

Abstract
Purpose
To characterize, directly and for the first time, the membrane transport and metabolism of pralatrexate, a new-generation dihydrofolate reductase inhibitor approved for the treatment of peripheral T-cell lymphoma.
Experimental Design
[3H]pralatrexate transport was studied in unique HeLa cell lines that express either the reduced folate carrier (RFC) or the proton-coupled folate transporter (PCFT). Metabolism to active polyglutamate derivatives was assessed by liquid chromatography. These properties were compared to those of methotrexate (MTX).
Results
The pralatrexate influx Kt, mediated by RFC, the major route of folate/antifolate transport at systemic pH, was 0.52 , 1/10th the MTX influx Ki. The electrochemical-potential of pralatrexate within HeLa cells far exceeded the extracellular level and was greater than for MTX. In contrast, MTX transport mediated by PCFT, the mechanism of folate/antifolate absorption in the small intestine, exceeded that for pralatrexate. After a 6h exposure of HeLa cells to 0.5 μM pralatrexate, 80% of intracellular drug was its active polyglutamate forms, predominantly the tetraglutamate, and was suppressed when cells were loaded with natural folates. There was negligible formation of MTX polyglutamates. The difference in pralatrexate and MTX growth inhibition was far greater after transient exposures (375-fold) than continuous exposure (25-fold) to the drugs.
Conclusion
Pralatrexate’s enhanced activity relative to MTX is due to its much more rapid rate of transport and polyglutamation, the former less important when the carrier is saturated. The low affinity of pralatrexate for PCFT predicts a lower level of enterohepatic circulation, and increased fecal excretion of the drug relative to MTX.
Keywords: Methotrexate, PCFT, polyglutamation, pralatrexate, RFC
INTRODUCTION
The antifolates were the first class of antimetabolites to enter the clinics with the introduction of aminopterin in 1947 and, shortly thereafter, methotrexate (MTX) [1,2]. An understanding of the mechanism of action of these agents evolved over the following decades, first with the identification of their target enzyme, dihydrofolate reductase (DHFR) [3], then elucidation of the competitive nature of the interaction of drug with this target within cells and clarification of the role of membrane transport mediated by the reduced folate carrier (RFC) as a determinant of cytotoxicity and drug resistance [4,5]. The metabolism of these agents to their polyglutamate forms was identified in 1973 [6] but their pharmacological importance clarified in the 1980s. These congeners are biologically active and are retained and accumulate to high levels in tumor cells to produce sustained suppression of DHFR [7]. This biochemical transformation was then shown to play a critical role in MTX selectivity occurring to a much lesser extent in intestinal and bone marrow progenitor cells than in tumor cells [5,7-10]. Finally, the polyglutamate derivatives of MTX were found to be direct inhibitors of thymidylate synthase and AICAR transformylase [11,12]. This provided an explanation, at the biochemical and cellular levels, for the selectivity of leucovorin “rescue”: Utilization of tetrahydrofolate cofactors generated from leucovorin is impaired in tumors cells that contain the MTX polyglutamate forms while utilization is not impaired, or is impaired to a lesser extent, in normal cells that generate much lower levels of these derivatives [5,13,14].
Recognition of the critical role of polyglutamation in drug action resulted in a search for antifolate analogs with enhancement of this property. This led to the identification and clinical evaluation of a number of antifolates with a spectrum of target enzyme sites ultimately yielding pemetrexed which, in its polyglutamate forms, is a direct inhibitor of thymidylate synthase and AICAR transformylase [15,16]. The search for more effective inhibitors of DHFR with both enhanced polyglutamation and transport was pursued methodically by Sirotnak and his collaborators, first with the identification of the 10-deaza- analog of aminopterin, then the 10-deaza-ethyl analog (edatrexate) and finally the 10-deaza propargyl derivative, pralatrexate [17-19]. This drug was reported to be a much better substrate than MTX for both RFC and folylpolyglutamate synthetase (FPGS) [18]. Pralatrexate was approved in 2009 for the treatment of peripheral T-cell lymphoma, the second antifolate, after pemetrexed in 2004, to be introduced in the United States for the treatment of cancer in 65 years [20,21]. We report on the first direct analysis of the cellular and biochemical pharmacology of pralatrexate using a tritiated derivative. The studies utilize unique HeLa cell lines developed in this laboratory that allow analysis of membrane transport mediated by the two major facilitative folate transporters, RFC and the proton coupled folate transporter (PCFT) [22,23], along with assessment of the metabolism of this drug to its polyglutamate derivatives, all within the context of a comparison of these properties for MTX.
MATERIALS AND METHODS
Reagents
Tritiated pralatrexate, pemetrexed (both generally labeled), and [3′,5′,7-3H]MTX were obtained from Moravek Biochemicals (Brea, CA). Pemetrexed and MTX purity was established and monitored by high-performance liquid chromatography (HPLC) as described previously [24]. Pralatrexate purity was monitored using a 5 mm OSD2 4.6 × 250 mm reversed phase high-performance liquid chromatography column (Waters Spherisorb), by isocratic elution with 100 mM sodium acetate pH 5.5 (solvent A) and 15% acetonitrile (solvent B). The mobile phase was delivered at 1 ml/min, reaching 100% solvent B in 30 min. Nonlabeled MTX was obtained from Sigma-Aldrich (St. Louis, MO), nonlabeled (6S)5-formyltetrahydrofolate (5-formylTHF) from Schircks Laboratories (Jona, Switzerland) and nonlabeled pemetrexed from LC Laboratories (Woburn, MA). Pralatrexate was obtained from Spectrum Pharmaceuticals (Irvine, CA).
Cell lines
HeLa cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (Gemini Bio-Products, Irvine, CA), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2. HeLa R1-PCFT cells, transfected to express high levels of PCFT, were derived from HeLa R1–11 cells (which lack endogenous expression of RFC and PCFT) and were maintained in 300 μg/ml hygromycin B (Calbiochem, San Diego, CA) as reported previously [25,26,27,28]. HeLa R1-PCFT4, lacking RFC but transfected to express PCFT at a level similar to that of wild-type HeLa cells, were derived from HeLa R1-11 cells and maintained in 100 μg/ml phleomycin (Zeocin; Invitrogen, Carlsbad, CA), as reported previously [26]. HeLa-RFC7 cells were derived from HeLa-R5 cells and express constitutive levels of PCFT and very high levels of RFC [29]. For growth inhibition studies, cells were maintained in folate-free RPMI 1640 medium supplemented with 10% dialyzed fetal bovine serum (Gibco, Grand Island, NY) and 25 nM 5-formylTHF as the folate source.
Transport measurements
Transport of tritiated substrates was measured in cells grown in monolayer culture at the bottom of glass vials for 3 days [30]. The medium was aspirated and cells washed twice in HBS buffer (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM dextrose; adjusted with 1 N NaOH to achieve a pH of 7.4). Then, 1 ml of HBS buffer was added, and the vials incubated in a 37°C water bath for 20 min. Buffer was then aspirated and the radiolabeled reagents added. Uptake at pH 7.4 was performed in HBS buffer; uptake at pH 6.0 utilized MBS buffer (20 mM MES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM dextrose, adjusted with 1 N HCl to achieve pH 6.0). Uptake was stopped by injection of 10 volumes of ice-cold HBS buffer at pH 7.4 after which the cells were washed three times in this buffer and then digested with 0.2 N NaOH at 65°C for 45 min. A portion (400 μl) was assessed for tritium on a liquid scintillation spectrometer or protein (10 μl) by the BCA assay (Thermo Fisher Scientific, Waltham, MA). Cellular antifolate is expressed as picomoles per milligram of protein.
Influx kinetics was determined by a nonlinear regression analysis of the relationship between pralatrexate influx as a function of the extracellular pralatrexate concentration based on the Michaelis-Menten equation using Prism software (version 5.0 for Windows; GraphPad Software Inc., San Diego, CA). To assess the level of free intracellular drug, cells were incubated with tritiated MTX or pralatrexate for 60 min following which one portion of cells was taken for measurement of intracellular drug. The other portion of cells was washed three times with 0°C HBS, then resuspended into drug-free buffer at 37°C, and intracellular drug measured until a constant level was achieved that represented the component bound to DHFR [31]. The difference between the initial intracellular drug level and the bound fraction was considered to be the level of free intracellular drug. The concentration of drug in the intracellular water (μM) was based upon the ratio of the intracellular water (μL) to protein (mg) determined to be 6 [28].
HPLC analysis of [3H]pralatrexate and its metabolites
HeLa cells were incubated in RPMI medium containing 0.5 M [3H]pralatrexate for 6h then washed twice in ice-cold HBS. One portion of cells was taken for assessment of total tritium and protein. The other portion was extracted with 1 ml of 10% TCA for 10 min at 0°C then neutralized by the addition of 0.2 ml of 1M NaH2PO4 and 0.1 ml of 6M NaOH. Liquid chromatographic analysis was performed on a reversed phase high-performance liquid chromatography column (Waters Spherisorb, 5 mm ODS2 4.6 3 250 mm-see above). Separation of the different polyglutamate forms was achieved by elution with 0.1 M sodium acetate, pH 5.5, for 5 min followed by two linear gradients of 0–30 and 30–50% acetonitrile in 0.1M sodium acetate over 35 and 20 min, respectively, and then 100% acetonitrile for 11 min. The flow rate was 1 ml/min; 1 ml fractions were collected in 13-ml vials and radioactivity was measured on a liquid scintillation spectrometer. While authentic standards of pralatexate polyglutamates are not available, the different peaks could be clearly delineated allowing discrimination of the various polyglutamate forms.
Growth inhibition assay
HeLa cells were seeded in 96-well plates at a density of 2 × 103 cells/well. The following day, pralatrexate or MTX was added to achieve a broad spectrum of concentrations for 6h followed by growth in drug-free medium for 6 days, or cells were exposed to drug continuously for 6 days. Cells were then assayed by sulforhodamine B staining. Absorbance was measured at 540 nm with the VERSAmax plate reader (GE Intelligent Platforms, Charlottesville, VA).
RESULTS
A comparison of the initial uptake rates of [3H]pralatrexate and [3H]MTX in HeLa cells
Figure 1A illustrates the time course of drug uptake at an extracellular concentration of 0.05 M at pH 7.4 in HeLa cells that express constitutive levels of RFC and PCFT. The increase in intracellular pralatrexate and MTX as a function of time was constant and the uptake slopes extrapolated close to the point of origin. Hence, uptake over this interval was unidirectional, and represented the initial rate (influx) of pralatrexate and MTX transport into the cells. Influx of pralatrexate was ~10 fold greater than that of MTX (0.62 ± 0.05 vs 0.06 ± 0.01 pmol · mg protein−1 min−1, respectively). To explore the relationship between pralatrexate and MTX transport over a broader concentration range, influx was assessed at an extracellular drug concentration 10-fold higher, 0.5 M, over a much shorter interval to capture initial rates (Figure 1B). Under these conditions unidirectional transport was sustained but pralatrexate influx exceeded that for MTX by a factor of only 2.5 (0.022 ± 0.001 vs 0.009 ± 0.001 pmol · mg protein−1 · min−1, respectively).
Fig. 1. Initial rates and kinetic analysis of [3H]pralatrexate and [3H[MTX uptake.

The time-course of drug uptake over 15 min at an extracellular concentration of 0.05 M (Panel a) or over 80 sec at an extracellular concentration of 0.5 M (Panel b) at pH 7.4. The data are the mean ± SEM from three independent experiments. Panel c: [3H]pralatrexate influx as a function of substrate concentration assessed over 1 min at pH 7.4 in presence or absence of 5 M non-labeled MTX. The lines are best-fit to the Michaelis-Menten equation (V = Vmax[S]/(Kt + [S])). Panel d: A double-reciprocal plot of the data (Lineweaver-Burk). Results are the mean ± SEM from four independent experiments.
Pralatrexate influx kinetics
Preliminary studies established that pralatrexate influx was unidirectional over at least 80 sec over a concentration range of 0.05 μM to 4 μM (data not shown). Influx kinetics was subsequently determined over 60 sec at pH 7.4 using [3H]pralatrexate concentrations within this range in the presence or absence of 5 μM non-labeled MTX (Figure 1, panels C, D). Based upon a nonlinear regression analysis and Michaelis-Menten kinetics, the pralatrexate influx Kt (concentration at which influx was one-half of the maximum rate) was computed to be 0.52 ± 0.2 μM, and maximum influx (Vmax) was 5.27 ± 0.6 pmol · mg protein−1 · min−1. The MTX influx Ki (an affinity constant based upon the inhibition of pralatrexate influx) was ~4.4 M, 8 fold greater than the pralatrexate influx Kt, consistent with a much higher affinity of pralatrexate for RFC relative to MTX and the large difference in initial uptake rates at a concentration of 0.05 M, one order magnitude below the pralatrexate influx Kt.
An analysis of antifolate transport mediated by PCFT
Pralatrexate was designed to have a high affinity for RFC [18]. At the time of its development the existence of PCFT, a transporter with optimal activity at low pH, was not known [23]. To assess the extent to which pralatrexate is a substrate for this carrier, transport was assessed in HeLa R1-PCFT cells that express very high levels of PCFT but lack expression of RFC. The high level of PCFT expression allows accurate transport measurements at neutral pH Figure 2 illustrates influx of tritiated antifolates at either pH 6.0 (A) or pH 7.4 (B) in these cells and in HeLa R1-11 cells that lack both transporters. It can be seen that there was essentially no uptake detected in cells that lack RFC and PCFT excluding transport of pralatrexate, or the other antifolates tested, mediated by other facilitative processes or passive diffusion over this interval. At pH 6.0, where the transporter function is near optimal, MTX influx was half that of pemetrexed (an antifolate with high affinity for PCFT) [32,26] and pralatrexate influx was one-third that of MTX. At pH 7.4 MTX influx mediated by PCFT was ~ 1/10th the rate for pemetrexed and pralatrexate influx half the rate of MTX. Hence, among the antifolates tested, pralatrexate was the poorest substrate for PCFT so that when transport is measured at physiological pH in HeLa cells that express both RFC and PCFT, RFC is the dominant transporter; PCFT does not contribute significantly to the transport of pralatrexate.
Fig. 2. Antifolate transport mediated by PCFT.

Influx of tritiated pralatrexate, MTX and pemetrexed at an extracellular concentration of 0.5 M in HeLa R1-PCFT cells that express only PCFT or HeLa R1-11 cells that lack both RFC and PCFT. The transport rates were assessed either at pH 6 (Panel A) or pH 7.4 (Panel B). Results are the mean ± SEM from three independent experiments.
An analysis of the net cellular uptake of pralatrexate, MTX and pemetrexed: impact of cellular folates
Figure 3 compares the time-course of uptake of [3H]pralatrexate, [3H]MTX and [3H]pemetrexed over 6h at an extracellular concentration of 0.5 M in HeLa cells previously grown in RPMI medium containing 2.0 folic acid (A) or in cells grown in RPMI supplemented with 100 M folic acid (B), a condition that markedly augments the intracellular folate pool [33]. It can be seen that loading cells with folates did not affect net [3H]MTX uptake, but markedly decreased uptake of [3H]pralatexate and [3H]pemetrexed. Indeed, in cells grown in 2.0 folic acid the net uptake of [3H]pralatrexate and [3H]pemetrexed exceeded that of [3H]MTX by 15 fold, at 6h. However, when cells were grown in 100 M folic acid, [3H]pralatrexate and [3H]pemetrexed accumulation was markedly suppressed, although still higher than that of [3H]MTX. These observations are consistent with the rapid formation of pralatrexate and pemetrexed polyglutamates retained within the cells, already demonstrated for the latter, a process suppressed by the presence of high levels of folates within the cells [24,33].
Fig. 3. An analysis of pralatrexate accumulation and metabolism in HeLa cells.

HeLa cells were grown in RPMI with the usual folic acid concentration of 2.0 μM (Panel A) or with 100 M folic acid (Panel B) for three days. Following this, the time-course of uptake of 0.5μM [3H]MTX, [3H]pralatrexate and [3H]pemetrexed was monitored in RPMI (2.0 μM folic acid) . The data are the mean ± SEM from three independent experiments. Panel C: HPLC analysis of intracellular tritiated constituents after exposure of cells grown in RPMI medium to [3H]pralatrexate with the usual folic acid concentration of 2 μM or, Panel D, with 100 μM folic acid. The extracellular [3H]pralatrexate concentration was 0.5 M; incubation time was 6h. The data is representative of three independent experiments.
This was confirmed by an analysis of the intracellular pralatrexate derivatives that accumulate under these conditions as illustrated in the lower two panels of Figure 3. HeLa cells grown in RPMI (C) or RPMI supplemented with 100 μM folic acid (D) for 3 days were exposed to 0.5 μM [3H]pralatrexate for 6h following which the cells were processed for HPLC. Under usual growth conditions with 2.0 μM folic acid, five peaks were identified, four preceeding the monoglutamate peak confirmed by a nonlabeled standard. Pralatrexate polyglutamates were not available to confirm the identity of the other peaks. However, they differentiated clearly into four peaks assumed to elute according to their molecular size, the largest of which likely represents the tetraglutamate. Based upon this analysis, the mono-, di- tri-, tetra, and penta- glutamates represented 21%, 15%, 12%, 50% and 2% of the total radioactivity on the column. When cells were grown in RPMI supplemented with 100 μM folic acid, all the peaks but the one corresponding to the monoglutamate were markedly suppressed. The suppression was not complete, consistent with the cellular uptake pattern in Figure 3, panels A and B.
A comparison of the free MTX and pralatrexate levels in HeLa cells
RFC is the dominant and, for all practical purposes, the sole route of pralatrexate transport into HeLa cells at pH 7.4. The free substrate levels achieved are determined by the net impact of RFC, an organic anion antiporter, opposed by the ATP binding cassette exporters that contribute to the efflux of antifolates (largely the monoglutamates) out of cells. To determine the free antifolate levels, cells grown with 100 μM folic acid to suppress polyglutamation were exposed to 0.05 μM [3H]MTX and [3H]pralatrexate over one hour when steady-state levels of free drugs were achieved. The total cellular drug level was measured in one portion of the cells, another portion was washed and resuspended into a large volume of drug-free medium to determine the level retained within the cells, bound to DHFR. The difference was considered to be the free intracellular drug level. In cells brought to steady-state with 0.05 μM antifolates the level of MTX and pralatrexate bound within the cells was comparable. The level of free intracellular pralatrexate (0.17 μM) exceeded the level of free MTX (0.05 μM) by a factor of 3.4 (p=0.04). It can be seen that free intracellular pralatrexate also exceeded the extracellular level by a factor of 3.4, consistent with an inward chemical gradient. There was no chemical gradient for MTX (Table 1).
Table 1. Comparison of the free intracellular pralatrexate and MTX levels.
Tritiated drug levels were assessed at extracellular concentrations of 0.05 and 0.5 M in HeLa cells grown in medium supplemented with 100 M folic acid to minimize formation of polyglutamate derivatives. At one hour total antifolate levels were measured following which the cells were washed in 0°C buffer, resuspended into antifolate-free HBS at 37°C, and the intracellular drug levels monitored. The difference between the total and DHFR-bound levels is considered the free intracellular drug concentrations. The gradient is the ratio between the extracellular and intracellular free drug concentration. Data are the mean ± SEM from four independent experiments.
| Extracellular concentration | 0.05 M | 0.5 M | ||
|---|---|---|---|---|
| pralatrexate | MTX | pralatrexate | MTX | |
| Total drug (μM) | 0.49 ± 0.07 | 0.39 ± 0.04 | 1.25 ± 0.10 | 1.04 ± 0.08 |
| DHFR-bound (μM) | 0.31 ± 0.05 | 0.33 ± 0.04 | 0.44 ± 0.02 | 0.59 ± 0.06 |
| Free intracellular drug (μM) | 0.17 ± 0.08 | 0.05 ± 0.04* | 0.81 ± 0.12 | 0.45 ± 0.10** |
| Free intracellular/extracellular drug | 3.4 | 1 | 1.6 | 0.9 |
P-value of 0.04,
P-value of 0.01, for the difference between pralatrexate and MTX.
When cells were brought to steady-state with 0.5 μM of the antifolates, the free pralatrexate level exceeded that for MTX by less than two and the chemical gradient for pralatrexate was half that observed at the lower extracellular concentration. Hence, the 10-fold higher unidirectional flux of pralatrexate relative to MTX at 0.05 μM translates into a ~3-fold difference in the free drug levels. At the higher concentration, a 2.5 difference in influx translated into a < 2 fold difference in free drug levels (Table 1). Hence, as the extracellular drug concentration increases and pralatrexate influx saturates, the differences in influx and free drug levels as compared to MTX decreases.
A comparison of pralatrexate and MTX growth inhibition in HeLa cells: continuous versus transient exposure
The pharmacological impact of pralatrexate and MTX was first evaluated by comparing the IC50 (concentration of drug that suppresses growth to 50% the rate in the absence of drug) in cells continuously exposed to the drugs. As indicated in in Figure 4A, the pralatrexate IC50 (7 nM) was ~ 1/25th that for MTX (180 nM). The difference in growth inhibition was much greater when exposure to the drugs was transient. As illustrated in Figure 4B, when cells were exposed to drug for six hours followed by growth in the absence of drug for six days, the pralatrexate IC50 increased ~ 10 fold to 80 nM but the MTX IC50 increased 170-fold to 30 M. Hence, with a six-hour exposure the IC50 for pralatrexate was ~ 1/375th that of MTX.
Fig. 4. A comparison of pralatrexate and methotrexate growth inhibition in HeLa cells.

HeLa cells were grown for 6 days continuously exposed to drug (Panel A) or pulsed for 6h with drug (Panel B) at the indicated concentrations. Growth in the absence of drug is indicated as 100%. Data are the mean ± SEM from three independent experiments. The vertical line intercepts the x axis at the concentration at which growth inhibition is 50% of the level of growth in the absence of drug (IC50).
Impact of a high level, or lack, of RFC expression on pralatrexate transport and growth inhibition
Figure 5 illustrates that RFC-mediated transport of pralatrexate is clearly a prerequisite for cytotoxic activity. HeLa-R1-PCFT4 cells, with constitutive levels of PCFT expression but a lack of RFC expression, were > 500 times less sensitive to pralatrexate compared with the wild-type cells (IC50 in HeLa cells and HeLa-R1-PCFT4 of 40 nM vs >20 M, respectively). This is also consistent with the very low affinity of pralatrexate for PCFT. However, in HeLa-RFC7 cells that express high levels of RFC (RFC mRNA is markedly increased [29], and influx is increased six-fold - 0.043 ± 0.008 vs 0.007 ± 0.001 pmol · mg protein−1 · min−1- data not shown), there was no significant increase in growth inhibitory activity as compared to wild-type HeLa cells (IC50 in HeLa cells and HeLa-RFC7 of 30 vs 40 nM - Figure 5).
Fig. 5. Impact of RFC expression on pralatrexate influx and growth inhibition.
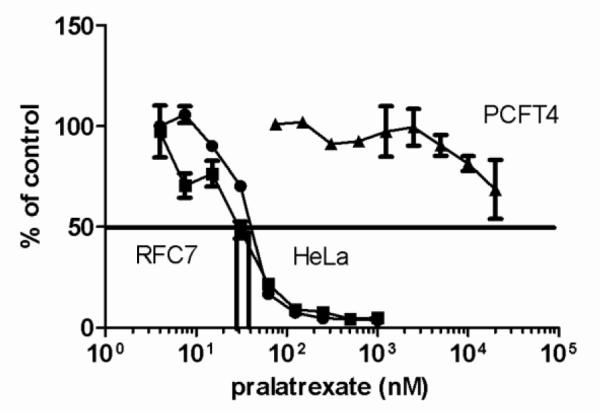
HeLa cells, HeLa-RFC7, that overexpress RFC, and HeLa-R1-PCFT4 that express constitutive levels of PCFT but do not express RFC, were exposed to pralatrexate for 6h at the indicated concentrations then grown for an additional 6 days in the absence of drug. Growth in the absence of pralatrexate is indicated as 100%. The vertical line intercepts the x axis at the concentration at which growth inhibition is 50% of the level of growth in the absence of drug (IC50). Data are the mean ± S.E.M from three independent experiments.
DISCUSSION
Pralatrexate is a new-generation DHFR inhibitor approved for the treatment of advanced-stage peripheral T-cell lymphoma [20,21] and is being evaluated for efficacy in other cancers. Pralatrexate was designed to have a high affinity for RFC [18], the main route of folate and antifolate transport into systemic tissues and tumor cells [34,22]. The drug also has a high affinity for FPGS, the enzyme that mediates the formation of folate and antifolate polyglutamate derivatives [18]. The current study represents the first direct characterization of the membrane transport and polyglutamation of tritiated pralatrexate in HeLa cell lines. The data confirms a much higher affinity for RFC ~10 fold greater than that for MTX, with a Kt somewhat higher than previously reported [18]. The current study also documents, for the first time, the very rapid rate of formation and accumulation of pralatrexate polyglutamate derivatives in cells comparable to what was observed for pemetrexed, another excellent substrate for FPGS [24]. While pralatrexate polyglutamate standards are not available, the chromatographic pattern of intracellular constituents, and the suppression of their formation by loading cells with folates, is consistent with the formation of polyglutamates of up to the pentaglutamate with the tetraglutamate the predominant form similar to what has been observed for pemetrexed [24].
Consistent with these superior pharmacological properties is pralatrexate’s greater potency relative to MTX. Hence, with continuous exposure to these drugs in vitro, pralatrexate was 25 times more potent in inhibiting the growth of HeLa cells than MTX. That difference was amplified 15-fold when exposure to the drugs was brief, exploiting the rapid rate of polyglutamation of pralatrexate relative to MTX and the intracellular accumulation of these derivatives that results in sustained inhibition of DHFR in the absence of extracellular drug [5]. Under the latter conditions growth inhibition by pralatrexate exceeded that for MTX by a factor of 375. What is intriguing about pralatrexate is its relative lack of toxicity in its clinical application despite its much greater potency relative to MTX. Pralatrexate can be administered weekly at doses (30 mg/m2) comparable to MTX (40 mg/m2) with only moderate mucositis which can be diminished, but not eliminated, by supplementation with low-dose folic acid [21].
A comparison between the pharmacological properties of pralatrexate and aminopterin is also informative. Aminopterin has a four-fold higher affinity for RFC than MTX and its catalytic activity mediated by FPGS is comparable to that of pralatrexate [35,18]. Yet, the MTD for aminopterin is 2 mg q-12 hrs for two doses, weekly, with leucovorin rescue [36]. Likewise, in mice, pralatrexate can be administered at doses greater than either MTX or aminopterin with substantially greater efficacy and a limited toxicity [37,18,38]. Hence, in mice there is a much superior therapeutic index for pralatrexate with an apparent lesser toxicity to bone marrow and intestinal cells. The basis for this is not clear; for MTX, at least, the accumulation of polyglutamate derivatives in intestinal cells and bone marrow progenitor cells is far less than in tumor cells [8-10]. It is possible that there may be even lesser formation of polyglutamate derivatives of pralatrexate in these cells. Pralatexate’s high therapeutic index relative to the other DHFR inhibitors is a phenomenon that certainly warrants further clarification.
The role that pralatrexate’s enhanced transport properties plays, relative to its enhanced polyglutamation, in its increased activity relative to MTX is not entirely clear. First, pralatrexate’s transport in terms of unidirectional transport into cells does not translate into a comparable increase in the free intracellular level which is the substrate for FPGS. This is the case because (i) RFC is a bidirectional transporter so that when influx increases, efflux increases as well although not in proportion. This was illustrated dramatically for MTX in that when RFC was transfected to high levels in L1210 leukemia cells, while there was a 10-fold increase in influx, efflux increased 5-fold and the steady-state free level only doubled [39]. (ii) A variety of ATP binding cassette exporters contribute to the efflux of antifolates and dissipate, in part, gradients achieved by RFC, an organic phosphate antiporter [22]. At the lowest level studied (50 nM), the free intracellular pralatrexate level exceeded the extracellular concentration by a factor of ~3. When the membrane potential of HeLa cells is considered, 40 mV at the low end of the measured spectrum [40,41], the expected intracellular to extracellular ratio for a bivalent anion if transport was passive is 0.05 based on the Nernst equation. In fact, the ratio of the concentration of pralatrexate in the intracellular to extracellular water exceeds the extracellular level. Hence, there is an enormous electrochemical-potential difference for this agent across the cell membrane consistent with RFC-mediated uphill transport.
Initially, after pralatrexate is administered intravenously, the blood level is high and the transporter is saturated (pralatrexate blood levels > 5 M) for at least 2-3h. By 12h the drug level has decreased to 0.1 M and by 24h to 0.05 M [42]. Hence, the enhanced pralatrexate transport properties relative to MTX will manifest largely long after the drug is administered when the blood level has fallen below the influx Kt. It is during that time that there may be continued synthesis of polyglutamates that expand and/or sustain the pralatrexate polyglutamate pool as these congeners are hydrolyzed to the monoglutamate which is free to leave the cells. Increasing expression of RFC beyond constitutive levels in most cells will minimally impact on activity, as was observed here for pralatrexate and reported earlier for MTX [39]. However, as RFC expression is decreased, influx will ultimately slow to a point in which transport and the free intracellular level becomes rate-limiting to the formation of polyglutamate derivatives and the inactivation of DHFR resulting in impaired drug action.
The other major folate transporter, PCFT, is the mechanism by which folates and antifolates are transported across the apical brush-border membrane of the proximal small intestine and across the basolateral membrane of choroid plexus ependymal cells [23,34,22]. Hence, the competency of this transporter and its affinity for its various substrates will determine the extent to which antifolates are re-absorbed during their enterohepatic circulation. The very low affinity of pralatexate for PCFT should accelerate its clearance from the blood and increase its fecal excretion relative to MTX. Consistent with a hepatic role in the excretion of MTX is the increased renal excretion that occurs with ligation of the bile duct in mice [43] and the decreased MTX clearance and increased toxicity associated with genetic variants of the liver-specific organic anion transporting polypeptide (OATP1B1) in children with acute lymphoblastic leukemia [44,45]. Any factors that accelerate the rate of pralatrexate clearance should decrease its toxicity to normal tissues while its antitumor activity is sustained by the polyglutamate derivatives that have been generated and retained in tumor cells. The low affinity of pralatrexate for PCFT is also indicated in these studies by the resistance to this agent in cells that express only constitutive levels of PCFT as compared to HeLa cells that express constitutive levels of both PCFT and RFC.
Acknowledgement
This study was supported by Spectrum Pharmaceutical (Irvine, CA) and the National Institutes of Health National Cancer Institute [Grant CA82621].
Abbreviations
- 5-formylTHF
(6S)5-formyltetrahydrofolate
- AICAR transformylase
phosphoribosylaminoimidazolecarboxamide formyltransferase
- DHFR
dihydrofolate reductase
- FPGS
folylpolyglutamate synthetase
- MTX
methotrexate
- PCFT
proton coupled folate transporter
- RFC
reduced folate carrier
Footnotes
Conflict of Interest No conflict to disclose.
Reference List
- 1.Bertino JR. Ode to methotrexate. J Clin Oncol. 1993;11:5–14. doi: 10.1200/JCO.1993.11.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Farber S, Diamond LK, Mercer RD, Sylvester RF, Wolff VA. Temporary remission in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl glutamic acid (aminopterin) N Engl J Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 3.Osborn MJ, Huennekens FM. Enzymatic reduction of dihydrofolic acid. J Biol Chem. 1958;233:969–974. [PubMed] [Google Scholar]
- 4.Visentin M, Zhao R, Goldman ID. The antifolates. Hematol Oncol Clin North Am. 2012;26:629–648. doi: 10.1016/j.hoc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao R, Goldman ID. Resistance to antifolates. Oncogene. 2003;22:7431–7457. doi: 10.1038/sj.onc.1206946. [DOI] [PubMed] [Google Scholar]
- 6.Baugh CM, Krumdieck CL, Nair MG. Polygammaglutamyl metabolites of methotrexate. Biochem Biophys Res Commun. 1973;52:27–34. doi: 10.1016/0006-291x(73)90949-2. [DOI] [PubMed] [Google Scholar]
- 7.Chabner BA, Allegra CJ, Curt GA, Clendeninn NJ, Baram J, Koizumi S, Drake JC, Jolivet J. Polyglutamation of methotrexate. Is methotrexate a prodrug? J Clin Invest. 1985;76:907–912. doi: 10.1172/JCI112088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koizumi S, Curt GA, Fine RL, Griffin JD, Chabner BA. Formation of methotrexate polyglutamates in purified myeloid precursor cells from normal human bone marrow. J Clin Invest. 1985;75:1008–1014. doi: 10.1172/JCI111761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poser RG, Sirotnak FM, Chello PL. Differential synthesis of methotrexate polyglutamates in normal proliferative and neoplastic mouse tissues in vivo. Cancer Res. 1981;41:4441–4446. [PubMed] [Google Scholar]
- 10.Fabre I, Fabre G, Goldman ID. Polyglutamylation, an important element in methotrexate cytotoxicity and selectivity in tumor versus murine granulocytic progenitor cells in vitro. Cancer Res. 1984;44:3190–3195. [PubMed] [Google Scholar]
- 11.Allegra CJ, Fine RL, Drake JC, Chabner BA. The effect of methotrexate on intracellular folate pools in human MCF-7 breast cancer cells. Evidence for direct inhibition of purine synthesis. J Biol Chem. 1986;261:6478–6485. [PubMed] [Google Scholar]
- 12.Allegra CJ, Chabner BA, Drake JC, Lutz R, Rodbard D, Jolivet J. Enhanced inhibition of thymidylate synthase by methotrexate polyglutamates. J Biol Chem. 1985;260:9720–9726. [PubMed] [Google Scholar]
- 13.Matherly LH, Barlowe CK, Goldman ID. Antifolate polyglutamylation and competitive drug displacement at dihydrofolate reductase as important elements in leucovorin rescue in L1210 cells. Cancer Res. 1986;46:588–593. [PubMed] [Google Scholar]
- 14.Matherly LH, Barlowe CK, Phillips VM, Goldman ID. The effects of 4-aminoantifolates on 5-formyltetrahydrofolate metabolism in L1210 cells. J Biol Chem. 1987;262:710–717. [PubMed] [Google Scholar]
- 15.Taylor EC, Kuhnt D, Shih C, Rinzel SM, Grindey GB, Barredo J, Jannatipour M, Moran RG. A dideazatetrahydrofolate analogue lacking a chiral center at C-6, N-[4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2, 3-d]pyrimidin-5-yl)ethyl]benzoyl]-L-glutamic acid, is an inhibitor of thymidylate synthase. J Med Chem. 1992;35:4450–4454. doi: 10.1021/jm00101a023. [DOI] [PubMed] [Google Scholar]
- 16.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009;69:5467–5474. doi: 10.1158/0008-5472.CAN-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piper JR, Johnson CA, Otter GM, Sirotnak FM. Synthesis and antifolate evaluation of 10-ethyl-5-methyl- 5,10-dideazaaminopterin and an alternative synthesis of 10-ethyl-10-deazaaminopterin (edatrexate) J Med Chem. 1992;35:3002–3006. doi: 10.1021/jm00094a011. [DOI] [PubMed] [Google Scholar]
- 18.Sirotnak FM, DeGraw JI, Colwell WT, Piper JR. A new analogue of 10-deazaaminopterin with markedly enhanced curative effects against human tumor xenografts in mice. Cancer Chemother Pharmacol. 1998;42:313–318. doi: 10.1007/s002800050823. [DOI] [PubMed] [Google Scholar]
- 19.Sirotnak FM, DeGraw JI, Schmid FA, Goutas LJ, Moccio DM. New folate analogs of the 10-deaza-aminopterin series. Further evidence for markedly increased antitumor efficacy compared with methotrexate in ascitic and solid murine tumor models. Cancer Chemother Pharmacol. 1984;12:26–30. [PubMed] [Google Scholar]
- 20.O’Connor OA, Horwitz S, Hamlin P, Portlock C, Moskowitz CH, Sarasohn D, Neylon E, Mastrella J, Hamelers R, Macgregor-Cortelli B, Patterson M, Seshan VE, Sirotnak F, Fleisher M, Mould DR, Saunders M, Zelenetz AD. Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol. 2009;27:4357–4364. doi: 10.1200/JCO.2008.20.8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, Lechowicz MJ, Savage KJ, Shustov AR, Gisselbrecht C, Jacobsen E, Zinzani PL, Furman R, Goy A, Haioun C, Crump M, Zain JM, Hsi E, Boyd A, Horwitz S. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol %20. 2011;29:1182–1189. doi: 10.1200/JCO.2010.29.9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao R, Diop-Bove N, Visentin M, Goldman ID. Mechanisms of Membrane Transport of Folates into Cells and Across Epithelia. Annu Rev Nutr. 2011;31:177–201. doi: 10.1146/annurev-nutr-072610-145133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917–928. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 24.Zhao R, Babani S, Gao F, Liu L, Goldman ID. The mechanism of transport of the multitargeted antifolate, MTA-LY231514, and its cross resistance pattern in cell with impaired transport of methotrexate. Clin Cancer Res. 2000;6:3687–3695. [PubMed] [Google Scholar]
- 25.Zhao R, Gao F, Hanscom M, Goldman ID. A prominent low-pH methotrexate transport activity in human solid tumor cells: Contribution to the preservation of methotrexate pharmacological activity in HeLa cells lacking the reduced folate carrier. Clin Cancer Res. 2004;10:718–727. doi: 10.1158/1078-0432.ccr-1066-03. [DOI] [PubMed] [Google Scholar]
- 26.Zhao R, Qiu A, Tsai E, Jansen M, Akabas MH, Goldman ID. The proton-coupled folate transporter (PCFT): impact on pemetrexed transport and on antifolate activities as compared to the reduced folate carrier. Mol Pharmacol. 2008;74:854–862. doi: 10.1124/mol.108.045443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diop-Bove NK, Wu J, Zhao R, Locker J, Goldman ID. Hypermethylation of the human proton-coupled folate transporter (SLC46A1) minimal transcriptional regulatory region in an antifolate-resistant HeLa cell line. Molecular Cancer Therapeutics. 2009;8:2424–2431. doi: 10.1158/1535-7163.MCT-08-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Visentin M, Zhao R, Goldman ID. Augmentation of Reduced Folate Carrier-mediated Transport of Folates/antifolates through an Antiport Mechanism with 5-aminoimidazole-4-carboxamide Riboside Monophosphate. Mol Pharmacol. 2012;82:209–216. doi: 10.1124/mol.112.078642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao R, Chattopadhyay S, Hanscom M, Goldman ID. Antifolate resistance in a HeLa cell line associated with impaired transport independent of the reduced folate carrier. Clin Cancer Res. 2004;10:8735–8742. doi: 10.1158/1078-0432.CCR-04-0932. [DOI] [PubMed] [Google Scholar]
- 30.Sharif KA, Goldman ID. Rapid determination of membrane transport parameters in adherent cells. BioTechniques. 2000;28:926–8. 930, 932. doi: 10.2144/00285st06. [DOI] [PubMed] [Google Scholar]
- 31.Goldman ID, Lichtenstein NS, Oliverio VT. Carrier-mediated transport of the folic acid analogue methotrexate, in the L1210 leukemia cell. J Biol Chem. 1968;243:5007–5017. [PubMed] [Google Scholar]
- 32.Zhao R, Hanscom M, Chattopadhyay S, Goldman ID. Selective preservation of pemetrexed pharmacological activity in HeLa cells lacking the reduced folate carrier; association with the presence of a secondary transport pathway. Cancer Res. 2004;64:3313–3319. doi: 10.1158/0008-5472.can-03-3953. [DOI] [PubMed] [Google Scholar]
- 33.Zhao R, Gao F, Goldman ID. Marked suppression of the activity of some, but not all, antifolate compounds by augmentation of folate cofactor pools within tumor cells. Biochem Pharmacol. 2001;61:857–865. doi: 10.1016/s0006-2952(01)00532-9. [DOI] [PubMed] [Google Scholar]
- 34.Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther. 2012;13:1355–1373. doi: 10.4161/cbt.22020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matherly LH, Voss MK, Anderson LA, Fry DW, Goldman ID. Enhanced polyglutamylation of aminopterin relative to methotrexate in the Ehrlich ascites tumor cell in Vitro. Cancer Res. 1985;45:1073–1078. [PubMed] [Google Scholar]
- 36.Ratliff AF, Wilson J, Hum M, Marling-Cason M, Rose K, Winick N, Kamen BA. Phase I and pharmacokinetic trial of aminopterin in patients with refractory malignancies. J Clin Oncol. 1998;16:1458–1464. doi: 10.1200/JCO.1998.16.4.1458. [DOI] [PubMed] [Google Scholar]
- 37.Khokhar NZ, She Y, Rusch VW, Sirotnak FM. Experimental therapeutics with a new 10-deazaaminopterin in human mesothelioma: further improving efficacy through structural design, pharmacologic modulation at the level of MRP ATPases, and combined therapy with platinums. Clin Cancer Res. 2001;7:3199–3205. [PubMed] [Google Scholar]
- 38.Wang ES, O’Connor O, She Y, Zelenetz AD, Sirotnak FM, Moore MA. Activity of a novel anti-folate (PDX, 10-propargyl 10-deazaaminopterin) against human lymphoma is superior to methotrexate and correlates with tumor RFC-1 gene expression. Leuk Lymphoma. 2003;44:1027–1035. doi: 10.1080/1042819031000077124. [DOI] [PubMed] [Google Scholar]
- 39.Zhao R, Seither R, Brigle KE, Sharina IG, Wang PJ, Goldman ID. Impact of overexpression of the reduced folate carrier (RFC1), an anion exchanger, on concentrative transport in murine L1210 leukemia cells. J Biol Chem. 1997;272:21207–21212. doi: 10.1074/jbc.272.34.21207. [DOI] [PubMed] [Google Scholar]
- 40.Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophys J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stein MA, Mathers DA, Yan H, Baimbridge KG, Finlay BB. Enteropathogenic Escherichia coli markedly decreases the resting membrane potential of Caco-2 and HeLa human epithelial cells. Infect Immun. 1996;64:4820–4825. doi: 10.1128/iai.64.11.4820-4825.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mould DR, Sweeney K, Duffull SB, Neylon E, Hamlin P, Horwitz S, Sirotnak F, Fleisher M, Saunders ME, O’Connor OA. A population pharmacokinetic and pharmacodynamic evaluation of pralatrexate in patients with relapsed or refractory non-Hodgkin’s or Hodgkin’s lymphoma. Clin Pharmacol Ther. 2009;86:190–196. doi: 10.1038/clpt.2009.80. [DOI] [PubMed] [Google Scholar]
- 43.Steinberg SE, Campbell CL, Bleyer WA, Hillman RS. Enterohepatic circulation of methotrexate in rats in vivo. Cancer Res. 1982;42:1279–1282. [PubMed] [Google Scholar]
- 44.Trevino LR, Shimasaki N, Yang W, Panetta JC, Cheng C, Pei D, Chan D, Sparreboom A, Giacomini KM, Pui CH, Evans WE, Relling MV. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27:5972–5978. doi: 10.1200/JCO.2008.20.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramsey LB, Panetta JC, Smith C, Yang W, Fan Y, Winick NJ, Martin PL, Cheng C, Devidas M, Pui CH, Evans WE, Hunger SP, Loh M, Relling MV. Genome-wide study of methotrexate clearance replicates SLCO1B1. Blood. 2013;121:898–904. doi: 10.1182/blood-2012-08-452839. [DOI] [PMC free article] [PubMed] [Google Scholar]