

Abstract
In mammalian species, the process of meiosis, in which genes are randomly assorted between parental chromosomes during formation of egg and sperm cells, occurs prenatally in females but postnatally in males. To understand sex-specific differences in signaling mechanisms that underlie fertility, many studies have focused on identifying factors that control meiotic induction. Studies in mice using genetic knockout of the transcriptional regulator Polycomb repressive complex–1 (PRC1) and pharmacological inhibition of retinoic acid (RA) signaling suggest that PRC1 prevents female meiotic induction until release of PRC1 repression by increased RA signaling in the ovary. However, genetic studies with mice lacking RA synthesis in reproductive tissues indicate that RA is required for male but not female meiosis, suggesting that RA functions as a male-specific inducer of meiosis and that another factor releases PRC1 repression to initiate female meiosis. Correct resolution of the molecular events governing female and male meiosis is important for treating infertility and devising improved birth control strategies.
A distinctive feature of primordial germ cell (PGC) maturation is induction of meiosis, which in mammals occurs prenatally in females and postnatally in males (1–3). Three isoforms of retinaldehyde dehydrogenase (RALDH1, RALDH2, RALDH3) play essential roles in synthesis of retinoic acid (RA), a vitamin A metabolite essential for reproduction. Pharmacological studies suggested that RA produced by RALDH2 in the mesonephros may function as a diffusible sex-specific factor, inducing meiosis in the adjacent prenatal ovary, but not in the prenatal testis, which has the cytochrome P450 enzyme CYP26B1 that degrades RA (4, 5). However, genetic studies with Raldh2−/− and Raldh2−/−;Raldh3−/− mouse embryos demonstrated that loss of RA activity in the mesonephros and developing gonad has no effect on female meiosis (6). The double knockout was examined because Raldh3 exhibits low expression in mesonephros; Raldh1 is unlikely to compensate because RA activity was undetectable with a reporter in Raldh2−/−;Raldh3−/− embryos in mesonephros and developing gonad (6).
Yokobayashi et al. (7) used gene knockout studies in mice to show that loss of Polycomb repressive complex–1 (PRC1) results in premature induction of female meiosis, indicating that PRC1 normally prevents female meiosis until this repression is released. Yokobayashi et al. (7) also reported that administration of an RALDH inhibitor prevents premature meiotic induction in PRC1-deficient ovary, and thus concluded that RA synthesis normally alleviates PRC1 repression to induce female meiosis. However, RALDH inhibitors have off-target effects, including inhibition of mitochondrial aldehyde dehydrogenase (8) and possibly other enzymes of this 20-member family that have functions other than RA synthesis. Thus, the pharmacological studies of Yokobayashi et al. (7) and others (4, 5) need to be carefully weighed against the genetic evidence demonstrating that elimination of RA synthesis does not prevent or delay female meiosis (6).
In mice, meiosis in the female gonad occurs at embryonic day 13.5 (E13.5). Yokobayashi et al. (7) propose that RA signaling increases in the ovary from E11.5 to E13.5 and that once it reaches a certain threshold, RA signaling overcomes PRC1 repression and allows meiotic induction. However, this conclusion conflicts with studies showing that RA activity monitored with the RARE-lacZ RA-reporter transgene does not increase in ovary between E12.0 and E13.5 (6). Indeed, RARE-lacZ expression is not detected in ovary during these stages, although it is easily detected in mesonephros and completely absent in the mesonephros of Raldh2−/−;Raldh3−/− mice (6). The sensitivity of RARE-lacZ had previously come into question (9), but recent studies have shown that RARE-lacZ is sensitive to 0.25 nM RA (10), lower than the 1 nM concentration shown to marginally induce meiosis in isolated PGCs and much lower than the 10 nM concentration needed for more complete meiotic induction in the dose-response curve (9).
Yokobayashi et al. (7) also reported that PRC1-deficient PGCs display low ectopic expression of Raldh2, but did not demonstrate that this leads to RA synthesis. Additionally, this is inconsistent with studies showing that PGCs are not the source of the factor that induces female meiosis; examination of isolated PGCs demonstrated that this factor is a diffusible extrinsic substance derived from mesonephric or ovarian somatic cells (3–6, 9). If a threshold level of RA is needed for female meiotic induction, as proposed by Yokobayashi et al. (7), then we would expect that loss of RALDH2 and 3 should reduce or delay this process even if RALDH1 plays a minor role, but the mouse genetic data do not support this model (6).
Stra8 encodes a cytoplasmic protein essential for both male and female meiosis, although its function in meiosis is unclear (4). Induction of Stra8 is governed by sex-specific intrinsic and extrinsic factors and serves as a marker of meiotic induction. In the prenatal ovary, Doublesex and mab-3 related transcription factor-1 (DMRT1) plus WNT4 and WNT5A are essential for Stra8 expression (1, 3), whereas in the prenatal testis, fibroblast growth factor 9 (FGF9) and CYP26B1 are essential to prevent Stra8 expression (2, 4, 5) (Fig. 1). CYP26B1 loss-of-function studies in mice suggested that this enzyme degrades mesonephric-derived RA in the prenatal testis to prevent Stra8 induction, which led to the assumption that mesonephric RA is required for Stra8 induction in the prenatal ovary because it lacks CYP26B1 activity (4, 5); however, this assumption is not supported by RALDH genetic loss-of-function studies (6). By contrast, PGCs of postnatal testis require RA for Stra8 induction as demonstrated by other RALDH genetic loss-of-function studies (11). Further, a role for RA in postnatal meiotic induction in males is supported by studies showing that PGCs in postnatal testis express Raldh2 and locally generate RA (11), unlike PGCs in prenatal ovary and prenatal testis, which do not (4, 5). Therefore, the previously reported female Stra8 response to pharmacological treatment with either RA (up-regulation) or RA receptor antagonists (down-regulation) (4, 5) may occur by invoking the RA-dependent male meiotic program (11); because genetic evidence indicates that female Stra8 induction does not normally require RA (6), this would constitute an off-target effect.
Fig. 1. Proposed model for sex-specific control of meiotic induction based on genetic findings.
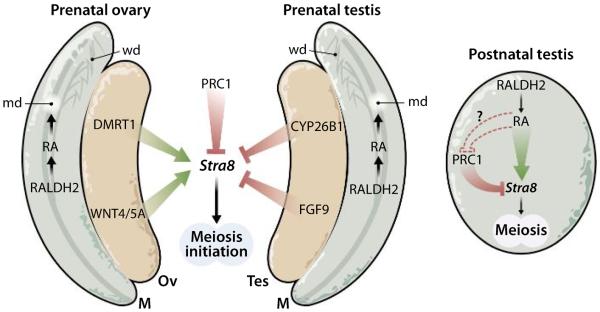
RA generated in the mesonephros acts locally to control differentiation of Müllerian (md) and Wolffian (wd) ducts (12), but diffusion of mesonephric RA to prenatal ovary or prenatal testis is insufficient to induce meiosis in prenatal PGCs that lack enzymes to locally generate RA (6). By contrast, RA is generated locally in PGCs of postnatal testis and is required to induce Stra8 for male meiotic induction (11); RA produced in postnatal male PGCs may directly or indirectly release PRC1 repression to induce Stra8. M, mesonephros; Ov, ovary; Tes, testis.
By taking into account the genetic studies (1–3, 5–7, 11), we propose that RA is a male-specific factor controlling meiotic induction (Fig. 1). Further, as Yokobayashi et al. (7) reported that PRC1 represses Stra8 not only in prenatal ovary but also in prenatal testis, we suggest that RA may release PRC1 repression of Stra8 in testis postnatally. Several unanswered questions remain: What releases PRC1 repression during female meiosis prenatally? Does RA release PRC1 repression during male meiosis postnatally, and does this occur directly on the Stra8 gene at the transcriptional level? Are some defects in human fertility due to abnormal RA signaling or aberrant PRC1 function? What are the implications for clinical strategies to alter fertility—either infertility or birth control? On the basis of our sex-specific model of RA action during meiosis, therapies designed to increase RA signaling might treat infertility only in males, and strategies for birth control that decrease RA signaling might also be male specific. With genetic studies guiding the way, further progress in understanding the molecular basis of fertility is on the horizon.
References
- 1.Krentz AD, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 promotes oogenesis by transcriptional activation of Stra8 in the mammalian fetal ovary. Dev. Biol. 2011;356:63–70. doi: 10.1016/j.ydbio.2011.05.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowles J, Feng CW, Spiller C, Davidson TL, Jackson A, Koopman P. FGF9 suppresses meiosis and promotes male germ cell fate in mice. Dev. Cell. 2010;19:440–449. doi: 10.1016/j.devcel.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Naillat F, Prunskaite-Hyyryläinen R, Pietilä I, Sormunen R, Jokela T, Shan J, Vainio SJ. Wnt4/5a signalling coordinates cell adhesion and entry into meiosis during presumptive ovarian follicle development. Hum. Mol. Genet. 2010;19:1539–1550. doi: 10.1093/hmg/ddq027. [DOI] [PubMed] [Google Scholar]
- 4.Koubova J, Menke DB, Zhou Q, Capel B, Griswold MD, Page DC. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2474–2479. doi: 10.1073/pnas.0510813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowles J, Knight D, Smith C, Wilhelm D, Richman J, Mamiya S, Yashiro K, Chawengsaksophak K, Wilson MJ, Rossant J, Hamada H, Koopman P. Retinoid signaling determines germ cell fate in mice. Science. 2006;312:596–600. doi: 10.1126/science.1125691. [DOI] [PubMed] [Google Scholar]
- 6.Kumar S, Chatzi C, Brade T, Cunningham TJ, Zhao X, Duester G. Sex-specific timing of meiotic initiation is regulated by Cyp26b1 independent of retinoic acid signalling. Nat. Commun. 2011;2:151. doi: 10.1038/ncomms1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokobayashi S, Liang CY, Kohler H, Nestorov P, Liu Z, Vidal M, van Lohuizen M, Roloff TC, Peters AH. PRC1 coordinates timing of sexual differentiation of female primordial germ cells. Nature. 2013;495:236–240. doi: 10.1038/nature11918. [DOI] [PubMed] [Google Scholar]
- 8.Hogarth CA, Evanoff R, Snyder E, Kent T, Mitchell D, Small C, Amory JK, Griswold MD. Suppression of Stra8 expression in the mouse gonad by WIN 18,446. Biol. Reprod. 2011;84:957–965. doi: 10.1095/biolreprod.110.088575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spiller CM, Bowles J, Koopman P. Regulation of germ cell meiosis in the fetal ovary. Int. J. Dev. Biol. 2012;56:779–787. doi: 10.1387/ijdb.120142pk. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham TJ, Zhao X, Sandell LL, Evans SM, Trainor PA, Duester G. Antagonism between retinoic acid and fibroblast growth factor signaling during limb development. Cell Reports. 2013;3:1503–1511. doi: 10.1016/j.celrep.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raverdeau M, Gely-Pernot A, Féret B, Dennefeld C, Benoit G, Davidson I, Chambon P, Mark M, Ghyselinck NB. Retinoic acid induces Sertoli cell paracrine signals for spermatogonia differentiation but cell autonomously drives spermatocyte meiosis. Proc. Natl. Acad. Sci. U.S.A. 2012;109:16582–16587. doi: 10.1073/pnas.1214936109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendelsohn C, Lohnes D, Décimo D, Lufkin T, LeMeur M, Chambon P, Mark M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–2771. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]