16):8468–76. doi: 10.1128/JVI.78.16.8468-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010 Mar;207(3):637–50. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010 Mar;207(3):651–67. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng WF, Hung CF, Chai CY, Hsu KF, He L, Ling M, et al. Tumor-specific immunity and antiangiogenesis generated by a DNA vaccine encoding calreticulin linked to a tumor antigen. J Clin Invest. 2001 Sep;108(5):669–78. doi: 10.1172/JCI12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim TW, Lee JH, Hung CF, Peng S, Roden R, Wang MC, et al. Generation and characterization of DNA vaccines targeting the nucleocapsid protein of severe acute respiratory syndrome coronavirus. J Virol. 2004 May;78(9):4638–45. doi: 10.1128/JVI.78.9.4638-4645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JW, Hung CF, Juang J, He L, Kim TW, Armstrong DK, et al. Comparison of HPV DNA vaccines employing intracellular targeting strategies. Gene Ther. 2004;11:1011–8. doi: 10.1038/sj.gt.3302252. [DOI] [PubMed] [Google Scholar]
- 19.Chen CH, Wang TL, Hung CF, Yang Y, Young RA, Pardoll DM, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res. 2000 Feb;60(4):1035–42. [PubMed] [Google Scholar]
- 20.Trimble C, Lin CT, Hung CF, Pai S, Juang J, He L, et al. Comparison of the CD8+ T cell responses and antitumor effects generated by DNA vaccine administered through gene gun, biojector, and syringe. Vaccine. 2003 Sep;21(25–26):4036–42. doi: 10.1016/s0264-410x(03)00275-5. [DOI] [PubMed] [Google Scholar]
- 21.Ji H, Wang TL, Chen CH, Hung CF, Pai S, Lin KY, et al. Targeting HPV-16 E7 to the endosomal/lysosomal compartment enhances the antitumor immunity of DNA vaccines against murine HPV-16 E7-expressing tumors. Hum Gene Ther. 1999;10(17):2727–40. doi: 10.1089/10430349950016474. [DOI] [PubMed] [Google Scholar]
- 22.Hung CF, Cheng WF, He L, Ling M, Juang J, Lin CT, et al. Enhancing major histocompatibility complex class I antigen presentation by targeting antigen to centrosomes. Cancer Res. 2003 May;63(10):2393–8. [PubMed] [Google Scholar]
- 23.Hung CF, Cheng WF, Hsu KF, Chai CY, He L, Ling M, et al. Cancer immunotherapy using a DNA vaccine encoding the translocation domain of a bacterial toxin linked to a tumor antigen. Cancer Res. 2001;61:3698–703. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.