

Abstract
Cancers of diverse origins exhibit marked glucose avidity and high rates of aerobic glycolysis. Increased understanding of this dysfunctional metabolism known as the Warburg effect has led to an interest in targeting it for cancer therapy. One promising strategy for such targeting is glycoconjugation, the linking of a drug to glucose or another sugar. This review summarizes the most salient examples of glycoconjugates, in which known cytotoxins or targeted anticancer therapeutics have been linked to glucose (or another glucose transporter substrate sugar) for improved cancer targeting and selectivity. Building on these examples, this review also provides a series of guidelines for the design and mechanistic evaluation of future glycoconjugates.
Keywords: cancer, anticancer agents, drug targeting, drug conjugation, Warburg effect, glucose, glycoside, glucose conjugate, glycoconjugate, glufosfamide
1. Introduction
Almost a century ago, the German scientist Otto Warburg observed that cancerous tissues consume large amounts of glucose compared to non-transformed tissue and have high rates of aerobic glycolysis [1, 2]. Today this phenomenon is known as the Warburg effect and is recognized as one of the hallmarks of cancer [3]. Glycolytic enzymes, as well as the insulin-independent glucose transporter GLUT-1, are widely overexpressed in human cancers [4, 5], and high expression levels of these proteins in tumor biopsy samples correlate with poor cancer prognosis, making them attractive therapeutic targets [6–9].
Targeting the Warburg effect as an anticancer strategy has garnered a great deal of interest in recent years. Most efforts have focused on using small molecules to inhibit the function of metabolic enzymes, as comprehensively reviewed elsewhere [10, 11]. This review focuses on the current progress and future directions of exploiting the Warburg effect by using glycoconjugation to selectively target anticancer therapeutics to cancer cells. This strategy was inspired by the widespread clinical use of 2-deoxy-2-(18F)fluoro-D-glucose (18F-FDG; Figure 1a), a radiolabeled glucose analog. 18F-FDG has been used to visualize tumors and their metastases due to the tendency of these cancerous tissues to take up glucose at a higher rate than most normal tissues (as shown in Figure 1b & 1c) [12, 13]. The synthesis and evaluation of sugar-conjugated anticancer drugs was first reported in the literature in 1995, and this field has grown markedly in recent years, with the first-in-class conjugate glufosfamide in advanced clinical trials and many others in development.
Figure 1.
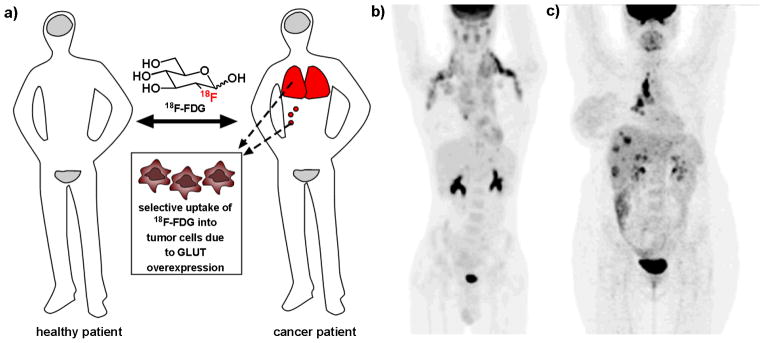
Positron emission tomography (PET) imaging using the radiolabeled glucose analog 18F-FDG is a widely used clinical tool for the diagnosis and staging of many types of cancer. a) In a healthy patient, 18F-FDG will be taken up only by tissues that constitutively consume glucose, such as the brain and bladder (grey). In a cancer patient, tumor cells will preferentially uptake 18F-FDG, allowing clinicians to identify sites of tumors and metastases (red), as well as stage cancer and monitor response to treatment. b) 18F-FDG PET scan of a patient with metastatic Hodgkin’s lymphoma. c) 18F-FDG PET scan of a patient with metastatic breast cancer. (Figure 1b and 1c are reprinted by permission of the Society of Nuclear Medicine from: Ben-Haim S and Ell P. 18F-FDG PET and PET/CT in the Evaluation of Cancer Treatment Response. J Nucl Med. 2009; 50(1): 88–99. Figures 1 and 3[13].)
2. Precedent for anticancer drug conjugates: antibody-drug conjugates and folate-drug conjugates
Conjugation of anticancer agents to molecules that allow for preferential delivery to cancer cells is well-precedented with several conjugates having clinical efficacy. One approach involves conjugating an established drug to an antibody targeting a specific tumor marker. Adcertis (brentuximab vedotin; Figure 2a) received US Food and Drug Administration (FDA) approval in August 2011, and European Union approval in October 2012, for the treatment of anaplastic large cell lymphoma and Hodgkin lymphoma. Adcertis is thought to enter cancer cells via CD30 receptor-mediated endocytosis and subsequently be trafficked to endosomes, where a combination of degradative enzymes and increased acidity lead to cleavage to the active tubulin inhibitor [14]. Another antibody-drug conjugate, Kadcyla (trastuzumab emtansine; Figure 2b), received US FDA approval in February 2013 for the treatment of HER2-positive breast cancer following the EMILIA clinical trial [15]. While these conjugates have substantial clinical promise, major drawbacks do exist, including the logistical and economic challenges associated with biologics as therapeutics.
Figure 2.

Antibody- and folate-conjugated anticancer agents. a) Adcertis (brentuximab vedotin) – CD30-targeting monoclonal antibody brentuximab (red) attached to the tubulin polymerization inhibitor monomethyl auristatin E (blue) via a modified peptide linker; b) Kadcyla (trastuzumab emtansine) – HER2-targeting monoclonal antibody trastuzumab (red) attached to the tubulin polymerization inhibitor mertansine (blue) via a linker; c) EC-145 – folate (red) coupled with a vinblastine-derived microtubule polymerization inhibitor (blue) via a modified peptide linker; d) EC-0225 – folate (red) conjugated to a vinblastine-derived microtubule polymerization inhibitor (blue) and the DNA alkylator mitomycin (green) via a modified peptide linker.
A second, well-precedented approach to drug conjugation for improved cancer selectivity and uptake is folate conjugation. Cancer cells prefer de novo nucleotide synthesis over the salvage pathway that is often favored by normal tissues [16]. This preference of cancer cells means that they must take up large amounts of folate, a cofactor needed in this process. For this reason, folate receptor α (FR-α) is overexpressed in an estimated 40% of human cancers (it is expressed only by certain types of epithelial cells in healthy individuals) [17, 18]. This overexpression makes FR-α an excellent drug target. The most clinically-advanced folate-conjugated drug to date is EC-145 (Figure 2c), which is currently in phase III trials (in combination with the established pharmaceutical Doxil) against ovarian cancer in the US. The peptide linker of EC-145 is expected to be cleaved by cellular cathepsins, releasing the cytotoxin intracellularly [19]. Another folate-conjugated drug in development is EC-0225 (Figure 2d. Both the disulfide and peptide bonds are expected to be cleaved in intracellular endosomal compartments [20].
3. Current progress in glucose conjugation as an anticancer strategy
(a) Prevalence of GLUT-1 overexpression in cancer
For glycoconjugation to be effective as a targeted anticancer strategy, glucose transporters must be overexpressed in cancer compared to normal tissues. Similar to FR-α overexpression in cancer, GLUT-1 has been demonstrated to be overexpressed in a large percentage of cancers from various tissues of origin (Table 1, see also a comprehensive review by Medina [5]).
Table 1.
GLUT-1 overexpression and relation to cancer prognosis in patient biopsy samples
| Tissue of origin | Fold-overexpression | Assessment method | Association with poor prognosis or survival? | Reference |
|---|---|---|---|---|
| Adrenal gland | Not quantified | Immunohistochemistry | Yes | [21] |
| Bladder | Not quantified | Immunohistochemistry | Yes | [22] |
| Breast | 2.8 | RT-PCR | Not determined | [23] |
| Head and neck | 2.1 | RT-PCR | Yes | [24] |
| Hepatocellular | ≤ 50 | RT-qPCR | Not determined | [25] |
| Oral | Not quantified | Immunohistochemistry | Yes | [7] |
| Ovarian | Not quantified | Immunohistochemistry | Yes | [26] |
| Prostate | ≤ 2 | RT-qPCR | Yes | [27] |
| Rectal | Not quantified | Immunohistochemistry | Yes | [28] |
| Renal | ≤ 10 | RT-qPCR | Not determined | [29] |
A second metric to determine which types of cancers would be good candidates for glycoconjugate targeting is to examine which cancers are clinically staged using 18F-FDG PET imaging, summarized in a recent review by Bensinger and Christofk [30]. Currently, lung, breast, colorectal, and endometrial carcinomas, as well as bone and soft tissue sarcomas and Hodgkin’s and non-Hodgkin’s lymphomas, are commonly staged based on their ability to preferentially uptake this radiolabeled glucose analog compared to non-cancerous tissues.
(b) Early efforts in evaluating sugar-conjugated drugs
Monosaccharide-conjugated analogs of diverse agents, designed to improve upon the water solubility, serum stability, and targeting of their aglycones, have been reported in the chemical literature since the early 1990s. In 1991, several glycopeptide analogs of the anti-hypertensive peptide renin, including a mannose and a 2-(acetylamino)-2-deoxyglucose linked to renin via a C1 amide, were shown to have increased serum half-life compared to their aglycones [31]. The synthesis of glucose and mannose phosphotriester conjugates of the anti-retroviral nucleotide analog 3′-azido-3′-deoxythymidine (AZT) was also reported in 1991[32]; these derivatives were shown to have comparable antiviral efficacy and enhanced targeting to the central nervous system (presumably via GLUT transporters) than their aglycones [32, 33]. However, none of these conjugates were explicitly queried as anticancer agents.
(c) The trailblazer for glycoconjugated anticancer agents: glufosfamide
Glufosfamide (Figure 3), initially reported by Wiessler and colleagues in 1995 [34], was the first sugar conjugate to be explicitly designed and evaluated as a cancer-targeting cytotoxic compound. Glufosfamide was developed to decrease the toxicity and increase the cancer selectivity of its aglycone, the DNA alkylating agent ifosfamide mustard. The potency of glufosfamide was comparable to that of its aglycone in cell culture. However, glufosfamide’s anticancer potency was markedly reduced upon co-treatment with 0.1 μM of the GLUT-1 transporter inhibitors phloretin and phlorizin, suggesting that the entry of glufosfamide into cells was at least partially GLUT receptor-mediated. Finally, glufosfamide matched the efficacy of its aglycone in increasing the length of survival of mice in two aggressive tumor models [34].
Figure 3.
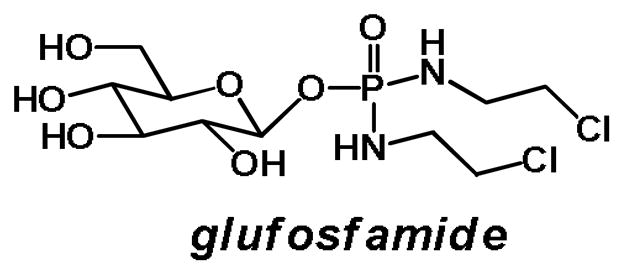
Glufosfamide: 1-β-D-glucose-conjugated ifosfamide mustard
When glufosfamide and ifosfamide were administered intravenously in mice and rats, glufosfamide was found to have a 4.5-fold greater LD50 value (was 4.5-fold less toxic) than its aglycone in rats (in mg/kg), and a 2.3-fold greater LD50 value than its aglycone in mice (in mg/kg) [34].
A pharmacokinetic and whole-body distribution study of glufosfamide in rats, published by Wiessler and colleagues in 1996, revealed that the drug had a short plasma half-life (32 minutes) with largely renal excretion [35]. Glufosfamide is a prodrug, requiring cleavage of glucose via spontaneous hydrolysis or glycosidase activity [36] to liberate the active drug. In this study, roughly 64% of total administered radiolabeled glufosfamide was excreted intact within 24 hours, while approximately 18% was excreted as either the liberated ifosfamide or two uncharacterized metabolites. When healthy rats were treated with radiolabeled glufosfamide, it was distributed among many of the tissues which rely on insulin-independent glucose transporters including the liver, kidneys, thyroid gland, thymus, and central nervous system. Treatment with glufosfamide generally did not lead to toxicity in healthy tissues including the liver, kidneys, and brain. Although a small degree of bone marrow toxicity was observed, it was less substantial than the bone marrow toxicity induced by the aglycone. Encouragingly, radiolabeled glufosfamide was shown to localize to the site of a tumor in rats with prostate cancer, with labeled drug remaining at the site of the tumor for at least 24 hours [35], suggesting that it may be selectively taken up and retained by cancerous cells.
The first human clinical trial on glufosfamide in Europe was initiated in 1997 by Briasoulis and coworkers, with results reported in 2000 [37]. This trial examined 20 patients with solid tumors of various origins treated intravenously with glufosfamide in 0.9% NaCl solution for 6 hours, with patients receiving between two and eight courses of glufosfamide 3 weeks apart. Through this trial, the human maximum tolerated dose (MTD) of glufosfamide was established at 6000 mg/m2,, or approximately 190 mg/kg. This dose resulted in reversible renal tubular acidosis in 2/6 patients [37], presumably due to toxic ifosfamide metabolites generated during renal processing of the drug [38]. Similar toxicity is observed for the aglycone, ifosfamide. While Briasoulis and coworkers did not include ifosfamide in their study, ifosfamide’s MTD has been reported as 17000 mg/m2, or approximately 530 mg/kg, when co-administered over 5 days with the adjuvant mesna to counteract renal adverse effects [39]. The plasma concentration of glufosfamide in patients peaked around 420 μM around 10 hours after intravenous administration of the 6000 mg/m2 dose (compared to approximately 500 μM several hours after intravenous infusion of 1000 mg/kg ifosfamide in a separate study [40]), and the median plasma half-life of glufosfamide was 2.3 hours, with only 34% of the total drug administered being excreted via the kidneys [37]. Eight patients showed tumor progression, 10 showed stable disease, and 2 showed objective response to treatment, with one pancreatic cancer patient experiencing complete remission for over 4 years [37].
Human clinical trials of glufosfamide have continued over the past decade, often accompanied with hydration in an attempt to counteract adverse renal effects. Encouraged by the complete remission of one pancreatic cancer patient in the initial trial [37], and bolstered by the observation that pancreatic cancer biopsy samples from a patients were found to overexpress GLUT-1 [41], several trials have examined the efficacy of glufosfamide against pancreatic tumors. Modest response was seen with glufosfamide alone in one phase II trial (2/35 patients with partial response, 11/35 with stable disease, 18/35 with disease progression) [42]. Combination of glufosfamide with the nucleoside analog gemcitabine – a standard-of-care chemotherapeutic in pancreatic cancer – yielded modest response in two trials on pancreatic adenocarcinoma patients [43, 44], with 4/29 patients treated with 4500 mg/m2 (approximately 140 mg/kg) glufosfamide experiencing nonreversible renal failure, 29/29 patients experiencing anemia, 27/29 patients experiencing thrombocytopenia, and 26/29 patients experiencing neutropenia in one study [43]. In a best supportive care phase III trial in terminal pancreatic cancer patients who had experienced disease progression in spite of gemcitabine treatment, treatment with 4500 mg/m2 glufosfamide plus best supportive care (analgesics, antibiotics, and other agents with no anti-tumor effects) led to an 18% increase in survival (a median of 105 days versus 84 days in the best supportive care only arm), which was not statistically significant [45].
European glufosfamide clinical trials in patients with glioblastoma multiforme [46] and non-small cell lung carcinoma [47] have shown glufosfamide to have very modest, if any, efficacy. In the glioblastoma study, researchers did not comment as to whether glufosfamide crossed the blood-brain barrier, although no adverse neurological effects in any of the 31 patients were observed following an intravenous infusion of 1000 mL of a 5000 mg/m2 (approximately 156 mg/kg) glufosfamide solution over the course of 60 minutes [46]. In the absence of both glufosfamide efficacy and neurological effects in this study, it is plausible that the glufosfamide administered was not blood-brain permeable.
The results of one phase I Japanese glufosfamide clinical trial in 13 patients with various solid tumors showed some promise (1/13 patients showed partial response, and 8/13 patients showed disease stabilization), [48] and phase II trials are ongoing. In the United States, 8 clinical trials of glufosfamide in various disease models (pancreatic, ovarian, and lung cancers; glioblastoma multiforme; soft tissue sarcoma) have been registered, with 6 completed and 2 terminated.
While clinical evaluation of glufosfamide is ongoing, its ultimate success may depend on whether administration methods can be devised to allow for delivery of effective therapeutic doses of glufosfamide without dose-limiting renal or hematological toxicity. It seems plausible that the renal adverse effects may be at least partly due to toxic metabolites of ifosfamide generated during renal metabolism [34], but the anemia observed may stem from the fact that erythrocytes express high levels of GLUT-1 [49, 50]. Clinical testing of future glucose conjugated drugs should be cognizant of this potential hemolytic phenotype.
(d) The molecular tools: glucose-conjugated paclitaxel, and other glycoconjugated taxoids
In 2001, Mikuni, Mandai, and coworkers reported the synthesis and cancer cell potency of several glycoconjugates of docetaxel, including conjugates with glucose, galactose, mannose, and xylose. These compounds were shown to have a 3- to 18-fold improvement in activity compared to the aglycone against B16 murine melanoma cells in culture [52], but neither mechanistic nor in vivo data was not reported. A 2008 follow-up publication highlighted the in vivo potency of galactose-conjugated docetaxel (Figure 4a) in a syngeneic P388 murine leukemia tumor model compared to the aglycone [51], but did not characterize the mode of entry or potential cleavage products of the conjugate. Further data validating the mechanism of cellular entry and in vivo pharmacokinetics of these glycoconjugates has not been reported.
Figure 4.
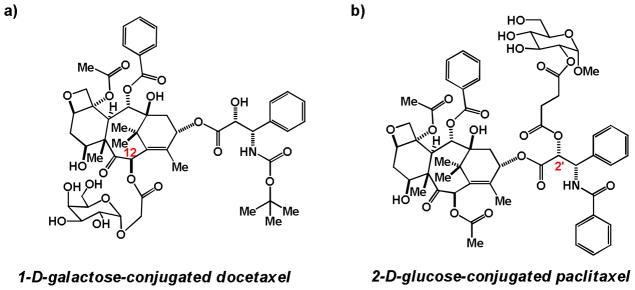
a) 1-α-D-galactose conjugated to position 12 of docetaxel via a short linker [51, 52] b) 1-methylglucose conjugated to position 2′ of paclitaxel via a short linker [53, 54].
In 2007 and 2008, Chen and coworkers reported the synthesis and cell culture evaluation of several glucose- and glucuronic acid-conjugates of paclitaxel [54] [53]. These key studies provided much mechanistic insight into how future glucose-conjugated drugs might be assessed. These glycoconjugates, were designed to improve upon the poor water solubility of paclitaxel, as well as to target it to cancer versus normal cells. Figure 4b depicts the most promising compound reported by Chen and coworkers – 2-D-glucose conjugated paclitaxel – which is comprised of 1-methylglucose conjugated to paclitaxel at position 2′ using a short linker. Position 2′ of paclitaxel is crucial for the drug’s interaction with microtubules [55], so this compound is presumed to be a prodrug that required activation in cells (in vitro assays to assess whether glycoconjugates were able to modulate tubulin polymerization were not reported). However, in vitro incubation for up to 24 hours of 2-D-glucose-conjugated paclitaxel with cell culture media and fetal bovine serum, which contains endogenous β-glucuronidase, failed to show cleavage of the glucose conjugate to the parent aglycone.
To determine whether this glycoconjugate was able to phenocopy paclitaxel’s method of inducing cell death, NPC-TW01 human nasopharyngeal carcinoma cells were treated with vehicle, paclitaxel, or 2-D-glucose-conjugated paclitaxel (Figure 4b) for 24 hours and stained for tubulin distribution and chromosome morphology. While vehicle-treated cells showed diffuse tubulin distribution (Figure 5a) and diffuse chromosomal distribution (Figure 5d), the cells treated with both paclitaxel and glucose-conjugated paclitaxel showed tubulin accumulation around cell nuclei and nuclear chromatin condensation after 24 hours, suggestive of apoptosis (Figure 5b–c, Figure 5e–f). However, in time course studies, cells treated with paclitaxel showed a more pronounced and rapid chromatin condensation than cells treated with glucose-conjugated paclitaxel [53].
Figure 5.

Fluorescent microscopy images of NPC-TW01 human nasopharyngeal carcinoma cells treated with paclitaxel and 2-D-glucose-conjugated paclitaxel (Figure 4b). Tubulin distribution is visualized above (a-c), and chromosome morphology below (d-f). Following 24 hour treatment with vehicle (a and d), tubulin was well-distributed throughout the cells and nuclear chromatin was diffuse, indicative of healthy cells. However, 24 hour treatment with either 1 μM paclitaxel (b and e) or 1 μM 2-D-glucose-conjugated paclitaxel (c and f) led to tubulin accumulation around the nucleus and nuclear chromatin condensation, suggestive of cells processing through apoptosis. (Reprinted with permission from Lin, et. al. J. Med. Chem. 51: 7428–7441. Figure 5a–c;g-i. [53]. Copyright 2008 American Chemical Society.)
Potential colocalization of glucose-paclitaxel with tubulin in cells was further assessed by appending a chromophore, m-aminobenzoic acid, to position 7 of paclitaxel on the glucose-ester conjugate (Figure 6a). Unlike the established unfavorability of substitution at position C2’ for microtubule interaction, substitution at position 7 is not known to interfere with paclitaxel’s interaction with microtubules [55]. Thus, Chen and coworkers used this compound to study whether the glucose substitution at C2’ would impede the compound from colocalizing with tubulin. Confocal microscopy studies examining tubulin distribution and compound fluorescence indicated that there was some convergence of tubulin and compound localization (Figure 6b). Additionally, it was found that this colocalization could be inhibited by co-treatment with the GLUT transport inhibitor phloretin, suggesting that translocation into the cells is at least partially GLUT transporter mediated [53]. However, it was not determined whether glucose was cleaved off prior to tubulin localization in cells.
Figure 6.
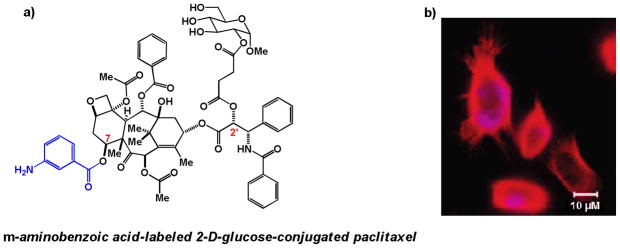
a) 2-D-glucose-conjugated paclitaxel appended at position 7 of paclitaxel with the chromophore m-aminobenzoic acid (blue) b) Merged confocal microscopy images showing the co-localization of m-aminobenzoic acid-labeled 2-D-glucose-conjugated paclitaxel (blue) with tubulin (red) in NPC-TW01 cells. (Figure 6b is reprinted with permission from Lin, et. al. J. Med. Chem. 51: 7428–7441. Figure 3c. [53]. Copyright 2008 American Chemical Society.)
Consistent with the microscopy study, it was found that the potency of several glycoconjugated paclitaxel compounds, assessed upon 72 hour treatment in seven immortalized cancer cell lines and two immortalized non-cancerous cell lines, was at least 5 times less than paclitaxel in both cancer and normal cell lines. The conjugate depicted in Figure 4b was the most potent of the all glycoconjugates tested [53, 54]. While no in vivo or further cell-based studies on these paclitaxel glycoconjugates have been reported to date, these initial studies highlight the need for cell-based mechanistic studies to validate future glycoconjugates before proceeding with in vivo studies.
(e) Sugar and mustard: monosaccharide-conjugated chlorambucil and other alkylator sugar conjugates
Perhaps inspired by the success of the glucose-conjugated DNA alkylator glufosfamide, several groups have reported the synthesis and preliminary evaluation of glycoconjugated analogs of other DNA alkylators, including chlorambucil (a commonly used agent for the treatment of chronic lymphocytic leukemia) and busulfan (a commonly used agent for the treatment of chronic myeloid leukemia). In 1996, Scherman and coworkers reported the synthesis of a series of glucose conjugates linked directly to chlorambucil via a C6 ester or amide bond. All compounds were able to competitively inhibit the uptake of radiolabeled glucose by GLUT-1 receptors in human erythrocytes in a dose-dependent manner. One ester-linked compound in the series, 6-D-glucose-conjugated chlorambucil (Figure 7a), was found to be 150-fold more active in inhibiting the entry of radiolabeled glucose than the native substrate, D-glucose [56].
Figure 7.
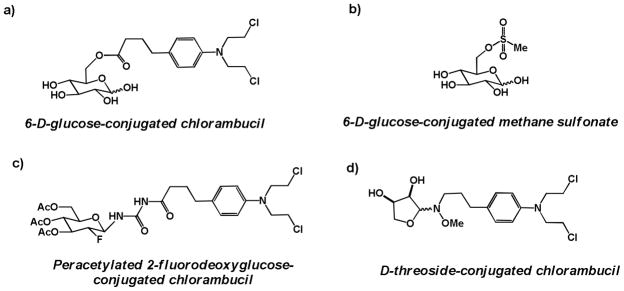
Glycoconjugates of DNA alkyators a) 6-D-glucose-conjugated chlorambucil; b) 6-D-glucose-conjugated methane sulfonate c) Peracetylated 2-fluorodeoxyglucose conjugated at C1 via an amide linkage to chlorambucil; d) D-threoside-conjugated chlorambucil
In a related 1997 study, seeking to make glucose-based analogs of the DNA alkylator busulfan, Scherman and coworkers synthesized and assessed a series of mono- and dimesylated glucose conjugates, with mesyl groups appended via a sulfonate to glucose at either C3, C4, C6, or a combination of the two. Several conjugates had comparable IC50 values to glucose in inhibiting the cellular uptake of radiolabeled glucose in human erythrocytes, suggesting they were substrates for GLUT-1 transporters. 6-D-glucose-conjugated methane sulfonate (Figure 7b) was only 3-fold less efficient at inhibiting radiolabeled glucose uptake than D-glucose [57], marking it as a candidate for future study.
In 2008, Weber and coworkers reported the synthesis and preliminary biological assessment of fluorodeoxyglucose-conjugated derivatives of chlorambucil [58], with preliminary in vivo results reported in 2011 [59]. Intriguingly, among the most promising fluorodeoxyglucose conjugates evaluated was a peracetylated glycoconjugate: peracetylated 2-fluorodeoxyglucose conjugated to chlorambucil via a C1 urea linkage; Figure 7c). This glycoconjugate proved to be more cytotoxic than its unacetylated sugar analog, and also showed enhanced cytotoxicity compared to unconjugated chlorambucil in a panel of normal and human cell lines, with up to a 25-fold enhancement in cytotoxicity compared to chlorambucil in human fibroblasts and MCF-7 human breast ductal carcinoma cells. In addition, peracetylated 2-fluorodeoxyglucose-conjugated chlorambucil was shown to demonstrate intracellular ROS production in L929 murine cells at 25 μM [58], suggestive of inducing DNA damage.
In mice, peracetylated 2-fluorodeoxyglucose-conjugated chlorambucil was shown to have a 6-fold increase in MTD on a mg/kg scale, or a 3-fold increase on a molar scale, compared to chlorambucil (90 mg/kg vs.15 mg/kg). This glycoconjugate was also shown to have efficacy in two different syngeneic mouse models of solid tumors at the 90 mg/kg dosage, while the parent compound chlorambucil was only modestly effective in one trial and ineffective in the second [59].
While peracetylted 2-fluorodeoxyglucose-conjugated chlorambucil was designed to have tumor-selective targeting, no experiments were reported indicating whether it is taken up through glucose transporters, cleaved intracellularly to liberate chlorambucil, or able to alkylate DNA in uncleaved form.
In a 2010 report, Goff and Thorson created a 63-member library of neoglycoside conjugates of chlorambucil, using a variety of amino-substituted monosaccharides common (glucose, mannose) and uncommon (galactose, xylose, gulose, lyxose) in mammalian metabolism. All library compounds were assessed for their ability to induce a decrease in growth proliferation in a panel of immortalized human cancer cell lines. The most promising compound in this assay was D-threoside conjugated to chlorambucil via a methoxyamine linker (Figure 7d), which reduced cell growth 8-fold compared to chlorambucil [60]. Neither the method of cellular uptake of these chlorambucil neoglycoside conjugates, nor the assessment of cleavage to the free alkylator or alkylating activity of the conjugate, was explored in this study.
(f) Diverse targets: preliminary studies on additional glucose conjugates as anticancer agents
Wiessler and coworkers published two reports in 2001 [61, 62] on glucose conjugates of the benzylguanine class of inhibitors against the human DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT), an enzyme that is involved in DNA repair upon damage induced by alkylating chemotherapeutics. β-D-glucose was linked via a C1 ether linkage to an alkyl chain of varying length attached to benzylguanine, and the glucose-conjugated complexes were assessed for their ability to inhibit MGMT activity in vitro and in cells. Most glucose conjugates had poorer inhibition compared to their aglycone, but 8-[O6-(4-bromothenyl)-guan-9-yl]-octyl-1-β-D-glucoside (Figure 8a) was shown to require only 2–4-fold higher concentrations than its aglycone, O6-(4-bromothenyl)-guanine, to attain the same inhibitory potential both in vitro and in cells. Furthermore, this glycoconjugate was shown to have enhanced growth inhibitory potential compared to its aglycone, although it was not cytotoxic at the concentrations required to ablate MGMT activity. Another glucose conjugate of this class was shown to be stable in cell culture media and pH 7.4 buffer for several days [62], although stability in serum or cell lysate was not reported. Experiments querying whether this glycoconjugate entered cells via GLUT transporters were not reported.
Figure 8.

a) 8-[O6-(4-bromothenyl)-guan-9-yl]-octyl-1-β-D-glucoside – a glucose-conjugated MGMT inhibitor; b) Azomycin with a 2-fluoropropyl linker conjugated via a C6 ether linkage to D-glucose; c) Clioquinol conjugated to β-D-glucose via a C1 ether linkage; d) 8-quinolnyl conjugated to β-D-glucose via a C1 ether linkage; e) D-lyxose conjugated warfarin; f) D-threoside-conjugated cyclopamine; g) 2-amino-2-deoxyglucose-conjugated adriamycin
The synthesis and preliminary biological evaluation of a series of glucose-azomycin adducts were recently reported in 2012 by Kumar and coworkers [63]. 2-nitroimidazole compounds have been studied in clinical trials as radiosensitizers. However, these trials failed because the compounds were found to have dose-limiting toxicities at concentrations lower than their therapeutic doses [64]. Kumar and coworkers attempted to overcome this toxicity via glucose-conjugates designed to have improved cancer cell selectivity. Compounds in this series were reported to have low millimolar cytotoxicity in a panel of immortalized murine and human cancer cell lines and to be modestly effective radiosensitizers. To assess whether the uptake of these glucose conjugates was partially GLUT receptor-mediated, glycoconjugates were tested at concentrations ranging from 100 μM to 10 mM for the ability to compete with 14C glucose uptake in Xenopus oocytes, which express GLUT-1. The compound depicted in Figure 8b, a diastereomeric mixture of α and β glucose conjugated at the 6 position to 3-carbon linker, off of which emerges a fluorine diagnostic arm and azomycin, was shown to be the strongest competitor for glucose uptake [63], suggesting that its cellular transport is at least partially GLUT-1 mediated. In vivo studies using glucoconjugated azomycins were not reported.
Vecchio, Viale, and coworkers reported in 2012 the synthesis and cell culture evaluation of glucose conjugates of a class of metal-binding compounds, the 8-hydroxyquinolines [65]. The 8-hydroxyquinolones, most notably clioquinol (5-chloro-7-iodo-8-hydroxyquinolone), have shown promise in early stage clinical trials as anticancer agents due to their scavenging of copper(II), a necessary co-factor in tumor growth [66]. 1-O-glucose conjugates of both clioquinol (GluCQ) (Figure 8c) and another 8-hydroxyquinolone, 8-quinolnyl (GluOHQ) (Figure 8d), were synthesized to improve targeting of these compounds to tumor cells. The 8-hydroxyquinolone glucose conjugates are prodrugs, unable to efficiently complex with metal ions, such that intracellular cleavage to free the quinolone moiety is necessary for activity. Both glucose conjugates were shown to be substantially cleaved by an almond β-glucosidase within a one hour incubation period; however, cleavage was not assessed in mammalian cells. When the antiproliferative ability of the glucose conjugates and their parent aglycones was assessed, the glucose conjugates were found to be less potent than the aglycones [65], perhaps due to insufficient cleavage of prodrugs to their aglycones in cells.
Thorson and coworkers reported in 2011 the synthesis of a library of 38 glycosylated conjugates of the anticoagulant drug warfarin. Like in Thorson’s earlier exploration of neoglycoside-conjugated chlorambucil [60], sugars selected for conjugation ranged from native mammalian metabolites such as glucose to unnatural sugars such as lyxose and xylose. While most sugar conjugates lost their ability to prevent blood coagulation, some did show cytotoxicity in cancer cells, especially the amino-substituted D- and L-xylosides and lyxosides (D-lyxose-conjugated warfarin, which showed the most anticancer potency, is depicted in Figure 8e) [67]. In a 2012 report, Goff and Thorson also prepared and evaluated neoglycoside conjugates of the alkaloid cyclopamine, which induces teratogenesis during embryonic development in many species. All neoglycoside conjugates synthesized were shown to have increased growth proliferation inhibition compared to their aglycone in NCI-H460 lung cancer cells, with D-threoside-conjugated cylopamine (Figure 8f) among the most potent of the set [68]. However, none of the glycoconjugated cyclopamine analogs were assessed for their mutagenic potential or ability to enter cells via GLUT transporters.
A 2013 report by Gu and coworkers presented the synthesis and cell culture and in vivo evaluation of a 2-amino-2-deoxyglucose conjugate of the topoisomeriase II inhibitor adriamycin (doxorubicin), linked using a succinic acid spacer (Figure 8g) [69]. This adriamycin glycoconjugate was designed to enhance the selectivity of adriamycin to cancer cells, and thus to counteract the dose-limiting toxicity of adriamycin treatment on healthy tissues (notably the heart, liver, and kidneys) [69]. In cell culture, 2-amino-2-deoxyglucose-conjugated adriamycin was found to have similar potency to its aglycone adriamycin against immortalized cancer cell lines, but the while adriamycin showed substantial toxicity to human embryonic lung fibroblast (HELF) cells at low nanomolar concentrations after 48 hours, 2-amino-2-deoxyglucose-conjugated adriamycin was not toxic to HELF cells at up to 50 nM for 48 hours [69], though the toxicity of higher concentrations of the glycoconjugate were not tested. The intrinsic fluorescence of adriamycin was used to monitor the uptake of 2-amino-2-deoxyglucose-conjugated adriamycin in cells by confocal microscopy. Using this technique, the entry of the glycoconjugate into HepG2 human liver carcinoma cells was shown to be substantially blocked by pretreatment of cells with high concentrations (25 or 50 mM) of 2-deoxyglucose [69], suggesting that the glycoconjugate’s cellular uptake was at least partially GLUT-mediated. A xenograft study examining the efficacy of adriamycin (6 mg/kg) versus 2-amino-2-deoxyglucose-conjugated adriamycin (6 mg/kg) versus saline control in nude mice bearing SKOV3 human ovarian carcinoma tumors demonstrated that both adriamycin and its glycoconjugate had similar efficacy in reducing tumor volume. Post-study histological examination of heart and kidney tissues from mice in all three treatment groups indicated that adriamycin-treated mice showed heart and kidney damage, while the heart and kidney sections of the 2-amino-2-deoxyglucose-conjugated adriamycin treated mice did not show overt damage and resembled the saline control [69], suggesting that the glycoconjugate may display enhanced tumor targeting or less toxicity to normal tissues. One drawback to this study is that while Gu and coworkers postulated that 2-amino-2-deoxyglucose-conjugated adriamycin was a prodrug [69], data querying the cleavage of the glycoconjugate in cells, in liver microsomes, or in vivo (pharmacokinetics) was not reported. Additionally, no data was reported on whether the glycoconjugate retained adriamycin’s ability to inhibit topoisomerase II in vitro or in cells.
Several other glucose or other monosaccharide-conjugated anticancer drugs have been reported but are still pending mechanistic evaluation. These include glucose, lactose, and galactose conjugates of the heat shock protein 90 inhibitor geldanamycin [70], glucose conjugates of the natural product cadalene [71], glycoconjugates of the DNA alkylator duocarmycin SA [72, 73], and glucose conjugates of the lipoxygenase inhibitor nordihydroguaiaretic acid [74].
Mechanistic considerations for the design and study of future glycoconjugates
For glucose-conjugated anticancer compounds to reach their full potential, several lingering issues must be addressed, and this would be aided by the development of various molecular tools.
(a) Which positions of glucose can be substituted?
In order to selectively target cancer cells, glycoconjugates should be substrates for the insulin-independent glucose transporter GLUT-1, which is overexpressed in a wide variety of solid tumors [6, 26, 75–78]. GLUT-1 is a passive, bidirectional glucose uniporter comprised of 12 transmembrane helical domains, 8 of which are assembled in a circular fashion from an exofacial (extracellular top-down) view.
The ability of substituted glucose analogues to be substrates for GLUT-1 has been investigated; a summary of findings is presented in Figure 9. Kinetic and computational modeling studies using glucose analogs suggest that the hydroxyl groups at positions 1 and 3, and the pyran oxygen 5 in glucose’s closed conformation, are involved in stabilizing hydrogen bonding interactions with amino acid residues within the transporter [79, 80]. Loss of hydrogen bond acceptors at these positions makes glucose analogs poor substrates for GLUT-1: both 1-deoxy-D-glucose and 3-deoxy-D-glucose, which have altered ring conformations, have been determined to have Ki values roughly 10-fold higher than D-glucose in competition assays in GLUT-1 transporters from human erythrocytes [79]. However, substitution of fluorine for the hydroxyl groups at position 1 (in the β conformation, in which the fluorine is in the equatorial position in the chair form) or position 3 yields Ki values comparable to glucose, which is not surprising given that fluorine has similar dipole properties to oxygen and is unlikely to change glucose’s preferred ring conformation. Interestingly, substitution of fluorine at position 1 in the α conformation results in a poor GLUT-1 substrate [79], suggesting that the β conformation is preferred for GLUT-1 transport. However, 2-D-glucose-conjugated paclitaxel (Figure 4b) contains glucose in the α conformation, and this compound was demonstrated to be partially inhibited from entering cells upon treatment with GLUT-1 transporter inhibitor phloretin [53]. Substitutions of C1 oxygen for C1 nitrogen have also been reported in FDG analogs; these analogs have been shown to compete with D-glucose for cellular entry, suggesting their uptake is GLUT-mediated [81].
Figure 9.
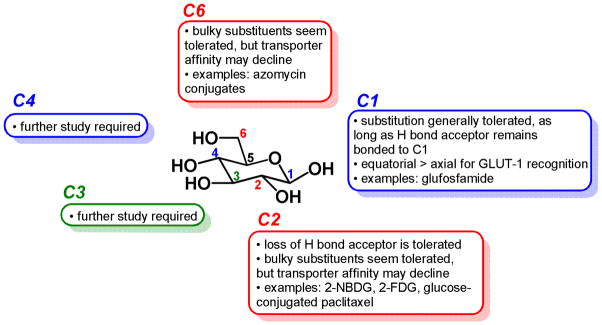
Structure-activity relationship of D-glucose as a substrate for the GLUT-1 transporter. Substitutions at C1, C2, and C6 have been most explored to date, demonstrating that some substitutions can be made with the resulting conjugate retaining affinity for GLUT-1. Substitutions at C3 and C4 require further study; to date, no C3- or C4-glucose-conjugated anticancer compounds have been reported.
Compounds with bulky substituents at C3, including 3-O-allyl-D-glucose and 3-O-(2′,3′-epoxypropyl)-D-glucose, appear to still be substrates for GLUT-1 [79], suggesting that this position may tolerate substitution well. In glucose’s most thermodynamically stable conformation, the closed chair form, C5 does not contain a hydroxyl substituent, and thus any glycoconjugate candidates substituted at C5 may not assume the chair configuration that is recognized by GLUT receptors.
Thus, these data suggest that for glucose conjugates to remain substrates for GLUT-1, compounds with hydrogen bond acceptors such as nitrogen or oxygen must be retained proximal to carbons 1 and 3, and substitutions at the 1 position may retain higher affinity for GLUT-1 if they are present in an equatorial conformation.
A large fraction of known glucose conjugates discussed in this review are conjugated to the anticancer agent at position 1, with the C1 oxygen intact and locked into equatorial (β) position. This review has described three such classes of glycoconjugates linked at glucose’s C1 position: glufosfamide (Figure 3), peracetylated 2-fluorodeoxyglucose-conjugated chlorambucil (Figure 7c), and glucose-conjugated cliquinol and quinolynl (Figure 8c and 8d). Of these, experiments were performed to indicate that glufosfamide’s cellular transport is at least partially GLUT-mediated, but no such experiments were published regarding the other two classes. No anticancer glycoconjugates that are linked to glucose at the C3 or C5 positions have been reported to date.
The hydroxyl groups of glucose at C2, C4, and C6 are not implicated in hydrogen bonding interactions with the GLUT-1 transporter and thus addition of bulk to these positions may be tolerated. As previously discussed, 2-18F-fluoro-2-deoxyglucose (18F-FDG) is a widely established clinical tool that is a substrate for GLUT-1 and GLUT-3 transporters [82], and 2-NBDG, in which the C2 hydroxyl is replaced by nitrogen and a bulky aromatic system, is a widely used fluorescent glucose bioprobe which has been shown to be taken into cells via GLUT transporters [83, 84]. Of the anticancer glucose conjugates discussed above, glucose-conjugated paclitaxel (Figure 4b) and 2-amino-2-deoxyglucose-conjugated adriamycin (Figure 8g) contain a C2 linkage and have been experimentally shown to enter cells via a GLUT-mediated mechanism [53, 69].
Some substituents at C4 of glucose, such as 4-O-propyl-D-glucose and cellobiose (the disaccharide consisting of 2 glucose molecules connected via a β 1→4 ether linkage), have been shown to have minimal difference compared to D-glucose on Ki in competition; however, lactose, in which glucose is conjugated via C4 to galactose, is not a substrate for GLUT-1 [79]. No anticancer glucose conjugates containing linkages at C4 have been reported to date.
Substituents at C6 of glucose have been reported to be comparable or slightly weaker substrates for GLUT-1 than unsubstituted glucose. 6-deoxy-D-glucose retains the same Ki value as D-glucose, while 6-deoxy-6-fluoro-D-glucose has five times higher inhibitory potential in competition studies than D-glucose [79]. Several reported anticancer glucose conjugates linked at C6 have been reported to date, including glucose-conjugated chlorambucil (Figure 7a), glucose-conjugated methane sulfonate (Figure 7b), and glucose-conjugated azomycin (Figure 8b), all three of which have been implicated as GLUT substrates through experimental evidence.
Importantly, although glucose-conjugated anticancer drugs have been in the literature for nearly 20 years, there has been no systematic study on the position of substitution for any given anticancer glycoconjugate.
(b) Are monosaccharides besides glucose efficient substrates for transport through GLUT-1?
As discussed above, currently explored glycoconjugates that have been demonstrated to be transported through the GLUT-1 receptor are limited to substituted glucose conjugates. Neoglycosides – amino-modified sugars – as well as peracetylated glucose conjugates, have been postulated to be taken up via GLUTs [60], but mechanistic studies have yet to be reported. However, it is known that other sugars besides D-glucose can be substrates for GLUT transporters, and thus can be considered candidates for a GLUT-targeting approach. Specifically, using light diffraction measurement in human erythrocytes that express GLUT-1 as their sole glucose transporter, 2-deoxy-D-glucose, D-mannose, D-galactose, D-mannose, D-xylose, 2-deoxy-D-galactose, L-arabinose, D-ribose, D-fucose, and D-lyxose, in order of decreasing affinity, were all found to be transported into cells in a transporter-mediated fashion. In contrast, D-arabinose, L-fucose, and L-rhammose were poor substrates, and the enantiomers of several GLUT substrates, L-glucose and L-xylose, were not GLUT substrates [85]. Thus, it seems that many sugars can be conjugated to anticancer agents to take advantage of GLUT-mediated cellular entry; the choice of which sugar to utilize may depend on the desired cleavage mechanism of the compound. For instance, D-galactose, the C4 epimer of glucose, has been reported to possess an equivalent affinity and uptake rate by GLUT-1 compared to glucose [86]. Galactose-conjugated drugs may be used to selectively target certain types of cancers known to highly express galactosidase enzymes, such as breast and colon cancers [87]. In the absence of substantial tumor galactosidase expression, galactose-conjugated prodrugs can be used in conjunction with tumor-selective monoclonal antibodies linked to galactosidases, which ideally cleave the inactive conjugate to its active form in tumor tissue [86].
(c) How can glycoconjugates be validated in vitro and in vivo?
This review has highlighted the glycoconjugates that have been most thoroughly mechanistically evaluated, to serve as a model for medicinal chemists and chemical biologists in their development of glycoconjugated drugs. Glycoconjugation is an exciting cancer targeting strategy, but caution must be taken before claiming that a glycoconjugate is cancer-selective and cancer-targeted. The following represent important questions that should be examined in the development of glycoconjugates, as well as guidelines for how to answer these questions based on previous work. Ideally, all assays should compare both the glycoconjugate and its aglycone in parallel.
Is entry of the glycoconjugate GLUT receptor-mediated?
A crucial mechanistic question in the development of glycoconjugates is whether their cellular entry is at least partially GLUT-mediated. There are many ways to address this question, and ideally any conjugate would be evaluated with multiple methods. To examine entry via GLUT-1, the transporter most commonly upregulated in cancer, human erythrocytes are a common model system since GLUT-1 is the only glucose transporter expressed in this cell type. Historically, competition with radiolabeled glucose [79] and changes in erythrocyte volume [85, 88] have been used in this model system.
A second approach is to compare the entry or efficacy of the glycoconjugate in the presence of a GLUT transporter inhibitor, either in erythrocytes or in a cultured cell line. A number of natural products have been demonstrated to inhibit GLUT transporters in all cell types, including the botanical phloretin (a GLUT-1 inhibitor [49]) and the fungally-produced cytochalasin B (a pan-GLUT transporter inhibitor [89, 90]). More recently, GLUT inhibitors have been discovered through screening and design studies, such as the GLUT-1 inhibitor fasentin [91] and the GLUT-1 inhibitor WZB117 [92]. Assays have been developed to determine whether glycoconjugate cellular entry and activity is lost when cells are co-treated with these inhibitors [53]. The general caveat to using GLUT inhibitors is that it may be difficult to meter the extent of the inhibitor’s interference with GLUT versus its off-target, potentially cytotoxic effects. Cytochalasin B is known to be relatively toxic with several reported targets, including actin [93]. However, newly-developed inhibitors may be more selective.
A third method to probe glycoconjugates’ mode of entry is to stably knock down or knock out GLUT expression. Both of these approaches were employed in a recent study by Anderson and colleagues [94]. In examining the entry of 3H-2-deoxyglucose into cells, it was reported that shRNA-mediated GLUT-1 knockdown in a murine cell line which expressed GLUT-1 as its predominant GLUT transporter led to only a 25% reduction in 3H-2-deoxyglucose entry compared to control shRNA cells [94], presumably due to an increase in expression of other GLUT transporters to allow knockdown cells to survive in culture. Anderson and colleagues also generated a GLUT-1 knockout cell line from a GLUT-1 knockout mouse; these cells showed close to a 50% reduction in entry of 3H-2-deoxyglucose compared to control cells [94]. If a GLUT-1 knockout cell line becomes widely available, evaluation of glycoconjugates’ entry, efficacy, and potency in such a cell line may become a valuable metric. Incidentally, humans born with a mutated, functionally-impaired GLUT-1 transporter experience a severe phenotype known as De Vivo disease which results in mental retardation, seizures, and a host of neurological deficits, underscoring the importance of GLUT-1 in supplying glucose to the nervous system [95]. To date, no live births of humans with a completely nonfunctional GLUT-1 transporter have been reported, so efforts to generate a human GLUT-1 knockout cell line must be relegated to the research laboratory.
A fourth approach is the use of fluorescently-labeled glucose bioprobes, as reviewed recently [96]. 2-NBDG [83, 84] (Figure 10) is perhaps the most well-known member of this class, but a growing interest in this area in recent years has led to the development of many new fluorescent probes which offer increased emission, slower photobleaching, and increased depth penetration to allow for imaging in vivo [96–98]. Competition assays can be conducted between glycoconjugates and these fluorescent probes, to determine whether increasing concentrations of glycoconjugate cause decreasing fluorescence in cells, using flow cytometry or fluorescent microscopy as a readout.
Figure 10.
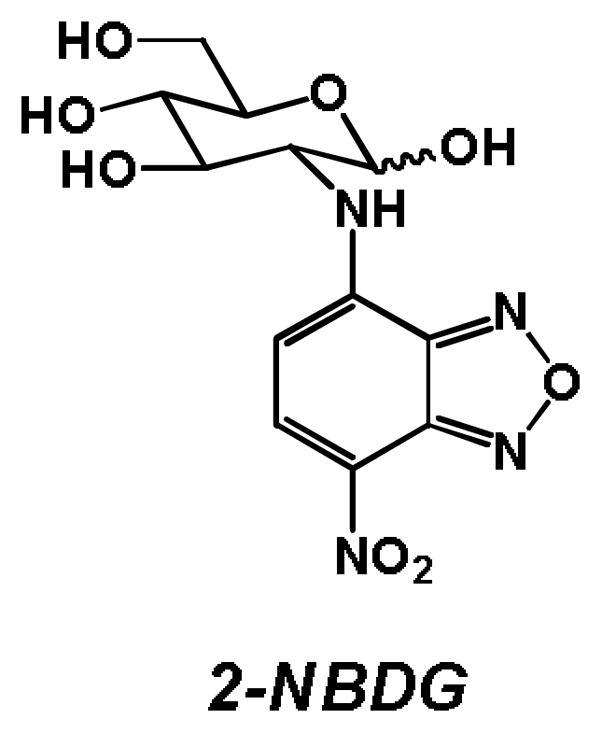
Structure of the fluorescent glucose bioprobe 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG).
Is the glycoconjugate directly able to inhibit its target, or is cleavage required?
Evaluation of a glycoconjugate should begin by discerning whether or not the glycoconjugate retains the activity of its aglycone, using appropriate kinetic evaluation for an enzyme, surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC) for binding assessment, or another comparable technique depending on the aglycone’s target. If the glycoconjugate is no longer active against the target and instead is a prodrug, its cleavage to its aglycone in cells or in vivo needs to be examined. For instance, chromatographic techniques can be used to detect the aglycone in cell lysate following treatment with the glycoconjugate, and serum stability or liver microsome analyses may be useful for discerning whether cleavage is likely to occur in vivo. If the glycoconjugate does not appear to be cleaved and no longer retains activity against its target, then the identity of its new target or targets should be examined using various biochemical and molecular biological approaches. Alternately, derivatives that are more likely to be cleaved in cells or in vivo can be synthesized and assessed.
What considerations should be taken for in vivo testing of glycoconjugates?
In vivo testing is a crucial proving ground for glycoconjugates. The extensive in vivo studies of glufosfamide in mice and rats should serve as a template for the testing of future drug conjugates. Particularly, it is important to establish whether the glycoconjugate is serum-stable and to discern its primary modes of metabolism and excretion. It is also critical to determine if the glycoconjugate is preferentially targeted to tumor tissue in animal models, to validate the hypothesis of glucose enabling preferential drug targeted to cancerous tissues. This might be accomplished by isotope labeling of the drug prior to administration, and whole-animal radiography following administration, as in the glufosfamide evaluation [35]. Appending a fluorophore to the glycoconjugate, such as a near-infared-emitting fluorophore to provide capability for in vivo imaging [99], may be useful, but only if this new construct is demonstrated to still be taken up by GLUT transporters using the molecular biology methods discussed above.
It is important to assess whether a glycoconjugate has off-target hematological or neurological adverse events, given that GLUT-1 is normally expressed in erythrocytes and in the endothelial cells of the blood-brain barrier. Human, but not mouse or rat, studies of glufosfamide demonstrated anemia due to hemolysis as a common adverse event [43]; for future glycoconjugates, hemolysis should be queried using in vitro assays before the initiation of studies in humans or other large mammals. Encouragingly, neurotoxicity has not been observed in the in vivo evaluation of any glycoconjugate to date; still, all future glycoconjugates should be evaluated for potential neurological adverse events.
Another potential off-target effect of glycoconjugated drugs is their uptake by healthy tissues expressing GLUT receptors other than GLUT-1. There are at least 12 classes of GLUT transporters (both insulin-dependent and –independent) which transport glucose or hexoses alone, as well as several in which the transport of glucose is coupled with sodium, as detailed by Medina [5]. While it is conceivable that these transporters expressed in normal tissues may also take up a glycoconjugate, the strongest evidence favoring the preferential targeting of a glycoconjugate to a tumor is the large body of work on 18F-FDG preferentially being localized to tumors, as evidenced in Figure 1. Most glycoconjugated drugs presented in this review have not been subjected to imaging studies to precisely localize the drug’s distribution in a whole organism or whole tumor-bearing organism. Conducting such an experiment, such as the rat imaging study reported by Wiessler with glufosfamide [35], would be useful, though caution should be taken in labeling the compound in such a way to not alter its ability to be taken up by GLUT transporters.
4. Outlook on this emerging field
Glycoconjugation generally offers improved water solubility and stability and, if the glycoside of choice is a GLUT substrate, the potential for selective targeting to cancer cells. Substantial strides in the field of glycoconjugation have been made, reaching as far as late stage human clinical trials. This field has a great deal of potential and tremendous opportunity for growth, but rigorous mechanistic testing at each stage of the drug development process, and carefully controlled, head-to-head experiments are required to determine the true utility of this strategy.
Acknowledgments
The authors would like to thank Dr. Matthew Brichacek and Betsy Parkinson for their helpful feedback on this manuscript. This work was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (award number R01GM098453). E.C.C. is an NIH Ruth L. Kirschstein National Research Service Award predoctoral fellow (1F30CA168323-01) and a UIUC Department of Biochemistry Herbert Carter fellow.
References
- 1.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8(6):519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014–20. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Medina RA, Owen GI. Glucose transporters: expression, regulation and cancer. Biol Res. 2002;35(1):9–26. doi: 10.4067/s0716-97602002000100004. [DOI] [PubMed] [Google Scholar]
- 6.Ohba S, Fujii H, Ito S, Fujimaki M, Matsumoto F, Furukawa M, Yokoyama J, Kusunoki T, Ikeda K, Hino O. Overexpression of GLUT-1 in the invasion front is associated with depth of oral squamous cell carcinoma and prognosis. J Oral Pathol Med. 2010;39(1):74–8. doi: 10.1111/j.1600-0714.2009.00814.x. [DOI] [PubMed] [Google Scholar]
- 7.Kunkel M, Moergel M, Stockinger M, Jeong JH, Fritz G, Lehr HA, Whiteside TL. Overexpression of GLUT-1 is associated with resistance to radiotherapy and adverse prognosis in squamous cell carcinoma of the oral cavity. Oral Oncol. 2007;43(8):796–803. doi: 10.1016/j.oraloncology.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Trarbach T, Folprecht G, Shi MM, Lebwohl D, Jalava T, Laurent D, Meinhardt G, Harris AL. Prognostic and Predictive Role of Lactate Dehydrogenase 5 Expression in Colorectal Cancer Patients Treated with PTK787/ZK 222584 (Vatalanib) Antiangiogenic Therapy. Clin Cancer Res. 2011;17(14):4892–4900. doi: 10.1158/1078-0432.CCR-10-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong L, Cui Z, Chen P, Han H, Peng J, Leng X. Reduced survival of patients with hepatocellular carcinoma expressing hexokinase II. Med Oncol. 2012;29(2):909–14. doi: 10.1007/s12032-011-9841-z. [DOI] [PubMed] [Google Scholar]
- 10.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 11.Granchi C, Minutolo F. Anticancer Agents That Counteract Tumor Glycolysis. ChemMedChem. 2012;7:1318–1350. doi: 10.1002/cmdc.201200176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrmann K, Benz MR, Krause BJ, Pomykala KL, Buck AK, Czernin J. 18F-FDG-PET/CT in evaluating response to therapy in solid tumors: where we are and where we can go. Q J Nucl Med Mol Imaging. 2011;55(6):620–32. [PubMed] [Google Scholar]
- 13.Ben-Haim S, Ell P. 18F-FDG PET and PET/CT in the Evaluation of Cancer Treatment Response. J Nucl Med. 2009;50(1):88–99. doi: 10.2967/jnumed.108.054205. [DOI] [PubMed] [Google Scholar]
- 14.Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol. 2012;30(7):631–7. doi: 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- 15.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N Engl J Med. 2012;367(19):1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visentin M, Zhao R, Goldman ID. The antifolates. Hematol Oncol Clin North Am. 2012;26(3):629–48. ix. doi: 10.1016/j.hoc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13(3):256–262. doi: 10.1016/j.cbpa.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 18.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338(2):284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 19.Dosio F, Milla P, Cattel L. EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers. Curr Opin Investig Drugs. 2010;11(12):1424–33. [PubMed] [Google Scholar]
- 20.Leamon CP, Reddy JA, Vlahov IR, Westrick E, Dawson A, Dorton R, Vetzel M, Santhapuram HK, Wang Y. Preclinical Antitumor Activity of a Novel Folate-Targeted Dual Drug Conjugate. Mol Pharmaceutics. 2007;4(5):659–667. doi: 10.1021/mp070049c. [DOI] [PubMed] [Google Scholar]
- 21.Fenske W, Völker HU, Adam P, Hahner S, Johanssen S, Wortmann S, Schmidt M, Morcos M, Müller-Hermelink HK, Allolio B, Fassnacht M. Glucose transporter GLUT1 expression is an stage-independent predictor of clinical outcome in adrenocortical carcinoma. Endocr Relat Cancer. 2009;16(3):919–928. doi: 10.1677/ERC-08-0211. [DOI] [PubMed] [Google Scholar]
- 22.Palit V, Phillips RM, Puri R, Shah T, Bibby MC. Expression of HIF-1alpha and Glut-1 in human bladder cancer. Oncol Rep. 2005;14(4):909–13. doi: 10.3892/or.14.4.909. [DOI] [PubMed] [Google Scholar]
- 23.Krzeslak A, Wojcik-Krowiranda K, Forma E, Jozwiak P, Romanowicz H, Bienkiewicz A, Brys M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol Oncol Res. 2012;18(3):721–8. doi: 10.1007/s12253-012-9500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou S, Wang S, Wu Q, Fan J, Wang Q. Expression of glucose transporter-1 and -3 in the head and neck carcinoma--the correlation of the expression with the biological behaviors. ORL J Otorhinolaryngol Relat Spec. 2008;70(3):189–94. doi: 10.1159/000124293. [DOI] [PubMed] [Google Scholar]
- 25.Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS, Stoeltzing O, Warnecke C, Schölmerich J, Oefner PJ, Kreutz M, Bosserhoff AK, Hellerbrand C. GLUT1 Expression Is Increased in Hepatocellular Carcinoma and Promotes Tumorigenesis. Am J Pathol. 2009;174(4):1544–1552. doi: 10.2353/ajpath.2009.080596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semaan A, Munkarah AR, Arabi H, Bandyopadhyay S, Seward S, Kumar S, Qazi A, Hussein Y, Morris RT, Ali-Fehmi R. Expression of GLUT-1 in epithelial ovarian carcinoma: correlation with tumor cell proliferation, angiogenesis, survival and ability to predict optimal cytoreduction. Gynecol Oncol. 2011;121(1):181–6. doi: 10.1016/j.ygyno.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 27.Stewart GD, Gray K, Pennington CJ, Edwards DR, Riddick AC, Ross JA, Habib FK. Analysis of hypoxia-associated gene expression in prostate cancer: lysyl oxidase and glucose transporter-1 expression correlate with Gleason score. Oncol Rep. 2008;20(6):1561–7. [PubMed] [Google Scholar]
- 28.Saigusa S, Toiyama Y, Tanaka K, Okugawa Y, Fujikawa H, Matsushita K, Uchida K, Inoue Y, Kusunoki M. Prognostic significance of glucose transporter-1 (GLUT1) gene expression in rectal cancer after preoperative chemoradiotherapy. Surg Today. 2012;42(5):460–469. doi: 10.1007/s00595-011-0027-2. [DOI] [PubMed] [Google Scholar]
- 29.Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Buttner M, Aigner M, Seliger B, Walter B, Schlosser H, Hartmann A, Andreesen R, Mackensen A, Kreutz M. Warburg phenotype in renal cell carcinoma: high expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int J Cancer. 2011;128(9):2085–95. doi: 10.1002/ijc.25543. [DOI] [PubMed] [Google Scholar]
- 30.Bensinger SJ, Christofk HR. New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol. 2012;23(4):352–361. doi: 10.1016/j.semcdb.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Fisher JF, Harrison AW, Bundy GL, Wilkinson KF, Rush BD, Ruwart MJ. Peptide to glycopeptide: glycosylated oligopeptide renin inhibitors with attenuated in vivo clearance properties. J Med Chem. 1991;34(10):3140–3143. doi: 10.1021/jm00114a026. [DOI] [PubMed] [Google Scholar]
- 32.Henin Y, Gouyette C, Schwartz O, Debouzy JC, Neumann JM, Huynh Dinh T. Lipophilic glycosyl phosphotriester derivatives of AZT: synthesis, NMR transmembrane transport study and antiviral activity. J Med Chem. 1991;34(6):1830–1837. doi: 10.1021/jm00110a011. [DOI] [PubMed] [Google Scholar]
- 33.Namane A, Gouyette C, Fillion MP, Fillion G, Huynh Dinh T. Improved brain delivery of AZT using a glycosyl phosphotriester prodrug. J Med Chem. 1992;35(16):3039–3044. doi: 10.1021/jm00094a018. [DOI] [PubMed] [Google Scholar]
- 34.Pohl J, Bertram B, Hilgard P, Nowrousian MR, Stuben J, Wiessler M. D-19575--a sugar-linked isophosphoramide mustard derivative exploiting transmembrane glucose transport. Cancer Chemother Pharmacol. 1995;35(5):364–70. doi: 10.1007/s002800050248. [DOI] [PubMed] [Google Scholar]
- 35.Stüben J, Port R, Bertram B, Bollow U, Hull WE, Schaper M, Pohl J, Wiessler M. Pharmacokinetics and whole-body distribution of the new chemotherapeutic agent β-D-glucosylisophosphoramide mustard and its effects on the incorporation of [methyl-3H]-thymidine in various tissues of the rat. Cancer Chemother Pharmacol. 1996;38(4):355–365. doi: 10.1007/s002800050495. [DOI] [PubMed] [Google Scholar]
- 36.Arafa HM. Possible contribution of beta-glucosidase and caspases in the cytotoxicity of glufosfamide in colon cancer cells. Eur J Pharmacol. 2009;616(1–3):58–63. doi: 10.1016/j.ejphar.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 37.Briasoulis E, Judson I, Pavlidis N, Beale P, Wanders J, Groot Y, Veerman G, Schuessler M, Niebch G, Siamopoulos K, Tzamakou E, Rammou D, Wolf L, Walker R, Hanauske A. Phase I trial of 6-hour infusion of glufosfamide, a new alkylating agent with potentially enhanced selectivity for tumors that overexpress transmembrane glucose transporters: a study of the European Organization for Research and Treatment of Cancer Early Clinical Studies Group. J Clin Oncol. 2000;18(20):3535–44. doi: 10.1200/JCO.2000.18.20.3535. [DOI] [PubMed] [Google Scholar]
- 38.Lee BS, Lee JH, Kang HG, Hahn H, Lee JH, Shin HY, Ha IS, Cheong HI, Ahn HS, Choi Y. Ifosfamide nephrotoxicity in pediatric cancer patients. Pediatr Nephrol. 2001;16(10):796–799. doi: 10.1007/s004670100658. [DOI] [PubMed] [Google Scholar]
- 39.Ellenhorn MJ, Schonwald S, Ordog G, Wasserberger J. Ellenhorn’s Medical Toxicology: Diagnosis and Treatment of Human Poisoning. Williams and Wilkins; Baltimore, MD: 1997. p. 1344. [Google Scholar]
- 40.Kerbusch T, Mathôt RAA, Keizer HJ, Kaijser GP, Schellens JHM, Beijnen JH. Influence of Dose and Infusion Duration on Pharmacokinetics of Ifosfamide and Metabolites. Drug Metab Dispos. 2001;29(7):967–975. [PubMed] [Google Scholar]
- 41.Reske SN, Grillenberger KG, Glatting G, Port M, Hildebrandt M, Gansauge F, Beger HG. Overexpression of glucose transporter 1 and increased FDG uptake in pancreatic carcinoma. J Nucl Med. 1997;38(9):1344–8. [PubMed] [Google Scholar]
- 42.Briasoulis E, Pavlidis N, Terret C, Bauer J, Fiedler W, Schoffski P, Raoul JL, Hess D, Selvais R, Lacombe D, Bachmann P, Fumoleau P. Glufosfamide administered using a 1-hour infusion given as first-line treatment for advanced pancreatic cancer. A phase II trial of the EORTC-new drug development group. Eur J Cancer. 2003;39(16):2334–40. doi: 10.1016/s0959-8049(03)00629-4. [DOI] [PubMed] [Google Scholar]
- 43.Chiorean EG, Dragovich T, Hamm J, Barrios CH, Gorini CF, Langmuir VK, Kroll S, Jung DT, Tidmarsh GT, Loehrer PJ. A phase 2 trial of glufosfamide in combination with gemcitabine in chemotherapy-naive pancreatic adenocarcinoma. Am J Clin Oncol. 2010;33(2):111–6. doi: 10.1097/COC.0b013e3181979204. [DOI] [PubMed] [Google Scholar]
- 44.Chiorean EG, Dragovich T, Hamm J, Langmuir VK, Kroll S, Jung DT, Colowick AB, Tidmarsh GF, Loehrer PJ. A Phase 1 dose-escalation trial of glufosfamide in combination with gemcitabine in solid tumors including pancreatic adenocarcinoma. Cancer Chemother Pharmacol. 2008;61(6):1019–26. doi: 10.1007/s00280-007-0559-8. [DOI] [PubMed] [Google Scholar]
- 45.Ciuleanu TE, Pavlovsky AV, Bodoky G, Garin AM, Langmuir VK, Kroll S, Tidmarsh GT. A randomised Phase III trial of glufosfamide compared with best supportive care in metastatic pancreatic adenocarcinoma previously treated with gemcitabine. Eur J Cancer. 2009;45(9):1589–96. doi: 10.1016/j.ejca.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 46.van den Bent MJ, Grisold W, Frappaz D, Stupp R, Desir JP, Lesimple T, Dittrich C, de Jonge MJ, Brandes A, Frenay M, Carpentier AF, Chollet P, Oliveira J, Baron B, Lacombe D, Schuessler M, Fumoleau P. European Organization for Research and Treatment of Cancer (EORTC) open label phase II study on glufosfamide administered as a 60-minute infusion every 3 weeks in recurrent glioblastoma multiforme. Ann Oncol. 2003;14(12):1732–4. doi: 10.1093/annonc/mdg491. [DOI] [PubMed] [Google Scholar]
- 47.Giaccone G, Smit EF, de Jonge M, Dansin E, Briasoulis E, Ardizzoni A, Douillard JY, Spaeth D, Lacombe D, Baron B, Bachmann P, Fumoleau P. Glufosfamide administered by 1-hour infusion as a second-line treatment for advanced non-small cell lung cancer; a phase II trial of the EORTC-New Drug Development Group. Eur J Cancer. 2004;40(5):667–72. doi: 10.1016/j.ejca.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu T, Okamoto I, Tamura K, Satoh T, Miyazaki M, Akashi Y, Ozaki T, Fukuoka M, Nakagawa K. Phase I clinical and pharmacokinetic study of the glucose-conjugated cytotoxic agent D-19575 (glufosfamide) in patients with solid tumors. Cancer Chemother Pharmacol. 2010;65:243–250. doi: 10.1007/s00280-009-1028-3. [DOI] [PubMed] [Google Scholar]
- 49.Lefevre PG, Marshall JK. The atachment of phloretin and analogues to human erythrocytes in connection with inhibition of sugar transport. J Biol Chem. 1959;234:3022–6. [PubMed] [Google Scholar]
- 50.London RE, Gabel SA. Fluorine-19 NMR studies of glucosyl fluoride transport in human erythrocytes. Biophys J. 1995;69(5):1814–8. doi: 10.1016/S0006-3495(95)80051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mikuni K, Nakanishi K, Hara K, Hara K, Iwatani W, Amano T, Nakamura K, Tsuchiya Y, Okumoto H, Mandai T. In Vivo Antitumor Activity of Novel Water-Soluble Taxoids. Biol Pharm Bull. 2008;31(6):1155–1158. doi: 10.1248/bpb.31.1155. [DOI] [PubMed] [Google Scholar]
- 52.Mandai T, Okumoto H, Oshitari T, Nakanishi K, Mikuni K, Hara K, Hara K, Iwatani W, Amano T, Nakamura K, Tsuchiya Y. Synthesis and Biological Evaluation of Water Soluble Taxoids Bearing Sugar Moieties. Heterocycles. 2001;54(2):561–566. [Google Scholar]
- 53.Lin YS, Tungpradit R, Sinchaikul S, An FM, Liu DZ, Phutrakul S, Chen ST. Targeting the delivery of glycan-based paclitaxel prodrugs to cancer cells via glucose transporters. J Med Chem. 2008;51(23):7428–41. doi: 10.1021/jm8006257. [DOI] [PubMed] [Google Scholar]
- 54.Liu DZ, Sinchaikul S, Reddy PVG, Chang MY, Chen ST. Synthesis of 2′-paclitaxel methyl 2-glucopyranosyl succinate for specific targeted delivery to cancer cells. Bioorg Med Chem Lett. 2007;17(3):617–620. doi: 10.1016/j.bmcl.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 55.Fu Y, Li S, Zu Y, Yang G, Yang Z, Luo M, Jiang S, Wink M, Efferth T. Medicinal chemistry of paclitaxel and its analogues. Curr Med Chem. 2009;16(30):3966–85. doi: 10.2174/092986709789352277. [DOI] [PubMed] [Google Scholar]
- 56.Halmos T, Santarromana M, Antonakis K, Scherman D. Synthesis of glucose-chlorambucil derivatives and their recognition by the human GLUT1 glucose transporter. Eur J Pharmacol. 1996;318(2–3):477–484. doi: 10.1016/s0014-2999(96)00796-0. [DOI] [PubMed] [Google Scholar]
- 57.Halmos T, Santarromana M, Antonakis K, Scherman D. Synthesis of O-methylsulfonyl derivatives of d-glucose as potential alkylating agents for targeted drug delivery to the brain. Evaluation of their interaction with the human erythrocyte GLUT1 hexose transporter. Carbohydr Res. 1997;299(1–2):15–21. doi: 10.1016/s0008-6215(96)00328-x. [DOI] [PubMed] [Google Scholar]
- 58.Reux B, Weber V, Galmier M-J, Borel M, Madesclaire M, Madelmont J-C, Debiton E, Coudert P. Synthesis and cytotoxic properties of new fluorodeoxyglucose-coupled chlorambucil derivatives. Bioorg Med Chem. 2008;16(9):5004–5020. doi: 10.1016/j.bmc.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 59.Miot-Noirault E, Reux B, Debiton E, Madelmont JC, Chezal JM, Coudert P, Weber V. Preclinical investigation of tolerance and antitumour activity of new fluorodeoxyglucose-coupled chlorambucil alkylating agents. Invest New Drugs. 2011;29(3):424–433. doi: 10.1007/s10637-009-9371-0. [DOI] [PubMed] [Google Scholar]
- 60.Goff RD, Thorson JS. Assessment of chemoselective neoglycosylation methods using chlorambucil as a model. J Med Chem. 2010;53(22):8129–8139. doi: 10.1021/jm101024j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reinhard J, Hull WE, von der Lieth CW, Eichhorn U, Kliem HC, Kaina B, Wiessler M. Monosaccharide-Linked Inhibitors of O6-Methylguanine-DNA Methyltransferase (MGMT): Synthesis, Molecular Modeling, and Structure–Activity Relationships. J Med Chem. 2001;44(24):4050–4061. doi: 10.1021/jm010006e. [DOI] [PubMed] [Google Scholar]
- 62.Reinhard J, Eichhorn U, Wiessler M, Kaina B. Inactivation of O6-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors. Int J Cancer. 2001;93(3):373–379. doi: 10.1002/ijc.1336. [DOI] [PubMed] [Google Scholar]
- 63.Kumar P, Shustov G, Liang H, Khlebnikov V, Zheng W, Yang XH, Cheeseman C, Wiebe LI. Design, synthesis, and preliminary biological evaluation of 6-o-glucose-azomycin adducts for diagnosis and therapy of hypoxic tumors. J Med Chem. 2012;55(13):6033–46. doi: 10.1021/jm2017336. [DOI] [PubMed] [Google Scholar]
- 64.Grigsby PW, Winter K, Wasserman TH, Marcial V, Rotman M, Cooper J, Keys H, Asbell SO, Phillips TL. Irradiation with or without misonidazole for patients with stages IIIb and IVa carcinoma of the cervix: final results of RTOG 80-05. Int J Radiat Oncol Biol Phys. 1999;44(3):513–517. doi: 10.1016/s0360-3016(99)00054-1. [DOI] [PubMed] [Google Scholar]
- 65.Oliveri V, Giuffrida ML, Vecchio G, Aiello C, Viale M. Gluconjugates of 8-hydroxyquinolines as potential anti-cancer prodrugs. Dalton Trans. 2012;41(15):4530–4535. doi: 10.1039/c2dt12371a. [DOI] [PubMed] [Google Scholar]
- 66.Schimmer AD, Jitkova Y, Gronda M, Wang Z, Brandwein J, Chen C, Gupta V, Schuh A, Yee K, Chen J, Ackloo S, Booth T, Keays S, Minden MD. A Phase I Study of the Metal Ionophore Clioquinol in Patients With Advanced Hematologic Malignancies. Clin Lymphoma Myeloma Leuk. 2012;12(5):330–336. doi: 10.1016/j.clml.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 67.Peltier-Pain P, Timmons SC, Grandemange A, Benoit E, Thorson JS. Warfarin Glycosylation Invokes a Switch from Anticoagulant to Anticancer Activity. ChemMedChem. 2011;6(8):1347–1350. doi: 10.1002/cmdc.201100178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goff RD, Thorson JS. Enhancement of cyclopamine via conjugation with nonmetabolic sugars. Org Lett. 2012;14(10):2454–2457. doi: 10.1021/ol300703z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao J, Cui S, Li S, Du C, Tian J, Wan S, Qian Z, Gu Y, Chen WR, Wang G. Targeted Cancer Therapy with a 2-Deoxyglucose-Based Adriamycin Complex. Cancer Res. 2013;73:1362–1373. doi: 10.1158/0008-5472.CAN-12-2072. [DOI] [PubMed] [Google Scholar]
- 70.Cheng H, Cao X, Xian M, Fang L, Cai TB, Ji JJ, Tunac JB, Sun D, Wang PG. Synthesis and Enzyme-Specific Activation of Carbohydrate–Geldanamycin Conjugates with Potent Anticancer Activity. J Med Chem. 2004;48(2):645–652. doi: 10.1021/jm049693a. [DOI] [PubMed] [Google Scholar]
- 71.Lee HY, Kwon JT, Koh M, Cho MH, Park SB. Enhanced efficacy of 7-hydroxy-3-methoxycadalene via glycosylation in in vivo xenograft study. Bioorg Med Chem Lett. 2007;17(22):6335–6339. doi: 10.1016/j.bmcl.2007.08.071. [DOI] [PubMed] [Google Scholar]
- 72.Tietze LF, von Hof JM, Krewer B, Muller M, Major F, Schuster HJ, Schuberth I, Alves F. Asymmetric synthesis and biological evaluation of glycosidic prodrugs for a selective cancer therapy. ChemMedChem. 2008;3(12):1946–55. doi: 10.1002/cmdc.200800250. [DOI] [PubMed] [Google Scholar]
- 73.Tietze LF, Major F, Schuberth I. Antitumor agents: Development of highly potent glycosidic duocarmycin analogues for selective cancer therapy. Angew Chem Int Ed Engl. 2006;45(39):6574–6577. doi: 10.1002/anie.200600936. [DOI] [PubMed] [Google Scholar]
- 74.Hwu JR, Hsu CI, Hsu MH, Liang YC, Huang RCC, Lee YC. Glycosylated nordihydroguaiaretic acids as anti-cancer agents. Bioorg Med Chem Lett. 2011;21(1):380–382. doi: 10.1016/j.bmcl.2010.10.137. [DOI] [PubMed] [Google Scholar]
- 75.Kobayashi M, Kaida H, Kawahara A, Hattori S, Kurata S, Hayakawa M, Hirose Y, Uchida M, Kage M, Fujita H, Hayabuchi N, Ishibashi M. The relationship between GLUT-1 and vascular endothelial growth factor expression and 18F-FDG uptake in esophageal squamous cell cancer patients. Clin Nucl Med. 2012;37(5):447–52. doi: 10.1097/RLU.0b013e31823924bb. [DOI] [PubMed] [Google Scholar]
- 76.Hussein YR, Bandyopadhyay S, Semaan A, Ahmed Q, Albashiti B, Jazaerly T, Nahleh Z, Ali-Fehmi R. Glut-1 Expression Correlates with Basal-like Breast Cancer. Transl Oncol. 2011;4(6):321–7. doi: 10.1593/tlo.11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brophy S, Sheehan KM, McNamara DA, Deasy J, Bouchier-Hayes DJ, Kay EW. GLUT-1 expression and response to chemoradiotherapy in rectal cancer. Int J Cancer. 2009;125(12):2778–82. doi: 10.1002/ijc.24693. [DOI] [PubMed] [Google Scholar]
- 78.Kunkel M, Reichert TE, Benz P, Lehr HA, Jeong JH, Wieand S, Bartenstein P, Wagner W, Whiteside TL. Overexpression of Glut-1 and increased glucose metabolism in tumors are associated with a poor prognosis in patients with oral squamous cell carcinoma. Cancer. 2003;97(4):1015–24. doi: 10.1002/cncr.11159. [DOI] [PubMed] [Google Scholar]
- 79.Barnett JE, Holman GD, Munday KA. Structural requirements for binding to the sugar-transport system of the human erythrocyte. Biochem J. 1973;131(2):211–21. doi: 10.1042/bj1310211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mueckler M, Makepeace C. Model of the exofacial substrate-binding site and helical folding of the human Glut1 glucose transporter based on scanning mutagenesis. Biochemistry. 2009;48(25):5934–42. doi: 10.1021/bi900521n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patt M, Sorger D, Scheunemann M, Stocklin G. Adduct of 2-[18F]FDG and 2-nitroimidazole as a putative radiotracer for the detection of hypoxia with PET: synthesis, in vitro- and in vivo-characterization. Appl Radiat Isot. 2002;57(5):705–12. doi: 10.1016/s0969-8043(02)00186-0. [DOI] [PubMed] [Google Scholar]
- 82.Marom EM, Aloia TA, Moore MB, Hara M, Herndon JE, 2nd, Harpole DH, Jr, Goodman PC, Patz EF., Jr Correlation of FDG-PET imaging with Glut-1 and Glut-3 expression in early-stage non-small cell lung cancer. Lung Cancer. 2001;33(2–3):99–107. doi: 10.1016/s0169-5002(00)00250-6. [DOI] [PubMed] [Google Scholar]
- 83.Yamada K, Nakata M, Horimoto N, Saito M, Matsuoka H, Inagaki N. Measurement of glucose uptake and intracellular calcium concentration in single, living pancreatic beta-cells. J Biol Chem. 2000;275(29):22278–83. doi: 10.1074/jbc.M908048199. [DOI] [PubMed] [Google Scholar]
- 84.Yoshioka K, Takahashi H, Homma T, Saito M, Oh KB, Nemoto Y, Matsuoka H. A novel fluorescent derivative of glucose applicable to the assessment of glucose uptake activity of Escherichia coli. Biochim Biophys Acta. 1996;1289(1):5–9. doi: 10.1016/0304-4165(95)00153-0. [DOI] [PubMed] [Google Scholar]
- 85.LeFevre PG. Sugar transport in the red blood cell: structure-activity relationships in substrates and antagonists. Pharmacol Rev. 1961;13(1):39–70. [PubMed] [Google Scholar]
- 86.Melisi D, Curcio A, Luongo E, Morelli E, Rimoli MG. D-Galactose as a vector for prodrug design. Curr Topics Med Chem. 2011;11(18):2288–2298. doi: 10.2174/156802611797183258. [DOI] [PubMed] [Google Scholar]
- 87.Devalapally H, Rajan KS, Akkinepally RR, Devarakonda RK. Safety, pharmacokinetics and biodistribution studies of a beta-galactoside prodrug of doxorubicin for improvement of tumor selective chemotherapy. Drug Dev Ind Pharm. 2008;34(8):789–95. doi: 10.1080/03639040701744202. [DOI] [PubMed] [Google Scholar]
- 88.Barros LF. Measurement of sugar transport in single living cells. Pflugers Arch. 1999;437(5):763–70. doi: 10.1007/s004240050843. [DOI] [PubMed] [Google Scholar]
- 89.Axelrod JD, Pilch PF. Unique cytochalasin B binding characteristics of the hepatic glucose carrier. Biochemistry. 1983;22(9):2222–2227. doi: 10.1021/bi00278a025. [DOI] [PubMed] [Google Scholar]
- 90.Griffin JF, Rampal AL, Jung CY. Inhibition of glucose transport in human erythrocytes by cytochalasins: A model based on diffraction studies. Proc Natl Acad Sci U S A. 1982;79(12):3759–63. doi: 10.1073/pnas.79.12.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, Klip A, Batey RA, Schimmer AD. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol Cancer Ther. 2008;7(11):3546–3555. doi: 10.1158/1535-7163.MCT-08-0569. [DOI] [PubMed] [Google Scholar]
- 92.Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, Hines J, Chen X. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In Vivo. Mol Cancer Ther. 2012;11(8):1672–1682. doi: 10.1158/1535-7163.MCT-12-0131. [DOI] [PubMed] [Google Scholar]
- 93.Theodoropoulos PA, Gravanis A, Tsapara A, Margioris AN, Papadogiorgaki E, Galanopoulos V, Stournaras C. Cytochalasin B may shorten actin filaments by a mechanism independent of barbed end capping. Biochem Pharmacol. 1994;47(10):1875–1881. doi: 10.1016/0006-2952(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 94.Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, Ilkun O, Pereira R, Abel ED, Anderson SM. Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PLoS One. 2011;6(8):e23205. doi: 10.1371/journal.pone.0023205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective Glucose Transport across the Blood-Brain Barrier as a Cause of Persistent Hypoglycorrhachia, Seizures, and Developmental Delay. New Engl J Med. 1991;325(10):703–709. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- 96.Kim WH, Lee J, Jung DW, Williams DR. Visualizing sweetness: increasingly diverse applications for fluorescent-tagged glucose bioprobes and their recent structural modifications. Sensors. 2012;12(4):5005–27. doi: 10.3390/s120405005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee HY, Lee JJ, Park J, Park SB. Development of fluorescent glucose bioprobes and their application on real-time and quantitative monitoring of glucose uptake in living cells. Chem Eur J. 2011;17(1):143–50. doi: 10.1002/chem.201002560. [DOI] [PubMed] [Google Scholar]
- 98.Park J, Lee HY, Cho MH, Park SB. Development of a cy3-labeled glucose bioprobe and its application in bioimaging and screening for anticancer agents. Angew Chem Int Ed Engl. 2007;46(12):2018–22. doi: 10.1002/anie.200604364. [DOI] [PubMed] [Google Scholar]
- 99.Zhou H, Luby-Phelps K, Mickey BE, Habib AA, Mason RP, Zhao D. Dynamic near-infrared optical imaging of 2-deoxyglucose uptake by intracranial glioma of athymic mice. PLoS One. 2009;4(11):e8051. doi: 10.1371/journal.pone.0008051. [DOI] [PMC free article] [PubMed] [Google Scholar]