

SUMMARY
The RAS-stimulated RAF-MEK-ERK pathway confers epithelial cells with critical motile and invasive capacities during embryonic development, tissue regeneration and carcinoma progression. Yet many mechanisms by which ERK exerts this control remain elusive. Here, we demonstrate that the ERK-activated kinase RSK is necessary to induce motility and invasive capacities in non-transformed epithelial cells and carcinoma cells. RSK is moreover sufficient to induce certain motile responses. Expression profiling analysis revealed that a primary role of RSK is to induce transcription of potent pro-motile/invasive gene program by FRA1-dependent and independent mechanisms. Strikingly, the program enables RSK to coordinately modulate the extracellular environment, the intracellular motility apparatus, and receptors mediating communication between these compartments to stimulate motility and invasion. These findings uncover a general mechanism whereby the RAS-ERK pathway controls epithelial cell motility by identifying RSK as a key effector, from which emanates multiple highly coordinate transcription-dependent mechanisms for stimulation of motility and invasive properties.
Keywords: RAS, ERK, RSK, epithelial, migration, invasion
INTRODUCTION
Individual cells of epithelial structures can undergo transformation to a motile and invasive mesenchymal phenotype. In this process, referred to as epithelial-mesenchymal transition (EMT), the epithelial cells down-regulate cell-cell junctions, lose apical-basolateral polarity, adopt a mesenchymal cell polarity with leading and trailing edges, and ultimately migrate and invade the surrounding tissue. Epithelial cell motility and invasiveness in the mesenchymal mode are essential for morphogenesis of many embryonic structures and for regenerative processes like wound healing. It also underlies several diseases, notably carcinoma development and progression (Friedl and Wolf, 2003; Grunert et al., 2003; Thiery and Sleeman, 2006; Yang and Weinberg, 2008).
The acquisition of mesenchymal, invasive capacities by epithelial cells requires critical gene expression changes. Proteases must be expressed for degradation of extracellular matrix (ECM) barriers and for proteolytic conversion of ECM proteins and latent growth factors into active forms that support motility and invasion. In this respect, matrix metalloproteinases (MMPs) and the blood-derived protease plasmin, activated by the cell-derived uPA-uPAR protease-receptor complex, constitute two major and collaborating protease systems, that are capable of processing most ECM proteins and latent motogens (Dano et al., 2005; Duffy, 2004; Egeblad and Werb, 2002). Furthermore, invading epithelial cells establish critical autocrine loops of ligands and/or receptors in order to elicit intracellular pro-motile signalling via the small GTPase Rac1 that is a master regulator of the mesenchymal mode of cell motility. Such loops include important and more general ones like the ECM protein laminin 332 acting via α6/β4 integrin/syndecan-1 receptors or uPAR ligated to the ECM protein vitronectin, as well as less ubiquitous loops, such as osteopontin acting via CD44 (Kjoller and Hall, 2001; Marinkovich, 2007; Mercurio et al., 2001; Shaw et al., 1997; Wilhelmsen et al., 2006). The various autocrine loops activate Rac1 by distinct mechanisms, but typically involving cooperation with tyrosine kinases and/or PI 3-kinase (Marinkovich, 2007; Shaw et al., 1997; Smith et al., 2008; Wilhelmsen et al., 2006). Once activated, Rac1 coordinates actin filament dynamics that drive membrane protrusions and migration through key mediators such as IQGAP1 (Noritake et al., 2005).
Upon leaving the epithelium, invasive epithelial cells lose essential survival cues from the basement membrane and cell-cell contacts. These must be replaced by alternative survival signals that will allow the invading epithelial cells to escape apoptosis. To this end, epithelial cells may employ above-mentioned autocrine loops that also elicit potent survival signals or establish more specialized autocrine survival loops, such as vascular endothelial growth factor A (VEGF-A) and Flt-1 receptor tyrosine kinase, epidermal growth factor (EGF) family members and receptors or TIMP1 and CD63 receptor (Bates et al., 2003; Jung et al., 2006; Schulze et al., 2001).
The growth factor-activated and RAS-dependent MAPK cascade composed of RAF, MEK and the effector kinase ERK, is a major signalling pathway to induce mesenchymal motility and invasiveness in epithelial cells (Fig. 1E). ERK exerts this function during development, as well as in carcinoma progression, often in cooperation with signals elicited by TGFβ family cytokines, whose expression may be induced by ERK (Grunert et al., 2003; Lehmann et al., 2000; Oft et al., 1996; Thiery and Sleeman, 2006; Yang and Weinberg, 2008). ERK stimulates most forms of epithelial, invasive motility, such as those occurring during scattering, multilayering, wound healing, tubulogenesis, EMT, malignant invasion and metastasis (Fisher et al., 2001; Grunert et al., 2003; Hansen et al., 2000; Janda et al., 2002; Lehmann et al., 2000; Matsubayashi et al., 2004; Thiery and Sleeman, 2006; Vial et al., 2003). However, while ERK is crucial for these processes, the mechanisms, whereby ERK controls motile and invasive capacities of epithelial cells are not well understood. ERK has hundreds of substrates, but very few direct and principal effectors of ERK have been identified with respect to induction of mesenchymal, invasive migration in epithelial cells.
Figure 1. RSK is a necessary, and partially sufficient, effector of the RAS-ERK pathway for induction of a motile, mesenchymal phenotype in epithelial cells.

(A) Native or RAF1:ER- or RAS:ER-expressing epithelial cell lines were exposed to 1 μM 4HT, 30 nM hepatocyte growth factor (HGF) or 10 μM fmk as indicated, and photographed after 24–48 h.
(B) Cells treated as in (A) were analysed by immunoblotting.
(C) BE cells were cultured in the absence or presence of fmk and photographed after 48 h.
(D) MDCK cells microinjected with plasmids expressing epitope-tagged, constitutively active (CA) or kinase-dead (KD) mutants of ERK2, RSK2 or MSK1 were photographed 0 h or 17 h after injection and thereafter subjected to immunostaining against the epitope tag.
(E) The RAS-activated RAF-MEK-ERK-RSK pathway and sites of action of kinase inhibitors used here. See Fig. S1 for details on RSK structure.
Experiments were conducted 3–6 times with similar results.
The ubiquitous 90 kDa Ribosomal S6 Kinases RSK1-RSK4 are activated by ERK (Hauge and Frodin, 2006). Numerous RSK substrates have been proposed among proteins that regulate differentiation, survival, growth and proliferation. However, any global gene repertoire controlled by RSK, which could indicate primary cellular functions of RSK, has not been identified. Any requirement or role for RSK in cell migration or invasion has not been established.
Here, we identify RSK1 and RSK2 as necessary, and partially sufficient effectors of the RAS-ERK pathway to induce a migratory, invasive mesenchymal phenotype in epithelial cells. These roles of RSK appear quite general, as they were observed in immortalized, but non-transformed, kidney, breast and thyroid epithelial cell lines, as well as in metastatic carcinoma cells from kidney, colon and prostate. In addition, RSK stimulated diverse forms of motility, including cell scattering, wound healing, cell multilayering, chemotaxis and 3D matrix invasion. A series of genome-wide mRNA expression analyses using Solexa sequencing technology revealed the basis of the pro-motile actions of RSK. Thus, out of 1089 genes regulated by ERK in kidney epithelial cells, RSK controlled the expression of 228 genes, 53 of these apparently via the transcription factor FRA1. Strikingly, among the RSK-regulated mRNAs, genes with established roles in motility and invasion comprised the largest functional group (~25%). Furthermore, the RSK-regulated genes were organized in functional clusters, including autocrine loops, through which RSK may induce motility and invasion in a highly coordinate manner. Thus, RSK induced the expression of all subunits of laminin 332 and its receptors α6/β4 integrin and syndecan-1, uPA and uPAR, VEGF-A and Flt-1, osteopontin and CD44, TIMP-1 and CD63, MMP-1 and its receptor α2 integrin, MMP-9 and its receptor CD44, and MMP-10, -13 and -25, as well as intracellular motility proteins, including α-actinins 1 and 4, RhoC and IQGAP1. Consistently, RSK mediated activation of Rac1 by ERK. Finally, we found RSK to stimulate a similar program in mammary epithelial cells and colon adenocarcinoma cells. These data reveal that RSK has the capacity to coordinately modulate the extracellular environment, the intracellular motility apparatus and the receptors mediating communication between these compartments to induce mesenchymal, invasive migration in epithelial cells. Concomitantly, RSK may establish autocrine loops that ensure epithelial cell survival during invasion.
In conclusion, our study reveals a key mechanism, whereby the RAS-ERK pathway induces motile and invasive capacities in epithelial cells by identifying RSK as a principal effector, from which emanates multiple, yet highly coordinate transcription-dependent mechanisms for stimulation of motility and invasiveness.
RESULTS
Activation of RSK is necessary and sufficient to induce a scattered, motile, mesenchymal phenotype in epithelial cells
To determine the mechanism whereby ERK controls epithelial cell motility, we first analysed immortalized, non-transformed MDCK kidney epithelial cells expressing conditionally active RAF1 fused to the hormone-binding domain of the estrogen receptor (MDCK-RAF1:ER). In MDCK-RAF1:ER islets, the estrogen analogue 4-hydroxytamoxifen (4HT) induces activation of ERK and elicits a motile, mesenchymal phenotype (Hansen et al., 2000). A possible involvement of RSK was tested using the highly selective RSK inhibitor fmk (Cohen et al., 2005; Cohen et al., 2007; Frodin, 2007). Fmk inhibits RSK via inactivation of its C-terminal kinase domain (CTK) that activates the substrate-phosphorylating N-terminal kinase domain (NTK) via autophosphorylation of S386 (Fig. 1E). Phosphorylation of RSK at S386 correlates well with NTK activity (Frodin et al., 2002).
Strikingly, fmk abrogated RAF1-induced scattering and migration of MDCK-RAF1:ER cells (Fig. 1A), consistent with inhibition of RSK activation, as assessed by immunoblotting for phospho(p)S386 (Fig. 1B). To establish a general requirement of RSK in scattering and migration of immortalized, but non-transformed cells, we generated and analysed mammary MCF10A and thyroid FRT cells expressing RAF1:ER and MDCK cells expressing H-RAS(G12V):ER. Furthermore, we analysed native MDCK and MCF10A cells treated with the physiological motogens EGF and hepatocyte growth factor (HGF). Strikingly, fmk greatly suppressed scattering and migration induced by these diverse stimuli in all the various epithelial cell types (Fig. 1A, Fig. S1A). Finally, we demonstrated that a requirement of RSK for cell scattering extends to cancer cells by analysing human BE colon carcinoma cells that harbour oncogenic mutations in K-RAS (G13D) and B-RAF (G463V). BE cells exhibit constitutive activation of RSK and have undergone complete transition into a scattered, invasive mesenchymal phenotype (Vial et al., 2003). However, treatment with fmk caused ~30% of BE cells to form islets of stationary, self-adherent, epithelial-like cells that were completely absent from control cultures (Fig. 1C).
Activation of RAF1 and MEK1 is sufficient to induce scattering and migration of MDCK cells, as demonstrated by expression of constitutively active (CA) mutants (Hansen et al., 2000; Schramek et al., 1997) (Fig. 1A). Using same approach, we showed that this is also true for ERK (Fig. 1D). Surprisingly however, given the multitude of ERK substrates other than RSK, we demonstrated that expression of a CA-RSK2 mutant is also sufficient to elicit scattering of MDCK cells (Fig. 1D), and render the cells highly motile (movie in Fig. S1D). The expression vector used (pMT2) yielded very low expression levels of CA-RSK2 that could not be detected by normal indirect immunofluoresence, but only by a very sensitive tyramide signal amplification protocol, indicating specificity of the response. Furthermore, a kinase-dead version of CA-RSK2 (CA-RSK2-KD) and a CA mutant of MSK1, the kinase most closely related to RSK in the human kinome, failed to elicit cell scattering (Fig. 1D).
RSK is required for several ERK-dependent motile capacities in non-transformed and cancerous epithelial cells
Surprisingly, RSK was required for many forms of ERK-stimulated epithelial cell motility. Thus, in MDCK-RAF1:ER cells, fmk blocked cell multilayering induced by RAF in tight and fully polarized MDCK monolayers (Fig. 2A), a cell autonomous motility process that is independent of cell proliferation (Hansen et al., 2000). Furthermore, fmk greatly suppressed native MDCK wound-healing migration (Fig. 2B), an ERK-driven process that occurs in the absence of significant cell proliferation (Matsubayashi et al., 2004).
Figure 2. RSK is required for several forms of ERK-stimulated motility in diverse immortalized or cancerous epithelial cell types.

(A) Confluent, polarized MDCK-RAF1:ER cells on filters were exposed to 1 μM 4HT or 10 μM fmk as indicated. After 24 h, the cells were analysed by phase contrast microscopy (arrows indicate areas with 2–4 cell thick multilayering) (A1) or by actin filament staining and confocal microscopy in Z-X (A2) sections or X-Y sections at the base of the cell layer (A3).
(B) MDCK cell monolayers were wounded in the absence or presence of 10 μM fmk and photographed at 0 h or 24 h.
(C) 3D organoids of LIM 1863 colon adenocarcinoma cells were exposed to 10 ng/ml TNFα, 2 ng/ml TGFβ, 10 μM fmk and 10 μM U0126 as indicated, and photographed after 24 h or analysed for chemotactic cell migration in Boyden chamber assays, using NIH-3T3 cell conditioned medium (CM) as chemoattractant.
(D) Various immortalized or transformed epithelial cell lines were treated, or not, for 24 h with 1 μM 4HT, 6 μM fmk or 10 μM BI-D1870 as indicated and then subjected to 3D invasion ssay through Matrigel basement membrane matrix, using indicated chemoattractants. After 24 h, cells that had invaded through the matrix were quantified and expressed as percent of maximum. Photographs of filters with invaded, crystal violet-stained cells from representative experiments are shown here and in Fig. S2.
(E) Cells treated as in (C) and (D) were analysed by immunoblotting. pS1798 blotting was performed on immunopurified TSC2.
Experiments were conducted 3–6 times with similar results. Data in (C) and (D) are mean ±SD of 3 experiments.
Human LIM 1863 colon adenocarcinoma cells grow as well-differentiated, suspended 3D organoids, exhibiting proper epithelial organization and polarity around a central lumen. Within 24 h of treatment with TGFβ+TNFα, LIM 1863 cells undergo ERK-dependent EMT, transforming into attached, migratory mesenchymal cell monolayers, capable of chemotactic cell motility (Bates et al., 2003). Strikingly, fmk completely abrogated the TGFβ+TNFα-induced 3D-organoid to 2D-migratory-cell-monolayer transition as well as the subsequent chemotactic cell migration (Fig. 2C, Fig. S2A).
Finally, we demonstrated that RSK is required for 3D invasive migration through Matrigel basement membrane matrix by a broad array of widely distinct immortalized epithelial cell lines or carcinoma cells. Thus, fmk greatly suppressed invasive migration of MDCK and MCF10A cells stimulated by RAF or RAS (Fig. 2D). Similarly, fmk or BI-D1870, another RSK inhibitor described below, greatly suppressed invasive migration of 786-0 or RCC10 renal clear cell carcinoma cells, PC3 prostate carcinoma cells and BE colon carcinoma cells, all derived from metastatic carcinomas (Fig. 2D). Invasion assays were performed using HGF, EGF or serum as chemoattractants. These factors also activates the ERK pathway, but much less sustained than e.g. ER:RAF or ER:RAS, and therefore caused little basal invasion under the present conditions. Since BI-D1870 is an inhibitor of RSK NTK, we could not use pS386 phosphorylation to control for inhibition of RSK in BI-D1870 experiments. Therefore, we blotted for the RSK phosphorylation sites S1798 and S428 in TSC2 and LKB1, respectively, and found phosphorylation of these sites to be greatly suppressed by BI-D1870, and to a somewhat lesser extent by fmk (Fig. 2E).
RSK activates a coordinate pro-motile and pro-invasive gene program in MDCK cells
To elucidate how RSK regulates motility and invasion we undertook the first genome-wide characterization of RSK-regulated mRNA expression, using Solexa tag sequencing technology, which allows very quantitative digital expression profiling. The experiment aimed to reveal the RAF-induced gene program that is regulated by ERK and the subprogram regulated by RSK, by analysing polarized MDCK-RAF1:ER cells left untreated or exposed to 4HT for 24 h in the absence or presence of U0126 or fmk. The experiment was performed twice and the highly similar data sets were pooled.
ERK regulated the levels of 1089 mRNAs (338 up-regulated, 523 down-regulated; Table S1), while RSK regulated 228 mRNAs (185 up-regulated, 43 down-regulated; Table S2). In 14 of 15 cases tested, the changes in mRNA levels were paralleled by similar changes in protein levels (Fig. 3A). Thus, the majority of RSK-regulated mRNAs identified by the expression profiling are likely to represent truly RSK-regulated proteins.
Figure 3. RSK induces the expression of a coordinate motility and invasion gene program in MDCK cells.
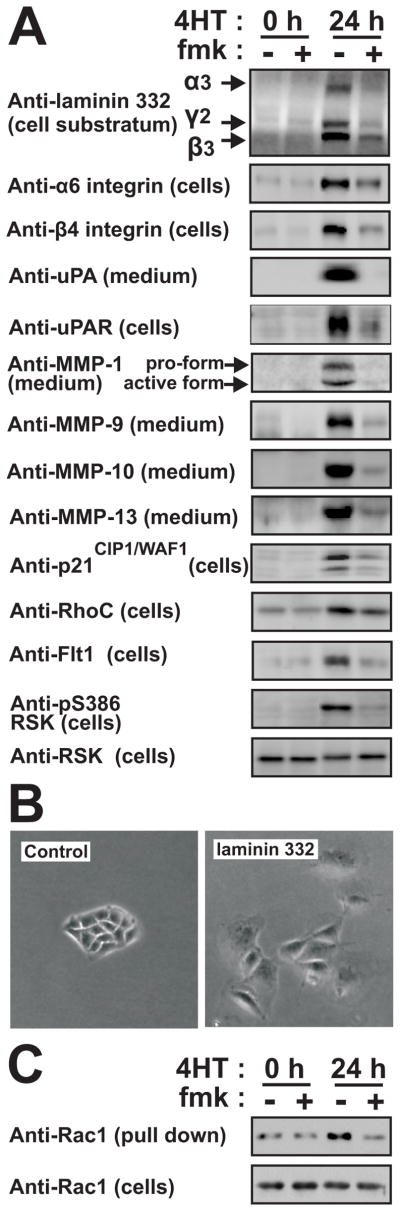
(A) Polarized MDCK-RAF1:ER cell monolayers were exposed to 1 μM 4HT and 6 μM fmk as indicated. After 24 h, the cells or the medium were analyzed by immunoblotting.
(B) Subconfluent MDCK cells were cultured in the absence or presence of 25 μM laminin 332 and photographed 48 h later.
(C) Polarized MDCK-RAF1:ER cell monolayers treated as in (A) were analyzed for active Rac1. All experiments were conducted 3–5 times with similar results.
Strikingly, among the RSK-regulated mRNAs, ~25% encoded pro-motile and pro-invasive proteins, thereby comprising the largest functional group (marked “M” and/or “I” in Table S2). Importantly, many RSK-activated genes constitute functional clusters, such as autocrine ligand-receptor loops. Furthermore, collectively the genes create a highly coordinate and structured program to support motility and survival of invading epithelial cells (illustrated in Fig. 6F). For instance, among the 11 mammalian laminin subunits, RSK selectively induced expression of the α3, β3 and γ2 chains of laminin 332 (Fig. 3A), that strongly stimulates MDCK cell motility (Fig. 3B) as well as both subunits of its α6/β4 integrin receptor and syndecan-1 (β4 integrin up-regulation was not identified in the screen, but by immunoblotting). Similarly, RSK stimulated expression of the ligand-receptor systems uPA and uPAR, VEGF-A and Flt-1, TIMP1 and CD63, osteopontin and CD44. Furthermore, RSK induced expression of a potent battery of ECM degrading/processing MMPs, like MMP-1 and its receptor α2 integrin, MMP-9 and its receptor CD44, MMP-10, MMP-13 and MMP-25 as well as ADAM-28. Furthermore, RSK stimulated mRNA expression of several intracellular proteins involved in transmitting or converting extracellular motility stimuli into altered actin dynamics underlying cell migration. These included α-actinin-1 and -4, RhoC and IQGAP1. RSK also induced expression of the cyclin-dependent kinase inhibitor p21CIP1/WAF1, thought to stimulate motility via modulation of Rho signalling. Among the down-regulated mRNAs were transcripts for uteroglobin, which inhibits epithelial cell migration and p0071, which stabilizes adherence junctions. Finally, RSK stimulated expression of markers or inducers of EMT, like fibronectin, BMP2 and BMP4. Consistently, we also detected RSK-dependent accumulation of TGF-β in the medium after 3 days of RAF-induction (Fig. S4).
Figure 6. RSK stimulates a pro-motile/invasive gene program in various non-transformed or cancerous epithelial cell types.

MCF10A-RAF1:ER (A), MCF10A (B) and LIM 1863 cells (C) were exposed to 1 μM 4HT, 20 nM EGF, 10 ng/ml TNFα, 2 ng/ml TGFβ, 10 μM BI-D1870, 6 μM fmk or 10 μM U0126, as indicated. After 24 h, the cells or medium were analyzed by immunoblotting.
(D) MDCK cells expressing conditionally active CA-RSK2 or vector Ctrl were exposed or not to 1 μM of the inducer Shield1. The cells were analysed after 24 h by immunoblotting or assessed for multilayering after 72 h, as described in legend to Fig. 2A.
(E) MCF10A-RAF1:ER cells were subjected to siRNA knockdown of RSK1-4 in the combinations indicated. After 48 h, the cells were analysed by immunoblotting or in invasion assays, as described in the legend to Fig. 2D.
(F) The model summarizes the results of the present study and illustrates that RSK may induce mesenchymal invasive capacities in epithelial cells by stimulating a coordinate gene program (green) via FRA1-dependent and -independent mechanisms.
We previously observed that RAF1 activates Rac1 in MDCK cells (Hansen et al., 2000). Here, we demonstrate, that this activation is mediated by RSK (Fig. 3C), most likely via RSK induction of the motility program, which contained multiple ligand-receptor systems or molecules previously shown to increase active Rac1 levels (uPA-uPAR, laminin 332-α6/β4 integrin, VEGF-A-Flt-1, IQGAP1, MMP-9, osteopontin-CD44, BMP2, protein C receptor and fibronectin).
A stringent inhibition protocol exploiting chemical genetic analysis confirms the pro-motile/invasive actions of RSK in epithelial cells
Fmk gains specificity via two residues in the ATP-binding site of RSK CTK: C436, covalently modified by fmk, and T493 (Cohen et al., 2005). In the human kinome, this combination exists only in RSK1, RSK2 and RSK4. Accordingly, fmk inhibited these RSKs, but not RSK3 (Fig. 4C), demonstrating the exquisite ability of fmk to discriminate between extremely homologous kinases.
Figure 4. Effects of a rigorous RSK inhibition protocol and an fmk-resistant RSK2 mutant on MDCK cell motility and expression of pro-motile/invasive proteins.

(A) MDCK-RAF1:ER cells were subjected to 6 μM fmk pulse (1 h fmk incubation followed by extensive washing), 10 μM BI-D1870, 100 μM SL0101, 5 μM GF109203X and 1 μM 4HT as indicated, and photographed after 24 h.
(B) Lysates or medium from cells treated as in (A) were analysed by immunoblotting.
(C) MDCK-RAF1:ER cells transfected with plasmid expressing HA-tagged RSK1, -2, -3 or -4 were exposed to 6 μM fmk and 1 μM 4HT, as indicated. After 24 h, the cells were analyzed by immunoblotting.
(D) MDCK-RAF1:ER cells stably expressing wild-type RSK2 or fmk-resistant RSK2-C436V mutant were analyzed in multilayering assays or by immunoblotting.
Experiments were conducted 3 times with similar results.
To validate our data, we first performed fmk pulse inhibition experiments, in which RSK remains inhibited by covalently bound fmk, but free fmk is washed out, precluding ATP-competitive inhibition of potential off-target kinases. Second, we used the two RSK NTK inhibitors, BI-D1870 and SL0101 that were remarkably selective when tested against a panel of 69 kinases (Bain et al., 2007; Sapkota et al., 2007; Smith et al., 2005; Table S3). Third, we used the semi-specific RSK NTK inhibitors GF109203X and Ro318020, which also inhibits certain RSK-related kinases, as well as the specific MEK inhibitor U0126 (Bain et al., 2007). The very few off-targets for the specific inhibitors are inhibited with 20–500 fold lower potency compared to RSK, only one of them, c-SRC (inefficiently inhibited by fmk) has been implicated in epithelial cell motility and, none of them are inhibited in an fmk pulse inhibition experiment (Bain et al., 2007; Cohen et al., 2005; Sapkota et al., 2007). Finally, we used siRNA knockdown of individual RSKs.
Importantly, in MDCK-RAF1:ER cells, all 6 direct and indirect inhibitors of RSK and the fmk pulse inhibition protocol greatly suppressed cell scattering, multilayering, wound healing, chemotactic migration and the motility/invasion gene program shown in Fig. 3A (Fig. 4A and B, Table S4). Inhibition of RAF1-induced multilayering and RSK activation by fmk showed similar IC50 values around 0.3 μM (Fig. S3A). In contrast, 10 μM fmk had no effect on the activity of c-SRC in MDCK cells (Fig. S3B). Furthermore, as described below, knockdown of RSK1 and RSK2 greatly suppressed invasive migration and expression of several components of the motility gene program in MCF10A-RAF1:ER cells (Fig. 6E).
We next performed chemical genetic validations by testing whether expression of an fmk-resistant RSK2 mutant (C436V) could eliminate the effects of fmk. MDCK-RAF1:ER cells stably expressing wild-type RSK2 or RSK2-C436V were generated. Expression of exogenous wild-type and C435V RSK2 was greatly induced by RAF1. However, only induction of wild-type RSK2 was inhibited by fmk, whereas the induction of RSK2-C436V was fmk-insensitive (Fig. 4D). The data suggest that RSK stimulates transcription from the promoter in the vector used. More importantly, in cells expressing RSK2-C436V, fmk failed to inhibit S386 phosphorylation, multilayering as well as expression of uPAR and other motility genes (Fig. 4D; Fig. S3C; data not shown). Similar results were obtained with the fmk-resistant RSK2-T493M mutant (data not shown). This demonstrates conclusively that fmk suppresses multilayering and motility gene expression via inhibition of RSK.
RSK induces motility genes and phenotypes in MDCK cells by FRA1-dependent and -independent mechanisms
According to the literature, RSK regulates >14 transcription factors. To establish mechanisms whereby RSK may regulate the motility program, the RSK-stimulated genes were analyzed bioinformatically for over-representation of any known transcription factor binding sites. The analysis showed selective over-representation of binding sites for poly_c, stat_q6, vmyb_01 and, most significantly AP1, which is composed of FOS/JUN family member dimers.
We pursued AP1 components, since c-FOS is known to be targeted by RSK and c-JUN was a RSK-induced gene (Table S2, gene 107). We first demonstrated that RSK contributes to induction of AP1 activity by RAF1 in MDCK cells by using luciferase reporter constructs containing either an artificial promoter or MMP-1 promoter sequence driven by AP1 binding site(s) (Fig. 5A). Surprisingly however, induction of c-FOS occurred in a largely RSK-independent manner (Table S1 gene 92; immunoblots not shown).
Figure 5. RSK induces the expression of FRA1 to promote induction of pro-motile/invasive genes and phenotypes in MDCK epithelial cells.

(A) MDCK-RAF1:ER cells transfected with AP1-luc or MMP1-luc reporter plasmids were exposed to 1 μM 4HT and 6 μM fmk as indicated, and analysed for reporter gene expression after 24 h.
(B) MDCK-RAF1:ER cells were exposed to 1 μM 4HT and 6 μM fmk as indicated, and analysed by immunoblotting.
(C) MDCK-RAF1:ER cells without (Ctrl) or with knockdown of FRA1 were exposed to 1 μM 4HT as indicated, and analysed by immunoblotting after 24 h.
(D) MDCK-RAF1:ER cells without (Ctrl) or with knockdown of FRA1 were transfected with AP1-luc or MMP1-luc reporter plasmids and analysed as described in (B).
(E) Graphical representation illustrating that the 53 fmk-sensitive genes that were also sensitive to FRA1 knockdown, quantitatively shows very similar sensitivity. Dot and diamond located at the same position of the X axis represent the same gene.
(F) MDCK-RAF1:ER cells with or without knockdown of FRA1 were exposed or not to 1 μM 4HT as indicated, and assessed for multilayering after 24 h, as described in legend to Fig. 2A.
(G) MDCK-RAF1:ER cells without (Ctrl) or with uPAR knockdown were treated and analyzed as described in (C).
(H) Wound healing assays on MDCK-RAF1:ER cells without (Ctrl) or with uPAR knockdown were performed as described in Fig. 2B.
Experiments were conducted 3–4 times with similar results. Data in (A) and (D) are mean ±SD of 3 independent experiments.
We therefore analysed the FOS homologue FRA1 that stimulates motility and invasion by various carcinoma cells (Eferl and Wagner, 2003; Ozanne et al., 2007; Vial et al., 2003). RAF1-induced FRA1 transcripts were not significantly affected by fmk (Table S1, gene 47), but RAF1-induced FRA1 protein levels were reduced by ~60% by fmk (Fig. 5B). We therefore established MDCK-RAF1:ER cell lines expressing short hairpin RNA constructs that reduced RAF1-induced FRA1 expression to roughly the same extent as did fmk (Fig. 5C). In these cells, RAF1-induced expression of luciferase from the AP1 reporter constructs was greatly reduced, demonstrating that FRA1 is a major RAF1/RSK-induced AP1 component in MDCK cells (Fig. 5D). We therefore performed a genome-wide identification of mRNAs dependent on FRA1 expression by subjecting wild-type and FRA1 knockdown MDCK-RAF1:ER cells to Solexa sequencing expression analysis. This analysis revealed that ~23% of the fmk-sensitive mRNAs were also sensitive to FRA1 knockdown (Table. S2). Strikingly, the quantitative effects of fmk treatment and FRA1 knockdown on mRNA expression were remarkably similar for the far majority of these genes, strongly suggesting that the expression of this set of ~50 genes is controlled by an ERK-RSK-FRA1 signaling cassette (Fig. 5E). At least 30% of the fmk-sensitive motility/invasion genes were also sensitive to FRA1 knockdown. We confirmed FRA1-dependent expression of laminins α3, β3 and γ2, uPAR and MMP-1 by immunoblotting (Fig. 5C). Interestingly, RAF1-induced cell multilayering was also greatly reduced by knocking down FRA1 (Fig. 5F). Finally, we generated MDCK-RAF1:ER cells with shRNA-mediated knockdown of uPAR expression, one of the RSK/FRA1-induced proteins, and found that multilayering and wound healing migration were substantially suppressed in these cells (Fig. 5G, H).
A RSK-dependent motility and invasion gene program is observed also in breast and colon epithelial cells
Immunoblotting revealed that the RSK-dependent motility and invasion program observed in MDCK cells was strikingly conserved in breast MCF10A and colon adenocarcinoma LIM 1863 cells challenged with conditional activation of RAF, EGF or TGFβ+TNFα. Thus, RSK-inhibitors significantly suppressed the stimulated and/or basal protein expression levels of laminin 332, β4 integrin, uPA, uPAR, MMP-1, MMP-9 and MMP-10 (Fig. 6A–C). RSK-dependent expression of uPA, uPAR and laminin was also observed in 786-0 and PC3 carcinoma cells by real-time quantitative RT-PCR (Fig. S2D). Furthermore, in all these cell lines and settings, FRA-1 was also induced in a RSK-dependent manner. Whereas RSK was found to induce the VEGF-A-Flt-1 survival loop in MDCK cells, RSK induced the EGF family members amphiregulin and HB-EGF in MCF10A cells, factors previously shown to underlie an essential RAF1-induced survival loop to suppress detachment-induced apoptosis (anoikis) in these cells (Schulze et al., 2001).
RSK1 and RSK2 are pro-motile/invasive RSK homologues
Finally, we further addressed the issue of RSK sufficiency as well as specific RSK homologue requirements in pro-motile/invasive signaling by the RAS-ERK pathway. First, we established MDCK cells expressing CA-RSK2 fused to a 12 kDa mutant of the FKBP protein (the destabilization domain, or DD). In MDCK-DD-CA-RSK2 cells, CA-RSK2 expression was found to be conditionally induced by addition of the small-molecule compound Shield1. Using these cells, we found that conditional induction of CA-RSK2 was sufficient to increase expression of certain laminin 332 chains, β4 integrin, uPA, uPAR and FRA1, but not sufficient to increase expression of certain other motility genes, including various MMPs (Fig. 6D; data not shown). Note, that the low, uninduced level of CA-RSK2 was sufficient to increase the expression of some of the proteins. Strikingly, conditional induction of CA-RSK2 was also sufficient to cause cell multilayering in fully confluent and polarized MDCK monolayers, albeit to a lower extent than conditional activation of RAF1 (Fig. 6D). Furthermore, conditional induction of RSK2 greatly accelerated wound healing migration by MDCK cells (data not shown).
Next, we identified RSK forms that may underlie our findings using siRNA-mediated knockdown. This analysis was performed in MCF10A cells, since poor knockdown was obtained with siRNA reagents in MDCK cells. Knockdown of RSK1-3 was confirmed by immunoblotting or quantitative RT-PCR (Fig. 6E; Fig. S2E). RSK4 expression could not be detected. Interestingly, knockdown of both RSK1 and RSK2 was required to substantially inhibit the induction of certain motility genes as well as invasive migration by RAF (Fig. 6E). For certain other motility genes, individual knockdown of RSK1 or RSK2 produced substantial effects. Thus, these data revealed that both RSK1 and RSK2 contribute to induce a pro-motile phenotype and gene program in epithelial cells.
Discussion
Regulation of epithelial cell motility and invasiveness by RSK
A stringent inhibition protocol enabled us to establish that stimulation of epithelial cell motility and invasive capacities is a cellular function of RSK that appears to be rather general, since it was observed in epithelial cell lines from 5 distinct tissues, i.e. kidney, breast, colon, thyroid and prostate. In addition, RSK was required for very diverse forms of epithelial cell motility, such as cell scattering, wound healing, cell multilayering, chemotaxis, 3D-organoid-to-2D migration and 3D ECM invasion and, apparently sufficient for cell scattering and multilayering motility. Finally, motility signalling may represent a major cellular function of RSK, since motility/invasion genes constituted by far the largest functional group among the RSK-regulated mRNAs.
Our study provides several mechanisms whereby RSK may stimulate epithelial cell motility in a highly organized and coordinate manner (Fig. 6F). Strikingly, RSK may establish autocrine loops to elicit intracellular signaling for mesenchymal, invasive migration and, at the same time elicit survival signaling essential for this mode of invasion: Thus, RSK coordinately induced all subunits of laminin 332, its processing enzymes and its receptors α6/β4 integrin and syndecan-1, which upon binding distinct sites on laminin 332 are thought to cooperate in a feed-forward loop for further deposition of laminin 332 and intracellular activation of Rac1 (Marinkovich, 2007). Furthermore, RSK induced expression of several other receptors and autocrine loops, such as uPA-uPAR and osteopontin-CD44 capable of activating Rac1, coordinate with RSK-induced expression of IQGAP1, a key effector of Rac1 in mesenchymal cell migration. Finally, RSK may establish yet further three PI3-kinase-based autocrine survival loops, namely VEGF-A/Flt-1 and TIMP-1/CD63, as observed in MDCK cells, and HB-EGF/amphiregulin loops, as observed in MCF10A cells. In our study, RSK inhibitors did not appreciably affect cell survival, likely because experiments were performed in the presence of exogenous survival stimuli like serum or growth factor. In conclusion, RSK orchestrates several mechanisms to cooperatively poise the intracellular survival and motility apparatus for mesenchymal, invasive migration by epithelial cells.
The ECM degrading proteases provide another example how RSK induces proteins that cooperate to promote motility and invasion: uPA requires binding to uPAR to activate plasmin. Alternatively, plasmin may be activated by MMP-9 bound to CD44. uPA/uPAR/plasmin and MMP-10 proteolytically activate MMP-1. Finally, uPA/uPAR/plasmin, MMP-1, MMP-9 and MMP-13 can activate MMPs outside the cluster, including MMP-2. RSK also enhanced expression of receptors for MMP-1 and MMP-9, i.e. α2 integrin and CD44, respectively. Importantly, these proteases not only activate each other, but also critically depend on each other for sequential and collaborative degradation of the many diverse components of ECM (Dano et al., 2005; Duffy, 2004; Egeblad and Werb, 2002). Furthermore, the RSK-stimulated proteases are known to activate or release ECM-bound pro-invasive cytokines like HGF and TGFβ, the latter found here to be induced by RSK (Fig. S4) and generate ECM fragments that stimulate motility. In conclusion, RSK controls an impressive proteolytic program that cooperatively modulates the ECM to support mesenchymal invasive migration in epithelial cells.
Our findings clearly demonstrate that RSK is required for induction of partial EMT. To determine whether RSK is necessary for complete EMT would necessitate analysis after 4–6 days (Grunert et al., 2003; Lehmann et al., 2000; Thiery and Sleeman, 2006), as opposed to the 1–2 day analysis performed here. Nevertheless, RSK stimulated the expression of EMT markers like fibronectin and EMT-inducers like MMP-9, BMP2, BMP4 and TGFβ, suggesting that RSK may contribute to induction of complete EMT in some epithelial cell types.
Global insight into transcriptional regulation by RSK
Our findings identify RSK as an important effector of ERK in global transcriptional regulation. In MDCK cells, RSK contributed to regulation of ~20% of mRNAs controlled by ERK, which may occur via direct mechanisms, such as phosphorylation of transcription factors and, more indirectly, via the RSK-induced autocrine loops. We identify FRA1 as a new transcription factor whose expression is stimulated by RSK. In MCF10A cells, RSK stimulated FRA1 expression at the mRNA level, as determined by real-time quantitative RT-PCR (data not shown), whereas in MDCK cells, RSK acted mainly at the post-transcriptional level. RSK may stimulate FRA1 protein expression in MDCK cells by phosphorylation of S252. The analogous site of c-FOS is phosphorylated by RSK and in FRA1, the site promotes FRA1 protein stability (Basbous et al., 2007). More importantly, our findings suggest that FRA1 is an important effector of RSK in transcriptional regulation in epithelial cells. Thus, FRA1 may mediate the expression of ~23% of the mRNAs controlled by RSK, including many motility and invasion genes. Notably, we also found that RSK stimulated the expression of c-JUN that might constitute the AP1 partner of FRA1 in our system. All together, the present demonstration that RSK controls FRA1 expression may be significant, since FRA1 and AP1 are considered key regulators of motility and invasion in development as well as in cancer (Eferl and Wagner, 2003; Ozanne et al., 2007).
It will be important to identify the RSK-regulated transcription factors underlying the FRA1-independent gene program. Further bioinformatics analysis of the present data sets of RSK-regulated genes may help identifying these factors.
RSK in regenerative and pathological epithelial cell invasion and potential as a drug target
Based on the present findings, it seems likely that future studies will reveal important roles of RSK in invasive migration of epithelial cells during wound healing and carcinoma progression. In various instances, these processes have been found to depend on several genes found here to be regulated by RSK. For instance, mouse studies have demonstrated that keratinocyte wound healing migration is partially impaired by individually interfering with uPA/uPAR/plasmin or MMP function and completely blocked by inhibition of both protease systems (D’Alessio et al., 2008; Lund et al., 1999). Furthermore, mice with deletion of the intracellular, RAC1-activating signaling domain of β4 integrin showed impaired wound healing (Nikolopoulos et al., 2005). Numerous studies have documented that a large fraction of the RSK-stimulated invasion/motility genes are over-expressed in carcinomas and promote cancer in animal models (genes marked “C” and “EC” in Table S2). Several of the genes are independent negative prognostic markers for various cancers, including laminin 332, uPA, uPAR, FRA1 and various MMPs (Dano et al., 2005; Duffy, 2004; Eferl and Wagner, 2003; Egeblad and Werb, 2002; Marinkovich, 2007; Nikolopoulos et al., 2005; Ozanne et al., 2007; Wilhelmsen et al., 2006). Furthermore, genes of the RSK-dependent program are often co-expressed in cancer, such as uPA, uPAR, MMP-9, MMP-13 and laminin 332 that are co-expressed in skin squamous carcinoma cells (Dano et al., 2005). Mouse studies have demonstrated essential roles of several of the RSK-stimulated genes in carcinoma progression. For instance, laminin 332 and α6/β4 integrin are required for RAS to induce squamous cell carcinomas (Dajee et al., 2003). Cancer studies demonstrating hyperactive RSK and concomitant up-regulation of the RSK-dependent motility/invasion genes identified here are lacking, presumably because RSK has not previously been linked to cancer cell invasiveness. However, the RAS-ERK pathway, including RSK, was recently shown to be hyper-activated in polycystic kidney disease, which is characterized by greatly expanded, hyper-proliferative and remodelled kidney epithelium (Shibazaki et al., 2008). Interestingly, ~35% of the RSK-induced genes identified here are also up-regulated in polycystic kidney disease (marked “PKD” in Table S2), providing the first correlative evidence that RSK induces these genes in disease.
Our findings suggest that RSK may constitute a new candidate drug target in certain invasive carcinomas and suggest mechanisms by which RSK inhibitors might be effective anti-carcinoma drugs. For instance, RSK inhibitors may suppress the expression of a potent battery of proteases with established roles in metastasis. Nevertheless, this may not be sufficient to abrogate invasion, since blocking extracellular protease systems may cause carcinoma cells to switch from a mesenchymal to an amoeboid, proteolysis-independent invasion mode (Friedl and Wolf, 2003). Importantly, however, we found that inhibition of RSK also suppressed expression of several autocrine loops with critical functions in survival of invading carcinoma cells. Thus, RSK inhibitors may not only carry the potential to prevent initiation of invasion, but also to kill carcinoma cells during the process of invasion.
Supplementary Material
Acknowledgments
We thank Dario Alessi for BI-D1870, Reider Albrechtsen for help with confocal microscopy, and HDDC Core facilities. This work was supported by grants from the Danish Cancer Society (DP06133), the Danish National Research Foundation and the Danish Cancer Research Foundation (47-07) to M.F., by US NIH R01 CA092354, Roy and Lynne Frank Foundation and Childrens’ Hospital, Boston grants to S.H.H and by US NIH R01 GM71434 grant to J.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbous J, Chalbos D, Hipskind R, Jariel-Encontre I, Piechaczyk M. Ubiquitin-independent proteasomal degradation of Fra-1 is antagonized by Erk1/2 pathway-mediated phosphorylation of a unique C-terminal destabilizer. Mol Cell Biol. 2007;27:3936–3950. doi: 10.1128/MCB.01776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates RC, Goldsmith JD, Bachelder RE, Brown C, Shibuya M, Oettgen P, Mercurio AM. Flt-1-dependent survival characterizes the epithelial-mesenchymal transition of colonic organoids. Curr Biol. 2003;13:1721–1727. doi: 10.1016/j.cub.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio S, Gerasi L, Blasi F. uPAR-deficient mouse keratinocytes fail to produce EGFR-dependent laminin-5, affecting migration in vivo and in vitro. J Cell Sci. 2008;121:3922–3932. doi: 10.1242/jcs.037549. [DOI] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Dano K, Behrendt N, Hoyer-Hansen G, Johnsen M, Lund LR, Ploug M, Romer J. Plasminogen activation and cancer. Thromb Haemost. 2005;93:676–681. doi: 10.1160/TH05-01-0054. [DOI] [PubMed] [Google Scholar]
- Duffy MJ. The urokinase plasminogen activator system: role in malignancy. Curr Pharm Des. 2004;10:39–49. doi: 10.2174/1381612043453559. [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Fisher CE, Michael L, Barnett MW, Davies JA. Erk MAP kinase regulates branching morphogenesis in the developing mouse kidney. Development. 2001;128:4329–4338. doi: 10.1242/dev.128.21.4329. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- Frodin M. A RSK kinase inhibitor reporting its selectivity in vivo. Nat Chem Biol. 2007;3:138–139. doi: 10.1038/nchembio0307-138. [DOI] [PubMed] [Google Scholar]
- Frodin M, Antal TL, Dummler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 2002;21:5396–5407. doi: 10.1093/emboj/cdf551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- Hansen SH, Zegers MM, Woodrow M, Rodriguez-Viciana P, Chardin P, Mostov KE, McMahon M. Induced expression of Rnd3 is associated with transformation of polarized epithelial cells by the Raf-MEK-extracellular signal-regulated kinase pathway. Mol Cell Biol. 2000;20:9364–9375. doi: 10.1128/mcb.20.24.9364-9375.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauge C, Frodin M. RSK and MSK in MAP kinase signalling. J Cell Sci. 2006;119:3021–3023. doi: 10.1242/jcs.02950. [DOI] [PubMed] [Google Scholar]
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KK, Liu XW, Chirco R, Fridman R, Kim HR. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006;25:3934–3942. doi: 10.1038/sj.emboj.7601281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjoller L, Hall A. Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J Cell Biol. 2001;152:1145–1157. doi: 10.1083/jcb.152.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K, Janda E, Pierreux CE, Rytomaa M, Schulze A, McMahon M, Hill CS, Beug H, Downward J. Raf induces TGFbeta production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev. 2000;14:2610–2622. doi: 10.1101/gad.181700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund LR, Romer J, Bugge TH, Nielsen BS, Frandsen TL, Degen JL, Stephens RW, Dano K. Functional overlap between two classes of matrix-degrading proteases in wound healing. EMBO J. 1999;18:4645–4656. doi: 10.1093/emboj/18.17.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovich MP. Tumour microenvironment: laminin 332 in squamous-cell carcinoma. Nat Rev Cancer. 2007;7:370–380. doi: 10.1038/nrc2089. [DOI] [PubMed] [Google Scholar]
- Matsubayashi Y, Ebisuya M, Honjoh S, Nishida E. ERK activation propagates in epithelial cell sheets and regulates their migration during wound healing. Curr Biol. 2004;14:731–735. doi: 10.1016/j.cub.2004.03.060. [DOI] [PubMed] [Google Scholar]
- Mercurio AM, Rabinovitz I, Shaw LM. The alpha 6 beta 4 integrin and epithelial cell migration. Curr Opin Cell Biol. 2001;13:541–545. doi: 10.1016/s0955-0674(00)00249-0. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Puri C, Tacchetti C, Giancotti FG. Targeted deletion of the integrin beta4 signaling domain suppresses laminin-5-dependent nuclear entry of mitogen-activated protein kinases and NF-kappaB, causing defects in epidermal growth and migration. Mol Cell Biol. 2005;25:6090–6102. doi: 10.1128/MCB.25.14.6090-6102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. IQGAP1: a key regulator of adhesion and migration. J Cell Sci. 2005;118:2085–2092. doi: 10.1242/jcs.02379. [DOI] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462–2477. doi: 10.1101/gad.10.19.2462. [DOI] [PubMed] [Google Scholar]
- Ozanne BW, Spence HJ, McGarry LC, Hennigan RF. Transcription factors control invasion: AP-1 the first among equals. Oncogene. 2007;26:1–10. doi: 10.1038/sj.onc.1209759. [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P, Alessi DR. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramek H, Feifel E, Healy E, Pollack V. Constitutively active mutant of the mitogen-activated protein kinase kinase MEK1 induces epithelial dedifferentiation and growth inhibition in madin-darby canine kidney-C7 cells. J Biol Chem. 1997;272:11426–11433. doi: 10.1074/jbc.272.17.11426. [DOI] [PubMed] [Google Scholar]
- Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001;15:981–994. doi: 10.1101/gad.191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, Louvi A, Velazquez H, Ishibe S, Cantley LG, Igarashi P, Somlo S. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008;17:1505–1516. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HW, Marra P, Marshall CJ. uPAR promotes formation of the p130Cas-Crk complex to activate Rac through DOCK180. J Cell Biol. 2008;182:777–790. doi: 10.1083/jcb.200712050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005;65:1027–1034. [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- Wilhelmsen K, Litjens SH, Sonnenberg A. Multiple functions of the integrin alpha6beta4 in epidermal homeostasis and tumorigenesis. Mol Cell Biol. 2006;26:2877–2886. doi: 10.1128/MCB.26.8.2877-2886.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.