

Abstract
A variant upstream of human leukocyte antigen C (HLA-C) shows the most significant genome-wide effect on HIV control in European Americans and is also associated with the level of HLA-C expression. We characterized the differential cell surface expression levels of all common HLA-C allotypes and tested directly for effects of HLA-C expression on outcomes of HIV infection in 5243 individuals. Increasing HLA-C expression was associated with protection against multiple outcomes independently of individual HLA allelic effects in both African and European Americans, regardless of their distinct HLA-C frequencies and linkage relationships with HLA-B and HLA-A. Higher HLA-C expression was correlated with increased likelihood of cytotoxic T lymphocyte responses and frequency of viral escape mutation. In contrast, high HLA-C expression had a deleterious effect in Crohn’s disease, suggesting a broader influence of HLA expression levels in human disease.
Variation within the human leukocyte antigen (HLA) class I genes of the major histocompatibility complex (MHC) has the greatest impact on outcome after HIV infection relative to the rest of the genome (1–3). A single-nucleotide polymorphism 35 kb upstream of the HLA-C locus (rs9264942) showed the most significant association with viral load (VL) control in a recent genome-wide association study (GWAS) (2) and the second most significant association in an earlier HIV GWAS where VL at set point was considered (3). We previously reported that rs9264942 genotype correlated with the level of HLA-C cell surface protein expression on primary T cells from European Americans (4). Whether rs9264942 is associated with the outcome of HIV infection because it marks HLA-C expression levels or individual HLA alleles that affect HIV remains unknown (4, 5).
If HLA-C expression levels have a direct influence on HIV control, then we expect to observe the effect across ethnic groups, despite their distinct HLA-C allele frequencies and linkage disequilibrium (LD) relationships with HLA-A and -B alleles (6). Unlike in European Americans, rs9264942 is in poor LD with HLA-C alleles in African Americans and does not mark expression levels of HLA-C or associate with HIV control in large studies (2, 7) (fig. S1). We determined the expression levels of individual HLA-C allotypes in African Americans, which showed a continuous distribution (Fig. 1), in order to test an effect of HLA-C expression level on HIV control.
Fig. 1. The distribution in expression levels of HLA-C allotypes present in African Americans.
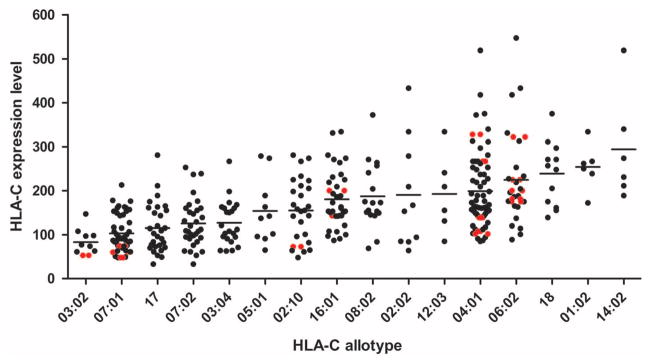
Peripheral blood CD3+ cells from 200 healthy donors were analyzed by flow cytometry for HLA-C expression level using the monoclonal antibody DT9. MFI of HLA-C staining is plotted twice for each donor (i.e., once for each HLA-C allele present), with HLA-C homozygous individuals marked in red. Expression level correlates significantly with HLA-C allotypes in analysis of variance (P = 5 × 10−21).
HLA-C alleles that are present in both African and European Americans show the same relative expression levels in the two populations (4) (Fig. 1). Indeed, a highly significant correlation was observed in both 50 European-American and 50 African-American donors when comparing the observed HLA-C expression levels with expected levels based on mean expression of each HLA-C allele observed in an independent group of 150 African Americans (fig. S2). After confirming that HIV infection does not alter HLA-C expression (fig. S2), we used the mean expression of individual HLA-C alleles measured in all 200 African-American donors to assign HLA-C expression levels to each HIV-infected subject based on their HLA-C genotypes (8). An effect of HLA-C expression level was tested, along with all individual HLA class I alleles, in chronically infected subjects using logistic regression with stepwise selection. HLA-C expression level showed a significant independent association with HIV control in an analysis of 2527 European-American patients [P = 1 × 10−7, odds ratio (OR) = 0.52] (Table 1), where OR for HLA-C expression represents the protection conferred by a difference of 100 higher median fluorescence intensity (MFI) expression units (Fig. 1). The average difference in expression of 224 units between Cw*07 and Cw*06 homozygotes, for example, would correspond to an OR of 0.24.
Table 1. HLA-C expression level affects control of HIV viral load in European and African Americans.
Antiretroviral therapy (ART)–naïve HIV patients were categorized as controllers (<2000 viral copies/ml plasma) or noncontrollers (>10,000 viral copies/ml plasma) on the basis of longitudinal mean VL in chronic infection. Effects of HLA-C expression level as a continuous variable and all individual HLA class I alleles with phenotypic frequency ≥2% were tested by logistic regression with stepwise selection. Significance (P), OR, and 95% confidence interval (CI) are reported for all covariates showing independently significant effects.
| European Americans (n = 2527)
|
African Americans (n = 1209)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Covariate | P | OR | 95% CI | Covariate | P | OR | 95% CI | ||
| B*57:01 | 1 × 10−19 | 0.13 | 0.09 | 0.21 | B*57:03 | 2 × 10−24 | 0.13 | 0.09 | 0.20 |
| C*04:01 | 8 × 10−14 | 3.12 | 2.32 | 4.21 | B*81 | 2 × 10−8 | 0.21 | 0.12 | 0.37 |
| A*01:01 | 2 × 10−10 | 2.21 | 1.73 | 2.82 | HLA-C expression | 8 × 10−6 | 0.61 | 0.49 | 0.76 |
| B*07:02 | 2 × 10−9 | 2.36 | 1.78 | 3.13 | B*35:01 | 1 × 10−5 | 2.92 | 1.80 | 4.74 |
| HLA-C expression | 1 × 10−7 | 0.52 | 0.41 | 0.67 | B*45:01 | 3 × 10−5 | 4.28 | 2.16 | 8.50 |
| A*02:01 | 4 × 10−7 | 1.68 | 1.38 | 2.06 | A*23:01 | 1 × 10−4 | 2.05 | 1.43 | 2.96 |
| B*27:05 | 2 × 10−5 | 0.48 | 0.34 | 0.67 | B*58:02 | 3 × 10−3 | 2.54 | 1.37 | 4.73 |
| C*06:02 | 2 × 10−5 | 2.65 | 1.69 | 4.15 | B*58:01 | 4 × 10−3 | 0.49 | 0.30 | 0.79 |
| B*13:02 | 4 × 10−4 | 0.39 | 0.23 | 0.65 | A*36:01 | 6 × 10−3 | 3.22 | 1.40 | 7.38 |
| C*16:01 | 6 × 10−4 | 2.12 | 1.38 | 3.26 | B*39:10 | 1 × 10−2 | 0.34 | 0.14 | 0.78 |
| A*24:02 | 9 × 10−4 | 1.58 | 1.20 | 2.07 | B*53:01 | 3 × 10−2 | 1.48 | 1.04 | 2.11 |
| C*01:02 | 3 × 10−3 | 1.90 | 1.24 | 2.91 | |||||
| B*52:01 | 4 × 10−3 | 0.49 | 0.30 | 0.79 | |||||
| A*26:01 | 4 × 10−3 | 1.80 | 1.20 | 2.70 | |||||
| B*40:01 | 6 × 10−3 | 1.69 | 1.16 | 2.45 | |||||
| B*38:01 | 7 × 10−3 | 2.02 | 1.22 | 3.34 | |||||
| C*05:01 | 1 × 10−2 | 1.43 | 1.09 | 1.89 | |||||
| B*18:01 | 1 × 10−2 | 1.56 | 1.10 | 2.23 | |||||
| B*58:01 | 2 × 10−2 | 0.49 | 0.27 | 0.90 | |||||
| B*14:02 | 3 × 10−2 | 0.67 | 0.46 | 0.96 | |||||
| B*51:01 | 4 × 10−2 | 1.47 | 1.01 | 2.13 | |||||
| B*55:01 | 5 × 10−2 | 2.07 | 1.02 | 4.21 | |||||
The protective effect of high HLA-C expression level in European Americans was mirrored in an analysis of 1209 African-American patients, where expression had the third most significant independent effect (P = 8 × 10−6, OR = 0.61) (Table 1) after HLA-B*57:03 and B*81, two well-documented protective alleles in cohorts of African descent (7, 9). Similar results were observed when mean VL was considered as a continuous variable (table S1). Although our African-American population is admixed to some extent with European genotypes, it is clearly distinct from our European-American cohorts (fig. S1). The effect of HLA-C expression level persisted when increasing the stringency at which covariates were considered to have independent effects, and permutation analyses indicated that the distribution of HLA-C allelic expression values observed are unlikely to correlate with HIV control by chance (8).
HLA-C expression level showed the most significant effect on progression from seroconversion to CD4 < 200 in a cohort of 1069 subjects [P = 1 × 10−6, hazard ratio (HR) = 0.67 for a difference of 100 MFI expression units] relative to all individual HLA alleles (Table 2). Thus, the continuous distribution of HLA-C allele expression levels correlates with multiple outcomes of HIV infection, tested in three distinct cohorts: longitudinal VL in European Americans, longitudinal VL in African Americans, and progression to CD4 < 200 in a separate cohort of European and African Americans combined (8). Use of regression models that include all HLA class I alleles as covariates, along with consistent effects across African and European Americans, strongly point to HLA-C expression level as a major independent contributor to control of HIV.
Table 2. HLA-C expression level affects progression to early AIDS outcomes.
HLA-C expression level as a continuous variable and all HLA class I alleles with phenotypic frequency ≥2% were tested by stepwise selection in a Cox model, using 1069 ART-naïve patients with known times for progression from seroconversion to CD4 < 200. Subjects comprised 783 European and 286 African Americans, with ethnicity used as a covariate. This was justified because stratification by ethnicity indicated a uniform strength of effects on progression across European and African Americans (table S2), which was also apparent in the larger longitudinal VL cohorts (Table 1). Independently significant covariates are reported with significance (P), HR, and 95% CI.
| Covariate | P | HR | 95% CI | |
|---|---|---|---|---|
| HLA-C expression | 1 × 10−6 | 0.67 | 0.55 | 0.74 |
| B*57:01 | 2 × 10−5 | 0.30 | 0.17 | 0.52 |
| C*06:02 | 7 × 10−4 | 1.75 | 1.26 | 2.41 |
| A*26:01 | 2 × 10−3 | 0.43 | 0.25 | 0.73 |
| B*35:03 | 2 × 10−3 | 2.11 | 1.3 | 3.41 |
| A*11:01 | 3 × 10−3 | 0.62 | 0.45 | 0.85 |
| A*74 | 4 × 10−3 | 0.26 | 0.11 | 0.65 |
| A*32:01 | 4 × 10−3 | 0.53 | 0.34 | 0.82 |
| A*31:01 | 3 × 10−2 | 0.56 | 0.33 | 0.95 |
| B*15:01 | 4 × 10−2 | 0.72 | 0.52 | 0.99 |
| C*04:01 | 4 × 10−2 | 1.30 | 1.01 | 1.69 |
A direct genetic effect of HLA-C expression level on HIV control predicts that more highly expressed HLA-C allotypes would confer greater immune pressure on the virus, which can be assessed by measuring the strength of association between a given HLA-C allele and corresponding mutations in the viral genome. HLA class I genotypes and nearly full-length HIV sequences were generated in a cohort of 1888 clade B chronically infected patients (the majority Caucasian) who were treatment naïve. A phylogenetically corrected logistic regression approach was used to estimate the probability of postinfection escape mutations and determine their independent associations with HLA alleles present (8, 10, 11). Viral sequence mutations showing significant independent associations with HLA-C were then filtered for predicted epitopes that fit the biochemical features of the HLA-C allele involved (12). Twenty-two mutations were identified, representing escape at 12 epitopes that associated with 7 HLA-C alleles (table S3). Median log odds ratios were calculated for mutations at all epitopes associated with the same HLA-C allele to determine the strength of selection conferred by each of these 7 HLA-C alleles (table S3). A strong positive correlation between HLA-C expression level and the strength of selection pressure conferred on the virus by the allele was observed (r = 0.87, P = 0.005) (Fig. 2A), indicating that higher HLA-C expression exerts greater selection on HIV. This finding is consistent with a study of Han Chinese that identified six viral mutations associated with HLA-C alleles that occurred more commonly in 99 patients with the rs9264942 genotype CC than in 63 patients with the TT genotype (13).
Fig. 2. HLA-C expression level correlates with frequency of viral escape mutation and HIV-specific CTL responses.
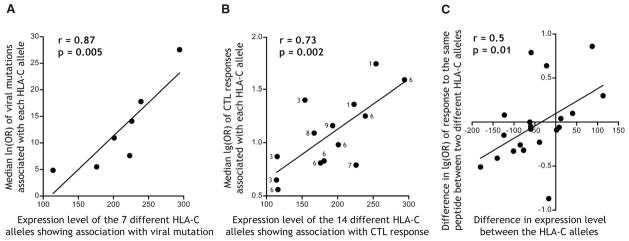
(A) Analysis of viral sequences from 1888 Clade B–infected individuals revealed 12 epitopes containing viral mutations that were associated independently with an HLA-C allele. Median ln(OR) for associations with each of the seven different HLA-C alleles involved correlated positively with the expression level of these alleles. (B) Clade C–infected Africans (n = 1010) were screened for CTL responses to overlapping peptides spanning the HIV proteome, and median log OR of all responses independently associated with each HLA-C allele are plotted. The number of peptide responses associated with each HLA-C allele is labeled next to each point (total number of CTL responses is 71). At least one HLA-C–restricted CTL response was detected in 71% of individuals in the population. (C) Differences in the odds of detecting a response to the same peptide when restricted by different HLA-C alleles correlate with the difference in expression level of these allelic pairs. Pearson coefficients are reported for each correlation.
We analyzed cytotoxic T lymphocyte (CTL) responses to overlapping peptides spanning the entire HIV clade C proteome, that were measured by enzyme-linked immunosorbent spot assay in 1010 HIV subtype C–infected South African subjects (14, 15). HLA-associated CTL responses were determined by testing the log OR of observing a response to each peptide in the presence versus absence of every HLA class I allele, using a decision tree with Fisher’s exact test to discriminate associations due to linked HLA alleles (8). CTL responses restricted by all of the 14 two-digit HLA-C alleles present in our population were observed, comprising 71 distinct associations between a given peptide and a given HLA-C allele (table S4). Median log ORs were calculated for responses to all peptides associating with the same HLA-C allele. A strong correlation between the level of HLA-C allele expression and average strength of association with CTL response to HIV was observed (r = 0.73, P = 0.002) (Fig. 2B), such that HIV peptides presented by higher expressed HLA-C alleles were more likely to elicit CTL responses than peptides presented by low expression HLA-C allotypes.
Of the total 60 peptides associated with HLA-C–restricted CTL responses, 8 peptides associated with responses restricted by two different HLA-C allotypes and 4 peptides associated with responses restricted by three different HLA-C allotypes. This permits a total of 20 separate comparisons of the strength of response to a single peptide between two HLA-C allotypes that differ in their level of expression (table S5). A significant correlation was observed between the difference in HLA-C expression level for allotype pairs and the difference in strength of association with CTL response to the same peptide when restricted by these two allotypes (r = 0.50, P = 0.01) (Fig. 2C). That is, the likelihood of initiating a CTL response to a given peptide correlated positively with a greater increase in expression between the two alleles compared. These results suggest that the protective mechanism conferred by increased HLA-C expression level occurs in part through an enhanced CTL-mediated immune response.
The effect of HLA-C expression level in HIV disease raises the possibility of its involvement in the risk of other human diseases. We tested this possibility in case-control cohorts of two inflammatory bowel diseases (IBD): Crohn’s disease (CD) and ulcerative colitis (UC). HLA-C expression as a continuous variable was considered in stepwise selection models along with all HLA class I and II alleles with phenotypic frequencies of ≥ 2%. We include class II alleles because they show the most consistent allelic effects and the strongest MHC signal in GWAS of IBD (16–18). HLA-C expression level showed the second most significant effect in CD (P = 3 × 10−7, OR = 1.35 for an increase of 100 MFI units) after the well-established risk allele DRB1*01:03 (19) (table S6). HLA-C expression level showed no effect in UC, although previously defined risk factors, such as HLA-DRB1*01:03, were observed (16) (P = 1 × 10−12, OR = 2.03) (table S6). A greater role of Th1 response in CD relative to UC may be related to the influence of HLA-C expression level in risk of CD but not UC (20).
It is a novel facet of human genetic variation that HLA class I alleles vary in their expression level between normal individuals to an extent that modulates the efficacy of an immune response. The importance of HLA expression level is well appreciated, however, because one of the most striking cellular changes in the inflammatory response is interferon-γ mediated up-regulation of HLA expression (21). As few as three peptide-MHC complexes can suffice for CTL killing in vitro (22), with increasing density improving efficiency of cytotoxicity and modulating the response to include secretion of inflammatory cytokines (23). Given the vast range of peptides bound by HLA class I allotypes, many individual peptides are likely present below the threshold required for CTL activation or at a level where increasing their density would influence the response (24). Thus, differences in expression of even twofold, which have been shown to improve the efficacy of CTL responses in vivo (25), could result in more beneficial control of HIV by more highly expressed HLA-C allotypes.
Unlike its protection in HIV infection, high expression increases the risk of CD. Contrasting effects of HLA-C expression level across human diseases may have contributed to the evolutionary history of the HLA-C locus, where expansion of higher expression HLA-C allotypes has occurred over the past 4 to 5 million years since their genesis, while maintaining lower expression allotypes (26). Thus, the complexity of HLA effects in disease pathogenesis go beyond peptide specificity to include the strength of immune responses as dictated by levels of HLA expression.
Supplementary Material
Acknowledgments
This study makes use of data generated by the Wellcome Trust Case Control Consortium (WTCCC); a full list of investigators who contributed to the generation of this data is available at www.wtccc.org.uk. The data reported in this paper are tabulated in the main paper and supplementary materials. Data and materials will be shared subject to institutional review board (IRB) provisions regarding patient privacy. Please contact the corresponding author. Those interested in obtaining Center for HIV/AIDS Vaccine Immunology (CHAVI) samples used in this study should contact M.C. and M.A.M. A Simple Letter Agreement for the Transfer of Materials will be executed between the institutions involved, and an IRB approval/exemption should be obtained by the recipient institution when appropriate. Clinical specimens from the Multicenter AIDS Cohort Study (MACS) are subject to the terms and conditions of the MACS Master Material Transfer Agreement for Non-Profit Institutions. The project has been funded in part with federal funds from the National Institutes of Health (NIH) under contracts HHSN261200800001E, N02-CP-55504, RO1-AI046995, R01-DA04334, R01-DA12568, and R01-AI060460. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services or the Department of Defense, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by grants from the Bill & Melinda Gates Foundation as part of the Collaboration for AIDS Vaccine Discovery and grant ID38599, the Mark and Lisa Schwartz Foundation, the Wellcome Trust, the NIH National Institute of Allergy and Infectious Diseases (NIAID) Center for HIV/AIDS Vaccine Immunology (U01-AI-067854), and the Australian National Health and Medical Research Council (Program Grant 384702). Further funding was from the National Institutes of Health grant K08AR057763 and the Creative and Novel Ideas in HIV Research Program through a supplement to the University of Alabama at the Birmingham Center For AIDS Research funding (P30 AI027767-24), made possible by collaborative efforts of the Office of AIDS Research, NIAID, and the International AIDS Society. We thank M. Trubey and A. Lara for cytometry assistance and all the patients and investigators contributing to samples studied in the WTCCC, CHAVI, Multicenter Hemophilia Cohort Study, San Francisco City Clinic Cohort, Massachusetts General Hospital Controller Cohort, United States Military HIV Natural History Study, DC Gay Cohort Study, British Columbia HOMER cohort, Western Australian HIV Cohort Study, US AIDS Clinical Trials Group, Durban, AIDS Linked to Intravenous Experience (supported by National Institutes of Health grants R01-DA-04334 and R01-DA-12568), Swiss HIV Cohort Study (supported by the Swiss National Science Foundation grant number 33CSC0-108787), Study on the Consequences of Protease Inhibitor Era (supported by grants RO1 AI087145, K24AI069994, P30 AI027763, UL1 RR024131, P30 MH62246, and R24 AI067039), and MACS cohorts (funded by NIAID, with supplemental funding from the National Cancer Institute and the National Heart, Lung, and Blood Institute [grants UO1-AI-35042, 5-MO1-RR-00722 (GCRC), UO1-AI-35043, UO1-AI-37984, UO1-AI-35039, UO1-AI-35040, UO1-AI-37613, and UO1-AI-35041].
Footnotes
References and Notes
- 1.Bashirova AA, Thomas R, Carrington M. Annu Rev Immunol. 2011;29:295. doi: 10.1146/annurev-immunol-031210-101332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International HIV Controllers Study. Science. 2010;330:1551. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fellay J, et al. Science. 2007;317:944. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas R, et al. Nat Genet. 2009;41:1290. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corrah TW, et al. J Virol. 2011;85:3367. doi: 10.1128/JVI.02276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR. Nucleic Acids Res. 2011;39(Database):D913. doi: 10.1093/nar/gkq1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelak K, et al. J Infect Dis. 2010;201:1141. doi: 10.1086/651382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Materials and methods are available as supplementary materials on Science Online
- 9.Wright JK, et al. J Virol. 2010;84:10820. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlson JM, et al. PLOS Comput Biol. 2008;4:e1000225. doi: 10.1371/journal.pcbi.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson JM, et al. J Virol. 2012;86:5230. doi: 10.1128/JVI.06728-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heckerman D, Kadie C, Listgarten J. J Comput Biol. 2007;14:736. doi: 10.1089/cmb.2007.R013. [DOI] [PubMed] [Google Scholar]
- 13.Blais ME, et al. J Immunol. 2012;188:4663. doi: 10.4049/jimmunol.1103472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiepiela P, et al. Nature. 2004;432:769. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 15.Kiepiela P, et al. Nat Med. 2007;13:46. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 16.Stokkers PC, Reitsma PH, Tytgat GN, van Deventer SJ. Gut. 1999;45:395. doi: 10.1136/gut.45.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wellcome Trust Case Control Consortium. Nature. 2007;447:661. [Google Scholar]
- 18.Anderson CA, et al. Nat Genet. 2011;43:246. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman B, et al. Am J Gastroenterol. 2004;99:306. doi: 10.1111/j.1572-0241.2004.04038.x. [DOI] [PubMed] [Google Scholar]
- 20.Bouma G, Strober W. Nat Rev Immunol. 2003;3:521. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 21.Koeffler HP, Ranyard J, Yelton L, Billing R, Bohman R. Proc Natl Acad Sci USA. 1984;81:4080. doi: 10.1073/pnas.81.13.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. Nat Immunol. 2004;5:524. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 23.Faroudi M, et al. Proc Natl Acad Sci USA. 2003;100:14145. doi: 10.1073/pnas.2334336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kageyama S, Tsomides TJ, Sykulev Y, Eisen HN. J Immunol. 1995;154:567. [PubMed] [Google Scholar]
- 25.Reits EA, et al. J Exp Med. 2006;203:1259. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Huigin C, et al. Am J Hum Genet. 2011;89:424. doi: 10.1016/j.ajhg.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.