

Summary
Amyloid-β 1–42 (Aβ42) oligomers are synaptotoxic for excitatory cortical and hippocampal neurons and might play a role in early stages of Alzheimer's disease (AD) progression. Recent results suggested that Aβ42 oligomers trigger activation of AMP-activated kinase (AMPK), and its activation is increased in the brain of patients with AD. We show that increased intracellular calcium [Ca2+]i induced by NMDA receptor activation or membrane depolarization activates AMPK in a CAMKK2-dependent manner. CAMKK2 or AMPK overactivation is sufficient to induce dendritic spine loss. Conversely, inhibiting their activity protects hippocampal neurons against synaptotoxic effects of Aβ42 oligomers in vitro and against the loss of dendritic spines observed in the human APPSWE,IND-expressing transgenic mouse model in vivo. AMPK phosphory-lates Tau on KxGS motif S262, and expression of Tau S262A inhibits the synaptotoxic effects of Aβ42 oligomers. Our results identify a CAMKK2-AMPK-Tau pathway as a critical mediator of the synaptotoxic effects of Aβ42 oligomers.
Introduction
Alzheimer's disease (AD) is the most prevalent form of dementia affecting more than 5 million people in the United States and over 25 million people worldwide. This neurodegenerative disease mainly affects people over 65 years old in its sporadic late-onset form but can affect younger individuals in its genetically inherited, early-onset form. AD is thought to be caused by the abnormal accumulation of a 40- to 42-amino acid-long amyloid-β (Aβ) peptide derived from cleavage of the transmem-brane protein amyloid precursor protein (APP). Amyloid-β 1-42 Aβ42 has a strong ability to oligomerize to form diffusible dimers and trimers as well as larger oligomers, which fibrillate to form insoluble amyloid plaques, a major hallmark of AD. Intracellular neurofibrillary tangles, the second histological hallmark of the disease, are composed of hyperphosphorylated microtubule-associated protein Tau. The molecular mechanisms linking Aβ to Tau hyperphosphorylation as well as their relative contribution to the pathophysiological mechanisms underlying AD progression are still poorly understood.
Reduction in density of excitatory synapses in the hippocampus and cortex is an early abnormality detected in the brain of patients with AD (Davies et al., 1987; Masliah et al., 2001; Moolman et al., 2004). Analyses of transgenic mice expressing mutations in APP found in families affected with early-onset AD support these findings. For example, the J20 transgenic mouse model (APPSWE,IND) shows clear signs of hyperexcitability, progressive loss of dendritic spines and excitatory synaptic connections (Jacobsen et al., 2006), and increased inhibitory synaptic connectivity before the appearance of amyloid plaques (Mucke et al., 2000; Palop et al., 2007). Soluble Aβ42 oligomers produced in vitro or extracted biochemically from the brains of patients with AD have been shown to induce acute and rapid synaptic loss (Jin et al., 2011; Lacor et al., 2004, 2007; Shankar et al., 2007, 2008). Current model proposes that abnormal accumulation of Aβ42 oligomers induces early synaptotoxic effects and progressive dendritic spine loss, whereas hyperphosphorylated Tau translocates from the axon to the dendrites and dendritic spines where it further reduces excitatory synaptic transmission by activating a Fyn-dependent pathway (Hoover et al., 2010; Ittner et al., 2010).
AMP-activated kinase (AMPK) is a heterotrimeric Serine/Threonine protein kinase composed of one catalytic subunit (encoded by α1 or α2 genes in mammals) and two regulatory subunits, β (an adaptor subunit) and γ (the AMP-binding sub-unit), which are encoded by (β1 or (β2 genes and γ1 γ2, or γ3 genes, respectively (Alessi et al., 2006; Hardie, 2007; Mihaylova and Shaw, 2011). AMPK is an important regulator of cellular metabolism and functions as a metabolic sensor (Mihaylova and Shaw, 2011). It is activated by various forms of metabolic stress involving lowering of the AMP:ATP ratio but can also be activated by other forms of cellular stress such as exposure to reactive oxygen species (ROS) (reviewed in Hardie, 2007). AMPK regulates a large number of biological responses, including cell polarity, autophagy, apoptosis, and cell migration (Williams and Brenman, 2008). Liver kinase B1 (LKB1, also called STK11 or Par4) is the main activator of AMPK in most cell types (Hawley et al., 2003; Shaw et al., 2004; Woods et al., 2003), acting by phosphorylating a single Threonine residue within the T-activation loop of the kinase domain of AMPK (residue T172). In addition to AMPK, LKB1 can activate a large family of AMPK-related kinases, including BRSK1/BRSK2 (for brain-specific kinases also known as SAD-B and SAD-A, respectively), NUAK1/NUAK2 (also known as ARK5 and SNARK, respectively), SIK1-SIK3 (for salt-induced kinases), MARK1–MARK4 (for microtubule affinity-regulated kinases), and SNRK (sucrose nonfermenting-related kinase). These kinases are all controlled by phosphorylation of the conserved T-activation loop Threonine residue, thereby making LKB1 a master kinase for the AMPK-like kinase family (Jaleel et al., 2005; Lizcano et al., 2004).
We previously reported that unlike in other cell types, LKB1 is not the major activator of AMPK in immature neurons because basal levels of activated AMPK remain unchanged in cortical neurons upon cortex-specific conditional deletion of LKB1 (Barnes et al., 2007). On the other hand, several lines of evidence suggest that in various neuronal subtypes, CAMKK2 can phosphorylate and activate AMPK (Anderson et al., 2008; Green et al., 2011). Recently, two reports provided biochemical evidence showing that Aβ42 oligomers can activate AMPK (Yoon et al., 2012) in a CAMKK2-dependent manner in neurons (Thornton et al., 2011). Furthermore, activated AMPK seems strongly enriched in tangle- and pretangle-bearing neurons in patients with AD (Vingtdeux et al., 2011b), suggesting that AMPK might play a role in AD progression (Salminen et al., 2011). However, the role of the CAMMK2-AMPK pathway in the etiology and/or the pathophysiology of AD is currently unknown, although some studies have suggested that AMPK activation in AD might provide protective effects by decreasing Aβ production/APP cleavage or increasing Aβ clearance (Vingtdeux et al., 2010, 2011a).
In the present study, we show that the CAMKK2-AMPK kinase pathway plays a major role in mediating the early synaptotoxic effects of Aβ42 oligomers both in vitro and in vivo. Our results suggest that the CAMKK2-AMPK kinase pathway represents a target for therapeutic approaches to treat AD.
Results
The CAMKK2-AMPK Kinase Pathway Is Required for the Synaptotoxic Effects of Aβ42 Oligomers In Vitro
To evaluate the function of the CAMKK2-AMPK pathway in AD, we first confirmed that application of amyloid-β 1–42 (Aβ42) oligomers (Figure S1A available online), but not a peptide of inverted sequence (INV42) on mouse cortical or hippocampal neurons, triggers rapid (within 15 min) and also prolonged (up to 24 hr) AMPK activation measured using the ratio between pT172-AMPK to total AMPK (Figures 1A, 1B, S1B, and S1C). The increase in AMPK activation triggered by Aβ42 oligomers is strongly attenuated by treatment with STO-609 (Figures 1A and 1B), a specific inhibitor of CAMKK2 at the concentration of 2.5 μM (Tokumitsu et al., 2002). Excitotoxicity due to overexcitation of NMDA receptors (NMDARs) and increased intra-cellular calcium levels have been implicated as a central mechanism by which Aβ42 oligomers induces synaptotoxicity (Shankar et al., 2007). A role of NMDARs in AD is further supported by the clinically beneficial effects of the partial NMDAR antagonist memantine (De Felice et al., 2007). Furthermore, application of Aβ42 oligomers is well documented to induce a rapid and prolonged increase in intracellular calcium levels through multiple mechanisms (Bezprozvanny and Mattson, 2008). Interestingly, we observed that extracellular signals triggering increase in [Ca2+]i such as membrane depolarization (which activates voltage-gated calcium channels, VGCCs) or NMDA (which activates calcium-permeable ionotropic glutamate NMDARs) both robustly activate AMPK, which can be blocked by using the CAMKK2 inhibitor STO-609 (Figures 1C–1F).
Figure 1. Aβ42-Dependent and Activity-Dependent Activation of the CAMKK2-AMPK Kinase Pathway in Cortical Neurons.
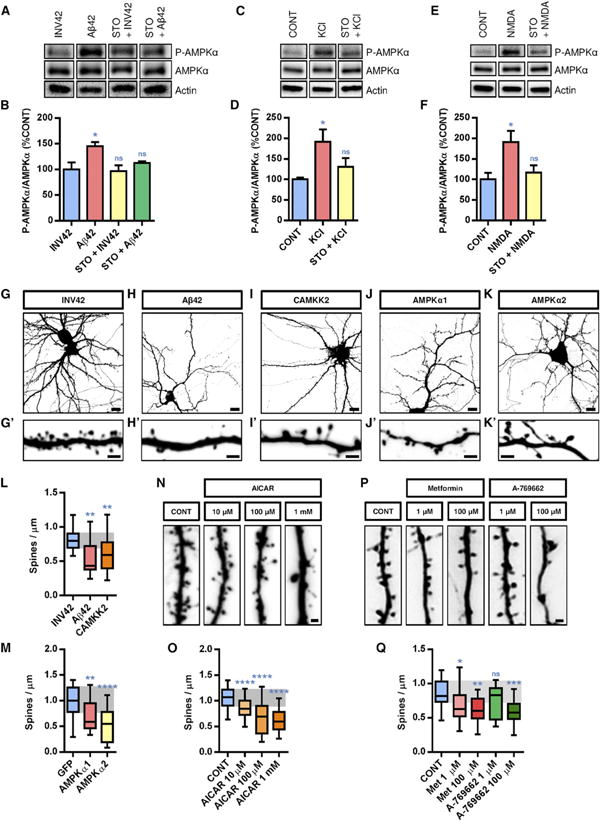
(A–F) Primary cortical neurons isolated from E18.5 mouse embryos were cultured for 21 DIV and then treated with Aβ42 oligomers (1μM monomer equivalent for 24 hr) or INV42 (1 μM for 24 hr), KCl (20 mM for 15 min), and NMDA (50 μM for 15 min), in absence (CONT, control vehicle only) or presence of STO-609 (2.5 hr prior to treatment; 2.5 μM). Activation of AMPK was assessed by western blotting using the phospho-specific T172-AMPKα antibody and total AMPKα antibody (n = 6 experiments). Histograms in (B), (D), and (F) represent mean with SEM.
(G–K′) Morphology of hippocampal neurons and their dendritic segments (secondary branches) treated at 21 DIV with Aβ42 oligomers (1 μM monomer equivalent for 24 hr) or INV42 (1 μM for 24 hr), or overexpressing CAMKK2, AMPKα1, or AMPKα2 (transfection at 15 DIV and visualization at 22 DIV).
(L and M) Application of Aβ42 oligomers induced a significant reduction in dendritic spine density compared to INV42. Overexpression of CAMKK2 (L), AMPKα1, or AMPKα2 (M) resulted in dendritic spine loss compared to control neurons expressing GFP only.
(N–Q) Hippocampal neurons treated at 21 DIV for 24 hr with compounds that activate AMPK such as AICAR, metformin, or A-769662 also induced rapid spine loss. Spine density was measured on 16–61 dendritic segments per condition.
Box plots in (L), (M), (O), and (Q) represent data distribution (box 25th–75th percentiles, bars 10th–90th percentiles with central bar representing median). Statistical analysis was performed using ANOVA test followed by Dunnett posttest in (B), (D), (F), and (O) and Kruskal-Wallis with Dunn's posttest in (L), (M), and (Q). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant p > 0.05. Scale bars, 5 μm (G–K) and 2 μm (G′, H′, I′, J′, K′, N, and P). See also Figures S1 and S2.
Based on these results, we tested if activating the CAMKK2-AMPK kinase pathway would mimic the cellular consequences of Aβ42 oligomer treatment in hippocampal and cortical neurons. As previously reported by Lacor et al. (2004, 2007), Shankar et al. (2007), and Wei et al. (2010), incubation of hippocampal neurons cultured for 21 days in vitro (DIV) with Aβ42 oligomers (1 μM) for 24 hr induced a significant reduction in dendritic spine density compared to control (neurons treated with INV42) (Figures 1G, 1H, and 1L). At this dose and duration, Aβ42 oligomers did not induce loss of neuronal viability (Figure S2), strongly arguing that the synaptotoxic effects are not a secondary consequence of impairing neuronal survival. Next, we tested if CAMKK2 and AMPKα overexpression was sufficient to mimic the synaptotoxic effects of Aβ42 oligomers. As shown in Figures 1l–1K′ and quantified in Figures 1L and 1M, our results show that the overexpression of CAMKK2, AMPKα1, or AMPKα2 induced a significant reduction in spine density of the same magnitude as Aβ42 oligomer application within 24 hr. Finally, application of the AMP-like small molecule AICAR, a potent activator of AMPK, induced a dose-dependent reduction in spine density within 24 hr on hippocampal neurons in culture (Figures 1N and 1O). A comparable synaptotoxic effect could also be observed upon activation of AMPK using metformin, which broadly activates AMPK by inducing a metabolic stress involving reduction of ATP level and conversely increase in ADP/AMP level (Hardie, 2006; Hawley et al., 2010) (Figures 1P and 1Q). Finally, application of a more specific AMPK activator, A-769662, induced a significant, dose-dependent decrease in spine density within 24 hr (Figures 1P and 1Q). Taken together, these experiments demonstrate that overactivation of CAMKK2 or AMPK is sufficient to mimic the synaptotoxic effects induced by Aβ42 oligomers.
We next tested if the CAMKK2-AMPK pathway is required for the synaptotoxic effects induced by Aβ42 oligomers in hippocampal neurons in vitro. We first took advantage of constitutive knockout (KO) mouse lines for CAMKK2 (Ageta-Ishihara et al., 2009) and AMPKα1 (Viollet et al., 2003) and treated dissociated neuronal cultures isolated from control (CAMKK2+/+ and AMPKα1+/+, respectively) or KO mice (CAMKK2−/− and AMPKα1−/−) at 21 DIV with INV42 or Aβ42 oligomers (1 μM for 24 hr) (Figures 2A and 2C). Quantitative analysis indicated that CAMKK2 null and AMPKα1 null neurons do not show a significant reduction of spine density following Aβ42 oligomer treatment (Figures 2B and 2D). Second, pharmacological inhibition of CAMKK2 activity using application of the inhibitor STO-609 in culture prevented the decrease of spine density induced by Aβ42 oligomer application in vitro (Figures 3A and 3B).
Figure 2. CAMKK2 and AMPKα1 Are Required for Aβ42 Oligomer-lnduced Spine Toxicity In Vitro.
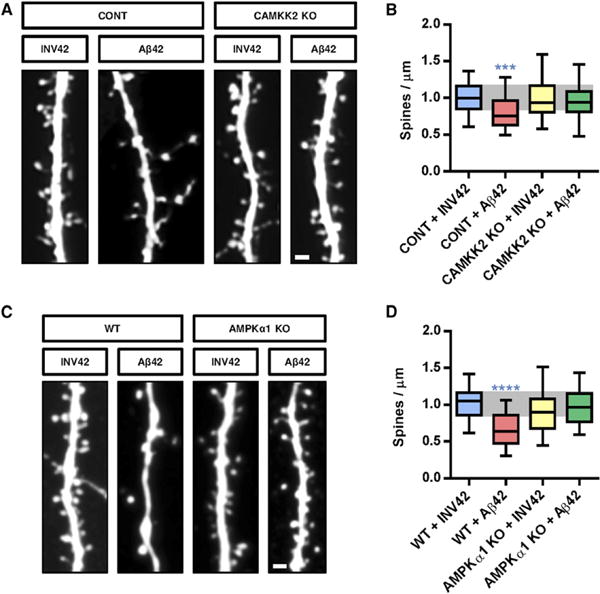
(A and C) Representative images of secondary dendritic segments of hippocampal neurons obtained from (A) control (CONT, CAMKK2+/+ or CAMKK2+/−) or CAMKK2 KO, and (C) WT (AMPKα1+/+) or AMPKα1 KO animals treated with Aβ42 oligomers or INV42 (1 μM for 24 hr). Hippocampal neurons were transfected with pCAG-EGFP plasmid at 11 DIV for spine visualization and treated at 20 DIV for 24 hr prior to fixation.
(B and D) Neurons deficient for CAMKK2 (B) or AMPKα1 (D) were resistant to spine loss induced by Aβ42 oligomer exposure. Spine density was measured on 37–58 dendritic segments per condition. Box plots represent data distribution (box 25th–75th percentiles, bars 10th–90th percentiles with central bar representing median).
Statistical analysis was performed using Kruskal-Wallis test followed by Dunn's posttest in (B) and (D). ***p < 0.001; ****p < 0.0001. Scale bars, 1 μm (A and C).
Figure 3. Inhibition of CAMKK2 or AMPK Activity Blocks the Synaptotoxic Effects of Aβ42 Oligomers in Hippocampal Neurons In Vitro.
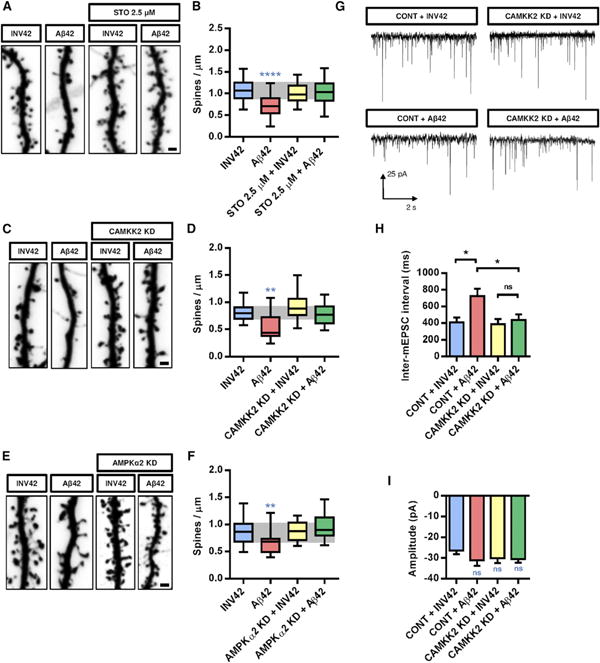
(A and B) Representative images of dendritic segments (A) and spine density quantification (B) of 21 DIV hippocampal neurons treated at 20 DIV with the CAMKK2 inhibitor STO-609 (2.5 μM) or DMSO (control) 2 hr prior to Aβ42 oligomer or INV42 application (1 μM for 24 hr). Neurons treated with STO-609 were resistant to spine loss induced by Aβ42 oligomers.
(C and D) Representative images of dendritic segments (C) and quantification of spine density (D) of hippocampal neurons at 21 DIV expressing a KD version of CAMKK2 (K194A, transfected at 15 DIV), which significantly reduces the synaptotoxic effects of Aβ42 oligomers.
(E and F) Representative images of dendritic segments (E) and quantification of spine density (F) of neurons that express a KD version of AMPKα2 (K45A, transfected at 15 DIV), which blocks the synaptotoxic effects of Aβ42 oligomers. Each condition is quantified from at least three independent experiments (19–151 dendritic segments quantified per condition).
(G) Representative traces of mEPSCs recorded from 18 to 19 DIV hippocampal neurons expressing CAMKK2 KD or control vector (transfected at 11 DIV) and treated with Aβ42 oligomers or INV42 (1 μM for 24 hr).
(H and I) Inter-mEPSC intervals (H) and mEPSC amplitudes (I) were quantified from 16 to 21 cells per condition (obtained from six independent experiments). Expression of CAMKK2 KD prevented the increase in inter-mEPSC interval induced by Aβ42 oligomers. There was no significant difference in mEPSC amplitude among the different conditions. Histograms represent mean with SEM.
Box plots in (B), (D), and (F) represent data distribution (box 25 th–75th percentiles, bars 10 th–90th percentiles with central bar representing median).
Statistical analysis was performed using ANOVA test followed by Dunnett's posttest in (B) and (F) and Kruskal-Wallis test followed by Dunn's posttest in (D), (H), and (I). *p < 0.05; **p < 0.01; ****p < 0.0001. Scale bars, 1 μm (A, C, and E).
Although the experiments presented above indicated that CAMKK2 and AMPKα kinases are required to mediate the synaptotoxic effects of Aβ42 in culture, they did not allow to conclude if CAMKK2 acts pre- or postsynaptically, or even indirectly by acting on nonneuronal cells such as astrocytes, which are critically important for synapse formation and maintenance (Eroglu and Barres, 2010). Therefore, we used a third approach where CAMKK2 function was inhibited in a cell-autonomous manner using low transfection efficiency of dominant-negative (kinase-dead, KD) forms of CAMKK2 (CAMKK2 KD) in wild-type (WT) hippocampal neuron cultures. This experiment revealed that cell-autonomous inhibition of CAMMK2 function prevents the reduction of spine density induced by Aβ42 oligomer application (Figures 3C and 3D). Similarly, cell-autonomous inhibition of AMPK catalytic activity by expression of a dominant-negative (KD) form of AMPKα (AMPKα2 KD) also abolished the reduction of spine density induced by Aβ42 oligomers (Figures 3E and 3F). Importantly, neither CAMKK2 KD nor AMPKα2 KD overexpression alone had any significant effect on spine density per se (Figures 3C–3F). These results strongly support the notion that the synaptotoxic effects of Aβ42 oligomers require activation of the CAMKK2-AMPK kinase pathway in hippocampal neurons.
We next assessed the protective effects of blocking CAMKK2 following Aβ42 oligomer application using a functional approach. To do this, we performed whole-cell patch-clamp recordings of pharmacologically isolated AMPA-type miniature excitatory postsynaptic currents (mEPSCs) in hippocampal cultures at 18 DIV. As previously shown by Shankar et al. (2007) and Wei et al. (2010), application of Aβ42 oligomers (1 μM for 24 hr) induced a significant reduction in mEPSC frequency (manifested as an increase in interevent intervals) compared to control (INV42) (Figures 3G and 3H). Importantly, overexpression of a KD version of CAMKK2 did not affect basal mEPSC frequency but abolished the decrease in mEPSC frequency induced by Aβ42 oligomer application (Figures 3G and 3H). None of the treatments had any significant effect on AMPA receptor-mediated mEPSC amplitude (Figure 3I). These results demonstrate that the CAMKK2-AMPK kinases are critical for the early structural and functional effects of Aβ42 oligomers on excitatory synaptic maintenance.
The CAMKK2-AMPK Kinase Pathway Is Required for the Dendritic Spine Loss in the APPSWE,IND Mouse Model In Vivo
Next, we tested the protective effects of inhibiting the CAMKK2-AMPK pathway in a context where neurons are exposed to Aβ42 oligomers derived from pathological human APP in vivo. We used a well-validated transgenic mouse model (J20 transgenic mice) overexpressing a pathological form of human APP carrying mutations present in familial forms of AD (APPSWE,IND) under PDGFβ promoter. These transgenic mice develop early signs of excitatory synaptotoxicity prior to amyloid plaque appearance (Mucke et al., 2000; Palop et al., 2007). We verified that this mouse model shows increased Aβ expression in the hippocampus (Figure 4A) and, in particular, increased APP and soluble Aβ both at 3 months (Figures 4B and 4C) and 8–12 months (Figure S3) compared to control littermates at the same ages. We could already detect a significant increase in activated pT172-AMPK in the cytosolic fraction of 4-month-old hippocampal tissue lysate from J20 transgenic mice compared to control littermates (Figures 4D and 4F). The increased AMPK activation is maintained in the hippocampus of older mice (8-12 months old; Figures 4E and 4G) compared to age-matched control littermates.
Figure 4. Inhibition of CAMKK2 or AMPK Activity Blocks the Synaptotoxic Effects of Aβ42 Oligomers in Hippocampal Neurons In Vivo.
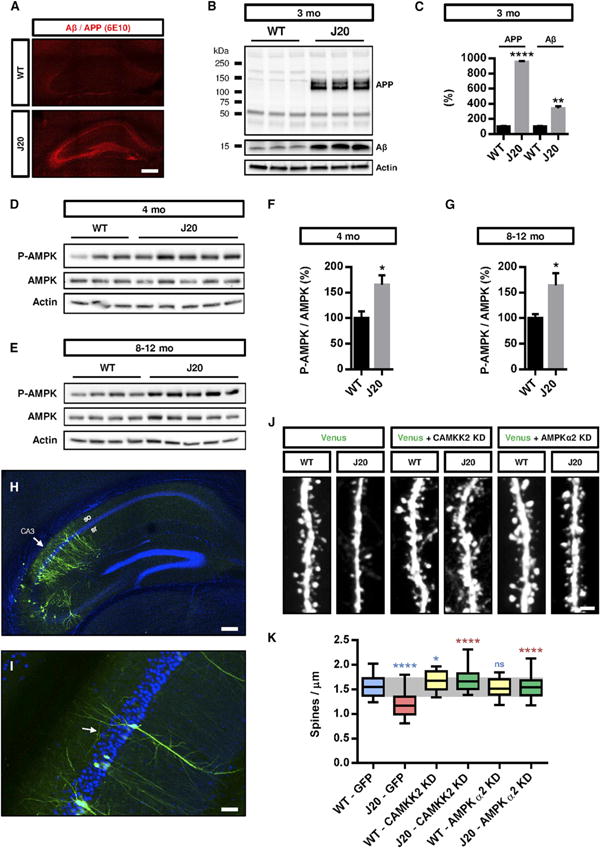
(A) Human APP and Aβ oligomers were detected by immunohistochemistry with the 6E10 antibody in the J20 transgenic mice (but not control littermates) as early as 3 months.
(B and C) The hippocampus from 3-month-old WT and J20 mice was dissected, lysed, and the crude homogenate was processed to assess human APP and Aβ levels by western blotting with the 6E10 antibody. The J20 mice presented increased APP and Aβ levels compared to WT littermates (n = 3 animals per genotype).
(D–G) The hippocampi of 4-month-old (D and F) and 8- to 12-month-old (E and G) WT and J20 mice were dissected and processed for cytosolic fractionation. AMPK activity for hippocampi isolated from individual mice was assessed by western blotting with pT172-AMPKα antibody, which was significantly increased in the cytosolic fraction of the J20 transgenic mice at both ages compared to littermate controls (n = 7–10 animals per genotype at both ages).
(H and I) Inhibition of CAMKK2 or AMPKα activity in hippocampal pyramidal neurons of the CA3 region (arrow in H) was performed by in utero electroporation of CAMKK2 KD and AMPKα2 KD encoding plasmids in E15.5 WT and J20 mouse embryos. Dendritic spine density was quantified in the collateral processes (arrow in I) of basal dendrites (in stratum oriens) of CA3 pyramidal neurons in 3-month-old transfected animals.
(J and K) Expression of CAMKK2 KD or AMPKα2 KD blocked the reduction of spine density observed in CA3 pyramidal neurons of 3-month-old J20 mice (33–53 dendritic segments from three to six animals per condition). Expression of CAMKK2 KD (but not AMPKα2 KD) induces a slight but significant increase in spine density in control mice. Box plots in (K) represent data distribution (box 25 th–75th percentiles, bars 10th–90th percentiles with central bar representing median).
Histograms in (C), (F), and (G) represent mean ± SEM. Statistical analysis was performed using Mann-Whitney U test in (C), (F), and (G). In (K), Mann-Whitney U test was used to compare specific groups to WT-GFP (in blue) and to J20-GFP (in red). *p < 0.05; **p < 0.01; ****p < 0.0001. Scale bars, 2 μm (J), 50 μm (I), 100 μm (H), and 200 μm (A). See also Figure S3.
In order to block the CAMKK2-AMPK signaling pathway in hippocampal neurons, we performed in utero electroporation at embryonic day (E)15.5, targeting specifically hippocampal pyramidal neurons located in CA1–CA3 regions of control or J20 transgenic mice (Figure 4H). Following long-term survival until 3 months postnatally, this approach allows optical isolation of single dendritic segments of pyramidal neurons in CA3 by confocal microscopy (Figure 4I) and to perform quantitative assessment of spine density. This analysis revealed that spine density of pyramidal neurons was already significantly decreased in the J20 mice at 3 months postnatally compared to control littermates (Figures 4J and 4K). Importantly, overex-pression of a KD version of CAMKK2 or a KD version of AMPKα2 prevented the reduction of spine density observed in CA3 pyramidal neurons of 3-month-old J20 transgenic mice without affecting spine density in the WT control mice (Figures 4J and 4K). These results demonstrate that the activation of the CAMKK2-AMPK kinase pathway is required to mediate the synaptotoxic effects observed in the APPSWE,IND mouse model in vivo.
AMPKα1 Phosphorylates Tau on S262 in Response to Aβ42 Oligomers
Plaques of Aβ and tangles formed by hyperphosphorylated forms of the microtubule-binding protein Tau are the two histo-pathological signatures found in the brains of patients with AD. Although both Aβ and Tau have been extensively studied independently with regard to their separate modes of toxicity, recent results have shed light on their possible interactions and synergistic effects during AD progression. For example, Tau-deficient mice are less susceptible to Aβ toxicity than control mice (Roberson et al., 2007). Recent results have shown that AMPK is a potent Tau kinase (Thornton et al., 2011). In order to reconstitute a biochemical pathway triggering AMPK activation, we expressed a GFP-tagged version of Tau and AMPKα in HeLa cells, which are naturally deficient for LKB1 (Hawley et al., 2003). In this model, AMPK can be specifically activated by reintroducing its upstream activator LKB1. This experiment confirmed that AMPK phosphorylates the well-characterized KxGS motif on Tau Serine 262 (S262) residue (Figure 5A). When coexpressed in cell lines, both LKB1 (coexpressed with its coactivator STRAD) and CAMKK2 are potent activators of AMPK, although we observed that CAMKK2 was significantly more potent in phosphorylating AMPK on T172 than LKB1 or CAMKK1 (Figure 5B). Furthermore, direct activation of AMPK using the AMP analog AICAR triggered a dose-dependent increase of Tau phosphorylation of S262 in cortical neurons (Figures 5C, 5D, and S4), a treatment that induces a dose-dependent reduction in spine density (Figures 1N and 1O). The microtubule-associated protein Tau is phosphorylated in multiple sites (Mandelkow and Mandelkow, 2012), and analysis of six well-characterized phosphorylation sites revealed that following 24 hr treatment with AICAR, phosphorylation of Tau on S262 is significantly increased in a dose-dependent manner but that other sites are either unchanged (for example, the other KxGS motif on S356, as well as S396, S422) or decreased (S202/T205, S404) (Figures S4A and S4B). This observation suggests that S262 is an important target of AMPK, and phosphorylation of this site might underlie AMPK-induced spine loss.
Figure 5. Phosphorylation of Tau on S262 Is Required for the Synaptotoxic Effects of Aβ Oligomers In Vitro and In Vivo.
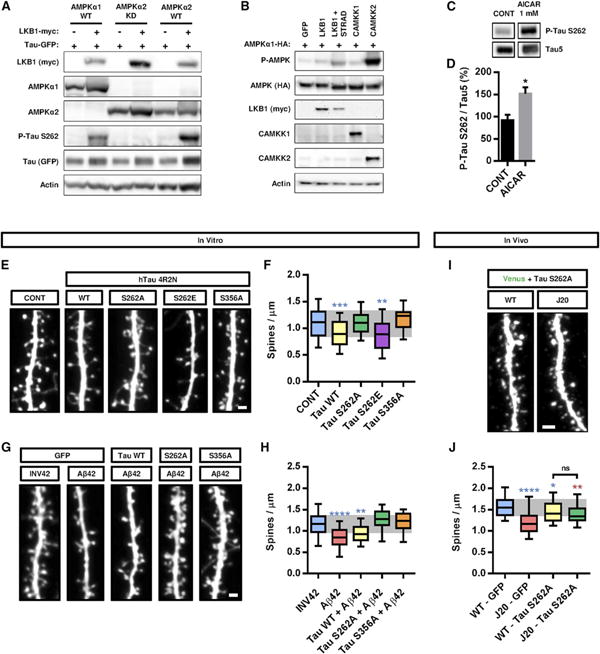
(A) Cotransfection of WT forms of AMPKα1 or α2 together with LKB1 is sufficient to trigger Tau phosphorylation on S262 residue in HeLa cells. Western blot on 25 μg of protein lysate with the indicated antibodies.
(B) Upstream kinases LKB1, CAMKK1, and CAMKK2 display various abilities to phosphorylate AMPK upon expression in HEK293T cells. Western blot on 25 μg of protein lysate with the indicated antibodies.
(C and D) AICAR treatment of cortical neurons at 21 DIV for 24 hr increased phosphorylation of Tau at epitope S262 compared to control (CONT, vehicle only). Histograms in (D) represent mean with SEM.
(E–H) Hippocampal neurons were transfected at 11 DIV with constructs for different versions of Tau, treated with Aβ42 oligomers or INV42 at 20 DIV (1 μM for 24 hr), and spine density was assessed at 21 DIV. (E and F) Overexpression of WT Tau or its phospho-mimetic S262E version decreased spine density, whereas the nonphosphorylatable forms S262A and S356A had no effect. (G and H) Overexpression of mutant Tau S262A or S356A blocked oligomer-induced spine toxicity, whereas the WT form of Tau had no protective effects (n = 16–71 segments per condition).
(I and J) Tau S262A was expressed in hippocampal pyramidal neurons of the CA3 region following in utero electroporation of E15.5 WT and J20 mouse embryos. Tau S262A expression induced a mild but significant decrease in spine density in WT animals but lessened the synaptotoxic effects of Aβ oligomers compared to J20 animals electroporated with control vector (19–53 dendritic segments from three to six animals per condition; compare figures with Figure 4J).
Box plots in (F), (H), and (J) represent data distribution (box 25 th–75th percentiles, bars 10 th–90th percentiles with central bar representing median). Statistical analysis was performed using Mann-Whitney U test in (C) and ANOVA test followed by Dunnett's posttest in (F) and (H). In (J), Mann-Whitney U test was used to compare specific groups to WT-GFP (in blue) and to J20-GFP (in red). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Scale bars, 1 μM (E and G) and 2 μrn (I). See also Figure S4.
Preventing Tau Phosphorylation on S262 Protects Hippocampal Neurons from the Synaptotoxic Effects of Aβ42 Oligomers In Vitro and the Dendritic Spine Loss Observed in the APPSWE,IND Mouse Model In Vivo
Previous studies in Drosophila suggested that overexpression of AMPK-related member PAR-1/MARK2 induced neurotoxicity through phosphorylation of Tau in the microtubule-binding domains on S262 and S356 and that phosphorylation of these sites played an initiator role in the pathogenic phosphorylation process of Tau (Nishimura et al., 2004). Given the importance of phosphorylation of S262 as a “priming” site (Biernat et al., 1993) and the recent implication of Tau in the synaptotoxic effects of Aβ42 oligomers (Ittner et al., 2010; Roberson et al., 2007), we wanted to test if expression of a form of Tau that cannot be phosphorylated on S262 could exert a protective effect in the context of Aβ42 oligomer-induced synaptotoxicity in cultured hippocampal neurons. Expression of Tau S262A abolished the loss of spines induced by Aβ42 oligomers (Figures 5E–5H), although its expression in control neurons did not have any effect on spine density. By contrast, expression of Tau WT or a phospho-mimetic version of Tau on S262 (Tau S262E) resulted in spine loss in control condition, and the WT form of Tau was unable to prevent the synaptotoxic effects of Aβ42 oligomers. Finally, the nonphosphorylatable form of Tau on S356 (S356A) displayed similar protective effects as Tau S262A mutant, indicating that the phosphorylation of these two serine residues in the microtubule-binding domains plays a critical role in mediating the synaptotoxic effects of Aβ42 oligomers.
To investigate the relevance of the phosphorylation of Tau on S262 in vivo, we performed in utero electroporation of Tau S262A construct in E15.5 WT and J20 embryos and analyzed spine density of CA3 hippocampal pyramidal neurons in the adult mice at 3 months (Figures 5I and 5J). Tau S262A slightly decreased spine density in WT animals compared to control vector, suggesting that phosphorylation of Tau on S262 plays a role in spine development. Nevertheless, Tau S262A administration was able to prevent spine loss induced by Aβ oligomers in the J20 animals to a level similar to WT animals electroporated with the same Tau mutant construct (Figure 5J). These results strongly suggest that phosphorylation of Tau on S262 mediates the synaptotoxic effects observed in the APPSWE,IND mouse model in vivo.
Preventing Tau Phosphorylation on S262 Protects Hippocampal Neurons from Spine Loss Induced by AMPKα1 Activation
To determine whether phosphorylation of Tau on S262 is required for AMPK-induced spine loss, we treated hippocampal neurons expressing Tau S262A mutant with the AMPK activators metformin or AICAR for 24 hr in vitro (Figures 6A and 6B). Although metformin and AICAR treatments resulted in a marked decrease in spine density, neurons expressing Tau S262A mutant were insensitive to metformin or AICAR treatment and did not show a significant decrease in spine density.
Figure 6. Phosphorylation of Tau on S262 by AMPKα1 Is Required for the Synaptotoxic Effects of Aβ42 Oligomers.
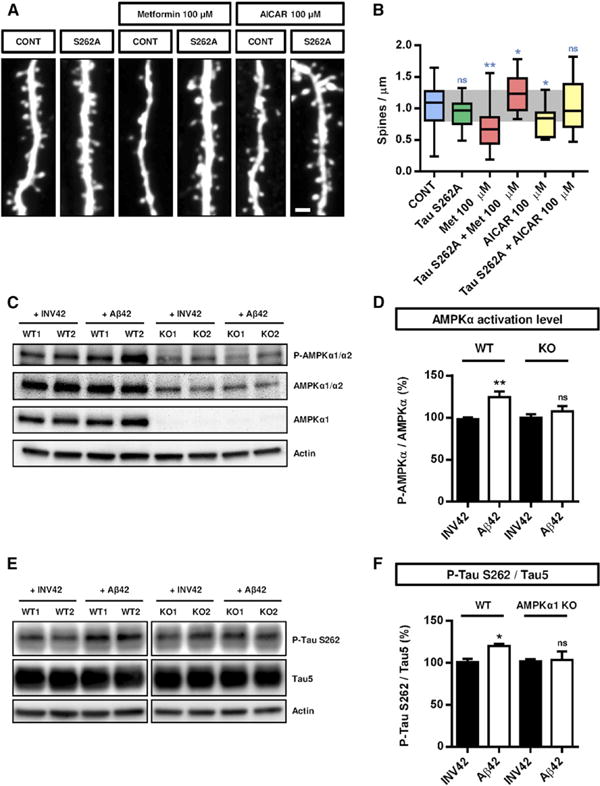
(A and B) Hippocampal neurons were transfected at 11 DIV with control (GFP) or Tau S262A construct, treated with AMPK activator metformin or AICAR (100 μM) at 20 DIV for 24 hr, and spine density was assessed at 21 DIV. Overexpression of Tau S262A prevented the loss of spines induced by AMPKα overactivation (16–28 dendritic segments per condition). Box plots in (B) represent data distribution (box 25 th–75th percentiles, bars 10 th–90th percentiles with central bar representing median).
(C-F) Cortical cells from WT or AMPKα1 KO animals were cultured for 20 DIV and then treated with Aβ42 oligomers or INV42 (1 μM for 24 hr). (C) AMPKα activation (phosphorylation of T172 of AMPKα1 and AMPKα2) and total AMPKα protein level were assessed by western blotting (n = 9–11 animals per genotype). (D) Aβ42 oligomers increased AMPK phosphorylation in WT cells, whereas oligomers had no effects in AMPKα1-deficient cells. (E and F) Phosphorylation of Tau on S262 and total Tau were assessed by western blotting (n = 9–10 animals per genotype). Aβ42 oligomers increased phosphorylation of Tau on S262 in WT cells, whereas no effect was detected in AMPKα1-deficient cells. Histograms in (D) and (F) represent mean with SEM.
Statistical analysis was performed using ANOVA test followed by Dunnett's post hoc test in (B), and paired t test in (D) and (F). *p < 0.05; **p < 0.01. Scale bar, 1 μm (A).
To further demonstrate the involvement of AMPK in Tau phosphorylation, we performed long-term cultures of cortical neurons isolated from individual AMPKα1+/+ and AMPKα1−/− mouse littermates, treated them with Aβ42 oligomers or INV42, and assessed Tau phosphorylation on S262. First, we could validate that Aβ42 oligomers increased AMPK activation detected by pT172-AMPK/total AMPK ratio (Figures 6C and 6D). AMPKα1 seems to be the major isoform of AMPKα responding to Aβ42 oligomers in cortical neurons because (1) AMPKα1 null neurons show a drastic reduction in total AMPK levels detected by an antibody recognizing both AMPKα1 and AMPKα2, and (2) AMPKα1 null neurons do not show increased AMPK activation following Aβ42 oligomer application (Figures 6C and 6D).
Finally, in the same neurons, treatment with Aβ42 oligomers led to a slight, albeit reproducible and significant, increase in Tau phosphorylation on S262 in control AMPKα1+/+ but not in AMPKα1 null hippocampal neurons (Figures 6E and 6F), suggesting that AMPKα1 mediates the phosphorylation of Tau on S262 induced by Aβ42 oligomers in hippocampal neurons.
Discussion
Loss of synapses begins during the early stages of AD and progressively affects neuronal network activity, leading to cognitive dysfunction (Coleman and Yao, 2003; Palop and Mucke, 2010; Terry et al., 1991). In vitro and in vivo studies have demonstrated that Aβ oligomers are contributing to early synapse loss (Hsia et al., 1999; Hsieh et al., 2006; Lacor et al., 2007; Mucke et al., 2000; Shankar et al., 2007), whereas recent studies support Tau as one of the mediators of Aβ toxicity in dendrites (Ittner et al., 2010; Roberson et al., 2007, 2011). However, our understanding of the molecular mechanisms linking Aβ oligomers and Tau synaptotoxicity in dendritic spines remains incomplete. Here, we report that (1) AMPK is overactivated in hippocampal neurons upon application of Aβ42 oligomers, and this activation is dependent on CAMKK2; (2) CAMKK2 or AMPK activation is sufficient to induce dendritic spine loss in hippocampal neurons in vitro and in vivo; (3) Aβ-mediated activation of AMPK induces the phosphorylation of Tau on residue S262 in the microtubule-binding domain; and (4) inhibition of either CAMKK2 or AMPK catalytic activity, or expression of a nonphosphorylatable form of Tau (S262A), blocks Aβ42 oligomer-induced synaptotoxicity in hippocampal neurons in vitro and in vivo.
AMPK is an important homeostatic regulator and is activated by various forms of cellular and metabolic stresses (Mihaylova and Shaw, 2011; Shaw et al., 2004). Oxidative stress such as elevation of ROS can activate AMPK through a mechanism that is still unclear (reviewed in Hardie, 2007). Because part of the neuronal toxicity induced by Aβ is thought to involve increased ROS production (Schon and Przedborski, 2011), future experiments should test if AMPK function during Aβ-mediated neurodegeneration requires the ability of ROS to activate AMPK.
In the brain, AMPK activity is increased in response to metabolic stresses such as ischemia, hypoxia, or glucose deprivation (Culmsee et al., 2001; Gadalla et al., 2004; Kuramoto et al., 2007; McCullough et al., 2005) and is abnormally elevated in several human neurodegenerative disorders, including AD and other tauopathies, amyotrophic lateral sclerosis, and Huntington's disease (Ju et al., 2011; Lim et al., 2012; Vingtdeux et al., 2011b). Whether activation of AMPK in these different pathological contexts has a neuroprotective or deleterious outcome in various neuronal subtypes remains controversial (Salminen et al., 2011). Here, we demonstrate that activation of AMPK, either pharmacologically or following overexpression of AMPKα, is sufficient to trigger dendritic spine loss in mature hippocampal neurons. Overexpression of CAMKK2 had a similar negative effect on spine density, presumably by increasing calcium sensitization and AMPK activity. The CAMKK2-AMPK pathway appears critical with regard to AD pathology since its blockade mitigates the synaptotoxic effects of Aβ oligomers in vitro and blocks the dendritic spine loss observed in the APPSWE,IND mouse model in vivo.
AMPK activity is increased in the hippocampus of the J20 transgenic mouse model as early as 4 months of age, a time when Aβ oligomer levels are high and signs of hippocampal network dysfunction already detectable (Palop et al., 2007). Similarly, AMPK activity is increased in the brain of other AD mouse models such as the double APP/PS2 or APPsw/PS1 dE9 mutants at 6 months (Lopez-Lopez et al., 2007; Son et al., 2012), supporting a link between Aβ oligomers and AMPK activation. In agreement with these results, we found that 1 μM Aβ42 oligomer exposure for 24 hr significantly increased AMPK activity in mature cortical cells, confirming previous studies by Thornton et al. (2011). Whether Aβ42 oligomers can activate other members of the AMPK-like family is still unclear, although recent studies report that acute treatment of Aβ42 oligomers does not activate BRSK2 or MARK3 in primary hippocampal neurons (Thornton et al., 2011).
Many kinases can act as direct upstream activators of AMPK, including LKB1 (Hawley et al., 2003; Shaw et al., 2004), CAMKK2, to a lesser extent CAMKK1 (Anderson et al., 2008; Green et al., 2011; Hawley et al., 2005; Hurley et al., 2005; Woods et al., 2005), and TAK1 (Momcilovic et al., 2006). We show that Aβ42 oligomer-induced activation of AMPK depends on CAMKK2 in mature synaptically active cortical cultures. Importantly, AMPK is the only member of the AMPK-like family known to be regulated by CAMKK2, whereas other related members of the family are presumably not (Bright et al., 2008; Fogarty et al., 2010). Thus, AMPK may represent the main member of this family that responds to increased intracellular calcium mediated by NMDAR activation and/or membrane depolarization. Aβ42 oligomer-induced activation of AMPK through CAMKK2 supports the hypothesis that Aβ oligomers may disrupt calcium homeostasis (Demuro et al., 2005; Mattson et al., 1992). Preferential targets of Aβ42 oligomers are dendritic spines (Lacor et al., 2004; Lacor et al., 2007), where they interfere with NMDAR signaling to trigger rise in cytoplasmic calcium (De Felice et al., 2007). Our results provide a mechanism whereby increased neuronal excitation activates the CAMKK2-AMPK pathway leading to Tau phosphorylation on S262 and compromises spine stability. In line with this hypothesis, (1) acute exposure of neuronal cultures to Aβ oligomers leads to local calcium level increase, hyperphosphorylation, and mislocalization of Tau into dendritic spines, which was associated with spine collapse (De Felice et al., 2008; Zempel et al., 2010); and (2) Tau phosphorylation mediates dendritic spine collapse upon overexpression of AMPK-related MARK/PAR-1 in hippocampal neurons (Yu et al., 2012).
Because of high similarity in their substrate specificity (Mihaylova and Shaw, 2011), most AMPK-related members might be able to directly phosphorylate Tau on S262 (Yoshida and Goedert, 2012). We have previously shown that BRSK1/BRSK2 (also called SAD-A/B) can potently phosphorylate Tau on S262 (Barnes et al., 2007). We now show that AMPK can robustly phosphorylate Tau, confirming a previous report by Thornton et al. (2011). Furthermore, AMPK is abnormally activated in tangle- and pretangle-bearing neurons in AD and several tauopathies in humans (Vingtdeux et al., 2011b), suggesting that AMPK may phosphorylate Tau in pathological conditions. We found that AMPK increased phosphorylation of Tau mainly on S262 in the microtubule-binding domain in primary mature neurons, whereas other sites such as S356, S396, and S422 were unaffected. Phosphorylation of other sites, S202/Thr205 and S404, was decreased, suggesting the implication of phosphatases or the negative regulation of the activity of other kinases by AMPK. Furthermore, preventing phosphorylation at Tau S262 prevented the toxic effects of Aβ oligomers in hippocampal neurons. Therefore, activation of the CAMKK2-AMPK pathway might converge on S262 of Tau to trigger deleterious effects on spine integrity. Alanine mutation of S262 in Tau has also been reported to be protective in a fly model of AD overexpressing human Aβ42 or MARK/PAR-1 kinase that can phosphorylate Tau at S262 (Chatterjee et al., 2009; Iijima et al., 2010; Nishimura et al., 2004). The mechanisms underlying Tau S262A protection against Aβ42-mediated synaptotoxicity are still unclear. There is growing recognition that Aβ42 oligomers induce Tau relocation from the axon to dendrites (Zempel et al., 2010), where it can act as a protein scaffold to facilitate the interaction of the Src kinase Fyn with NMDAR. This stabilizes NMDAR to the postsynaptic density and couples the receptor to excitotoxic downstream signaling, representing a potential mechanism by which phosphorylated Tau could mediate Aβ42 oligomer synaptotoxicity (Ittner et al., 2010). Removing Tau or preventing Tau/Fyn interaction would uncouple excitotoxic downstream signaling (Ittner et al., 2010; Roberson et al., 2007, 2011). Tau phosphorylation of its KxGS motifs (S262 and S356) in the microtubule-binding domains is thought to act as a priming site for other phosphorylation sites and globally controls Tau solubility by decreasing microtubule affinity (Waxman and Giasson, 2011).
According to our results, impinging on the CAMKK2-AMPK pathway may be of therapeutic value to lessen the synaptotoxic effects of Aβ42 oligomers. A previous study already targeted this pathway in the hypothalamus to efficiently protect mice from high-fat diet-induced obesity using intraventricular infusion of the CAMKK2 inhibitor STO-609 (Anderson et al., 2008). It will be of interest to determine if such treatment would protect neurons from Aβ toxicity in mouse models of AD and determine if these protective effects ameliorate long-term behavioral outcomes in the context of spatial learning for example.
Epidemiological and clinical studies identified type 2 diabetes as a major risk factor for developing AD (Hassing et al., 2002; MacKnight et al., 2002). Metformin is a widely prescribed insulin-sensitizing drug and a potent activator of AMPK (Hundal et al., 2000; Zhou et al., 2001). A recent study suggested that metformin increases the generation of Aβ40 and Aβ42 through upregulation of β secretase activity in an AMPK-dependent manner (Chen et al., 2009). The authors also reported that a small but significant amount of metformin crosses the blood-brain barrier when administered to the drinking water in rodents. Together with our present observations, long-term metformin treatments could potentially have deleterious effects on AD progression in the central nervous system. Future investigations should examine the effects of long-term metformin treatments on symptom progression in various AD and obesity/type 2 diabetes mouse models in vivo.
Experimental Procedures
Animals
Mice were used according to protocols approved by the Institutional Animal Care and Use Committee at Scripps Research Institute and in accordance with National Institutes of Health guidelines. 129/SvJ, C57BI/6J nontransgenic mice and hemizygous transgenic mice from line J20 (hereafter referred as J20) (The Jackson Laboratory) were maintained in a 12 hr light/dark cycle. J20 mice express human APP carrying the Swedish and Indiana mutations under PDGFβ promoter (Mucke et al., 2000; Palop et al., 2007). Constitutive AMPKα1 KO mice (Prkaa1tm1Vio) (Viollet et al., 2003) were a kind gift from Dr. Benoit Viollet (INSERM, Institut Cochin, Paris). Constitutive CAMKK2 KO mice (Ageta-Ishihara et al., 2009) were obtained from Dr. Talal Chatila (Harvard Medical School, Boston). Timed-pregnant females were obtained by overnight breeding with males of the same strain. Noon following breeding was considered as E0.5.
Aβ42 Oligomer Preparation
Aβ42 (rPeptide) was processed to generate Aβ42 oligomers as described previously by Klein (2002). Briefly, Aβ42 was dissolved in hexafluoro-2-propanol (HFIP; Sigma–Aldrich) for 2 hr to allow monomerization. HFIP was removed by speed vacuum, and Aβ42 monomers were stored at −80°C. Aβ42 monomers were dissolved in anhydrous DMSO to make a 5 mM solution, then added to cold phenol red-free F12 medium (Invitrogen) to make a 100 μM solution. This solution was incubated at 4°C for 2 days and then centrifuged at 14,000 × g for 15 min in order to discard fibrils. The supernatant containing Aβ42 oligomers was assayed for protein content using the BCA kit (Pierce). For control, a peptide corresponding to the inverted sequence of Aβ42 (INV42; Bachem) was used and processed as for Aβ42 oligomerization. Oligomerization of Aβ42 was monitored by western blotting using 16.5% Tris-Tricine gels (Bio-Rad) and the previously characterized 6E10 antibody (Chromy et al., 2003).
Primary Neuronal Culture and Magnetofection
Cortices and hippocampi from E17.5 to E18.5 embryos were dissected in Hank's balanced salt solution (HBSS) supplemented with HEPES (10 mM) and glucose (0.66 M; Sigma-Aldrich). Tissues were dissociated in papain (Worthington) supplemented with DNase I (100 mg/ml; Sigma-Aldrich) for 20 min at 37°C, washed three times, and manually triturated in plating medium. Cells were then plated at 565 cells/mm2 on glass-bottom dishes coated with poly-D-lysine (1 mg/ml; Sigma-Aldrich) and cultured in neurobasal medium supplemented with 2.5% fetal bovine serum (Gemini), B27 (1 ×), L-glutamine (2 mM), and penicillin (2.5 U/ml)-streptomycin (2.5 mg/ml) (Invitrogen). At 5 DIV, half of the medium was replaced with serum-free medium, and one-third of the medium was then changed every 5 days. At 7 DIV, 5-Fluoro-5′-deoxyuridine (Sigma-Aldrich) was added to the culture medium at a final concentration of 5 μM to limit glia proliferation. Cells were maintained at 37°C in 5% CO2 for 18–22 days. Neurons were transfected at 11 or 15 DIV by magnetofection using NeuroMag (OZ Bioscience), according to manufacturer's instructions. Co-transfections were performed at a 1:1 ratio (w/w). Briefly, cDNA (2 μg final) was incubated with NeuroMag in neurobasal medium for 15 min at room temperature and then the mixture was applied dropwise on culture cells. Cultures were placed on a magnet for 20 min for transfection (see Supplemental Information).
Electrophysiology
Cell recordings were performed using a multiclamp 700B amplifier (Axon Instruments). Neurons were recorded in a bath solution containing 140 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 10 mM HEPES, 2 mM CaCl2, and 10 mM glucose. The whole-cell internal solution contained 135 mM CsCl2, 10 mM HEPES, 1 mM EGTA, 4 mM Na-ATP, and 0.40 mM Na-GTP. Spontaneous mEPSCs were isolated by adding 0.2 mM picrotoxin and 0.1 mM tetrodotoxin in the recording bath solution and sampled in voltage-clamp configuration using pClamp 10 (Axon Instruments). Analyses were done offline using Clampfit 10 (Axon Instruments) and Excel (Microsoft). For illustration purpose, traces were filtered at 200 Hz to remove noise. There were no differences in membrane capacitance (Cm) or input resting membrane resistance (Rm) among experimental groups: CONT (control, EGFP only), Cm = 71.12 ± 5.9 pF and Rm = 117.62 ± 7.2 MΩ (n = 16); CONT+Aβ42, Cm = 68.50 ± 4.4 pF and Rm = 105.28 ± 7.8 MΩ (n = 21); CAMKK2 KD, Cm = 67.66 ± 4.0 pF and Rm = 103.31 ± 8.2 MΩ (n = 18); and CAMKK2 KD+Aβ42, Cm = 83.95 ± 7.0 pF and Rm = 113.01 ± 8.0 MΩ (n = 16).
In Utero Electroporation
In utero electroporation was performed as previously described by Yi et al. (2010) with slight modifications in order to target the embryonic hippocampus (see Supplemental Information).
Image Acquisition and Analyses
Images were acquired in 1,024 × 1,024 resolution with a Nikon Ti-E microscope equipped with the A1R laser-scanning confocal microscope using the Nikon software NIS-Elements (Nikon, Melville, NY, USA). We used the following objective lenses (Nikon): 10× PlanApo; NA 0.45 (for images of cortical slices), 60× Apo TIRF; NA 1.49 (for analyses of spine densities in cultured neurons), 100× H-TIRF; and NA 1.49 (for analyses of spine densities in brain slices). Dendritic spine density was quantified on secondary dendritic branches that were proximal to the cell body, on z projections for cultured neurons and in the depth of the z stack for slices, using FiJi software (ImageJ; NIH) (see Supplemental Information).
Cell and Tissue Lysis and Western Blotting
Cultured cells or brain tissues were lysed in RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl in 50 mM Tris buffer [pH 8]) supplemented with benzonase (0.25 U/μl of lysis buffer; Novagen), and cocktails of protease (Roche) and phosphatase (Sigma-Aldrich) inhibitors. Equal amounts of lysates (20–50 μg) were loaded on a Mini-Protean TGX (4%–20%) SDS-PAGE (Bio-Rad). The separated proteins were transferred onto polyvinylidene difluoride membranes (Amersham). For phospho-specific antibodies, the membranes were blocked for 1 hr with blocking buffer containing 5% BSA in Tris-buffered saline solution and Tween 20 (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.05% Tween 20; TBS-T). For other antibodies, membranes were blocked for 1 hr with blocking buffer containing 5% fat-free dry milk in TBS-T. Membranes were then incubated overnight at 4°C with different primary antibodies diluted in the same blocking buffer. Incubations with HRP-conjugated secondary antibodies were performed for 1 hr at room temperature, and visualization was performed by quantitative chemiluminescence using Fluorochem Q imager (ProteinSimple). Signal intensity was quantified using AlphaView software (ProteinSimple). Antibodies were the following: anti-phospho-T172-AMPKα (40H9, 1:1,000; Cell Signaling); AMPKα1/2 (1:1,000; Cell Signaling); AMPKα1 (1:1,000; Abcam); CAMKK2 (1:1,000; Santa Cruz Biotechnology); phospho-Tau (S262, S356, S396, S404, and S422, 1:1,000; Invitrogen); phospho-PHF-Tau (S202/Thr205, AT8, 1:1,000; Pierce); Tau5 (1:1,000; Invitrogen); mouse monoclonal anti-GFP (1:2,000; Roche); and anti-Myc (1:5,000, 9E10; Cell Signaling). Human APP and Aβ were detected by western blotting using 12% Tris-Glycine and 16.5% Tris-Tricine gels (Bio-Rad), respectively, and the anti-human APP/Aβ 6E10 antibody (1:1,000; Covance). To control for loading, blots were stripped and reprobed with mouse monoclonal anti-actin (1:5,000; Millipore).
Statistics
Statistical analyses were performed with Prism 6 (GraphPad Software). The statistical test applied for data analysis is indicated in the corresponding figure legend. The normality of the distributions of values obtained for each group/experimental treatment was determined using the Kolmogorov-Smirnov test. Experimental groups where all distributions were Gaussian/normal were assessed using the unpaired t test for two-population comparison, or oneway ANOVA with Dunnett's post hoc test for multiple comparisons. Nonpara-metric tests including the Mann-Whitney U test for two-population comparison and Kruskal-Wallis with Dunn's posttest for multiple comparisons were applied when distributions were not Gaussian. Unless otherwise noted, data are expressed as mean ± SEM. For dendritic spine analysis, all data were obtained from at least three independent experiments or at least three individual mice. The test was considered significant when p < 0.05. For all analyses, the following apply: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant p > 0.05. For clarity purpose, the color of the marker (asterisk [*] or “ns”) refers to the corresponding condition used for statistical comparison.
Supplementary Material
Acknowledgments
We would like to thank members of the F.P. lab for discussions, Dr. Reuben Shaw (Salk Institute, La Jolla, CA, USA) for the AMPK constructs, Dr. Pascal Lacor (Northwestern University School of Medicine, Chicago, IL, USA) for initial advice on Aβ oligomer production, Dr. Benoit Viollet (INSERM, Institut Cochin, Paris, France) for providing AMPKα1 knockout mice, and Dr. Talal Chatila (Harvard Medical School, Boston, MA, USA) for providing CAMKK2 knockout mice. This work was partially supported by NIH RO1 AG031524 (to F.P.) and ADI Novartis funds (to F.P.).
Footnotes
Supplemental Information: Supplemental Information includes four figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2013.02.003.
References
- Ageta-Ishihara N, Takemoto-Kimura S, Nonaka M, Adachi-Morishima A, Suzuki K, Kamijo S, Fujii H, Mano T, Blaeser F, Chatila TA, et al. Control of cortical axon elongation by a GABA-driven Ca2+/calmodulin-dependent protein kinase cascade. J Neurosci. 2009;29:13720–13729. doi: 10.1523/JNEUROSCI.3018-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, Witters LA, Kemp BE, Means AR. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008;7:377–388. doi: 10.1016/j.cmet.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- Bright NJ, Carling D, Thornton C. Investigating the regulation of brain-specific kinases 1 and 2 by phosphorylation. J Biol Chem. 2008;283:14946–14954. doi: 10.1074/jbc.M710381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Sang TK, Lawless GM, Jackson GR. Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum Mol Genet. 2009;18:164–177. doi: 10.1093/hmg/ddn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, et al. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty S, Hawley SA, Green KA, Saner N, Mustard KJ, Hardie DG. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J. 2010;426:109–118. doi: 10.1042/BJ20091372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- Green MF, Anderson KA, Means AR. Characterization of the CaMKKβ-AMPK signaling complex. Cell Signal. 2011;23:2005–2012. doi: 10.1016/j.cellsig.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology. 2006;131:973. doi: 10.1053/j.gastro.2006.07.032. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hassing LB, Johansson B, Nilsson SE, Berg S, Pedersen NL, Gatz M, McClearn G. Diabetes mellitus is a risk factor for vascular dementia, but not for Alzheimer's disease: a population-based study of the oldest old. Int Psychogeriatr. 2002;14:239–248. doi: 10.1017/s104161020200844x. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al. Tau mislocal-ization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF, Landau BR, Shulman GI. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Iijima K, Gatt A, Iijima-Ando K. Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer's disease. Hum Mol Genet. 2010;19:2947–2957. doi: 10.1093/hmg/ddq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, McBride A, Lizcano JM, Deak M, Toth R, Morrice NA, Alessi DR. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 2005;579:1417–1423. doi: 10.1016/j.febslet.2005.01.042. [DOI] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directlyinduce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ, Sun CP, Tao MH, Tu PH, Chang C, et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington's disease. J Cell Biol. 2011;194:209–227. doi: 10.1083/jcb.201105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein WL. Abeta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, et al. Phosho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, et al. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MA, Selak MA, Xiang Z, Krainc D, Neve RL, Kraemer BC, Watts JL, Kalb RG. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci. 2012;32:1123–1141. doi: 10.1523/JNEUROSCI.6554-10.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez C, Dietrich MO, Metzger F, Loetscher H, Torres-Aleman I. Disturbed cross talk between insulin-like growth factor I and AM P-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of Alzheimer's disease. J Neurosci. 2007;27:824–831. doi: 10.1523/JNEUROSCI.4345-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer's disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord. 2002;14:77–83. doi: 10.1159/000064928. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan FR, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP-activated protein kinase: a potential player in Alzheimer's disease. J Neurochem. 2011;118:460–474. doi: 10.1111/j.1471-4159.2011.07331.x. [DOI] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I. Aβ-induced formation of autophagosomes is mediated by RAGE-CaMKKβ-AMPK signaling. Neurobiol Aging. 2012;33:1006.e11–23. doi: 10.1016/j.neurobiolaging.2011.09.039. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid β-peptide exposure. Biochem J. 2011;434:503–512. doi: 10.1042/BJ20101485. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285:9100–9113. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Chandakkar P, Zhao H, d'Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J. 2011a;25:219–231. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer's disease and other tauopathies. Acta Neuropathol. 2011b;121:337–349. doi: 10.1007/s00401-010-0759-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jørgensen SB, Perrin C, Flamez D, Mu J, Wojtaszewski JF, Schuit FC, Birnbaum M, Richter E, et al. Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem Soc Trans. 2003;31:216–219. doi: 10.1042/bst0310216. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31:7604–7618. doi: 10.1523/JNEUROSCI.0297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Brenman JE. LKB1 and AMPK in cell polarity and division. Trends Cell Biol. 2008;18:193–198. doi: 10.1016/j.tcb.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Barnes AP, Hand R, Polleux F, Ehlers MD. TGF-beta signaling specifies axons during brain development. Cell. 2010;142:144–157. doi: 10.1016/j.cell.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, Lim TH, Pastorino L, Kunwar AJ, Walton JC, et al. JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron. 2012;75:824–837. doi: 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Goedert M. Phosphorylation of microtubule-associated protein tau by AMPK-related kinases. J Neurochem. 2012;120:165–176. doi: 10.1111/j.1471-4159.2011.07523.x. [DOI] [PubMed] [Google Scholar]
- Yu W, Polepalli J, Wagh D, Rajadas J, Malenka R, Lu B. A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Aβ on synapses and dendritic spines. Hum Mol Genet. 2012;21:1384–1390. doi: 10.1093/hmg/ddr576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.