

SUMMARY
ATP-binding cassette transporters protect cells via efflux of xenobiotics and endogenous byproducts of detoxification. While the cost of this ATP-dependent extrusion is known at the molecular level, i.e. the ATP used for each efflux event, the overall cost to a cell or organism of operating this defense is unclear, especially as the cost of efflux changes depending on environmental conditions. During prolonged exposure to xenobiotics, multidrug transporter activity could be costly and ineffective because effluxed substrate molecules are not modified in the process and could thus undergo repeated cycles of efflux and re-entry. Here we use embryos of the purple sea urchin, Strongylocentrotus purpuratus, as a model to determine transport costs and benefits under environmentally relevant xenobiotic concentrations. Strikingly, our results show that efflux transporter activity costs less than 0.2% of total ATP usage, as a proportion of oxygen consumption. The benefits of transport, defined as the reduction in substrate accumulation due to transporter activity, depended largely, but not entirely, on the rate of passive flux of each substrate across the plasma membrane. One of the substrates tested exhibited rapid membrane permeation coupled with high rates of efflux, thus inducing rapid and futile cycles of efflux followed by re-entry of the substrate. This combination significantly reduced transporter effectiveness as a defense and increased costs even at relatively low substrate concentrations. Despite these effects with certain substrates, our results show that efflux transporters are a remarkably effective and low-cost first line of defense against exposure to environmentally relevant concentrations of xenobiotics.
KEY WORDS: ABC transporters, environmental toxicology, P-GP, MRP, Strongylocentrotus purpuratus
INTRODUCTION
Survival in an unpredictable and changing environment requires defenses to mitigate or prevent cellular damage. These defenses entail costs, however, which can be viewed as trade-offs for survival. It is presumed that one mechanism by which these adaptations to stress impact fitness is through their energetic costs. A portion of this cost is allocated towards synthesis of proteins responsible for the protection. Once this protein infrastructure is present, there can be additional costs associated with utilizing these defenses to maintain homeostasis.
This study examines the energy cost and overall effectiveness of a primary defense against foreign chemicals, the multidrug efflux transporter. These transmembrane proteins utilize the energy of ATP hydrolysis to power the efflux of a structurally diverse range of moderately hydrophobic compounds against their concentration gradients (Choudhuri and Klaassen, 2006; Epel et al., 2008). Multidrug transporters are constitutively active in embryos (Hamdoun and Epel, 2007; Hamdoun et al., 2004) and in adult barrier tissues, such as the placenta (Behravan and Piquette-Miller, 2007), and the blood–brain barrier (Begley, 2004).
These multidrug efflux transporters are members of the ABCB, ABCC and ABCG subfamilies of the ATP-binding cassette (ABC) superfamily of proteins. Transporter activity can prevent xenobiotic access to the cytoplasm by binding chemicals in solution in the membrane and effluxing them in an unmodified form before they permeate into the cytosol (Greenberger et al., 1991; Pleban et al., 2005). Thus they act as a first line of defense against xenobiotics. Chemicals that get past the transporter defense and enter the cell are frequently modified by cytochrome oxidases and/or conjugated to small polar molecules, such as glutathione, and the resultant detoxified compounds can be effluxed out of the cell by several different ABCC family transporters (Higgins, 2007).
In this study we focus on determining the cost in terms of ATP used for the efflux of unmodified xenobiotics by ABCB and ABCC transporters. Based on kinetic studies of transporter activity, the efflux of a substrate molecule appears to be powered by the hydrolysis of one to two molecules of ATP (Aänismaa and Seelig, 2007; Al-Shawi et al., 2003; Sauna and Ambudkar, 2000). These kinetic studies, combined with detailed crystal structure of transporters in multiple states and from multiple organisms, from bacteria to vertebrates, indicate an alternating catalytic cycle of ATP hydrolysis (Aller et al., 2009; Dawson and Locher, 2006; Patzlaff et al., 2003; Rosenberg et al., 2003). In this model, one ATP is hydrolyzed successively at each of the two nucleotide binding domains to efflux a single substrate molecule. Alternating cycles of ATP binding and hydrolysis power conformational changes to the transporter that open the substrate binding pocket to the extracellular environment, allowing substrate efflux, and a second ATP hydrolysis to reset the original state that allows substrate binding from the membrane. There is high uniformity in transporter structure and function across a wide span of evolutionary distances (Davidson et al., 2008; Gökirmak et al., 2012; Rees et al., 2009; Ricardo and Lehmann, 2009). Similar mechanisms of activity have been observed for ABCB transporters across organisms (Patzlaff et al., 2003; Seeger and van Veen, 2009), and are thus expected for sea urchins as well.
While it is reasonable to postulate that two molecules of ATP are hydrolyzed per efflux event, knowing this singular cost does not predict total costs of transporter activity in an organism exposed to substrates for longer durations, as would occur in the environment. Total costs to the organism could be much larger than the ATP utilized in the primary efflux event because the expelled, unmodified chemicals can simply diffuse back across the membrane (Holland and Blight, 1999). Indeed, this is probable because the efflux action of transporters sets up a gradient in substrate concentration across the plasma membrane, thus favoring re-entry of external molecules into the cell. Multiple iterations of substrate efflux and re-entry could occur, with each cycle using additional ATP, yet still leave the compound as a potential threat. Ultimately, the chemical gradient created and maintained by substrate efflux without modification causes costs to accrue indefinitely, until the molecule diffuses away into the environment or makes it into the cell. Such active mechanisms that utilize energy to create and maintain equilibrium gradients are common in biology, but have the potential to become quite costly. Note, for example, the Na+/K+-ATPase, which can account for up to 80% of total ATP consumption in sea urchin embryos (Leong and Manahan, 1997).
Costs of transporter activity to the organism can be compounded because multidrug transporters recognize a wide range of substrates on criteria of hydrophobicity and size rather than toxicity (Pawagi et al., 1994). In a worst-case scenario, non-toxic substrates could hijack the system and be effluxed repeatedly, costing the cell energy for each efflux cycle. In addition to increasing the ATP cost, benign substrates can act as chemosensitizing agents that utilize some portion of the finite efflux capacity of transporters, thus diminishing their effectiveness as a defense (Smital et al., 2004).
Initial attempts to measure costs of transporter activity used drug-resistant cell lines or proteoliposomes, and indicated high costs associated with efflux transporter activity (Bains and Kennedy, 2005; Broxterman et al., 1988; Broxterman et al., 1989; Hildebrand et al., 2009). However, the actual cost of this defense has not previously been determined in a naturally occurring system under environmentally relevant conditions.
Sea urchin embryos, used in this study, are an ideal natural model system to examine transporter activity and effectiveness in the environment. The sea urchin genome contains at least 65 ABC transporters, more than 80% of which are expressed in the embryo (Sea Urchin Genome Sequencing Consortium et al., 2006). Efflux transporter activity in these embryos is capable of preventing the accumulation of up to 5 μmol l−1 vinblastine (Hamdoun et al., 2004), and as we show in this paper, is similarly effective against other substrates. Using low concentrations of three model substrates, calcein-AM (CAM), calcein red/orange-AM (CROAM) and bodipy-FL verapamil (BFLV) (Essodaigui et al., 1998; Hamdoun et al., 2004), we have quantitatively measured the efflux rate and calculated the costs of transporter activity. We find that efflux transporter activity comprises just 0.2 to 0.5% of total cellular energy consumption. Despite these low costs, transporter activity can still be highly effective at preventing accumulation of low micromolar concentrations of substrates. These results have direct implications for understanding the role of transporters as a defense against environmental contaminants, and may inform efforts to overcome drug resistance in chemotherapy and pharmacology.
MATERIALS AND METHODS
Embryo culture and counting
Adult Strongylocentrotus purpuratus (Stimpson 1857) were injected with 0.5 ml of 0.5 mol l−1 potassium chloride to induce spawning. Eggs were spawned directly into seawater and sperm were collected dry and stored at 4°C (for details, see Hamdoun et al., 2004). Eggs were fertilized, and fertilization percentage was determined by scoring the presence of a fertilization envelope under 20× magnification. Batches of eggs were only used if they had >95% fertilization. Embryo density was determined by counting 4×50 μl aliquots of fertilized embryos under 20× magnification. The embryo culture was subsequently diluted to a final density of 2000 embryos ml−1, and held at 16°C while being suspended via continuous mixing on an orbital shaker.
Exposure to inhibitors, embryo lysis and fluorescence measurements
Ninety to 120 min exposures to inhibitors and substrates were carried out at 16°C in 15 ml conical tubes containing 7 ml of embryo suspension, with continuous mixing on a rocker. After exposure, embryos were washed three times by low-speed centrifugation with a hand-crank centrifuge (to prevent damage to embryos), the supernatant was decanted, and embryos were re-suspended in fresh seawater. After the final wash, the embryo pellet was suspended in 1 ml of hypotonic lysis buffer [10 mmol l−1 KCl, 1.5 mmol l−1 MgCl2, 10 mmol l−1 Tris HCl, pH 7.4 (Morris et al., 1991)] and sonicated on ice with a Branson Cell Disruptor 200 (St Louis, MO, USA) until visual inspection revealed complete lysis (10–15 s). Four aliquots of 200 μl each of the resulting lysate were loaded into a 96-well plate. Fluorescence measurements were made using the Perspective Biosystems Cytoflour 2 (Perseptive Biosystems Cytofluor 2, Framingham, MA, USA) using 485 nm excitation and 530 nm emission for BFLV and CAM experiments; 580 nm excitation and 610 nm emission was used for CROAM. To prevent CROAM from adhering to the tube walls, 15 ml conical tubes were pre-incubated with a 2% bovine serum albumin solution, which was removed prior to adding embryos.
To convert raw fluorescence values into numbers of molecules, we compared fluorescence with a standard curve made with 2000 lysed embryos per milliliter (the same density as substrate exposures) and known quantities of calcein, BFLV or calcein-orange. Because each well contained a known number of embryos, we could calculate the number of fluorescent molecules per embryo. The difference in accumulated substrate molecules between maximally inhibited and uninhibited embryos (see below for information on maximal inhibition) is the number of substrate molecules kept out of the embryo because of transporter activity. These numbers of intracellular substrate molecules were then converted into cytosolic concentration within the embryos using a cellular volume of 0.382 nl (calculated based on a radius of 45 μm) or were converted into ATP cost of transport by multiplying by 2 (Aänismaa and Seelig, 2007; Al-Shawi et al., 2003; Sauna and Ambudkar, 2000).
Respiration
To measure total oxygen consumption, 17 ml of a 2000 embryos ml−1 solution of two-cell (120 min post-fertilization) embryos were placed in a sealed chamber filled with constant 16°C seawater. Thorough mixing was achieved during measurements with a small magnetic stirbar underneath a slotted false bottom to the measurement chamber. Embryos were allowed to equilibrate, then O2 consumption was measured using a Strathkelvin 781 oxygen meter and 1302 electrode (North Lanarkshire, UK) calibrated for 100% saturation using seawater that was mixed for several minutes, and 0% using 2 g l−1 of sodium sulfite. The steady-state rate of O2 consumption was determined over ~2 h. These values were converted into ATP regeneration by multiplying O2 molecules consumed by 6.
Reagents and materials
CAM, CROAM, MK571, BFLV and PSC833 were dissolved in 100% DMSO and then diluted to the indicated final concentrations. The final DMSO concentration in all experiments was less than 0.5%. CAM, CROAM and BFLV were obtained from Invitrogen Life Technologies (Carlsbad, CA, USA). MK571 was obtained from Cayman Chemical (Ann Arbor, MI, USA). PSC833 was generously provided by Novartis (Basel, Switzerland) through a material transfer agreement.
Data analysis and experimental replication
Unless stated otherwise, all error bars are ±1 s.e.m. These error measurements are based on five to eight experimental replicates. Each individual replicate is an average of four fluorescence measurements taken at each time point and/or inhibitor/substrate dose. This value for each of the five to eight replicates is then averaged and displayed with the standard error as the final result.
RESULTS
Principle of efflux transport activity assay
To determine the cost of transport, we first quantified the rate of substrate accumulation in control embryos with unmodified transporter activity. This rate was then compared with the rate of accumulation in embryos treated with sufficient concentrations of inhibitors to maximally block transport activity. The difference in number of substrate molecules accumulated under these two conditions (maximally inhibited versus uninhibited) represents the molecules effluxed by multidrug transporters. PSC833 is an inhibitor of PGP-type transporters (ABCB family) (Boesch et al., 1991) and MK571 primarily inhibits activity of MRP-type transporters (ABCC family) (Gekeler et al., 1995). The inhibitor combination presumably blocks the activity of both families.
In this assay we quantitatively measured the accumulation rate over a wide range of conditions for three fluorescent transporter substrates: CAM, BFLV and CROAM. All of these chemicals are substrates of both ABCB and ABCC transporters, as evidenced by their increased accumulation in the presence of transporter inhibitors (Figs 1, 2). The regression lines for the rate of intracellular accumulation of substrate over time, shown in Fig. 2, are displayed as a single rate for any given substrate and inhibitor concentration combination in all subsequent figures.
Fig. 1.

(A) Eight-cell Strongylocentrotus purpuratus embryos exposed to the indicated probes [calcein-AM (CAM), bodipy-FL verapamil (BFLV) and calcein red/orange-AM (CROAM)] exhibit increased fluorescence in the presence of maximally effective levels of the transporter inhibitors PSC833 and MK571. Scale bar, 20 μm. (B) The same embryos under brightfield imaging.
Fig. 2.

Intracellular concentrations of the transporter substrates CAM (A), BFLV (B) and CROAM (C) in the presence of the indicated concentrations of transporter inhibitors. In all cases, embryos were exposed to 0.25 μmol l−1 extracellular substrate concentration. All regressions are linear with the x-intercept set to the origin, as there is no fluorescence expected at 0 min, after normalization for any background. Regression lines for all inhibitor exposures have R2 values greater than 0.9.
CAM is a non-fluorescent chemical that, upon entering a cell, is rapidly hydrolyzed by cytosolic esterases that remove the acetoxymethyl esters (AM). This hydrolysis produces the fluorescent molecule calcein, which is not membrane-permeable and has greatly reduced affinity as a substrate of PGP-type transporters, thus effectively trapping it within the cell (Essodaigui et al., 1998). CROAM, an AM-ester of the fluorescent molecule bodipy, is structurally unlike CAM, despite the similar name. As with CAM, CROAM is trapped within cells following -AM cleavage by cytosolic esterases. BFLV is also trapped upon entering cells, but trapping is in lysosomes and other acidic vesicles (Crowley et al., 2003). All substrates were verified to remain trapped intracellularly for at least 60 min after washing exposed embryos out of the substrate solution and into clean seawater (data not shown). Intracellular modification of CAM and CROAM by esters and vesicular accumulation of BFLV all result in cellular trapping of any substrate molecules that get past the transporter defense system and into the cytoplasm, thus creating an infinite disequilibrium gradient, and allowing accurate assessment of their rate of membrane flux.
For this assay to accurately measure efflux rates, it is crucial that this disequilibrium be fueled by a constant supply of substrate molecules outside of the cell. With such a continuous extracellular pool, substrate molecules are always present and able to permeate into the membrane where they can be effluxed by transporters. Thus every time one molecule is removed from the equilibrium system by esterases or vesicular trapping, it is always replaced from the external milieu by another substrate molecule.
We tested whether this condition was met in our assay and found that in almost all cases this intracellular accumulation rate remains linear over the time course of the exposure, indicating that this infinite disequilibrium is intact. Fig. 2 shows the accumulation rate over time for each substrate under various levels of transporter inhibition. The only instance where accumulation began to slow over the assay was observed with BFLV in the presence of inhibitors, and this is likely indicative of either internal saturation or depletion of extracellular supply. In this case, only the linearly accumulating phase, shown in Fig. 2, was used for analysis of transporter efflux and effectiveness rates.
Kinetics of accumulation
Efflux transporters exhibited large differences in their effectiveness at reducing substrate accumulation rates over an environmentally relevant range of substrate concentrations (Fig. 3). This represents the baseline effectiveness of transporters at reducing accumulation of these substrates, i.e. the number of substrate molecules that accumulate within the cell despite efflux transporter activity. The range of substrate concentrations used (0.1 to 5 μmol l−1) was chosen to reflect the measured concentrations of many environmental contaminants in polluted freshwater (Kolpin et al., 2002) and marine environments (Anderson et al., 2010), as well as the serum concentrations of certain therapeutic agents used in treatment of a variety of diseases (Rodríguez Robledo and Smyth, 2008).
Fig. 3.

Uninhibited flux rates (i.e. passive permeation across the membrane) for each substrate over a series of concentrations given in 106 substrate molecules embryo−1 s−1 (means ± s.d.). While both CROAM and BFLV overload transport capacity and accumulate rapidly at higher doses, CAM accumulation in embryos with active transporters is negligible at all concentrations tested.
As can be seen in Fig. 3, there was almost no increase in CAM accumulation at any substrate concentration tested, indicating that CAM transport was never saturated over the tested concentrations. Accumulation of BFLV and CROAM increased substantially at higher concentrations, indicating saturation of efflux capacity as the dose increased. Each individual point in this and all subsequent figures represents a linear substrate accumulation rate measured over 90–120 min, such as those shown in Fig. 2.
Exposing embryos to the transporter inhibitors PSC833 or MK571 individually resulted in substantially increased substrate accumulation rates (Fig. 4). Fig. 5 shows the accumulation rates of all three substrates when exposed to a combination of both inhibitors.
Fig. 4.

Rate of substrate accumulation in 106 substrate molecules embryo−1 s−1 (means ± s.d.) for transporter substrates CAM (diamonds) and BFLV (squares). (A) Accumulation with the PGP inhibitor PSC833. (B) Accumulation with the MRP inhibitor MK571.
Fig. 5.

Substrate accumulation rate in 106 substrate molecules embryo−1 s−1 (means ± s.d.) for the transporter substrates CAM (diamonds), BFLV (squares) and CROAM (circles). In addition to the indicated MK571 dosage, CAM and CROAM experiments were co-incubated with 2.0 μmol l−1 PSC833 and BFLV experiments were co-incubated with 1.0 μmol l−1 PSC833.
Calculation of the intracellular substrate concentration shows that there is considerable accumulation/trapping of all three substrates in the cell when efflux transporters are inhibited. With an extracellular substrate concentration of 0.25 μmol l−1, intracellular concentrations can reach 180 μmol l−1 within 90 min (Fig. 2). This hyper-accumulation indicates that when CAM and CROAM are hydrolyzed the resultant molecule is no longer in equilibrium with the extracellular pool of substrate. Similarly, for BFLV this indicates that trapping in vesicles is robust enough to remove vesicular BFLV from equilibrium with the extracellular environment.
Maximal inhibition of efflux transport
Maximal inhibition is defined here as the inhibitor concentration above which higher inhibitor levels do not increase substrate accumulation. At this point we assume that the majority of target transporter activity is blocked. When both inhibitors are combined at their maximally effective dose (Fig. 5), then all activity of ABCB and ABCC transporters is presumably blocked.
To quantify the cost of efflux transport, we first determined the point of maximal inhibition for MK571, PSC833 and with both inhibitors combined (Figs 4, 5). As can be seen in Fig. 4A,B, maximal inhibition required between 2 and 5 μmol l−1 PSC833 for CAM and BFLV. Maximal inhibition with MK571 occurred at 40 μmol l−1 for CAM and between 20 and 40 μmol l−1 for BFLV. In Fig. 5 we show that when both inhibitors are combined, maximal accumulation of CAM and CROAM occurs at 2.0 μmol l−1 PSC833 with 20 μmol l−1 MK571. Maximal accumulation of BFLV requires only 1.0 μmol l−1 PSC833 with 5 μmol l−1 MK571. With a 0.25 μmol l−1 extracellular substrate concentration, maximal inhibition results in CAM, BFLV and CROAM accumulation rates in millions of molecules per embryo per second of 3±0.4, 5±0.2 and 5.4±0.2, respectively.
At what substrate concentrations do transporters provide effective protection?
Transporters were significantly more effective at preventing cellular accumulation of CAM than other substrates (Fig. 3). To better characterize these differences we measured both passive (maximally inhibited) and uninhibited flux across a series of substrate concentrations. In Fig. 6A we show the ratio of accumulation rates between inhibited and control (solvent-exposed) embryos. This ratio is a measure of transport effectiveness. For all substrates this ratio is nearly 1 at low concentrations (below 10 nmol l−1), presumably because passive diffusion into the embryo is low regardless of transport activity. With increasing extracellular substrate concentrations the ratio increases to some maximum value, which is the substrate concentration where transporters provide the greatest net benefit, and then begins to decrease as the transporter efflux capacity is overwhelmed by the increasing flux of substrate across the membrane.
Fig. 6.

Effectiveness of efflux transporters at preventing accumulation of transporter substrates across a concentration series (means ± s.d.). The results for concentrations of CAM and CROAM that exceed esterase capacity, 1 and 2.5 μmol l−1, respectively (see Fig. 7 for absolute accumulation rates), underestimate the actual values. (A) The ratio of fluorescence for embryos exposed to maximally effective doses of the inhibitors MK571 and PSC833 or to a solvent control. (B) The same data set from Fig. 3A, but displayed as a percent instead of a ratio, where percent effectiveness is calculated as: 100×(AI–AU)/AI, where AI and AU indicate the rate of substrate accumulation in inhibited and uninhibited embryos, respectively.
The ratio curves in Fig. 6A differ in several aspects. For CAM, the ratio increases steadily from nanomolar concentrations to a maximum of 32 at 1 μmol l−1. This is the highest dose of CAM that we were accurately able to measure because of limitations to the esterase capacity (see below). Unlike CAM, the ratio for BFLV spikes rapidly from 1.3 at 0.01 μmol l−1 to a maximum of only 5.4 at 0.1 μmol l−1, then drops back to 2.1 at 1.0 μmol l−1. A maximum ratio of 15 for CROAM also occurred at 0.1 μmol l−1.
In Fig. 6B we show these same data expressed as percent effectiveness – the percent that total substrate accumulation is diminished by efflux transporter activity. Expressing the data in this manner shows that transporters are highly effective against CAM at substrate concentrations ranging from 0.1 to 5 μmol l−1, a remarkably broad range. CROAM transport exhibits a peak effectiveness of 93% at 0.1 μmol l−1, which decreases rapidly at higher or lower doses. Transporters are much less effective against BFLV. Maximum effectiveness for BFLV transport is 81%, and occurs at 0.1 μmol l−1. At 1.0 μmol l−1, transport of CAM is greater than 98% effective, whereas transport effectiveness for BFLV and CROAM is ~50%. Based on the results shown in Fig. 3, we can conclude that transporters are nearly 100% effective at preventing CAM accumulation even up to 5 μmol l−1, while at that same dose they are only 19.6% effective at preventing accumulation of BFLV. In all, transporters are effective against CROAM and BFLV at lower concentrations than CAM and across a narrower concentration range.
Esterase limitations
For both CAM and CROAM, the esterase that cleaves the -AM esters has a maximum capacity (Essodaigui et al., 1998). Esterase activity is vital to this assay because -AM cleavage produces the fluorescent molecule that is trapped within the cell. As such, we are unable to measure flux rates of CAM and CROAM that exceed the esterase capacity. This limit is observable in the presence of transporter inhibitors, such as shown in Fig. 7A,C, where flux rates for both chemicals reach a plateau. It is important to note that this is not simply concentration-dependent quenching, because fluorescence continues to increase linearly over time at each concentration (as in Fig. 2), but the rate of that increase remains constant at higher CAM concentrations. For CAM, the esterase has a maximum capacity of ~4×106 molecules embryo−1 s−1, which begins to limit measurement of both passive flux and energy costs at concentrations above 1 μmol l−1. The esterase(s) that act on CROAM have a greater capacity, ~15×106 molecules embryo−1 s−1, which limits measurement of passive flux and energy cost above 2.5 μmol l−1 (Fig. 7).
Fig. 7.

Cost (106 molecules ATP embryo−1 s−1) and flux (106 substrate molecules embryo−1 s−1) (means ± s.d.) for the transporter substrates CAM (A), BFLV (B) and CROAM (C). The regression line in A is a linear extrapolation of the first three CAM concentrations.
Substrates are presumably diffusing across the membrane at a faster rate than we are able to measure for concentrations that exceed the esterase capacity. This only impacts conclusions for CAM, which is kept out of cells at levels as high as 10 μmol l−1, as shown in Fig. 3. This finding has ramifications for the calculations of maximal cost and overall transport efficiency for CAM. Esterases are not rate limiting for either of the other two substrates at low concentrations, nor is this limit detectable in the absence of inhibitors, except at the highest dose tested of CROAM (Fig. 3). This allows accurate measurement of flux and energy costs at lower concentrations and, in the case of CAM, extrapolation of cost at the levels that exceed esterase limitations.
Energy cost of efflux activity
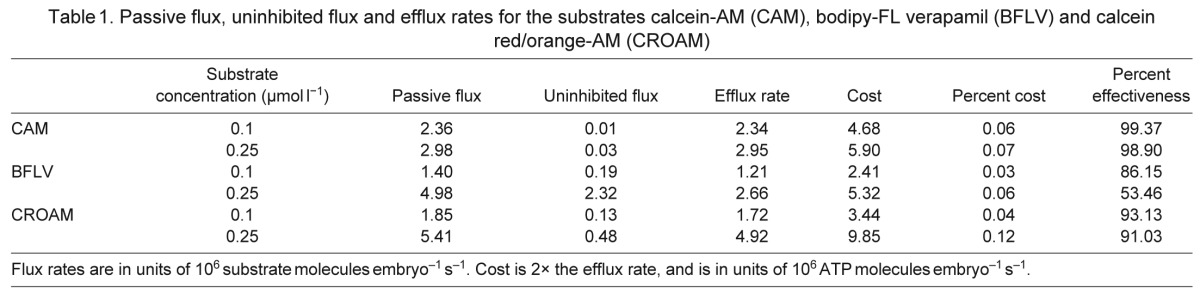
From the efflux rates of each substrate we calculated the approximate cost of using multidrug transporters by assuming an expenditure of two molecules of ATP hydrolyzed per substrate molecule effluxed (Aänismaa and Seelig, 2007; Al-Shawi et al., 2003; Sauna and Ambudkar, 2000). The efflux rate and effectiveness values for 0.1 and 0.25 μmol l−1 are reported in Table 1. Transport costs for each substrate, alongside passive flux across the membrane, are displayed for 0.1 to 5 μmol l−1 for each substrate in Fig. 7. CAM transport costs increased from 4.7×106±0.6×106 molecules ATP embryo−1 s−1 at 0.1 μmol l−1 to 7.9×106±0.4×106 at 1 μmol l−1, the highest measurable cost before reaching the esterase limit. Costs for CROAM transport increased from 3.4×106±0.3×106 molecules ATP embryo−1 s−1 at 0.1 μmol l−1 to a maximum of 15×106±0.5×106 at 1.0 μmol l−1. As mentioned previously, the plateau in passive flux of CAM and CROAM at the highest substrate concentrations is probably due to esterase limitations (Essodaigui et al., 1998). Cost for BFLV transport was 2.4×106±0.4×106 molecules ATP embryo−1 s−1 at 0.1 μmol l−1, lower than both CAM and CROAM. At higher BFLV concentrations, transport costs approach a maximum of 15×106±4.5×106 molecules ATP embryo−1 s−1.
Table 1.
Passive flux, uninhibited flux and efflux rates for the substrates calcein-AM (CAM), bodipy-FL verapamil (BFLV) and calcein red/orange-AM (CROAM)
How do these transport costs relate to the total energy budget of the embryo? The oxygen consumption rate was determined, and was converted into ATP generation by multiplying O2 molecules consumed by 6 (Fig. 8). Average oxygen consumption in this assay was found to be 8.1±0.9 pmol embryo−1 h−1, corresponding to an ATP generation rate of ~8.5×109 molecules embryo−1 s−1. This oxygen consumption rate is similar to previously reported values for S. purpuratus embryos, which range from 4.7 pmol embryo−1 h−1 for 10-h-old embryos (Leong and Manahan, 1997) to 7.7 pmol embryo h−1 for 1-day-old larvae (Shilling and Manahan, 1990) and 7.9 pmol embryo−1 h−1 for 3-day-old larvae (Marsh and Manahan, 1999).
Fig. 8.

Percent oxygen saturation (diamonds) for a representative suspension of four to 16 cell sea urchin embryos held at 16°C (squares) over time. The indicated regression equation for percent O2 was used to calculate the rate of ATP regeneration. This rate of decrease in percent oxygen saturation corresponds to an O2 consumption rate of 8.1±0.9 pmol embryo−1 h−1.
Calculated cost for transport of all substrates examined was less than 0.2% of developmental ATP usage. BFLV transport costs increased from 0.03% of developmental respiration at 0.1 μmol l−1 to 0.17% at 5 μmol l−1. CROAM costs approached a similar maximum of 0.18% of developmental ATP consumption. Above 1 μmol l−1, esterase-limited passive flux prevented measurement of the actual CAM flux rate. To approximate actual transport costs for this substrate, we extrapolated a linearly increasing CAM flux rate based on the three lowest doses (Fig. 7A). Extrapolated CAM transport costs approach 0.5% of ATP consumption at the highest substrate concentration examined. If we use the lowest measured rate of oxygen consumption (listed above) of 4.7 pmol embryo−1 h−1, then the relative values for maximal costs of transporter activity would only increase from 0.2–0.5% of respiration up to 0.3–0.8%. At 5 μmol l−1, where CAM efflux is still nearly 100% effective, BFLV and CROAM transport at the same dose are just ~20 and ~30% effective, respectively (Fig. 6).
DISCUSSION
In this study we have quantitatively examined the cost, efficiency and effectiveness of protection against xenobiotics by ABC efflux transporters in sea urchin embryos. We find that the cost of transport varies with substrate but represents only a fraction of a percent of total respiratory energy consumption. Our finding of such a low cost to transporter activity was unexpected, based on earlier studies of transporters that indicated much higher costs to the protection afforded by transporter activity. Unlike our study using sea urchin embryos, these early studies used highly derived or modified systems such as drug-resistant cell lines in culture or reconstituted proteoliposomes. Drug-resistant tumor cell lines overexpressing efflux transporters showed a significant decrease in ATP levels when exposed to the substrate verapamil (Broxterman et al., 1988); however, detection of this decrease required inhibition of respiration and associated ADP phosphorylation. Additionally, studies on reconstituted proteoliposomes often showed high rates of ATP hydrolysis by transporters that were uncoupled from substrate translocation (Shapiro and Ling, 1995). Such leakage is now seen as an artifact of the system or assay conditions (Patzlaff et al., 2003) as this uncoupling does not occur under all conditions.
Several studies on freshly isolated suspensions of fish hepatocytes also reported high costs associated with transporter activity. These studies showed an 18–25% increase in respiration in the presence of the transporter substrate rhodamine 123 (R123) (Bains and Kennedy, 2005), and a similar percent decrease in steady-state concentrations of ATP in the presence of 125 μmol l−1 doxorubicin (Hildebrand et al., 2009). However, Bow et al. have reported that freshly isolated hepatocytes exhibit rapid internalization of membrane-bound ABCB1 and ABCC2 transporter proteins, causing the authors to conclude that suspended hepatocytes ‘give an inaccurate assessment of true apical elimination’ (Bow et al., 2008), and thus also of transporter costs.
There are additional costs associated with the usage of transporters as a xenobiotic defense that were not examined as part of the present study, and could thus increase the low values reported here. For example, we only examined the cost of substrate efflux by ABCB and ABCC transporters at the plasma membrane. Additional costs associated with transporter protein translation, translocation and membrane maintenance will certainly add to the total pool of cellular resources used for this defense, as would the protective action of efflux transporters in the ABCG subfamily. There is also the consideration of costs associated with other cellular defenses against xenobiotics, including cytochrome P450 oxidization, conjugation to glutathione and other carrier molecules, as well as the costs for extrusion of these conjugated chemicals by ABCC transporters.
A potential confounding factor in the energy estimates presented here is transport of substrates by organic anion transporters (OATs) (Kalliokoski and Niemi, 2009; Niemi, 2007). The assay used here determines cost based on the difference in accumulation rates in the presence and absence of specific ABC transporter inhibitors. As a result, any transport of substrates by OATs should be identical on both sides of the equation, unless the inhibitors used impacted OAT activity. Such an impact has not been reported at the concentrations used here, and is deemed unlikely because increased accumulation of each substrate became saturated at some dose of PSC833 and MK571, a finding that is consistent with efflux transporter activity comprising the majority of substrate movement.
In sea urchin embryos we find that transporter efflux capacity saturates at an extracellular substrate concentration in the range of 1–5 μmol l−1, for the substrates examined. Maximal transport costs, however, will vary as a function of transporter protein titer in different organisms and under different environmental conditions. There is a distinct and important trade-off here. Increased protective capacity can directly lead to an increase – potentially a large increase – in total transport costs. If that protective capacity were raised to 5 mmol l−1, for example, then maximal transport costs would similarly be 1000-fold higher, which would eclipse all other sources of embryonic ATP hydrolysis. Such elevated costs would also require exposure to millimolar substrate concentrations. Environmental contaminants are typically present only at or below micromolar concentrations, except in highly polluted environments, making this range of effectiveness for transporter activity well-suited to defend against most environmental conditions without causing exorbitantly high costs. In fact, capping maximal costs via limitation of transporter protein titer is most likely an important factor in the evolution of all organisms, and would be based on historic xenobiotic exposure levels.
Relating costs in sea urchin embryos to other organisms and life stages
Developmental life stages, such as the sea urchin embryos used here, are typically considered to have greater metabolic rates than later life stages (Leong and Manahan, 1997). If true, this would increase the relative proportion of total ATP consumption allocated to transporter activity as development progresses and total metabolic rates are reduced. However, the synthetic activity in the early cleavage stage embryos used in this study is actually quite low because there are large stores of membrane precursor lipids (Schmell and Lennarz, 1974) and DNA precursors (Mathews, 1975), and there are only a few cells during these stages. The bulk of the sea urchin embryo mass is in yolk and the synthetically active cell is actually quite small until the embryo has reached the 500-cell blastula stage at 16 h after fertilization; our measurements were made at the two- to eight-cell stage. As cell number increases through development, these maternally loaded resources are depleted and respiration rate increases (Leong and Manahan, 1997). As development progresses into these later stages, this would presumably result in a decreasing relative cost of transporter activity even further below what we report here, assuming the total transport capacity remains constant throughout development.
Another question is how the rate of oxygen consumption and relative transporter costs in urchin embryos relates to other organisms. Direct comparisons are difficult to make because of the large variety in cell sizes; however, 10-h-old sea urchin embryos are reported to consume oxygen at a rate of just 2 nmol mg−1 protein min−1 (Leong and Manahan, 1997), as compared with cultured trout hepatocytes, which are reported to have an oxygen consumption rate of 7 nmol mg−1 protein min−1 (Pannevis and Houlihan, 1992). This suggests that respiration rates, and presumably also relative transporter costs, in early urchin embryos are similar to, if not lower than, that found in other species and life stages.
Transport effectiveness and substrate flux
Transport effectiveness refers to the percent reduction in substrate accumulation due to transporter activity. Many aspects of our findings on effectiveness agree with previous work (Eytan et al., 1996b; Eytan et al., 1997; Regev et al., 2007; Tran et al., 2005; von Richter et al., 2009) reporting that the passive flux of a substrate, i.e. the rate of permeation into and across the plasma membrane, is the major factor governing transport effectiveness. As with these studies, we find that raising the extracellular substrate concentration increases the flux rate and decreases transporter effectiveness (Fig. 6). However, our data also indicate that flux alone does not explain all differences in transport effectiveness of these substrates. For example, transporters are 91% effective at preventing accumulation of CROAM when the flux was 5.4×106 molecules s−1, but only 53% effective against a slightly slower BFLV flux of 4.98×106 molecules s−1 (Table 1).
If not flux, then what else could explain these differences in effectiveness? Tran et al. (Tran et al., 2005) report that two distinct rates govern efflux kinetics by human ABCB1: the rate that a substrate diffuses into the membrane (i.e. flux), and the off-rate for substrate release from the transporter into the extracellular environment. It follows that the rate-limiting step for transport of slow flux substrates should, generally speaking, be the rate of substrate diffusion into the membrane (Raub, 2006), consistent with our observations. Transporters should be more effective at preventing cellular accumulation of these slow-flux substrates because the rate-limiting step of efflux is on the side of supplying substrate to the transporter. Conversely, efflux of rapidly permeating substrates would be limited by the off-rate for substrate release into the extracellular space, indicating that substrates should be supplied at a faster rate than they can be effluxed. The large difference in transport effectiveness between BFLV and CROAM – despite their similar passive flux rates – could then arise from differences in this off-rate from the substrate-binding domain. Because flux depends on concentration, a slower off-rate could result in overloading transport capacity at a lower concentration and decrease effectiveness of the protection at all concentrations, as we observe for BFLV.
Support for this theory comes from studies of verapamil, the parent compound for BFLV. Verapamil exhibits rapid flux compared with other transporter substrates (Eytan et al., 1996a) and a relatively high affinity to the substrate-binding domain of PGP (Döppenschmitt et al., 1999). The end result of this combination of characteristics is rapid, futile cycling of efflux immediately followed by fast permeation of substrate back into the membrane (Eytan and Kuchel, 1999). Such futile cycling of verapamil is so effective at overloading transporter capacity that it can be used as a chemosensitizing agent. Verapamil increases accumulation of other transporter substrates as though it were inhibiting transporter activity when it is, in fact, stimulating the transporters. Such futile and costly transporter activity is not unique to verapamil and its derivatives, such as BFLV. Other studies have identified membrane-permeable substrates that similarly stimulate the transporter ATPase but rapidly accumulate in cells even at low concentrations (Polli et al., 2001; Shapiro and Ling, 1995; von Richter et al., 2009). Further research into the conditions governing futile transport could be instrumental in overcoming drug resistance in cancer chemotherapy, an important application in which transporter inhibitors have generally not proven effective (Szakács et al., 2006).
Potential for decreased fitness
Despite their low costs relative to total metabolism, there is a potential for decreased fitness associated with efflux transporters. One potential fitness cost is from futile cycling, such as what occurs with BFLV and verapamil. Continual cycling of such substrates into and out of the cell is wasteful, especially if these substrates ultimately accumulate in the cell despite the energy being used for efflux. We have shown in embryos that even futile cycling is unlikely to represent a significant cost as a result of reaching a maximal transporter capacity at sufficient substrate concentrations. However, if transporter titer is elevated, such as in ABCB overexpressing drug-resistant cells, exposure to typically benign chemicals that are transporter substrates can result in cell death (Pluchino et al., 2012). Referred to as collateral sensitivity, this most likely arises through secondary effects of transporter activity such as excess production of reactive oxygen species (Hall et al., 2009). In another case of collateral sensitivity of drug-resistant cells, a transporter substrate rapidly induces depletion of cellular ATP via simultaneous stimulation of the transporter ATPase and disruption of mitochondrial membranes, thus using cellular ATP stores and impeding recovery of depleted ATP (Kabanov et al., 2003).
A different type of futile transport can also ensue because transporters recognize a structurally diverse array of substrates based on hydrophobicity, not toxicity. This is a good general strategy for dealing with a broad and variable range of membrane-permeable foreign chemicals. However, wide substrate specificity can result in transporters being hijacked into effluxing non-toxic substrates (Smital et al., 2004). These chemosensitizers can reduce the effectiveness of the toxicant protection system and add further costs with no benefit.
Obligate versus discretionary costs in an energy budget
A final factor in contextualizing the cost of protection by efflux transporters is that of obligatory versus discretionary energy costs. Examples of obligatory costs are maintaining the Na+/K+ ratio, regulating cell pH and maintaining a constant level of ATP. Specialized cells also have to maintain necessary activities such as nerve conduction, muscle activity in the heart and so on. Energy from these functions cannot be diverted for long without deleterious consequences, and can represent a large portion of metabolism, though this energy would vary by tissue. As noted earlier, the energy for maintaining ion gradients in marine organisms tends to be particularly high; 50–80% of the energy of the developing sea urchin embryo is used by the Na+/K+-ATPase (Leong and Manahan, 1997; Leong and Manahan, 1999). Examples of discretionary costs are those associated with growth and reproduction, which can be postponed or reduced until nutrients become available (Ramirez Llodra, 2002). In barrier tissues, where transporters are used as a constitutively active first-line of defense, costs of xenobiotic efflux would fall under the category above of ‘obligatory costs in specialized cells’. The ability of the blood–brain barrier to effectively efflux substrates is a necessary function; without transporter activity, organisms can become more susceptible to neurotoxins (Bard and Gadbois, 2007).
It is useful to consider obligatory and discretionary activities in the context of available energy. While an additional expense, such as effluxing a xenobiotic, may seem small in comparison to the entire energy budget, the more realistic comparison is to the energy resources available at that moment. Even though the energy use for xenobiotic protection by efflux transporters is a small percentage of total aerobic respiration, it could become a significant drain if food is limiting and/or transporter expression is elevated. A rapid increase in mitochondrial respiration for fueling transporter activity at the onset of exposure to a substrate could be particularly deleterious because it utilizes short-term energy and simultaneously increases reactive oxygen species production by forcing mitochondria into peak levels of activity (Hall et al., 2009; Pluchino et al., 2012). This would all be exacerbated for futile substrates.
These views enlarge the cost to fitness from exposure to xenobiotics. It is generally assumed that any cost arises from toxic effects of the chemical, but under stress conditions such as resource limitation, a fitness decrease could also ensue from this increased energy expenditure (or byproducts thereof) towards cellular defenses. Future research separating these distinct but related costs will further a more accurate understanding of trade-offs involved in the evolution and ecology of mitigating environmental stress.
In all, this study demonstrates that multidrug transporters are a low-cost but effective first line of defense against xenobiotics at concentrations likely to be found in the environment. By limiting the total efflux capacity, sea urchin embryos prevent costs from ballooning out of control while still protecting against unpredictable exposure to foreign chemicals in the environment.
ACKNOWLEDGEMENTS
We would like to acknowledge and thank Gabrielle Winters for her volunteer efforts on this research. We would also like to thank Dr Gary Cherr for help revising the manuscript.
FOOTNOTES
COMPETING INTERESTS
No competing interests declared.
FUNDING
Supported by grant 0446384 to D.E. from the National Science Foundation. B.C. was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) T32 HD007249. A.H. was supported by NICHD 050870. Deposited in PMC for release after 12 months.
REFERENCES
- Aänismaa P., Seelig A. (2007). P-Glycoprotein kinetics measured in plasma membrane vesicles and living cells. Biochemistry 46, 3394-3404 [DOI] [PubMed] [Google Scholar]
- Al-Shawi M. K., Polar M. K., Omote H., Figler R. A. (2003). Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 278, 52629-52640 [DOI] [PubMed] [Google Scholar]
- Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., et al. (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718-1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson B. S., Phillips B. M., Hunt J. W., Clark S. L., Voorhees J. P., Tjeerdema R. S., Casteline J., Stewart M., Crane D., Mekebri A. (2010). Evaluation of methods to determine causes of sediment toxicity in San Diego Bay, California, USA. Ecotoxicol. Environ. Saf. 73, 534-540 [DOI] [PubMed] [Google Scholar]
- Bains O. S., Kennedy C. J. (2005). Alterations in respiration rate of isolated rainbow trout hepatocytes exposed to the P-glycoprotein substrate rhodamine 123. Toxicology 214, 87-98 [DOI] [PubMed] [Google Scholar]
- Bard S. M., Gadbois S. (2007). Assessing neuroprotective P-glycoprotein activity at the blood-brain barrier in killifish (Fundulus heteroclitus) using behavioural profiles. Mar. Environ. Res. 64, 679-682 [DOI] [PubMed] [Google Scholar]
- Begley D. J. (2004). ABC transporters and the blood-brain barrier. Curr. Pharm. Des. 10, 1295-1312 [DOI] [PubMed] [Google Scholar]
- Behravan J., Piquette-Miller M. (2007). Drug transport across the placenta, role of the ABC drug efflux transporters. Expert Opin. Drug Metab. Toxicol. 3, 819-830 [DOI] [PubMed] [Google Scholar]
- Boesch D., Gavériaux C., Jachez B., Pourtier-Manzanedo A., Bollinger P., Loor F. (1991). In vivo circumvention of P-glycoprotein-mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Res. 51, 4226-4233 [PubMed] [Google Scholar]
- Bow D. A., Perry J. L., Miller D. S., Pritchard J. B., Brouwer K. L. (2008). Localization of P-gp (Abcb1) and Mrp2 (Abcc2) in freshly isolated rat hepatocytes. Drug Metab. Dispos. 36, 198-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxterman H. J., Pinedo H. M., Kuiper C. M., Kaptein L. C., Schuurhuis G. J., Lankelma J. (1988). Induction by verapamil of a rapid increase in ATP consumption in multidrug-resistant tumor cells. FASEB J. 2, 2278-2282 [DOI] [PubMed] [Google Scholar]
- Broxterman H. J., Pinedo H. M., Kuiper C. M., Schuurhuis G. J., Lankelma J. (1989). Glycolysis in P-glycoprotein-overexpressing human tumor cell lines. Effects of resistance-modifying agents. FEBS Lett. 247, 405-410 [DOI] [PubMed] [Google Scholar]
- Choudhuri S., Klaassen C. D. (2006). Structure, function, expression, genomic organization, and single nucleotide polymorphisms of human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) efflux transporters. Int. J. Toxicol. 25, 231-259 [DOI] [PubMed] [Google Scholar]
- Crowley K. S., Phillion D. P., Woodard S. S., Schweitzer B. A., Singh M., Shabany H., Burnette B., Hippenmeyer P., Heitmeier M., Bashkin J. K. (2003). Controlling the intracellular localization of fluorescent polyamide analogues in cultured cells. Bioorg. Med. Chem. Lett. 13, 1565-1570 [DOI] [PubMed] [Google Scholar]
- Davidson A. L., Dassa E., Orelle C., Chen J. (2008). Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72, 317-364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R. J., Locher K. P. (2006). Structure of a bacterial multidrug ABC transporter. Nature 443, 180-185 [DOI] [PubMed] [Google Scholar]
- Döppenschmitt S., Langguth P., Regårdh C. G., Andersson T. B., Hilgendorf C., Spahn-Langguth H. (1999). Characterization of binding properties to human P-glycoprotein: development of a [3H]verapamil radioligand-binding assay. J. Pharmacol. Exp. Ther. 288, 348-357 [PubMed] [Google Scholar]
- Epel D., Luckenbach T., Stevenson C. N., Macmanus-Spencer L. A., Hamdoun A., Smital T. (2008). Efflux transporters: newly appreciated roles in protection against pollutants. Environ. Sci. Technol. 42, 3914-3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essodaigui M., Broxterman H. J., Garnier-Suillerot A. (1998). Kinetic analysis of calcein and calcein-acetoxymethylester efflux mediated by the multidrug resistance protein and P-glycoprotein. Biochemistry 37, 2243-2250 [DOI] [PubMed] [Google Scholar]
- Eytan G. D., Kuchel P. W. (1999). Mechanism of action of P-glycoprotein in relation to passive membrane permeation. Int. Rev. Cytol. 190, 175-250 [DOI] [PubMed] [Google Scholar]
- Eytan G. D., Regev R., Assaraf Y. G. (1996a). Functional reconstitution of P-glycoprotein reveals an apparent near stoichiometric drug transport to ATP hydrolysis. J. Biol. Chem. 271, 3172-3178 [DOI] [PubMed] [Google Scholar]
- Eytan G. D., Regev R., Oren G., Assaraf Y. G. (1996b). The role of passive transbilayer drug movement in multidrug resistance and its modulation. J. Biol. Chem. 271, 12897-12902 [DOI] [PubMed] [Google Scholar]
- Eytan G. D., Regev R., Oren G., Hurwitz C. D., Assaraf Y. G. (1997). Efficiency of P-glycoprotein-mediated exclusion of rhodamine dyes from multidrug-resistant cells is determined by their passive transmembrane movement rate. Eur. J. Biochem. 248, 104-112 [DOI] [PubMed] [Google Scholar]
- Gekeler V., Ise W., Sanders K. H., Ulrich W. R., Beck J. (1995). The leukotriene LTD4 receptor antagonist MK571 specifically modulates MRP associated multidrug resistance. Biochem. Biophys. Res. Commun. 208, 345-352 [DOI] [PubMed] [Google Scholar]
- Gökirmak T., Campanale J. P., Shipp L. E., Moy G. W., Tao H., Hamdoun A. (2012). Localization and substrate selectivity of sea urchin multidrug (MDR) efflux transporters. J. Biol. Chem. 287, 43876-43883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberger L. M., Lisanti C. J., Silva J. T., Horwitz S. B. (1991). Domain mapping of the photoaffinity drug-binding sites in P-glycoprotein encoded by mouse mdr1b. J. Biol. Chem. 266, 20744-20751 [PubMed] [Google Scholar]
- Hall M. D., Handley M. D., Gottesman M. M. (2009). Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 30, 546-556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdoun A., Epel D. (2007). Embryo stability and vulnerability in an always changing world. Proc. Natl. Acad. Sci. USA 104, 1745-1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdoun A. M., Cherr G. N., Roepke T. A., Epel D. (2004). Activation of multidrug efflux transporter activity at fertilization in sea urchin embryos (Strongylocentrotus purpuratus). Dev. Biol. 276, 452-462 [DOI] [PubMed] [Google Scholar]
- Higgins C. F. (2007). Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749-757 [DOI] [PubMed] [Google Scholar]
- Hildebrand J. L., Bains O. S., Lee D. S. H., Kennedy C. J. (2009). Functional and energetic characterization of P-gp-mediated doxorubicin transport in rainbow trout (Oncorhynchus mykiss) hepatocytes. Comp. Biochem. Physiol. 149, 65-72 [DOI] [PubMed] [Google Scholar]
- Holland I. B., Blight M. A. (1999). ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol. 293, 381-399 [DOI] [PubMed] [Google Scholar]
- Kabanov A. V., Batrakova E. V., Alakhov V. Y. (2003). An essential relationship between ATP depletion and chemosensitizing activity of Pluronic block copolymers. J. Control. Release 91, 75-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliokoski A., Niemi M. (2009). Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 158, 693-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpin D. W., Furlong E. T., Meyer M. T., Thurman E. M., Zaugg S. D., Barber L. B., Buxton H. T. (2002). Pharmaceuticals, hormones, and other organic wastewater contaminants in U.S. streams, 1999-2000: a national reconnaissance. Environ. Sci. Technol. 36, 1202-1211 [DOI] [PubMed] [Google Scholar]
- Leong P., Manahan D. (1997). Metabolic importance of Na+/K+-ATPase activity during sea urchin development. J. Exp. Biol. 200, 2881-2892 [DOI] [PubMed] [Google Scholar]
- Leong P. K., Manahan D. T. (1999). Na+/K+-ATPase activity during early development and growth of an Antarctic sea urchin. J. Exp. Biol. 202, 2051-2058 [DOI] [PubMed] [Google Scholar]
- Marsh A. G., Manahan D. T. (1999). A method for accurate measurements of the respiration rates of marine invertebrate embryos and larvae. Mar. Ecol. Prog. Ser. 184, 1-10 [Google Scholar]
- Mathews C. K. (1975). Giant pools of DNA precursors in sea urchin eggs. Exp. Cell Res. 92, 47-56 [DOI] [PubMed] [Google Scholar]
- Morris D. I., Speicher L. A., Ruoho A. E., Tew K. D., Seamon K. B. (1991). Interaction of forskolin with the P-glycoprotein multidrug transporter. Biochemistry 30, 8371-8379 [DOI] [PubMed] [Google Scholar]
- Niemi M. (2007). Role of OATP transporters in the disposition of drugs. Pharmacogenomics 8, 787-802 [DOI] [PubMed] [Google Scholar]
- Pannevis M. C., Houlihan D. F. (1992). The energetic cost of protein synthesis in isolated hepatocytes of rainbow trout (Oncorhynchus mykiss). J. Comp. Physiol. B 162, 393-400 [DOI] [PubMed] [Google Scholar]
- Patzlaff J. S., van der Heide T., Poolman B. (2003). The ATP/substrate stoichiometry of the ATP-binding cassette (ABC) transporter OpuA. J. Biol. Chem. 278, 29546-29551 [DOI] [PubMed] [Google Scholar]
- Pawagi A. B., Wang J., Silverman M., Reithmeier R. A., Deber C. M. (1994). Transmembrane aromatic amino acid distribution in P-glycoprotein. A functional role in broad substrate specificity. J. Mol. Biol. 235, 554-564 [DOI] [PubMed] [Google Scholar]
- Pleban K., Kopp S., Csaszar E., Peer M., Hrebicek T., Rizzi A., Ecker G. F., Chiba P. (2005). P-glycoprotein substrate binding domains are located at the transmembrane domain/transmembrane domain interfaces: a combined photoaffinity labeling-protein homology modeling approach. Mol. Pharmacol. 67, 365-374 [DOI] [PubMed] [Google Scholar]
- Pluchino K. M., Hall M. D., Goldsborough A. S., Callaghan R., Gottesman M. M. (2012). Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 15, 98-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polli J. W., Wring S. A., Humphreys J. E., Huang L., Morgan J. B., Webster L. O., Serabjit-Singh C. S. (2001). Rational use of in vitro P-glycoprotein assays in drug discovery. J. Pharmacol. Exp. Ther. 299, 620-628 [PubMed] [Google Scholar]
- Ramirez Llodra E. (2002). Fecundity and life-history strategies in marine invertebrates. Adv. Mar. Biol. 43, 87-170 [DOI] [PubMed] [Google Scholar]
- Raub T. J. (2006). P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol. Pharm. 3, 3-25 [DOI] [PubMed] [Google Scholar]
- Rees D. C., Johnson E., Lewinson O. (2009). ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 10, 218-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev R., Katzir H., Yeheskely-Hayon D., Eytan G. D. (2007). Modulation of P-glycoprotein-mediated multidrug resistance by acceleration of passive drug permeation across the plasma membrane. FEBS J. 274, 6204-6214 [DOI] [PubMed] [Google Scholar]
- Ricardo S., Lehmann R. (2009). An ABC transporter controls export of a Drosophila germ cell attractant. Science 323, 943-946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez Robledo V., Smyth W. F. (2008). A study of the analytical behaviour of selected new molecular entities using electrospray ionisation ion trap mass spectrometry, liquid chromatography, gas chromatography and polarography and their determination in serum at therapeutic concentrations. Anal. Chim. Acta 623, 221-230 [DOI] [PubMed] [Google Scholar]
- Rosenberg M. F., Kamis A. B., Callaghan R., Higgins C. F., Ford R. C. (2003). Three-dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J. Biol. Chem. 278, 8294-8299 [DOI] [PubMed] [Google Scholar]
- Sauna Z. E., Ambudkar S. V. (2000). Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc. Natl. Acad. Sci. USA 97, 2515-2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmell E., Lennarz W. J. (1974). Phospholipid metabolism in the eggs and embryos of the sea urchin Arbacia punctulata. Biochemistry 13, 4114-4121 [DOI] [PubMed] [Google Scholar]
- Sea Urchin Genome Sequencing Consortium. Sodergren E., Weinstock G. M., Davidson E. H., Cameron R. A., Gibbs R. A., Angerer R. C., Angerer L. M., Arnone M. I., Burgess D. R., et al. (2006). The genome of the sea urchin Strongylocentrotus purpuratus. Science 314, 941-952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M. A., van Veen H. W. (2009). Molecular basis of multidrug transport by ABC transporters. Biochim. Biophys. Acta 1794, 725-737 [DOI] [PubMed] [Google Scholar]
- Shapiro A. B., Ling V. (1995). Reconstitution of drug transport by purified P-glycoprotein. J. Biol. Chem. 270, 16167-16175 [DOI] [PubMed] [Google Scholar]
- Shilling F. M., Manahan D. T. (1990). Energetics of early development for the sea urchins Strongylocentrotus purpuratus and Lytechinus pictus and the crustacean Artemia sp. Mar. Biol. 106, 119-128 [Google Scholar]
- Smital T., Luckenbach T., Sauerborn R., Hamdoun A. M., Vega R. L., Epel D. (2004). Emerging contaminants – pesticides, PPCPs, microbial degradation products and natural substances as inhibitors of multixenobiotic defense in aquatic organisms. Mutat. Res. 552, 101-117 [DOI] [PubMed] [Google Scholar]
- Szakács G., Paterson J. K., Ludwig J. A., Booth-Genthe C., Gottesman M. M. (2006). Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5, 219-234 [DOI] [PubMed] [Google Scholar]
- Tran T. T., Mittal A., Aldinger T., Polli J. W., Ayrton A., Ellens H., Bentz J. (2005). The elementary mass action rate constants of P-gp transport for a confluent monolayer of MDCKII-hMDR1 cells. Biophys. J. 88, 715-738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Richter O., Glavinas H., Krajcsi P., Liehner S., Siewert B., Zech K. (2009). A novel screening strategy to identify ABCB1 substrates and inhibitors. Naunyn Schmiedebergs Arch. Pharmacol. 379, 11-26 [DOI] [PubMed] [Google Scholar]