

Insulin-dependent translocation of Glut4 to the plasma membrane of fat and skeletal muscle cells plays the key role in postprandial clearance of blood glucose. In undifferentiated cells, insulin responsiveness of Glut4 depends on the presence of sortilin, whereas sortilin responds to insulin regardless of Glut4 expression.
Abstract
Insulin-dependent translocation of glucose transporter 4 (Glut4) to the plasma membrane of fat and skeletal muscle cells plays the key role in postprandial clearance of blood glucose. Glut4 represents the major cell-specific component of the insulin-responsive vesicles (IRVs). It is not clear, however, whether the presence of Glut4 in the IRVs is essential for their ability to respond to insulin stimulation. We prepared two lines of 3T3-L1 cells with low and high expression of myc7-Glut4 and studied its translocation to the plasma membrane upon insulin stimulation, using fluorescence-assisted cell sorting and cell surface biotinylation. In undifferentiated 3T3-L1 preadipocytes, translocation of myc7-Glut4 was low regardless of its expression levels. Coexpression of sortilin increased targeting of myc7-Glut4 to the IRVs, and its insulin responsiveness rose to the maximal levels observed in fully differentiated adipocytes. Sortilin ectopically expressed in undifferentiated cells was translocated to the plasma membrane regardless of the presence or absence of myc7-Glut4. AS160/TBC1D4 is expressed at low levels in preadipocytes but is induced in differentiation and provides an additional mechanism for the intracellular retention and insulin-stimulated release of Glut4.
Adipocytes, skeletal muscle cells, and some neurons respond to insulin stimulation by translocating intracellular glucose transporter 4 (Glut4) to the plasma membrane. In all these cells, the insulin-responsive pool of Glut4 is localized in small membrane vesicles, the insulin-responsive vesicles (IRVs; Kandror and Pilch, 2011; Bogan, 2012). The protein composition of these vesicles has been largely characterized (Kandror and Pilch, 2011; Bogan, 2012). The IRVs consist predominantly of Glut4, insulin-responsive aminopeptidase (IRAP), sortilin, low-density-lipoprotein receptor–related protein 1 (LRP1), SCAMPs, and VAMP2. Glut4, IRAP, and sortilin physically interact with each other, which might be important for the biogenesis of the IRVs (Shi and Kandror, 2007; Shi et al., 2008). In addition, the IRVs compartmentalize recycling receptors, such as the transferrin receptor and the IGF2/mannose 6-phosphate receptor, although it is not clear whether these receptors represent obligatory vesicular components or their presence in the IRVs is explained by mass action (Pilch, 2008), inefficient sorting, or other reasons.
Deciphering of the protein composition of the IRVs is important because it is likely to explain their unique functional property: translocation to the plasma membrane in response to insulin stimulation. Even if we presume that IRV trafficking is controlled by loosely associated peripheral membrane proteins, the latter should still somehow recognize the core vesicular components that create the “biochemical individuality” of this compartment. In spite of our knowledge of the IRV protein composition, however, the identity of the protein(s) that confer insulin sensitivity to these vesicles is unknown.
Insulin responsiveness of the IRVs was associated with either IRAP or Glut4. Thus it was shown that Glut4 interacted with the intracellular anchor TUG (Bogan et al., 2003, 2012), whereas IRAP associated with other proteins implemented in the regulation of Glut4 translocation, such as AS160 (Larance et al., 2005; Peck et al., 2006), p115 (Hosaka et al., 2005), tankyrase (Yeh et al., 2007), and several others (reviewed in Bogan, 2012). Results of these studies, or at least their interpretations, are not necessarily consistent with each other, as the existence of multiple independent anchors for the IRVs is, although possible, unlikely.
Ablation of the individual IRV proteins has also led to controversial data. Thus knockout of IRAP decreases total protein levels of Glut4 but does not affect its translocation in the mouse model (Keller et al., 2002). On the contrary, knockdown of IRAP in 3T3-L1 adipocytes has a strong inhibitory effect on translocation of Glut4 (Yeh et al., 2007). In yet another study, knockdown of IRAP in 3T3-L1 adipocytes did not affect insulin-stimulated translocation of Glut4 but increased its plasma membrane content under basal conditions (Jordens et al., 2010). By the same token, total or partial ablation of Glut4 had various effects on expression levels, intracellular localization, and translocation of IRAP (Jiang et al., 2001; Abel et al., 2004; Carvalho et al., 2004; Gross et al., 2004; Yeh et al., 2007). Knockdown of either sortilin or LRP1 decreased protein levels of Glut4 (Shi and Kandror, 2005; Jedrychowski et al., 2010).
One model that might explain these complicated and somewhat inconsistent results is that depletion of either major integral protein of the IRVs disrupts the network of interactions between vesicular proteins and thus decreases the efficiency of protein sorting into the IRVs (Kandror and Pilch, 2011). Correspondingly, the remaining IRV components that cannot be faithfully compartmentalized in the vesicles are either degraded (Jiang et al., 2001; Keller et al., 2002; Abel et al., 2004; Carvalho et al., 2004; Shi and Kandror, 2005; Yeh et al., 2007; Jedrychowski et al., 2010) or mistargeted (Jiang et al., 2001; Jordens et al., 2010), depending on experimental conditions and types of cells used in these studies. In other words, knockdown of any major IRV component may decrease vesicle formation along with insulin responsiveness. Thus, in spite of a large body of literature, the identity of protein(s) that confer insulin responsiveness to the IRVs is unknown.
Here we used a gain-of-function approach to address this question. Specifically, we attempted to “build” functional IRVs in undifferentiated 3T3-L1 preadipocytes by forced expression of the relevant proteins. Undifferentiated preadipocytes do not express Glut4 or sortilin and lack IRVs (ElJack et al., 1999; Shi and Kandror, 2005; Shi et al., 2008). Correspondingly, IRAP, which is expressed in these cells, shows low insulin response (Ross et al., 1998; Shi et al., 2008). We found that ectopic expression of increasing amounts of Glut4 in undifferentiated preadipocytes does not lead to its marked translocation to the plasma membrane upon insulin stimulation. On the contrary, sortilin expressed in undifferentiated preadipocytes was localized in the IRVs and was translocated to the plasma membrane in response to insulin stimulation. Moreover, upon coexpression with Glut4, sortilin dramatically increased its insulin responsiveness to the levels observed in fully differentiated adipocytes. Thus sortilin may represent the key component of the IRVs, which is responsible not only for the formation of vesicles (Shi and Kandror, 2005; Ariga et al., 2008; Hatakeyama and Kanzaki, 2011), but also for their insulin responsiveness. It is worth noting that sortilin levels are significantly decreased in obese and diabetic humans and mice (Kaddai et al., 2009). We thus suggest that sortilin may be a novel and important target in the fight against insulin resistance and diabetes.
Our experiments also demonstrate that undifferentiated preadipocytes lack a mechanism for the full intracellular retention of Glut4 that can be achieved by ectopic expression of AS160/TBC1D4.
RESULTS
Ectopically expressed sortilin shows insulin responsiveness in 3T3-L1 preadipocytes
In differentiating 3T3-L1 adipocytes, sortilin and Glut4 are induced on days 3 and 4, respectively (Shi and Kandror, 2005). Immediately upon induction, Glut4 is incorporated in the preexisting IRVs (ElJack et al., 1999; Shi and Kandror, 2005). Of importance, the IRVs do not exist in undifferentiated preadipocytes but are formed on day 3 of differentiation simultaneously with induction of sortilin (Shi and Kandror, 2005). On the basis of these results, we hypothesized that sortilin played the key role in the formation of insulin-responsive vesicles in differentiating adipocytes (Shi and Kandror, 2005). To further clarify the role of this protein, we stably expressed sortilin tagged with myc/histidine (His) epitopes at the C-terminus in 3T3-L1 cells, which are named S cells. Note that expression of sortilin-myc/His in undifferentiated S preadipocytes is equivalent to expression of endogenous sortilin in differentiated adipocytes (Figure 1A). Using sucrose gradient centrifugation, we showed that, in undifferentiated preadipocytes, ectopically expressed sortilin is localized in the IRV-like vesicles (Figure 1B; see also Shi and Kandror, 2008).
FIGURE 1:

Sortilin ectopically expressed in undifferentiated 3T3-L1 preadipocytes is localized in the IRV-like vesicles and is translocated to the cell surface in response to insulin stimulation. (A) Undifferentiated and differentiated 3T3-L1 cells were homogenized, and total cell lysates (40 μg) were analyzed by Western blotting with antibodies against sortilin. Cyclophilin A was used as a loading control. Dotted line indicates that irrelevant lanes have been spliced out. Representative result of three independent experiments. (B) Undifferentiated S preadipocytes and differentiated wild-type 3T3-L1 adipocytes were homogenized and centrifuged at 27,000 × g for 35 min, and supernatants (1 mg of total protein) were separated by continuous sucrose gradient centrifugation. The arrow indicates the direction of sedimentation. Representative result of at least 10 independent experiments. (C) Undifferentiated S preadipocytes were biotinylated with sulfo-NHS-S-S-biotin, and biotinylated proteins were isolated from 100–140 μg of total cell lysates using streptavidin-agarose and analyzed by Western blotting along with total lysates and unbound material (40 μg each). Representative result of four independent experiments. (D) Undifferentiated empty vector (EV)–infected and S preadipocytes were incubated with 7 nM [3H]neurotensin for the indicated periods of time, washed as described in Materials and Methods, and counted in the scintillation counter. Normalized mean values ± SEM of three experiments.
We showed that cell surface biotinylation of sortilin-myc/His in S preadipocytes is increased by insulin (2.00 ± 0.23)-fold, suggesting that this protein is translocated to the plasma membrane (Figure 1C). We decided to support this result by an independent approach based on the uptake of radioactive neurotensin. It was reported that sortilin binds neurotensin and, in fact, may represent neurotensin receptor in neurons (Mazella et al., 1998). We used this property of sortilin to study its response to insulin stimulation. As is shown in Figure 1D, internalization of radioactive neurotensin is increased by insulin in S cells but not in control 3T3-L1 preadipocytes, suggesting that sortilin is translocated to the plasma membrane in response to insulin.
Sortilin confers insulin responsiveness to Glut4
Insulin responsiveness of Glut4 ectopically expressed in undifferentiated cells is a controversial issue. Earlier studies (Haney et al., 1991; Hudson et al., 1992), as well as our subsequent experiments (Shi and Kandror, 2005), showed that Glut4 expressed in undifferentiated fibroblasts is not insulin responsive. McGraw's group, however, reported that Glut4 can undergo insulin-dependent translocation to the cell surface even in undifferentiated cells (Kanai et al., 1993; Lampson et al., 2000; Sadacca et al., 2013). These studies raise a possibility that translocation of Glut4 in undifferentiated cells has not been observed due to technical reasons, such as low levels of its expression and/or low stability of the transporter (Shi and Kandror, 2005; Liu et al., 2007). Given that sortilin is known to stabilize Glut4 in preadipocytes (Shi and Kandror, 2005; Hatakeyama and Kanzaki, 2011), this effect alone might account for the effect of sortilin on translocation of Glut4 in response to insulin.
To test this possibility, we used the retroviral expression system to prepare two lines of stably transfected 3T3-L1 preadipocytes, one with low (low G) and one with high (high G) content of Glut4 tagged with seven consecutive myc epitopes in the first luminal loop (myc7-Glut4). Then we stably expressed sortilin-myc/His in high-G cells with the help of the lentivirus carrying a different selection marker. This cell line was named GS. Figure 2A shows expression of various proteins in all three cell lines. Note that, in agreement with our earlier report (Shi and Kandror, 2005), expression of sortilin in high-G cells causes a small but significant increase in the total myc7-Glut4 content (Figure 2B).
FIGURE 2:

Compartmentalization of ectopically expressed myc7-Glut4 in undifferentiated preadipocytes. (A) Undifferentiated and differentiated cells expressing low levels of myc7-Glut4 (LG), high levels of myc7-Glut4 (HG), or both myc7-Glut4 and sortilin-myc/His (GS) were homogenized, and total cell lysates were analyzed by Western blotting. Cellugyrin and glyceraldehyde 3-phosphate dehydrogenase were used as a loading control. (B) Quantification of myc7-Glut4 signal shown in A, based on three independent experiments. *p < 0.05, **p < 0.01. (C) Undifferentiated LG, HG, and GS preadipocytes were homogenized and centrifuged at 27,000 × g for 35 min. Supernatants were pelleted by centrifugation at 200,000 × g for 90 min and analyzed by Western blotting along with aliquots of 27,000 × g pellets. Representative result of three independent experiments. (D) Undifferentiated HG and GS preadipocytes were homogenized and centrifuged at 27,000 × g for 35 min. Supernatants were analyzed by continuous sucrose gradient centrifugation for 55 min in a SW55 rotor at 48,000 rpm. The arrow indicates direction of sedimentation. Fractions with myc7-Glut4 from HG and GS preadipocytes were transferred to the same membrane and processed simultaneously and therefore are directly comparable to each other. Representative result of at least four independent experiments.
In the next experiment, we separated the total lysate of low-G, high-G, and GS preadipocytes by centrifugation into two fractions: a vesicular fraction (27,000 × g supernatant) and a heavy membrane fraction (27,000 × g pellet). As shown in Figure 2C, expression of sortilin-myc/His increases myc7-Glut4 content specifically in the vesicular fraction. Then we analyzed the intracellular compartmentalization of myc7-Glut4 in the vesicular fraction with the help of sucrose gradient centrifugation. Figure 2D demonstrates that the presence of myc7-Glut4 in small vesicles is increased in GS preadipocytes in comparison to high G cells. Further analysis shows that the sedimentational properties of Glut4 vesicles formed in undifferentiated cells by ectopic expression of myc7-Glut4 and sortilin-myc/His are close to those of “classic” IRVs from fat and skeletal muscle cells (compare Figures 2D and 1B).
Using fluorescence-assisted cell sorting, we found that myc7-Glut4 is translocated to the cell surface in both low-G and high-G cells, but only to a small degree (Figure 3, A, B, and F). Note that a fourfold difference in the total content of myc7-Glut4 between these two cell lines does not lead to corresponding changes in translocation of the transporter in these cells. Then we compared insulin responsiveness of myc7-Glut4 in high-G and GS preadipocytes that express close amounts of the transporter. We found that the mean insulin responsiveness of myc7-Glut4 in GS preadipocytes is markedly higher than in G cells (Figure 3 B, C, and G).
FIGURE 3:

Translocation of ectopically expressed myc7-Glut4 in undifferentiated and differentiated 3T3-L1 cells. Undifferentiated (Fb) and differentiated for 5–7 d (Ad) cells were treated (+) or not treated (–) with insulin for 15 min. (A–E) Immunofluorescence staining of representative cells with anti-myc antibody. (F–I) Translocation of myc7-Glut4 analyzed by fluorescence-assisted cell sorting as described in Materials and Methods. Each is a representative result of at least two independent experiments.
To compare the maximal insulin response of GS preadipocytes with that of differentiated adipocytes, we performed fluorescence-activated cell sorting (FACS) analysis of myc7-Glut4 translocation in undifferentiated GS preadipocytes and differentiated high-G adipocytes that express sortilin endogenously (Figure 1A). Figure 3, C, D, and H, shows that insulin response of myc7-Glut4 in GS preadipocytes is even higher than in high-G adipocytes. The explanation for this “overshoot” is not known. It might be attributed to the fact that, in differentiated cells, myc7-Glut4 is “diluted” by endogenously expressed Glut4. At the same time, undifferentiated cells have significantly more myc7-Glut4 at the plasma membrane under the basal conditions than differentiated cells. Thus GS preadipocytes may have the same amount of the IRVs as differentiated adipocytes but, in agreement with results in Figure 3, B, C, and G, do not have a mechanism for the efficient intracellular sequestration and/or basal retention of Glut4. Apparently the latter mechanism is independent of sortilin-driven biogenesis of the IRVs (see next section).
In the next experiment, we directly demonstrated that intracellular sequestration of myc7-Glut4 is achieved upon differentiation of preadipocytes. Indeed, Figure 3, C, E, and I, shows that basal GS preadipocytes have much more myc7-Glut4 at the plasma membrane than differentiated cells in spite of the fact that the latter express significantly more myc7-Glut4 (Supplemental Figure S1). The latter observation suggests that activity of lentiviral promoters that drive the expression of myc7-Glut4 and sortilin-myc/His is markedly stimulated upon adipocyte differentiation.
We supported data shown in Figure 3 by manual analysis of cells using ImageJ software, with essentially the same results (Supplemental Figure S2). Note that because of differentiation-induced changes in the shape of the cell, preadipocytes and adipocytes have to be analyzed separately.
We decided to confirm the effect of sortilin on translocation of Glut4 by an independent approach, unrelated to immunofluorescence staining, that is, by cell surface biotinylation. Although wild-type Glut4 has but one lysine in its extracellular regions and thus cannot be efficiently biotinylated, every myc epitope has a lysine residue, so that myc7-Glut4 represents a good target for biotinylation. As is shown in Figure 4A, cell surface biotinylation of myc7-Glut4 in high-G cells is not increased by insulin (1.01 ± 0.22). We believe that the sensitivity of this approach is not sufficient to detect a relatively small change in the plasma membrane myc7-Glut4 caused by insulin administration in high-G cells. On the contrary, biotinylation of myc7-Glut4 and sortilin-myc/His is clearly increased by insulin in GS preadipocytes (2.44 ± 0.26 and 1.95 ± 0.38, respectively; Figure 4B), suggesting that both proteins are translocated to the cell surface in response to insulin stimulation.
FIGURE 4:

Translocation of ectopically expressed myc7-Glut4 and sortilin-myc/His in undifferentiated preadipocytes by cell surface biotinylation. Undifferentiated preadipocytes were biotinylated with sulfo-NHS-biotin, and myc-tagged proteins were isolated by immunoprecipitation and analyzed by Western blotting. Representative result of three independent experiments.
Of importance, insulin-stimulated increase in sortilin biotinylation is similar in GS and S preadipocytes (Figure 4B). Thus Glut4 does not contribute much in terms of insulin responsiveness of corresponding vesicles, and the protein that renders this compartment insulin sensitive could be sortilin.
Intracellular sequestration of the IRVs during differentiation depends on the expression of AS160/TBC1D4
As shown in Figure 3 and Supplemental Figure S2, differentiation of high-G and GS preadipocytes is associated with efficient intracellular sequestration of myc7-Glut4. Two mechanisms for such sequestration have been described. One depends on the expression of TUG (Bogan et al., 2003) and another on AS160/TBC1D4 together with its target Rab10 (Sano et al., 2003, 2007, 2008; Eguez et al., 2005; Chen et al., 2012). As is shown in Figure 5A, all of these proteins are expressed in both undifferentiated and differentiated cells; however, AS160/TBC1D4 is strongly induced upon differentiation (Figure 5A and Supplemental Figure S3). Low expression of AS160/TBC1D4 in preadipocytes may explain our observation that the intracellular retention of myc7-Glut4 in these cells is not complete. Indeed, a substantial amount of myc7-Glut4 in preadipocytes is localized at the plasma membrane and is sequestered inside the cell upon differentiation (Figure 3). To determine whether the latter effect is associated with the induction of AS160/TBC1D4, we overexpressed this protein in undifferentiated GS preadipocytes. cDNA for green fluorescent protein (GFP) was used to visualize transfected cells. Immunofluorescence staining demonstrates that expression of AS160/TBC1D4 leads to an efficient intracellular sequestration of myc7-Glut4 in preadipocytes (Figure 5, B and C). Of importance, treatment of transfected preadipocytes with insulin leads to phosphorylation of ectopically expressed AS160/TBC1D4 (Figure 5C) and reconstitutes PM localization of myc7-Glut4 (Figure 5B). In agreement with our results, Sadacca et al. (2013) recently found that AS160/TBC1D4 increases intracellular retention and insulin responsiveness of Glut4 ectopically expressed in CHO cells.
FIGURE 5:
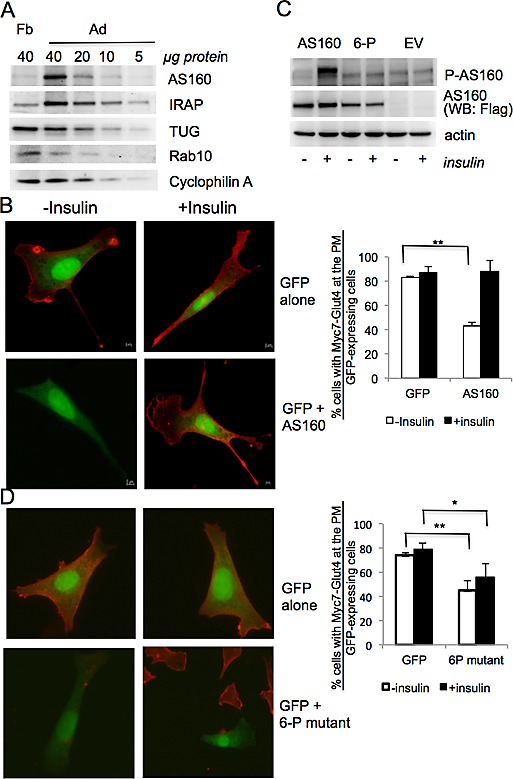
AS160/TBC1D4 is induced upon differentiation of 3T3-L1 cells and is required for intracellular sequestration of myc7-Glut4. (A) Expression of proteins in undifferentiated and differentiated 3T3-L1 cells. Representative results of two independent experiments. (B) GS preadipocytes were electroporated with either GFP cDNA alone or a mixture of cDNAs for GFP and AS160/TBC1D4 as indicated. After 48 h, cells were treated or not treated with insulin for 15 min and immunostained with antibody against the myc epitope without permeabilization. Left, representative cells. Right, percentage of myc-positive cells among randomly chosen GFP-positive cells (35–56 cells for each field). Representative results of three experiments. (C) GS preadipocytes were transfected with cDNA for AS160/TBC1D4 and 6-P mutant using Lipofectamine 2000. After 48 h, cells were treated or not treated with insulin for 15 min and harvested, and total cell lysates (40 μg) were analyzed by Western blotting. Representative result of three independent experiments. (D) GS preadipocytes were transfected with either GFP cDNA alone or with the mixture of cDNAs for GFP and 6-P mutant as indicated using Lipofectamine 2000. After 48 h, cells were treated or not treated with insulin for 15 min and immunostained with antibody against the myc epitope without permeabilization. Left, representative cells. Right, percentage of myc-positive cells among randomly chosen GFP-positive cells. Representative results of three experiments, with 286–299 insulin-treated and untreated cells counted. *p < 0.05, **p < 0.01.
In the next experiment, we transfected preadipocytes with the cDNA for the unphosphorylated 6-P mutant of AS160/TBC1D4, which also provides efficient intracellular retention for myc7-Glut4 in basal cells (Figure 5D). This protein, however, is not phosphorylated in response to insulin stimulation (Figure 5C) and blocks the effect of insulin on translocation of myc7-Glut4 (Figure 5D).
DISCUSSION
Sortilin and other members of the sortilin family represent multiligand protein receptors in mammalian cells with various functions in protein sorting and signaling (Hermey, 2009). We (Shi and Kandror, 2005; Kim and Kandror, 2012) and others (Ariga et al., 2008; Hatakeyama and Kanzaki, 2011) found that sortilin is essential for the acquisition of insulin-stimulated glucose uptake in cells, although the mechanism of sortilin action is uncertain. According to one model, sortilin is directly involved in the formation of the IRVs on the perinuclear “donor” membranes that represent a specialized domain of the trans-Golgi network (TGN; Shewan et al., 2003), recycling endosomes (Karylowski et al., 2004), or both. It is also feasible that sortilin is involved in stabilization of Glut4 (Shi and Kandror, 2005) by, for example, facilitating its retrieval from early endosomes to the TGN (Hatakeyama and Kanzaki, 2011). In agreement with the latter hypothesis, it was shown that sortilin cycles between endosomes and the TGN (Nielsen et al., 2001) and is abundant in endosome-to-TGN transport vesicles (Mari et al., 2008).
To characterize the mechanism of sortilin action, we prepared and compared four stably transfected lines of 3T3-L1 cells: with low Glut4 expression (low-G cells), high Glut4 expression (high-G cells), cells expressing both Glut4 and sortilin (GS cells), and cells expressing only sortilin (S cells). We found that Glut4 has a small degree of insulin responsiveness in undifferentiated cells (Figure 3F and Supplemental Figure S3). Increase in levels of Glut4 protein per se, however, is not sufficient to confer full insulin responsiveness to the transporter in undifferentiated preadipocytes. Indeed, a fourfold up-regulation of myc7-Glut4 expression in high-G cells in comparison to low-G cells results in only a minute increase in translocation of the transporter (Figure 3, A, B, and F, and Supplemental Figure S2). At the same time, expression of sortilin results in a relatively small increase in total myc7-Glut4, whereas its insulin responsiveness is increased to the maximal level observed in differentiated adipocytes (Figure 3, B–D, G, and H). This result suggests that sortilin not only stabilizes myc7-Glut4 but also directly facilitates formation of the IRVs, a notion supported by the sucrose gradient analysis of Glut4-containing vesicles in preadipocytes (Figure 2, C and D).
According to our model of IRV biogenesis, the cytoplasmic tails target Glut4, IRAP, and sortilin to the perinuclear compartment where the IRVs are formed. In the lumen of this compartment, the Vps10p domain of sortilin interacts with the first luminal loop of Glut4 and the luminal domains of IRAP and, likely, LRP1. The heteromeric complex consisting of the major IRV proteins is then distributed from the donor membranes to the IRV as a single entity with the help of GGA adaptors, which bind to the cytoplasmic tail of sortilin, and ACAP1, which interacts with the central loop of Glut4 (reviewed in Kandror and Pilch, 2011).
On ectopic expression in undifferentiated preadipocytes that do not express sortilin, targeting of Glut4 (this study) and IRAP (Shi et al., 2008) to small vesicles is inefficient, and plasma membrane translocation of these proteins is low. On the contrary, sortilin ectopically expressed in these cells is recovered in small, IRV-like vesicles (Figure 1B), shows considerable insulin responsiveness (Figures 1, C and D, and 4B), and facilitates recruitment of both Glut4 and IRAP into the IRVs (Figure 2D; Shi et al., 2008). We suggest, therefore, that sortilin not only plays the key role in vesicle biogenesis but may also represent the long-sought IRV component responsible for their insulin sensitivity. The mechanism of this effect is unknown. We were unable to detect phosphorylation of sortilin in response to insulin stimulation (G.H. and K.V.K., results not shown). We believe that the search for sortilin-binding proteins in adipocytes may help to find an answer to this question.
Of importance, acquisition of insulin-stimulated glucose uptake in differentiating adipocytes requires not only formation of the IRVs, but also efficient sequestration of glucose transporters from the plasma membrane under basal conditions. Previous studies showed that in the process of differentiation, 3T3-L1 cells acquire a mechanism for the efficient sequestration of Glut1 (Yang et al., 1992) and IRAP (Ross et al., 1998). Here we show that the same is true for Glut4, as preadipocytes have significantly more myc7-Glut4 at the plasma membrane under basal conditions than differentiated adipocytes (Figure 3). We also show that complete sequestration of myc7-Glut4 is associated with an increase in expression of AS160/TBC1D4 (Figure 5).
Still, undifferentiated GS cells possess a significant pool of intracellular IRVs (Figure 2). Thus low endogenous levels of either AS160/TBC1D4 or TUG, which is abundant in preadipocytes, may be responsible for intracellular retention and insulin-stimulated release of the IRVs. Of note, massive translocation of myc7-Glut4 in GS preadipocytes that express low levels of AS160/TBC1D4 (Figure 3, G–I) implies that the latter protein may not regulate the principal insulin-sensitive event in Glut4 exocytosis, which is consistent with previous studies (Eguez et al., 2005; Bai et al., 2007; Brewer et al., 2011). Once AS160/TBC1D4 is expressed, however, it provides an additional mechanism for the intracellular retention and insulin-regulated release of Glut4 (Figure 5B).
MATERIALS AND METHODS
Reagents and antibodies
Insulin, bovine serum albumin, and other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). Bovine serum and fetal bovine serum (FBS) were from Atlanta Biologicals (Lawrenceville, GA). DMEM, Opti-MEM, D-PBS, Slowfade antifade solution, and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). Alexa 488–conjugated transferrin was from Molecular Probes (Carlsbad, CA). Sulfo-NHS-biotin, sulfo-NHS-S-S-biotin, streptavidin-agarose, and horseradish peroxidase–conjugated streptavidin were from Thermo Scientific (Rockford, IL). [3,11-tyrosyl-3,5-3H(N)]-Neurotensin and [3H]2-deoxyglucose were purchased from PerkinElmer (Waltham, MA). Monoclonal and polyclonal antibodies against myc epitope and rabbit polyclonal antibody against the phosphorylated form of AS160 (Thr-642) were from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibody against sortilin was from BD Bioscience PharMingen (San Diego, CA). Rabbit monoclonal antibody against LRP1 was from Abcam (Cambridge, MA). Rabbit polyclonal antibody against cellugyrin (Ac-CQNVETTEGYQPPPVY-OH) was raised and affinity purified by BioSource International (Camarillo, CA; Xu and Kandror, 2002). Cy3-conjugated anti-mouse immunoglobulin G (IgG) was obtained from Jackson ImmunoResearch (West Grove, PA). Rabbit polyclonal antibody against TUG was a kind gift of Jonathan Bogan (Yale Medical School, New Haven, CT). Chicken polyclonal antibody against the C-terminus of AS160/TBC1D4 (HPTNDKAKAGNKP), generated by Quality Controlled Biochemicals (Hopkinton, MA), was a kind gift of Michael Czech (University of Massachusetts Medical School, Worcester, MA). cDNA for Flag-tagged AS160/TBC1D4 and Flag-tagged unphosphorylated 6-P mutant (Sano et al., 2003) with Ser-318, Ser-341, Ser-570, Ser-588, Thr-642, and Thr-751 replaced for Ala was a kind gift of Gustav Lienhard (Dartmouth Medical School, Hanover, NH) and Takahiro Nagase (Kazusa DNA Research Institute, Chiba, Japan).
Stable cell lines
Preparation, culturing, and differentiation of 3T3-L1 cells stably transfected with mLNCX2 (EV cells), mLNCX2-sortilin-myc/His (S cells), and pBabe-myc7-Glut4 (G cells), as well as 3T3-L1 double transfected with pBabe-myc7-Glut4 and mLNCX2-sortilin-myc/His (GS cells), were described previously (Shi and Kandror, 2005). Cells were grown in DMEM containing 10% calf bovine serum. Two days after confluence, cells were transferred to the differentiation medium (DMEM with 10% FBS, 1.67 μM insulin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine). After 48 h, differentiation medium was replaced with DMEM containing 10% FBS.
Transient transfection of undifferentiated 3T3-L1 preadipocytes
3T3-L1 cells were trypsinized, washed with D-PBS twice, and resuspended in 500 μl of electroporation buffer with 60 μg of cDNA in a Gene Pulser cuvette with 0.4-cm electrode gap (Bio-Rad, Hercules, CA). Electroporation was performed with a Gene Pulser MXcell Electroporation System at 950 μF, 0.16 kV. After electroporation, 1 ml of DMEM containing 10% FBS was added to the cuvette, and cells were left to recover for 10 min at room temperature and replated on collagen IV–coated cover slips (Fisher Scientific, Pittsburgh, PA). Alternatively, 3T3-L1 cells were transfected with Lipofectamine 2000 according to the manufacturer's instructions using 0.2 μg of cDNA/well of a 24-well plate or 2 μg/60-mm dish. After 48 h, cells were analyzed by immunofluorescence and Western blotting.
Immunofluorescence
Undifferentiated and differentiated 3T3-L1 cells were grown on coverslips coated with collagen IV (Sigma Aldrich). Serum-starved cells were treated with either insulin (100 nM) or carrier (5 mM HCl) for 15 min and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS; pH 7.4) for 20 min. Fixed cells were stained overnight at 4ºC with mouse monoclonal anti-myc antibody or nonspecific mouse IgG, followed by incubation with Cy3-conjugated anti-mouse IgG for 1 h at room temperature. Antifade solution was used for mounting cells on slides. Slides were examined with an Axio Observer Z1 fluorescence microscope equipped with C10600/ORCA-R2 digital camera (Hamamatsu, Hamamatsu, Japan) and AxioVision 4.8.1 (Carl Zeiss, Thornwood, NY). Quantitative analysis of immunostaining was carried out with ImageJ software (National Institutes of Health, Bethesda, MD).
Fluorescence-assisted cell sorting
Fluorescence-activated cell sorter analysis was performed as described previously (Shi and Kandror, 2008) with minor modifications. In brief, 3T3-L1 cells were grown in six-well plates. Before the experiment, cells were incubated in serum-free DMEM for 2 h, and insulin (100 nM) was administered for 15 min. Cells were then cooled to 4°C and washed with cold PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2 (PBS++). All subsequent steps were carried out at 4°C. Cells were incubated with anti-myc (1:1000) antibody or nonspecific mouse IgG in PBS++ containing 5% BSA and 5% donkey serum (1 ml/well) for 1 h and washed twice with PBS++ for 5 min each time. Cells were then incubated with 5 μg/ml phycoerythrin-conjugated donkey F(ab)2 anti-mouse IgG (Jackson ImmunoResearch) for 1 h. At the end of the incubation, cells were rinsed twice with PBS++ and additionally washed three times with PBS++ for 10 min each wash. Cells were incubated with 1 ml of 0.25% trypsin and 0.5 mg/ml collagenase in PBS without calcium and magnesium (to decrease cell adhesion) at 37°C for 3–5 min until most cells detached from the plate. Detached cells were washed once by centrifugation at 300 × g for 4 min and resuspended in 1 ml of PBS. Immediately before sorting, cells were passed through a 40-μm cell strainer (BD Biosciences, San Jose, CA). Data were acquired using a FACScan (BD Biosciences) maintained by the Boston University Medical Campus Flow Cytometry Core Facility. At least 20,000 cells were counted in each sample. Specific phycoerythrin fluorescence signal was determined by subtracting the signal from transfected cells treated under the same conditions as the experimental group and stained with nonspecific IgG and phycoerythrin-conjugated secondary antibody. Mean fluorescence intensities were used for quantification.
Cell surface biotinylation
Serum-starved cells in KRP buffer (12.5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 120 mM NaCl, 6 mM KCl, 1.2 mM MgSO4, 1.0 mM CaCl2, 0.6 mM Na2HPO4, 0.4 mM NaH2PO4, 2.5 mM d-glucose, pH 7.4) were pretreated with 100 nM insulin or carrier for 2 min at 37°C, and sulfo-NHS-biotin or sulfo-NHS-S-S-biotin was added to cells to final concentration 0.5 mg/ml for 15 min. The reaction was stopped by adding quenching buffer (50 mM Tris, 10 mM EDTA, 150 mM NaCl, 1 μM aprotinin, 2 μM leupeptin, 1 μM pepstatin, 5 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride [PMSF], pH 7.4) for 15 min at 4°C, followed by two washes with the same buffer. Cells were then lysed in 400 μl of quenching buffer with 1% Triton X-100, and cell lysates were incubated with 15–30 μl of monoclonal anti-myc antibody or nonspecific mouse IgG-conjugated agarose beads overnight at 4°C. Alternatively, biotinylated proteins were isolated using streptavidin-agarose. The beads were washed three times with 1% Triton X-100 in quenching buffer, and elution was carried out with Laemmli sample buffer at room temperature for 30 min.
Neurotensin uptake
EV or S cells were grown in 35-mm dishes. Cells were starved in serum-free DMEM for 2 h and treated with 100 nM insulin or carrier along with 7 nM [3,11-tyrosyl-3,5-3H(N)]-neurotensin for indicated times at 37°C. Cells were then rinsed once with ice-cold PBS and washed with ice-cold stripping buffer (0.15 M NaCl, 50 mM [2-(N-morpholino)] ethanesulfonate, pH 5.0) for 2 min, followed by two washes with PBS. Cells were then lysed with 400 μl of 1% SDS in KRH buffer (Krebs Ringer HEPES buffer; 121 mM NaCl, 12 mM HEPES, 4.9 mM KCl, 1.2 ml MgSO4, 0.33 mM CaCl2) without glucose, and 300-μl aliquots were used for determination of radioactivity by liquid scintillation counter (LKB-Wallac, Bromma, Sweden). Protein concentration was determined using the bicinchoninic acid protein assay kit and was used to normalize counts.
Subcellular fractionation of 3T3-L1 cells
3T3-Ll adipocytes or preadipocytes were washed three times with serum-free DMEM warmed to 37°C and starved in the same media for 2 h. Cells were treated with 100 nM insulin or carrier (5 mM HCl at 1000× dilution) in DMEM for 15 min at 37°C. Cells were then washed three times with cold HES buffer (250 mM sucrose, 20 mM HEPES, 1 mM EDTA, pH 7.4, 1 μM aprotinin, 2 μM leupeptin, 1 μM pepstatin, 5 mM benzamidine, and 1 mM PMSF) and harvested in the same buffer (0.3–1 ml/10-cm dish). Homogenization was performed in a ball-bearing homogenizer (Isobiotec, Heidelberg, Germany) with a 12-μm clearance by 10 strokes for adipocytes and 15 strokes for preadipocytes. Homogenates were centrifuged at 27,000 × g for 35 min. In some experiments, membrane vesicles in supernatants were concentrated by pelleting at 200,000 × g for 90 min. Supernatants (or thoroughly resuspended 200,000 × g pellets) were loaded onto a 4.6-ml linear 10–30% (wt/vol) sucrose gradient in HES buffer without sucrose and centrifuged for 55 min in a SW55 rotor (Beckman Coulter, Fullerton, CA) at 48,000 rpm. Each gradient was separated into 22–26 fractions starting from the bottom of the tube. The fractions were further analyzed by SDS–PAGE and Western blotting.
Gel electrophoresis and Western blotting
Proteins were separated in SDS–polyacrylamide gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA) in 25 mM Tris and 192 mM glycine. After transfer, the membrane was blocked with 10% BSA in PBS with 0.5% Tween 20 for 1 h. Blots were probed overnight with specific primary antibodies at 4°C, followed by 1-h incubation at room temperature with horseradish peroxidase–conjugated secondary antibodies (Sigma-Aldrich). Protein bands were detected with the enhanced chemiluminescence substrate kit (PerkinElmer Life Sciences, Boston, MA) using a Kodak Image Station 440CF (Eastman Kodak, Rochester, NY).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Research Grants DK52057 and AG039612, Research Award 7-11-BS-76 from the American Diabetes Association, and a Research Award from the Allen Foundation to K.V.K.
Abbreviations used:
- ACAP1
ArfGAP with coiled-coil, ankyrin repeat, and PH domains 1
- Ad
adipocyte
- AS160
Akt substrate of 160 kDa
- BS
bovine serum
- BSA
bovine serum albumin
- EV
empty vector
- FACS
fluorescence-activated cell sorting
- Fb
fibroblast
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- GGA
Golgi-localizing gamma-adaptin ear homology ARF-binding protein
- HRP
horseradish peroxidase
- IRAP
insulin-responsive aminopeptidase
- IRVs
insulin responsive vesicles
- LRP1
low-density-lipoprotein receptor–related protein 1
- SCAMP
secretory carrier membrane protein
- TBC1D4
TBC1 family member D4
- TGN
trans-Golgi network
- TUG
tether containing UBX domain for Glut4
- VAMP
vesicle-associated membrane protein
- Vps10p
vacuolar protein sorting 10 protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-10-0765) on August 21, 2013.
*These authors contributed equally to this study.
Present addresses: †Biogen Idec, Cambridge, MA 02142.
‡VU University Medical Center, 1081BT Amsterdam, Netherlands.
§Novartis Institutes for Biomedical Research, Cambridge, MA 02139.
¶University of California School of Medicine, San Diego, La Jolla, CA 92093.
REFERENCES
- Abel ED, Betuing S, Pham M, Reay P, Kandror V, Haney N, Kupriyanova TA, Xu Z, Kandror KV. Glut4 is required for the formation of the insulin responsive vesicle compartment in cardiac muscle. Mol Endocrinol. 2004;18:2491–2501. doi: 10.1210/me.2004-0175. [DOI] [PubMed] [Google Scholar]
- Ariga M, Nedachi T, Katagiri H, Kanzaki M. Functional role of sortilin in myogenesis and development of insulin-responsive glucose transport system in C2C12 myocytes. J Biol Chem. 2008;283:10208–10220. doi: 10.1074/jbc.M710604200. [DOI] [PubMed] [Google Scholar]
- Bai L, Wang Y, Fan J, Chen Y, Ji W, Qu A, Xu P, James DE, Xu T. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab. 2007;5:47–57. doi: 10.1016/j.cmet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem. 2012;81:507–532. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- Bogan JS, Hendon N, McKee AE, Tsao TS, Lodish HF. Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature. 2003;425:727–733. doi: 10.1038/nature01989. [DOI] [PubMed] [Google Scholar]
- Bogan JS, Rubin BR, Yu C, Loffler MG, Orme CM, Belman JP, McNally LJ, Hao M, Cresswell JA. Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J Biol Chem. 2012;287:23932–23947. doi: 10.1074/jbc.M112.339457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer PD, Romenskaia I, Kanow MA, Mastick CC. Loss of AS160 Akt substrate causes Glut4 protein to accumulate in compartments that are primed for fusion in basal adipocytes. J Biol Chem. 2011;286:26287–26297. doi: 10.1074/jbc.M111.253880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho E, Schellhorn SE, Zabolotny JM, Martin S, Tozzo E, Peroni OD, Houseknecht KL, Mundt A, James DE, Kahn BB. GLUT4 overexpression or deficiency in adipocytes of transgenic mice alters the composition of GLUT4 vesicles and the subcellular localization of GLUT4 and insulin-responsive aminopeptidase. J Biol Chem. 2004;279:21598–21605. doi: 10.1074/jbc.M312269200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Zhang J, Deng Y, Jiang L, Song E, Wu XS, Hammer JA, Xu T, Lippincott-Schwartz J. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J Cell Biol. 2012;198:545–560. doi: 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- ElJack A, Kandror KV, Pilch PF. Formation of an insulin-responsive vesicular compartment is an early event in 3T3-L1 adipocyte differentiation. Mol Biol Cell. 1999;10:1581–1594. doi: 10.1091/mbc.10.5.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DN, Farmer SR, Pilch PF. Glut4 storage vesicles without Glut4: transcriptional regulation of insulin-dependent vesicular traffic. Mol Cell Biol. 2004;24:7151–7162. doi: 10.1128/MCB.24.16.7151-7162.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney PM, Slot JW, Piper RC, James DE, Mueckler M. Intracellular targeting of the insulin-regulatable glucose transporter (GLUT4) is isoform specific and independent of cell type. J Cell Biol. 1991;114:689–699. doi: 10.1083/jcb.114.4.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama H, Kanzaki M. Molecular basis of insulin-responsive GLUT4 trafficking systems revealed by single molecule imaging. Traffic. 2011;12:1805–1820. doi: 10.1111/j.1600-0854.2011.01279.x. [DOI] [PubMed] [Google Scholar]
- Hermey G. The Vps10p-domain receptor family. Cell Mol Life Sci. 2009;66:2677–2689. doi: 10.1007/s00018-009-0043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka T, Brooks CC, Presman E, Kim SK, Zhang Z, Breen M, Sztul E, Pilch PF. p115 Interacts with the GLUT4 vesicle protein, IRAP, and plays a critical role in insulin-stimulated GLUT4 translocation. Mol Biol Cell. 2005;16:2882–2890. doi: 10.1091/mbc.E05-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson AW, Ruiz ML, Birnbaum MJ. Isoform-specific subsellular targeting of glucose transporters in mouse fibroblasts. J Cell Biol. 1992;116:785–797. doi: 10.1083/jcb.116.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrychowski MP, Gartner CA, Gygi SP, Zhou L, Herz J, Kandror KV, Pilch PF. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J Biol Chem. 2010;285:104–114. doi: 10.1074/jbc.M109.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Li J, Katz EB, Charron MJ. GLUT4 ablation in mice results in redistribution of IRAP to the plasma membrane. Biochem Biophys Res Commun. 2001;284:519–525. doi: 10.1006/bbrc.2001.4994. [DOI] [PubMed] [Google Scholar]
- Jordens I, Molle D, Xiong W, Keller SR, McGraw TE. IRAP is a key regulator of GLUT4 trafficking by controlling the sorting of GLUT4 from endosomes to specialized insulin-regulated vesicles. Mol Biol Cell. 2010;21:2034–2044. doi: 10.1091/mbc.E10-02-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddai V, Jager J, Gonzalez T, Najem-Lendom R, Bonnafous S, Tran A, Le Marchand-Brustel Y, Gual P, Tanti JF, Cormont M. Involvement of TNF-alpha in abnormal adipocyte and muscle sortilin expression in obese mice and humans. Diabetologia. 2009;52:932–940. doi: 10.1007/s00125-009-1273-3. [DOI] [PubMed] [Google Scholar]
- Kanai F, Nishioka Y, Hayashi H, Kamohara S, Todaka M, Ebina Y. Direct demonstration of insulin-induced GLUT4 translocation to the surface of intact cells by insertion of a c-myc epitope into an exofacial GLUT4 domain. J Biol Chem. 1993;268:14523–14526. [PubMed] [Google Scholar]
- Kandror KV, Pilch PF. The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic. 2011;12:665–671. doi: 10.1111/j.1600-0854.2011.01175.x. [DOI] [PubMed] [Google Scholar]
- Karylowski O, Zeigerer A, Cohen A, McGraw TE. GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol Biol Cell. 2004;15:870–882. doi: 10.1091/mbc.E03-07-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SR, Davis AC, Clairmont KB. Mice deficient in the insulin-regulated membrane aminopeptidase show substantial decreases in glucose transporter GLUT4 Levels but maintain normal glucose homeostasis. J Biol Chem. 2002;277:17677–17686. doi: 10.1074/jbc.M202037200. [DOI] [PubMed] [Google Scholar]
- Kim J, Kandror KV. The first luminal loop confers insulin responsiveness to the glucose transporter 4. Mol Biol Cell. 2012;23:910–917. doi: 10.1091/mbc.E11-10-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Racz A, Cushman SW, McGraw TE. Demonstration of insulin-responsive trafficking of Glut4 and vpTR in fibroblasts. J Cell Sci. 2000;113:4065–4076. doi: 10.1242/jcs.113.22.4065. [DOI] [PubMed] [Google Scholar]
- Larance M, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- Liu LB, Omata W, Kojima I, Shibata H. The SUMO conjugating enzyme Ubc9 is a regulator of GLUT4 turnover and targeting to the insulin-responsive storage compartment in 3T3-L1 adipocytes. Diabetes. 2007;56:1977–1985. doi: 10.2337/db06-1100. [DOI] [PubMed] [Google Scholar]
- Mari M, Bujny MV, Zeuschner D, Geerts WJ, Griffith J, Petersen CM, Cullen PJ, Klumperman J, Geuze HJ. SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic. 2008;9:380–393. doi: 10.1111/j.1600-0854.2007.00686.x. [DOI] [PubMed] [Google Scholar]
- Mazella J, et al. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. Biol J Chem. 1998;273:26273–26276. doi: 10.1074/jbc.273.41.26273. [DOI] [PubMed] [Google Scholar]
- Nielsen MS, Madsen P, Christensen EI, Nykjaer A, Gliemann J, Kasper D, Pohlmann R, Petersen CM. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001;20:2180–2190. doi: 10.1093/emboj/20.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck GR, Ye S, Pham V, Fernando RN, Macaulay SL, Chai SY, Albiston AL. Interaction of the Akt substrate, AS160, with the glucose transporter 4 vesicle marker protein, insulin-regulated aminopeptidase. Mol Endocrinol. 2006;20:2576–2583. doi: 10.1210/me.2005-0476. [DOI] [PubMed] [Google Scholar]
- Pilch P. The mass action hypothesis: formation of Glut4 storage vesicles, a tissue specific, regulated exocytic compartment. Acta Physiol. 2008;192:89–101. doi: 10.1111/j.1748-1716.2007.01788.x. [DOI] [PubMed] [Google Scholar]
- Ross SA, Keller SR, Lienhard GE. Increased intracellular sequestration of the insulin-regulated aminopeptidase upon differentiation of 3T3-L1 cells. Biochem J. 1998;330:1003–1008. doi: 10.1042/bj3301003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadacca LA, Bruno J, Wen J, Xiong W, McGraw TE. Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Mol Biol Cell. 2013;24:2544–2557. doi: 10.1091/mbc.E13-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 2007;5:293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J. 2008;411:89–95. doi: 10.1042/BJ20071318. [DOI] [PubMed] [Google Scholar]
- Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in Syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell. 2003;14:973–986. doi: 10.1091/mbc.E02-06-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Huang G, Kandror KV. Self-assembly of Glut4 storage vesicles during differentiation of 3T3-L1 adipocytes. J Biol Chem. 2008;283:30311–30321. doi: 10.1074/jbc.M805182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Kandror KV. Sortilin is essential and sufficient for the formation of Glut4-storage vesicles in 3T3-L1 adipocytes. Dev Cell. 2005;9:99–108. doi: 10.1016/j.devcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Shi J, Kandror KV. The luminal Vps10p domain of sortilin plays the predominant role in targeting to insulin-responsive Glut4-containing vesicles. J Biol Chem. 2007;282:9008–9016. doi: 10.1074/jbc.M608971200. [DOI] [PubMed] [Google Scholar]
- Shi J, Kandror KV. Study of glucose uptake in adipose cells. Methods Mol Biol. 2008;456:307–315. doi: 10.1007/978-1-59745-245-8_23. [DOI] [PubMed] [Google Scholar]
- Xu Z, Kandror KV. Translocation of small preformed vesicles is responsible for the insulin activation of glucose transport in adipose cells. Evidence from the in vitro reconstitution assay. J Biol Chem. 2002;277:47972–47975. doi: 10.1074/jbc.C200486200. [DOI] [PubMed] [Google Scholar]
- Yang J, Clark AE, Kozka IJ, Cushman SW, Holman GD. Development of an intracellular pool of glucose transporters in 3T3-L1 cells. J Biol Chem. 1992;267:10393–10399. [PubMed] [Google Scholar]
- Yeh TY, Sbodio JI, Tsun ZY, Luo B, Chi NW. Insulin-stimulated exocytosis of GLUT4 is enhanced by IRAP and its partner tankyrase. Biochem J. 2007;402:279–290. doi: 10.1042/BJ20060793. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.