

Abstract
Progression of premalignant lesions is restrained by oncogene-induced senescence. Oncogenic Ras triggers senescence in many organs, including the lung, which exhibits high levels of the angiogenesis inhibitor thrombospondin-1 (TSP-1). The contribution of TSP-1 upregulation to the modulation of tumorigenesis in the lung is unclear. Using a mouse model of lung cancer, we have shown that TSP-1 plays a critical and cell-autonomous role in suppressing Kras-induced lung tumorigenesis independent of its antiangiogenic function. Overall survival was decreased in a Kras-driven mouse model of lung cancer on a Tsp-1–/– background. We found that oncogenic Kras–induced TSP-1 upregulation in a p53-dependent manner. TSP-1 functioned in a positive feedback loop to stabilize p53 by interacting directly with activated ERK. TSP-1 tethering of ERK in the cytoplasm promoted a level of MAPK signaling that was sufficient to sustain p53 expression and a senescence response. Our data identify TSP-1 as a p53 target that contributes to maintaining Ras-induced senescence in the lung.
Introduction
Numerous studies have shown that the progression of benign or premalignant lesions is controlled in part by activation of a permanent form of growth arrest, also known as cellular senescence (1). Such senescence, which is typically triggered by the same activated oncogenes that drove the initial tumorigenesis (2), is thought to serve as an innate restraint to tumorigenesis that has been referred to as cell-intrinsic tumor suppression. Senescence has been demonstrated in multiple mouse cancer models, including lymphoma (3) as well as mammary (4), lung (5, 6), and prostate carcinoma (7), and is evident in benign lesions of the skin (8), prostate (7), and neurofibromas (9) in humans associated with oncogenic BRAF, PTEN inactivation, and NF1 mutations. Senescence is an especially frequent outcome of oncogenic activation of Ras or its effectors in numerous cell types and tissues in vitro and in vivo (10). However, initial studies of Ras-induced senescence involved the ectopic expression of non–physiologically high levels of oncogenic Ras in primary mouse and human cells in vitro, which led to uncertainty about the relevance of this phenomenon as a bona fide cancer-suppressing mechanism in vivo (11–13). More recently, however, senescence induced by precocious signaling of the Ras network has been observed in vivo, reinforcing its physiologic relevance as a tumor-suppressive mechanism.
Published studies suggest that thresholds of Ras activation may be important in determining when oncogenic Ras–induced senescence is triggered. Indeed, a study using a Kras-driven mouse mammary tumor model in which the level of oncogenic Ras activity can be titrated has confirmed that low levels of activated Ras drive proliferation and hyperplasia and only high levels of Ras activation trigger irreversible, p16INK4a-p19ARF–dependent cellular senescence (4). On the other hand, another transgenic mouse study indicted that low-level expression from only a single allele of oncogenic Ras is sufficient to engage a senescence response in lung epithelium (14). Taken together, such observations suggest that both oncogenic Ras activity level and tissue type are likely important determinants of whether oncogenic Ras triggers cell proliferation versus replicative senescence.
The molecular basis for oncogene-induced senescence remains ill defined. Nonetheless, it clearly involves multiple mechanisms that differ with cell and tissue type and specific oncogenic mutation. Furthermore, oncogene-induced senescence involves both cell-autonomous processes, mediated by the key p16INK4a/Rb and/or p19ARF/p53 tumor suppressor pathways (2, 15, 16), and a variety of cell-extrinsic signals mediated by chemokines, cytokines, and adhesion molecules (17–21). For example, induction of senescence in melanocytes by oncogenic BRAFV600E, a downstream effector of Ras, is dependent on both p16INK4a and a number of secreted proteins that mediate oncogenic BRAF–induced senescence by both autocrine and paracrine mechanisms (19, 21). Similarly, oncogenic Hras–induced senescence in a mouse model of liver carcinoma is critically and continuously dependent on p53 and rapidly triggered when p53 function is restored to established tumors (22). Inflammatory cytokines are then required for the clearance of the resulting premalignant senescent cells by infiltrating immune cells (17). Such secreted proteins are members of a growing list of factors that together comprise the senescence-associated secretory phenotype, originally thought to represent mere consequences of senescence, but now implicated in a causal role. Whether analogous secreted factors underlie Kras-induced senescence in lung tumor progression is unclear.
Sporadic activation of oncogenic KrasG12D in the bronchioalveolar lung epithelium of LSL-KrasG12D mice by intranasal inhalation of adenovirus–Cre recombinase (Ad-Cre) triggers initiation and evolution of tumors that closely resemble human non–small cell lung cancers (23). The consistency of the kinetics and pathology of tumor evolution in this model makes it an especially useful system with which to interrogate the effect of novel candidate tumor suppressors and cooperating oncogenes.
The lung exhibits very high expression of thrombospondin-1 (TSP-1), a secreted matricellular glycoprotein with multiple domains (24) that is a well-characterized endogenous angiogenesis inhibitor. Since many other potential functions have been ascribed to TSP-1, we analyzed the role of TSP-1 in modulating lung tumorigenesis using the LSL-KrasG12D mouse model. In this model, focal hyperplasias/adenomas appear by 4–6 weeks after Kras activation, with adenocarcinomas developing between 16 and 25 weeks (23), depending on the genetic background of the mice (25, 26). It is proposed that the protracted latency in adenocarcinoma development is due, at least in part, to Kras-induced senescence stalling progression in premalignant adenomas: only when clones evolve that are refractory to oncogenic Ras–induced senescence can tumors progress toward malignancy (14). However, the mechanisms that mediate oncogenic Ras–induced senescence in the lung remain largely undefined.
Oncogenic Ras–induced senescence is mediated by the Raf/MEK/MAPK pathway. Oncogenic activation of the MAPK pathway in fibroblasts leads to the induction of p19ARF, which is required for initiation and maintenance of p53-dependent cellular senescence (27, 28). The net outcome of MAPK signaling depends on both its magnitude and duration, and recent studies suggest that oncogenic Ras activation of MAPK signaling necessitates its activation over a certain threshold to switch from a proliferative to a senescent phenotype (29, 30). Activation of Raf/MEK by oncogenic Ras expression leads to ERK phosphorylation (pERK) and translocation of pERK from the cytoplasm to the nucleus, a requirement for activation of proliferation-dependent genes. However, studies have also shown that, upon establishment of a senescent phenotype in fibroblasts, ERKs remains phosphorylated but relocalize back to the cytoplasm (31).
In the present study, we demonstrated that Tsp-1 status had a potent effect on survival of LSL-KrasG12D mice after KrasG12D activation in lung epithelium. The absence of TSP-1 markedly accelerated Kras-driven tumor progression in the lung. Collectively, our data suggest a model whereby TSP-1 is upregulated after oncogenic Ras activation in a p53-dependent manner, which then positively feeds back into the Ras-MAPK signaling pathway, directly interacting with pERK and tethering it to the cytosol, thus altering the substrates of activated ERK. Hence, TSP-1 may play an important role in regulating the biological outcome of oncogenic Ras signaling via the MAPK pathway, as TSP-1–mediated localization of pERK in the cytosol appears to be necessary to maintain a senescent phenotype as a consequence of oncogenic Ras activation in the lung.
Results
Loss of Tsp-1 accelerates the progression of KrasG12D-induced lung tumorigenesis.
The lung exhibited high expression of TSP-1 (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI67465DS1), a secreted matricellular glycoprotein that is a well-characterized angiogenesis inhibitor (24). To examine the role of TSP-1 in the progression of lung tumorigenesis, we generated LSL-KrasG12D mice (23) on a Tsp-1–/– background (referred to herein as KrasG12D×Tsp-1–/– mice; Supplemental Figure 1B). After intranasal inhalation of Ad-Cre to trigger KrasG12D expression, we monitored overall survival of both KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– mice. Survival of KrasG12D×Tsp-1–/– mice was significantly decreased by an average of 11 weeks compared with KrasG12D×Tsp-1+/+ mice (P < 0.01; Figure 1A). Histological analysis of lungs isolated at various times after KrasG12D activation confirmed the substantially more rapid progression to lung adenocarcinoma in KrasG12D×Tsp-1–/– mice (Figure 1B and Supplemental Figure 2). Oncogenic Kras–driven adenocarcinomas were evident in lungs of KrasG12D×Tsp-1–/– mice after only 12 weeks, compared with 20 weeks in KrasG12D×Tsp-1+/+ animals. On average, 60% of KrasG12D×Tsp-1–/– mice were dead from lung adenocarcinoma at 20 weeks, whereas only approximately 15% of KrasG12D×Tsp-1+/+ mice harbored adenocarcinoma lesions at death (Supplemental Figure 3).
Figure 1. Loss of TSP-1 leads to decreased survival and rapid progression of KrasG12D-mediated lung tumorigenesis.

(A) Overall KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– mouse survival after induction of KrasG12D expression via intranasal instillation of Ad-Cre. Data represent mean ± SEM. P = 0.0064. (B) H&E images of lungs at the indicated times after oncogenic Kras activation. Lungs were harvested, stained with H&E, and scored for tumor pathology. (C) TSP-1 expression in oncogenic KrasG12D-driven lung lesions at the indicated times after Ad-Cre infection. Isolated KrasG12D×Tsp-1+/+ lungs were examined for TSP-1 expression by immunofluorescence. KrasG12D×Tsp-1–/– lung tumors were immunostained for TSP-1 as a negative control. Corresponding H&E images of tumors are shown below. AAH, atypical adenomatous hyperplasia. (D) Time course analysis of TSP-1 expression upon oncogenic Kras activation in primary murine adult lung fibroblasts. KrasG12D×Tsp-1+/+ lung fibroblasts were infected with Ad-Cre, harvested at the indicated time points, and analyzed for TSP-1 expression by immunoblotting. KrasG12D×Tsp-1–/– fibroblasts served as a negative control. (E) Representative images of TSP-1 expression by immunofluorescence in normal human lungs and lung adenocarcinomas at different stages. Scale bars: 1 mm (B, ×40); 50 μm (B, ×400; C; and E).
To determine whether oncogenic Kras regulates TSP-1 expression, we assayed for TSP-1 after KrasG12D activation in lungs in vivo and isolated primary adult lung fibroblasts in vitro (Figure 1, C and D). TSP-1 expression was initially upregulated after oncogenic Ras activation, but then decreased during tumor progression, becoming virtually undetectable in late-stage adenocarcinomas (Figure 1C). Similarly, TSP-1 expression in primary lung fibroblasts was initially upregulated, then subsequently downregulated beginning approximately 31 days after KrasG12D activation (Figure 1D).
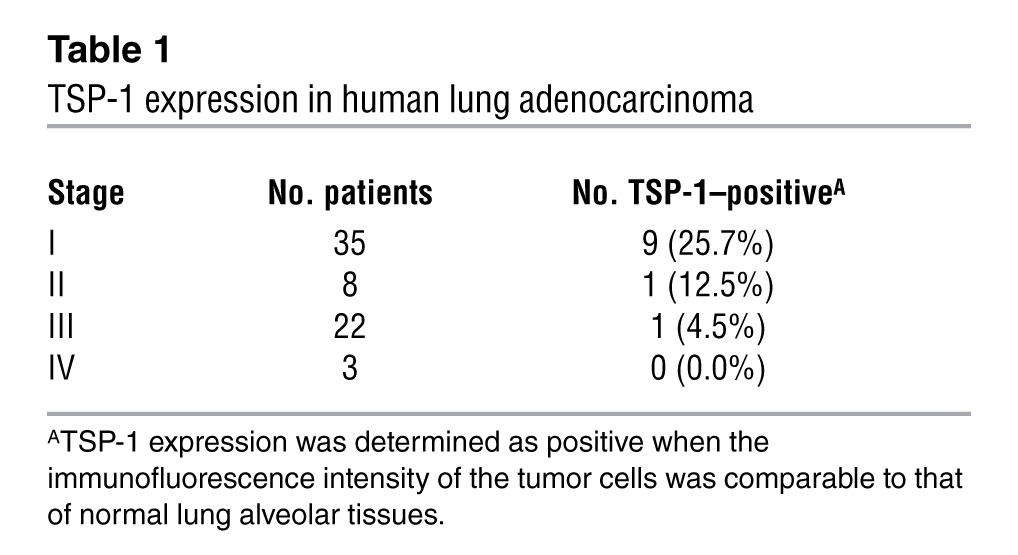
Our data indicated that upregulation of TSP-1 expression occurs after oncogenic Ras activation in premalignant adenomas in the lung. However, upon progression of these benign lesions to malignant adenocarcinoma, TSP-1 expression was markedly diminished. To investigate whether deregulation of this pathway is involved in human lung cancers, TSP-1 expression was assessed by immunofluorescence analysis of a panel of 14 normal lung samples and 68 lung adenocarcinomas. TSP-1 expression was high in normal lung alveolar tissues and epithelial cells, but absent in lung adenocarcinomas and lung adenocarcinoma cell lines (Figure 1E, Supplemental Figure 4, and Table 1).
Table 1.
TSP-1 expression in human lung adenocarcinoma
KrasG12D-induced senescence is attenuated in the absence of Tsp-1.
Taken together, these data suggest that TSP-1 can function to suppress lung tumorigenesis and, like a classical tumor suppressor, may be lost in the course of tumor evolution. However, since the lung is a highly vascularized organ, and no association is found between TSP-1 expression and tumor angiogenesis in non–small cell lung cancer patients (32), the ability of TSP-1 to inhibit angiogenesis seems unlikely as the mechanism limiting lung tumorigenesis. Indeed, we found that TSP-1 status had no effect on microvessel density in large lung adenomas (Figure 2A), reinforcing the notion that the tumor-suppressive effect of TSP-1 is not a consequence of its antiangiogenic activity. To ascertain whether TSP-1 status specifically affects tumor initiation, we quantified the incidence of early, hyperproliferative lesions in the lung 4 weeks after Ad-Cre activation of KrasG12D (Figure 2B). Absence of TSP-1 moderately increased the multiplicity of early hyperplastic lesions, which is unlikely to account for the significant decrease in survival we observed in KrasG12D×Tsp-1–/– mice (Figure 1A).
Figure 2. KrasG12D-induced senescence during lung tumor progression is compromised in the absence of TSP-1.

(A) Microvessel density in large lung adenomas of similar size from KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– mice 21 weeks after oncogenic Kras activation. Tumor microvessels were visualized by immunostaining for CD31 and quantified by measuring microvessel density per high-powered field (MVD/hpf). Arrows indicate representative tumor microvessels. (B) Hyperplastic lesions in lungs 4 weeks after Ad-Cre inhalation. Lungs were isolated (n = 4 per group), and the number of lesions with atypical adenomatous hyperplasia or epithelial hyperplasia (EH) was counted after H&E staining. Arrows indicate hyperplastic lesions. (C) H&E staining, BrdU uptake, and SA–β-gal staining of serial sections from lung tumors harvested 21 weeks after Ad-Cre infection. (D) Proliferation in lung tumor lesions, as assessed by immunostaining for Ki-67, 21 weeks after Ad-Cre infection. (E) KrasG12D-induced senescence of primary adult lung epithelial cells isolated in vitro, as assessed by SA–β-gal positivity, at the indicated times after oncogenic Kras activation. (F) PML expression 21 days after oncogenic Kras activation in primary adult lung epithelial cells, determined by immunostaining. (G) Proliferation of primary adult lung fibroblasts isolated in vitro at the indicated times after oncogenic Kras activation. (H) KrasG12D-induced senescence of primary adult lung fibroblasts in vitro, measured by SA–β-gal positivity, at the indicated times after oncogenic Kras activation. SA–β-gal staining was quantified 35 days after Kras activation. Scale bars: 50 μm (A; B, insets; C; and D); 1 mm (B); 200 μm (E and H); 100 μm (F). Data represent mean ± SEM.
Endogenous oncogenic Kras expression in the lungs has previously been shown to elicit cellular senescence in premalignant lung adenomas (14). This is consistent with the idea that Kras-induced senescence in the lungs is an important barrier to tumor progression and is responsible for the long latency in progression to malignant adenocarcinoma in this model (14). To determine whether Kras-induced senescence was influenced by Tsp-1 status, serial lung sections were analyzed for both BrdU incorporation and expression of senescence-associated β-galactosidase (SA–β-gal). Whereas early lung lesions from KrasG12D×Tsp-1+/+ mice were negative for BrdU uptake and stained positive for SA–β-gal, comparably sized lesions from KrasG12D×Tsp-1–/– lungs stained positive for BrdU and negative for SA–β-gal (Figure 2C and Supplemental Figure 5A). Quantification demonstrated an increased number of SA–β-gal lesions in KrasG12D×Tsp-1+/+ versus KrasG12D×Tsp-1–/– lungs harvested at similar times (Figure 2C and Supplemental Figure 5A). Consistent with their senescent status, the proliferative indices, as measured by Ki67 positivity of comparably sized KrasG12D-driven lesions, were lower in KrasG12D×Tsp-1+/+ than KrasG12D×Tsp-1–/– mice (Figure 2D). Notably, costaining for the macrophage marker CD68 demonstrated no overlap with SA–β-gal (Supplemental Figure 5B), which confirmed that SA–β-gal–positive cells were not infiltrating macrophages.
Lung epithelial cells were isolated from both KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– mice, their epithelial identities were confirmed (Supplemental Figure 5C), and then cells were examined for senescence after KrasG12D activation in vitro. While few cells were found to undergo senescence in KrasG12D×Tsp-1+/+ lung epithelial cells infected with mock adenovirus (Supplemental Figure 5D), KrasG12D×Tsp-1+/+ lung epithelial cells exhibited decreased proliferation and increased SA–β-gal reactivity relative to KrasG12D×Tsp-1–/– lung epithelial cells 7 days after Ad-Cre infection. By 28 days, KrasG12D×Tsp-1+/+ lung epithelial cells were completely growth arrested, and SA–β-gal reactivity was widespread throughout the cultures (Figure 2E). To further validate oncogenic Ras–induced senescence in these cells, we immunostained lung epithelial cells for expression of promyelocytic leukemia (PML) protein. PML has been shown to be upregulated during Ras-induced senescence and to localize to PML nuclear bodies (33). Indeed, PML bodies were abundant in KrasG12D×Tsp-1+/+ versus KrasG12D×Tsp-1–/– lung epithelial cells 21 days after oncogenic Ras activation (Figure 2F).
To validate the generality of the obligate role for TSP-1 in oncogenic Ras–induced senescence, we next used easily accessible KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– adult primary lung fibroblasts as a model system with which to dissect the mechanism by which TSP-1 is involved. KrasG12D expression was triggered by exposure of cells to Ad-Cre, and cellular proliferation was then monitored. Upon Ad-Cre infection, proliferation was substantially decreased in KrasG12D×Tsp-1+/+ versus KrasG12D×Tsp-1–/– primary lung fibroblasts (Figure 2G). Notably, 28–35 days after Ad-Cre infection, a majority of the KrasG12D×Tsp-1+/+ lung fibroblasts displayed the enlarged, flattened morphology typical of senescent cells and expressed SA–β-gal. In contrast, KrasG12D×Tsp-1–/– primary lung fibroblasts exhibited minimal SA–β-gal reactivity (Figure 2H), which suggests that oncogenic Ras–induced senescence is abrogated in the absence of TSP-1 and contributes to the differences in growth between KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– lung fibroblasts.
Primary fibroblasts undergo senescence within 10–12 days after ectopic overexpression of oncogenic HrasV12 (2). HrasV12-induced senescence was also diminished in the absence of TSP-1, as demonstrated by decreased SA–β-gal reactivity and continuous proliferation after 12 days (Figure 3, A and B, and Supplemental Figure 6A). We propose that the comparatively slow onset of senescence in the KrasG12D×Tsp-1+/+ epithelial cells and fibroblasts, relative to its rapid induction by ectopic overexpression of oncogenic Hras, reflects the much lower levels of oncogenic KrasG12D in these cells, expressed as it is from a single, endogenous Kras2 allele. Finally, to confirm the requirement for TSP-1 in Ras-induced senescence, we restored expression of TSP-1 in Tsp-1–/– primary fibroblasts (Figure 3C). TSP-1 expression restored both replicative arrest and SA–β-gal reactivity to an extent comparable to that of Tsp-1+/+ fibroblasts expressing HrasV12 (Figure 3D and Supplemental Figure 6, B and C).
Figure 3. Rescue of oncogenic Ras–induced senescence in Tsp-1–/– cells by ectopic expression of TSP-1.

(A) SA–β-gal staining of Tsp-1+/+ and Tsp-1–/– lung fibroblasts at the indicated days after retroviral HrasV12 or mock infection. Percent SA–β-gal–positive cells relative to total number of cells was quantified by counting 300 cells. (B) Proliferation of Tsp-1+/+ and Tsp-1–/– lung fibroblasts upon HrasV12 expression in vitro. (C) Restoration of TSP-1 expression was confirmed by immunoblotting Tsp-1–/– primary lung fibroblasts after retroviral TSP-1 infection. β-actin served as loading control. (D) Oncogenic Hras–induced senescence in Tsp-1+/+ and Tsp-1–/– fibroblasts after ectopic TSP-1 expression, measured by SA–β-gal staining. Tsp-1+/+ and Tsp-1–/– lung fibroblasts expressing TSP-1 or GFP were retrovirally infected with HrasV12 or mock infected, then stained for SA–β-gal activity to detect senescent cells. Data represent mean ± SEM. Scale bars: 200 μm.
Aberrant activation of PI3K/AKT as well as Raf/MEK/MAPK signaling induces cellular senescence. To determine whether TSP-1 was required for senescence due to the Raf/MEK/MAPK pathway, we induced senescence by enforced MAPK signaling through overexpression of oncogenic BRAFV600E in both Tsp-1+/+ and Tsp-1–/– adult lung fibroblasts and found that SA–β-gal reactivity was significantly decreased in the absence of Tsp-1 (Supplemental Figure 7A). In contrast, senescence induced by hyperactivation of PI3K/AKT signaling via Pten knockdown was unaffected by Tsp-1 status (Supplemental Figure 7B). Thus, TSP-1 appears to be important specifically for induction of senescence by oncogenic Kras via the MAPK pathway.
Since TSP-1 is a secreted protein, we investigated whether addition of TSP-1 to the conditioned media could also rescue oncogenic Ras–induced senescence in Tsp-1–/– fibroblasts. Platelets contain the highest levels of TSP-1 of all cell types; therefore, we activated platelets isolated from Tsp-1+/+ and Tsp-1–/– mice and added their contents to cultures of HrasV12-expressing Tsp-1–/– fibroblasts. However, exogenously supplemented TSP-1 failed to restore Ras-induced senescence (Supplemental Figure 8A), which suggests that TSP-1 mediates Ras-induced senescence via a cell-intrinsic mechanism. To explore further the cell-autonomous ability of TSP-1 to mediate oncogene-induced senescence, we examined the influence of TSP-1 host status on lung tumor senescence. Adenomas were harvested from KrasG12D×Tsp-1+/+ or KrasG12D×Tsp-1–/– lungs 20 weeks after Ad-Cre infection, dissociated, then inoculated via tail vein into Tsp-1+/+ or Tsp-1–/– mice. Examination of lungs 38 days later revealed the number of lesions in lungs to be significantly greater in both Tsp-1+/+ and Tsp-1–/– mice inoculated with KrasG12D×Tsp-1–/– tumor cells compared with KrasG12D×Tsp-1+/+ tumor cells (Supplemental Figure 8B), which indicates that the growth of these lung tumors is dictated by Tsp-1 expression in tumor cells and not due to host Tsp-1 expression.
Oncogenic Ras–induced upregulation of TSP-1 is p53 dependent.
Previous studies suggested that p53 may regulate Tsp-1 expression (34). To confirm this, we examined levels of TSP-1 in lung tissue harvested from p53+/+, p53+/–, and p53–/– mice. Basal TSP-1 expression was reduced in p53+/– lung tissue and lower still in p53–/– lung tissue (Figure 4A). To confirm that TSP-1 is specifically upregulated by p53 after oncogenic Ras activation, we initially examined primary adult lung fibroblasts from KrasG12D×Tsp-1+/+, KrasG12D×p53fl/fl, and KrasG12D×Tsp-1–/– mice. The early wave of TSP-1 expression induced by oncogenic Kras was markedly abrogated in the absence of p53 (Figure 4B). We next investigated TSP-1 expression in large lung adenomas harvested from KrasG12D×p53fl/fl and KrasG12D×Tsp-1+/+ mice. TSP-1 was dramatically downregulated in lung adenomas lacking p53 (Supplemental Figure 9), further supporting a model whereby oncogenic Ras induces TSP-1 expression in a p53-dependent manner. Interestingly, while p53 was initially upregulated after Kras activation in the absence of TSP-1, p53 protein levels fell thereafter (Figure 4B), suggestive of a requirement for TSP-1 in maintaining p53 expression. Given that TSP-1 induction is downstream of p53, one possible scenario is that TSP-1, once induced, may then function in a positive feedback loop to stabilize p53 levels. To further establish the effect of TSP-1 loss on oncogenic Kras–mediated p53 activation and stability, we assayed levels of both p53 and its target, p21cip1, in Tsp-1+/+ versus Tsp-1–/– cells at various times after KrasG12D activation in lung fibroblasts (Figure 4C). Again, we observed that p53 was initially induced in KrasG12D×Tsp-1–/– fibroblasts at levels comparable to those in KrasG12D×Tsp-1+/+ cells. At later times, however, p53 was progressively downregulated in KrasG12D×Tsp-1–/– fibroblasts, even though oncogenic Kras expression was sustained (Figure 4C). Consistent with studies showing that p21cip1 induction, while necessary, is not sufficient for oncogene-induced senescence (28, 35, 36), p21cip1 was induced after oncogenic Ras activation — but at a substantially lower level in the absence of TSP-1 — in lung fibroblasts, lung epithelial cells, and lung tumors (Figure 4, C and D, and Supplemental Figure 10, A and B).
Figure 4. Oncogenic Kras activation triggers TSP-1 upregulation in a p53-dependent manner.

(A) TSP-1 protein expression in lungs harvested from p53+/+, p53+/–, and p53–/– mice. Isolated lung tissue was probed for TSP-1 expression by immunoblotting. Tsp-1–/– lung tissue served as negative control. TSP-1 expression relative to β-actin (loading control) was quantified by densitometry. (B) TSP-1 expression in the absence of p53 after KrasG12D activation in primary lung fibroblasts. KrasG12D×Tsp-1+/+, KrasG12D×p53fl/fl, and KrasG12D×Tsp-1–/– lung fibroblasts were isolated, infected with Ad-Cre to activate oncogenic Kras and delete p53, harvested at the indicated time points, and probed for TSP-1 expression by immunoblotting. (C) Western blot of p53 and p21cip1 expression in KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– primary lung fibroblasts isolated at the indicated times after KrasG12D activation. β-actin served as loading control. (D) Immunohistochemistry for p21cip1 in KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– lung sections isolated 21 weeks after KrasG12D activation. Arrows indicate p21cip1-positive cells. (E) pp53Ser15 after oncogenic Hras activation. Tsp-1+/+ and Tsp-1–/– lung fibroblasts were infected with HrasV12 retrovirus, harvested at the indicated days, and probed for pp53Ser15, p53, p21cip1, and β-actin by immunoblotting. Uninfected lung fibroblasts served as negative controls. pp53Ser15 expression relative to total p53 was quantified by densitometry. (F) Nutlin-3 treatment restored HrasV12-induced senescence in Tsp-1–/– lung fibroblasts. Lung fibroblasts were infected with HrasV12 or mock retrovirus, treated with 5 μM nutlin-3 or DMSO for 9 days, then stained for SA–β-gal to detect senescent cells. Scale bars: 50 μm (D); 200 μm (F).
Oncogenic Ras has been shown to trigger phosphorylation of p53 at serine 15 (pp53Ser15) to stabilize its expression (28). Since TSP-1 expression appears necessary to sustain p53 expression after oncogenic Ras activation, we directly assessed the effect of Tsp-1 status on oncogenic Ras–induced pp53Ser15 (Figure 4E). In Tsp-1+/+ fibroblasts, pp53Ser15 was dramatically increased upon oncogenic Hras expression followed by sustained p53 expression. However, in Tsp-1–/– cells, oncogenic Ras activation failed to induce pp53Ser15, with lower expression of both p53 and its target, p21cip1. Notably, MDM2 expression was unaffected throughout (Supplemental Figure 10C). These observations are consistent with the notion that TSP-1 is critical for p53 stability and function (Figure 4E and Supplemental Figure 10D). To confirm this, we expressed HrasV12 in Tsp-1+/+ and Tsp-1–/– primary fibroblasts and treated the cells with nutlin-3 to pharmacologically stabilize p53 (37) and bypass any requirement for TSP-1. We then assayed for senescence by measuring SA–β-gal reactivity. At 9 days after oncogenic Hras expression, both Tsp-1+/+ and Tsp-1–/– primary fibroblasts treated with nutlin-3 showed comparable levels of SA–β-gal staining (Figure 4F), which indicates that enforced p53 expression is sufficient to restore Ras-induced senescence in the absence of TSP-1. These results suggest the existence of a positive feedback loop within the oncogenic Ras–mediated p53 activation pathway that is centrally regulated by TSP-1.
Oncogenic KrasG12D signaling is modified in the absence of Tsp-1.
Continuous oncogenic activation of the MAPK pathway in murine fibroblasts leads to the induction of p19ARF, which is required for the initiation and maintenance of p53-dependent cellular senescence (27, 38). Our present studies indicated that TSP-1 is also necessary for oncogenic Ras–induced senescence; however, TSP-1’s role in this outcome was associated both with its induction by p53 and, once induced, its maintenance of p53 expression. These results suggest that TSP-1 may play a key role in a positive feedback loop within an oncogenic Ras–mediated p53 activation pathway. To investigate the molecular mechanism by which TSP-1 maintains p53 activation, we examined the role of each component of the Ras-MAPK pathway. First, we established that expression of oncogenic KrasG12D is not itself effected by Tsp-1 status: levels of KrasG12D were similar in tumors derived from both KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– mice (Supplemental Figure 11A). Next, we assayed levels of pMEK, pERK, p19ARF, and p16INK4a after KrasG12D activation in primary lung fibroblasts. In the absence of TSP-1, pERK induction by KrasG12D expression was downregulated (Figure 5A and Supplemental Figure 11B). Loss of TSP-1 also decreased pERK triggered by oncogenic Kras in lung epithelial cells (Supplemental Figure 11C). While p16INK4a levels were unaffected by TSP-1 status, p19ARF expression was markedly reduced in the absence of TSP-1, in both lung fibroblasts and lung tumors harvested from KrasG12D×Tsp-1–/– mice (Figure 5, A and B, and Supplemental Figure 11B). These data demonstrated that ERK activation downstream of oncogenic KrasG12D is downregulated in the absence of TSP-1. In contrast, activation of the upstream ERK regulator MEK was not affected by TSP-1 loss (Figure 5C), which suggests that TSP-1 plays a role specifically in regulating ERK activity. To further confirm that p19ARF upregulation and ERK activation were impaired during oncogenic Ras signaling in the absence of TSP-1, we expressed HrasV12 in either Tsp-1+/+ or Tsp-1–/– primary lung fibroblasts and probed for p19ARF and pERK by Western blot. In Tsp-1+/+ primary fibroblasts, oncogenic Ras activation induced sustained pERK up to 7 days, followed by a substantial increase in p19ARF expression. However, Tsp-1–/– primary fibroblasts showed only transient ERK activation and modest p19ARF upregulation upon oncogenic Ras expression (Figure 5D). Since our data imply a dependence on TSP-1 for oncogenic Ras–induced senescence via the MAPK pathway, we investigated whether normal Ras signaling also requires TSP-1. Tsp-1+/+ and Tsp-1–/– fibroblasts were treated with EGF and probed for TSP-1 upregulation and ERK activation. TSP-1 expression did not increase after EGF treatment of Tsp-1+/+ fibroblasts, and pERK expression was unaffected by the loss of TSP-1 after EGF treatment (Figure 5E), implicating a requirement for TSP-1 specifically during oncogenic Ras activation, but not during normal Ras signaling.
Figure 5. Sustained oncogenic Kras signaling is attenuated in the absence of TSP-1.

(A) Western blot of ERK, p19ARF, and p16INK4a expression after KrasG12D activation in KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– primary adult lung fibroblasts isolated at the indicated times after oncogenic Kras activation. β-actin served as loading control. (B) Immunofluorescence for p19ARF in KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– lung sections isolated 21 weeks after KrasG12D activation. Hoechst stain indicates nuclei. Scale bars: 50 μm. (C) KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– primary lung fibroblasts were infected with Ad-Cre, harvested at the indicated times, and probed for pMEK and total MEK by immunoblotting. β-actin served as loading control. (D) Tsp-1+/+ and Tsp-1–/– primary lung fibroblasts were infected with HrasV12 or mock retrovirus, harvested at the indicated times, and probed for TSP-1, pERK, total ERK, and p19ARF expression by immunoblotting. Uninfected lung fibroblasts were used as negative controls; β-actin served as loading control. (E) Tsp-1+/+ and Tsp-1–/– primary lung fibroblasts were treated with 10 nM EGF, harvested at the indicated times, and probed for TSP-1, pERK, and total ERK expression by immunoblotting. β-actin served as loading control.
TSP-1 tethers pERK in the cytosol.
Because our data suggested that TSP-1 may be important for specifically regulating ERK activity, we investigated whether TSP-1 directly interacts with pERK. Many studies have indicated that scaffolding proteins are critical for efficient ERK signaling and for tight regulation of their multiple downstream functions (39). TSP-1 is a large multimeric glycoprotein with multiple domains and has been shown to function as a scaffold protein in other cell types (40). Thus, we tested whether ERK and TSP-1 directly interact (Figure 6A). Endogenous TSP-1 specifically coprecipitated with endogenous pERK, but not with inactive ERK, total ERK, or MEK, after oncogenic Ras activation (Figure 6A). To confirm the interaction of TSP-1 with pERK in vivo, we used the Duolink method in KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– lung sections 21 weeks after KrasG12D activation (Figure 6B). Duolink yields a single signal if 2 different antibody epitopes are within 40 nm of each other (41). Using antibodies against pERK and TSP-1, we confirmed positive signals throughout KrasG12D×Tsp-1+/+ lung sections, but not KrasG12D×Tsp-1–/– lung sections (Figure 6B).
Figure 6. TSP-1 coprecipitates with pERK, and its loss alters pERK subcellular localization.

(A) Endogenous TSP-1 coprecipitated with endogenous pERK after HrasV12 expression. Top: WT lung fibroblasts were infected with HrasV12 retrovirus, harvested 10 days after infection, and subjected to IP with isotype-matched IgG and TSP-1 or pERK antibodies. IP samples were then probed with antibodies for TSP-1 and pERK. Bottom: WT lung fibroblasts with HrasV12 retrovirus were also subjected to IP with TSP-1, ERK, pERK, and MEK-1/2 antibodies probed for MEK-1/2, ERK, and pERK. (B) Interaction of TSP-1 with pERK in vivo was assessed by Duolink immunostaining for TSP-1 and pERK in KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– lung tumors 21 weeks after Ad-Cre infection. (C) Subcellular localization of pERK, determined by immunofluorescence staining for pERK in KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– lung tumors 21 weeks after Ad-Cre infection. (D) Subcellular localization of pERK, assessed by cell fractionation followed by immunoblotting for pERK, in HrasV12-infected Tsp-1+/+ and Tsp-1–/– lung fibroblasts. Lamin B1 and α-tubulin served as markers for nuclear (N) and cytosolic (C) compartments, respectively. Nuclear relative to cytosolic gel density was quantified by densitometry. (E) pp90RSK and pELK-1, assessed by immunoblotting, in Tsp-1+/+ and Tsp-1–/– lung fibroblasts after retroviral HrasV12 infection. β-actin served as loading control. Gel density of phospho- relative to total substrates was quantified by densitometry. (F) TSP-1 coprecipitated with pERK, even in the presence of neutralizing TSP-1 antibody in the culture media. WT primary lung fibroblasts were infected with HrasV12 retrovirus and treated with 10 μg/ml neutralizing TSP-1 antibody (nAb) or an isotype-matched control (IgG) for 10 days, followed by IP with anti–TSP-1 and immunoblot for TSP-1 and pERK. Scale bars: 100 μm (B); 50 μm (C); 25 μm (C, insets).
Sustained activation of ERK by oncogenic Ras expression leads to translocation of pERK from the cytoplasm to the nucleus, a requirement for activation of proliferation-dependent genes (42–44). However, studies have shown that, upon establishment of a senescent phenotype in fibroblasts, ERKs remain phosphorylated but relocalize back to the cytoplasm (31). To examine whether pERK localization was altered by the absence of Tsp-1, we examined pERK localization in lung tumors harvested from KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– mice by immunofluorescence (Figure 6C). Strikingly, pERK was predominantly nuclear in KrasG12D×Tsp-1–/– lung tumors, as indicated by purple nuclei caused by the overlap of red pERK and blue Hoechst staining, whereas in KrasG12D×Tsp-1+/+ tumors, pERK was localized in the cytoplasm, as evidenced by immunostaining showing distinct red (pERK) in the cytosol and blue (Hoechst) nuclei (Figure 6C). We next expressed HrasV12 in either Tsp-1+/+ or Tsp-1–/– primary fibroblasts, fractionated the cytoplasm and nuclei from these cells, and examined each fraction for expression of pERK. The purity of cytoplasmic and nuclear cellular fractions was confirmed by the restricted expression of the nuclear protein lamin B1 to the nuclear fraction and the cytoskeletal protein α-tubulin to the cytoplasmic fraction. Confirming the nuclear localization of pERK in KrasG12D×Tsp-1–/– lung tumors, we found a substantial increase in pERK levels in the nuclear fraction in HrasV12-infected Tsp-1–/– fibroblasts, in contrast to its predominantly cytoplasmic expression in HrasV12-infected Tsp-1+/+ fibroblasts (Figure 6D).
To assess the functional significance of differential pERK subcellular localization, we explored the extent of activation of cytoplasmic and nuclear ERK after oncogenic Ras expression in the presence and absence of TSP-1 by assaying the phosphorylation status of known pERK substrates. Compared with Tsp-1+/+ cells, we observed a marked decrease in the level of phosphorylation of the cytoplasmic ERK substrate p90RSK in Tsp-1–/– cells after oncogenic Ras expression. Similarly, we found increased phosphorylation of the nuclear pERK substrate Elk-1 in Tsp-1–/– cells (Figure 6E). Collectively, our data suggest that TSP-1 directly interacts with pERK and preferentially tethers it to the cytosol, disabling pERK-mediated phosphorylation of nuclear substrates necessary for cell proliferation. Since TSP-1 is prototypically a secreted protein, we asked whether its coprecipitation with pERK could be inhibited by addition of neutralizing TSP-1 antibody to the cell culture media to sequester secreted TSP-1 (45). However, such addition of anti–TSP-1 neutralizing antibody had no inhibitory effect on endogenous TSP-1–pERK coprecipitation (Figure 6F), which indicates that oncogenic Ras–induced TSP-1–pERK interaction occurs intracellularly in a cell-intrinsic manner.
MAPK signaling is regulated by many different mechanisms, since the outcome of MAPK signaling is controlled by both its magnitude and its duration. For example, recent studies indicate that oncogenic Ras activation of MAPK signaling requires its activation over a certain threshold to switch from a proliferative to a senescent phenotype (29, 30). Hence, loss of TSP-1 could prevent the transition from proliferation to senescence either by attenuation of pERK signal intensity or by the absence of pERK cytoplasmic relocalization. To determine whether decreased pERK signaling is sufficient to diminish a senescence response, we used a pharmacologic approach to dampen MAPK signaling. We examined the ability of oncogenic Ras to activate senescence, as indicated by SA–β-gal reactivity and by expression of p19ARF, p53, and p21cip1 in the presence of increasing concentrations of the well-characterized MEK inhibitor U0126 (Figure 7A and ref. 46). Upon increasing MEK inhibition, diminished ERK activation led to decreased senescence as well as decreased p19ARF, p53, and p21cip1 expression, even in the presence of persistent oncogenic Ras expression (Figure 7, A and B).
Figure 7. Pharmacologic attenuation of ERK activity suppresses oncogenic Ras–induced senescence.

(A) WT adult lung fibroblasts were infected with HrasV12 retrovirus, treated with the MEK inhibitor U0126 at the indicated concentrations, and stained for SA–β-gal activity. Percent SA–β-gal–positive cells was also quantified. Data represent mean ± SEM. Scale bars: 200 μm. (B) WT lung fibroblasts infected with HrasV12 retrovirus were treated with U0126 at the indicated concentrations for 3 days and probed for pERK, ERK, p19ARF, p53, and p21cip1 by immunoblotting. (C) A p53-dependent positive feedback role for TSP-1 in the maintenance of oncogenic Ras–induced senescence by tethering pERK in the cytoplasm.
Finally, to verify that our in vitro findings with oncogenic Hras were relevant to oncogenic Kras, we monitored the induction of senescence in Tsp-1+/+ and Tsp-1–/– adult lung fibroblasts lentivirally transduced with KrasG12D. Our data showed a similar reduction in the SA–β-gal–positive population in the absence of TSP-1 (Supplemental Figure 12A). Additionally, pERK after oncogenic KrasG12D expression was also compromised in Tsp-1–/– fibroblasts (Supplemental Figure 12B). These data indicate that TSP-1 is required for both oncogenic Hras– and oncogenic Kras–induced senescence through similar mechanisms.
Collectively, our data indicate that TSP-1 plays a critical role in sustained oncogenic Ras signaling, as measured by pERK expression, p19ARF upregulation, and p53 expression, to mediate an oncogenic Ras–dependent senescence response. Our findings also suggest that sustained oncogenic Ras signaling is mediated, at least in part, by a p53–TSP-1–dependent positive feedback loop directly interacting with activated pERK, tethering it to the cytosol and attenuating the ability of pERK to activate pro-proliferative nuclear targets (Figure 7C).
Discussion
Oncogenic Ras–induced replicative senescence has been proposed to underlie the long latency between the progression of premalignant adenomas in the lung to the appearance of frank adenocarcinoma (14). Due to its high expression in the lung, we examined the role of the endogenous angiogenesis inhibitor TSP-1 in limiting lung tumor progression. Unexpectedly, we identified TSP-1 as a critical component mediating oncogenic Ras–induced senescence in the lung. Survival of KrasG12D×Tsp-1–/– versus KrasG12D×Tsp-1+/+ mice was significantly diminished by approximately 3 months. Our data showed that TSP-1 expression was necessary for the maintenance of Ras-induced senescence and that TSP-1 upregulation was a consequence of p53 activation after oncogenic Ras expression. While activation of the MAPK pathway and p19ARF and p53 expression were initially observed after Kras activation, in the absence of TSP-1, we found substantially diminished pERK, p19ARF, and p53 expression. These data suggest a model whereby TSP-1 functions in a feedback loop to stabilize an oncogenic Ras–mediated senescence response in the lung. It was previously reported that the expression of endogenous KrasG12D is insufficient to activate ERK and induce senescence in mouse embryonic fibroblasts (MEFs) from LSL-KrasG12D mice upon Ad-Cre infection (11, 12). However, in the adult lung fibroblasts used in our study, we showed that endogenous KrasG12D increased pERK and induced premature cell senescence. The discrepancy between MEFs and adult lung fibroblasts in response to the expression of endogenous KrasG12D may be due to cell-intrinsic differences (47–51). We also demonstrated that TSP-1 interacted directly with pERK, implicating the necessity of TSP-1 for a MAPK-dependent senescence response. Re-expression of TSP-1 in Tsp-1–/– cells restored the ability of oncogenic Ras to induce senescence, indicative of the central role TSP-1 plays in mediating Ras-induced senescence. Furthermore, our data showed that TSP-1 expression was also lost in lung adenocarcinoma patient samples, implicating its physiological relevance in human lung cancer progression.
Since TSP-1 is a well-characterized secreted protein, its mechanism of action has been generally assumed to occur in a cell-extrinsic manner via receptor engagement and subsequent activation of intracellular signaling pathways. Our present data demonstrated that TSP-1 functions in a cell-intrinsic manner in mediating oncogenic Ras–induced senescence, as addition of soluble TSP-1 to the media of Tsp-1–/– cells did not restore a senescence response after Hras expression. Similarly, the growth of transplanted KrasG12D×Tsp-1–/– lung tumor cells in vivo was not affected by TSP-1 host status. The present study illustrated that the absence of TSP-1 promotes precocious progression of lung lesions to diffuse adenocarcinomas, seemingly bypassing benign adenoma formation observed in the lungs of KrasG12D mice and decreasing survival.
It has been well established that oncogenic Ras induces a senescence response in primary cells involving the tumor suppressors p53, p16INK4a, p19ARF, and p15INKb (2, 15, 16, 52). While expression of oncogenic Ras initially triggers a mitogenic response in primary cells, constitutive Ras activity eventually leads to acquisition of characteristic senescence morphology. Oncogenic Ras activates a series of effector proteins, including Raf-1 and PI3K, each activating its own distinct targets that produce different aspects of phenotypes attributed to enforced oncogenic Ras expression (53). Oncogene-induced senescence has been shown to occur as a consequence of either constitutive Raf/MEK/MAPK pathway activation by oncogenic Ras (8) or PI3K/AKT pathway hyperactivation due to PTEN loss (7), with each pathway requiring its own set of effectors. Oncogenic BRAF–induced senescence has been shown to require a number of secreted factors, referred to as the inflammatory secretome, which function through autocrine and paracrine pathways to mediate its effects (19, 21). Here we showed that TSP-1 was specifically required for oncogenic BRAF–induced senescence, but had no effect on senescence induced by PTEN knockdown. These data, together with our studies showing decreased pERK expression and increased nuclear localization in the absence of TSP-1, suggest that TSP-1 may function to regulate MAPK signaling. Our data illustrated a specific requirement for TSP-1 during oncogenic Ras activation, but not during normal Ras signaling, as pERK expression was not affected in Tsp-1–/– fibroblasts after EGF treatment.
Oncogenic Ras activates the p19ARF-p53 pathway to initiate cellular senescence (15, 38). Numerous studies have shown that initiation of oncogenic Ras–induced senescence is dependent on functional p53. Although p53 is critical for the initiation of a senescence response, it appears to be less important for the maintenance of senescence, as withdrawal of p53 after several days of p53-dependent senescence did not allow cells to re-enter the cell cycle (28). While oncogene-induced senescence is thought to be largely irreversible, p53 targets that are necessary and sufficient to maintain oncogene-induced cellular senescence have not yet been identified. p53 is a key effector of cellular responses to various stresses and coordinates gene expression as a consequence of specific stimuli. p53 activity is primarily modulated by protein stability and posttranslational modifications (54). We showed here that levels of p53 as well as pp53Ser15 were downregulated in KrasG12D×Tsp-1–/– versus KrasG12D×Tsp-1+/+ mice. While the p53 target p21cip1 was necessary for cell cycle arrest, its expression alone in the absence of p53 was insufficient to trigger replicative senescence (28). p53 has been shown to directly transactivate TSP-1 (our unpublished observations and ref. 34). TSP-1 was originally identified as a potent angiogenesis inhibitor, and its expression in tumor cells has been attributed to its role in antiangiogenic surveillance (24). Our present findings indicate that loss of TSP-1 has no effect on tumor angiogenesis in the lung and suggest that TSP-1 plays a key role in the maintenance of an oncogenic Ras–induced senescence response in this tissue.
The data presented here suggest that TSP-1, while transactivated by p53, may also be necessary to stabilize p53 expression and subsequent function. The mechanism by which TSP-1 stabilizes p53 expression appears to occur via a feedback loop through a direct interaction with pERK. Our studies demonstrated that pERK expression decreased after Kras activation in KrasG12D×Tsp-1–/– fibroblasts and that TSP-1 specifically coprecipitated with pERK, but not with unphosphorylated ERK, pMEK, or p19ARF. Furthermore, we showed that in the absence of TSP-1, pERK was predominantly localized in the nucleus during oncogenic Ras signaling, with a corresponding increase in the activation of pERK nuclear versus cytoplasmic targets. Collectively, these data suggest that TSP-1 may function to tether pERK in the cytosol. Since ERK signaling has highly pleiotropic effects, it is tightly regulated by numerous mechanisms, including magnitude of signaling, duration of signaling, and nucleo-cytoplasmic localization (41, 55–59). Previous studies have demonstrated that oncogenic Ras–induced senescence requires relocalization of pERK from the nucleus to the cytosol to suppress the activation of pro-proliferative targets in the nucleus, thus permitting cells to switch from a proliferative to a senescent phenotype (31). Hence, we propose that loss of TSP-1 could prevent the transition from proliferation to senescence either by attenuation of pERK signal intensity or by the absence of pERK cytoplasmic retention.
Our data provide new insight into how oncogenic Ras activation promotes senescence during lung tumorigenesis. TSP-1 influences the biological outcome of oncogenic Ras signaling via the MAPK pathway by regulating the localization of pERK, thus altering the substrates of activated ERK. Sequestration of activated ERK in the cytosol by TSP-1 appears to be necessary to maintain a senescent phenotype. We propose a model whereby sustained oncogenic Ras signaling is mediated in part by a p53–TSP-1–dependent positive feedback loop, with TSP-1 directly interacting with pERK and promoting senescence by maintaining expression of p19ARF, p53, and p21cip1. Hence, upregulation of TSP-1 represents a critical component of a more general host tumor-suppressor response and offers new molecular insights into oncogenic Ras–induced senescence “addiction” to TSP-1 during lung tumorigenesis.
Several studies have reported that TSP-1 expression has a positive correlation with survival of lung cancer patients (60). It was also recently reported that tumor expression of TSP-1 is a promising prognostic factor for overall survival of patients with lung adenocarcinomas treated with a docetaxel-gemcitabine regimen (61). Interestingly, chronic cigarette smoke exposure increases TSP-1 expression, apoptotic cell numbers, and parenchymal cell loss, which are characteristics of emphysema development (62). In addition, Rezzani and colleagues have shown that nicotine upregulates TSP-1 expression in rat aortas, which suggests that TSP-1 can be a biomarker for smoking cessation (63). Our present findings revealed a novel role of TSP-1 as a tumor suppressor mediating oncogenic Ras–induced senescence with potentially significant clinical implications.
Methods
Animals.
LSL-KrasG12D mice (i.e., KrasG12D×Tsp-1+/+ WT) and LSL-KrasG12D×p53fl/fl mice on a 129SvEv background were provided by T. Jacks (MIT Center for Cancer Research, Cambridge, Massachusetts, USA) and backcrossed onto a C57BL/6 background. KrasG12D×Tsp-1–/– mice were generated by mating LSL-KrasG12D with Tsp-1–/– mice (provided by J. Lawler, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA) on a C57BL/6 background.
Vectors.
Retroviral and lentiviral infections were performed as previously described (64). HrasV12 was in a pBabe retroviral vector backbone. GFP, murine Tsp-1, and inducible estrogen receptor–BRAFWT (BRAFWT:ER) and –BRAFV600E (BRAFV600E:ER) were in pWZLBlast retroviral vector. The pLKO.1 lentiviral vector containing shRNA for PTEN (5′-GCTAGAACTTATCAAACCCTTCTC-3′) was purchased from Open Biosystems. Upon infection with retroviruses from pBabe and pLKO.1 or pWZLBlast retroviral vectors, cells were selected with 2 μg/ml puromycin or 12.5 μg/ml blasticidine, respectively, for 3 days and used for further experiments.
Ad-Cre infection.
Recombinant adenovirus encoding Cre recombinase (Ad-Cre) was purchased from the University of Iowa Gene Transfer Vector Core. For in vivo studies, approximately 4- to 6-week-old mice were infected intranasally with 5 × 106 PFU Ad-Cre as described previously (23). Briefly, mice were anesthetized with isofluorane and inoculated intranasally with Ad-Cre/CaPi coprecipitates prepared as previously described (65). For in vitro experiments, primary lung fibroblasts from LSL-KrasG12D and KrasG12D×Tsp-1–/– mice were infected with Ad-Cre at 500 MOI overnight.
Isolation of primary lung fibroblasts and epithelial cells.
Primary lung fibroblasts were isolated from approximately 3- to 4-week-old mice as previously described (66). Briefly, lungs were minced and digested in calcium- and magnesium-free HBSS containing 10 mg/ml type II collagenase (Worthington) and 20 μg/ml DNase I (Sigma-Aldrich) for 30 minutes at 37°C with shaking. Cell suspensions were filtered through progressively smaller cell strainers (100 and 40 μm), centrifuged, and plated in culture media (1:1 [v/v] DMEM/Ham’s F-12 containing 10% FBS, 15 mM HEPES, 2 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin). After 1 hour, adherent fibroblasts were rinsed with HBSS and cultured in media.
Primary lung epithelial cells were isolated from approximately 3- to 4-week-old mice and cultured as previously described (67, 68). Briefly, lungs were digested in HBSS containing 10 mg/ml type II collagenase and 20 μg/ml DNase I at 37°C for 30 minutes. Cell suspensions were strained sequentially through a 100-μm and a 40-μm strainer, and collagenase activity was quenched with equal volume FBS. Cells were washed once with HBSS, resuspended with culture media (1:1 [v/v] DMEM/Ham’s F-12 supplemented with 5% FBS, 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 120 IU/ml ITS [BD Biosciences]), and incubated at 37°C for 1 hour in culture dishes to remove nonepithelial cells. The nonadherent cells were collected with centrifugation at 200 g for 5 minutes, washed 3 times with HBSS, resuspended with culture media, plated on 0.1% gelatin-coated dishes, and cultured for 3 days. On day 4, the culture media was replaced with differentiation media (1:1 [v/v] DMEM/Ham’s F-12 containing 2% NuSerum [BD Biosciences], 50 U/ml penicillin, and 50 μg/ml streptomycin). Epithelial cell purity was determined by immunostaining for the lung epithelial cell marker SP-C.
SA–β-gal staining.
Lungs were harvested from mice at the indicated times, perfused with OCT solution, rapidly frozen in liquid nitrogen, and mounted onto glass slides. SA–β-gal staining was then performed on 8-μm cryosections using a commercial kit (Sigma-Aldrich) according to the manufacturer’s instructions and observed under bright field at ×100–×200 magnification. 3 × 105 primary lung fibroblasts were seeded in 100-mm plates and infected with viruses as described above. 5 × 104 infected cells were then seeded in triplicate in 6-well tissue culture dishes, fixed, and stained with SA–β-gal staining kit at the indicated time points. For nutlin-3 treatment, 5 × 104 infected cells were plated in 6-well tissue culture plates, incubated overnight, and treated with 5 μM nutlin-3 (Sigma-Aldrich) for 9 days.
Proliferation.
After infection with viruses, 5 × 104 lung fibroblasts were plated in triplicate in 6-well tissue culture plates. Cells were then counted and reseeded at a density of 5 × 104 every 3 or 4 days.
Immunoblotting and IP.
For immunoblot analysis, 3 × 105 primary lung fibroblasts were plated in 100-mm plates, infected with virus, harvested at the indicated times, and lysed with RIPA buffer supplemented with a protease inhibitor cocktail (Roche). Whole cell lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the following antibodies: TSP-1 (NeoMarkers); pERK, total ERK, p53, pp53Ser15, pMEK1, total MEK1, pElk-1, total Elk-1 (Cell Signaling Technology); p16INK4a, p21cip1, MDM2 (Santa Cruz Biotechnologies); p19ARF (Novus); pp90RSKThr573, total p90RSK (Assay Biotechnology); β-actin (Sigma-Aldrich). To measure relative gel densities, band densities were quantified with densitometric analysis using ImageJ software (NIH) and then normalized to β-actin.
For IP experiments, primary lung fibroblasts cells were plated in 150-mm plates, infected with virus, harvested, and lysed with 25 mM Tris-HCl (pH 7.4) buffer supplemented with 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 1 mM DTT, 1 mM EDTA, 1% NP-40, 0.5% SDS, 5% glycerol, 1 mM PMSF, and protease inhibitor cocktail. After preclearing with normal IgG and protein A/G–conjugated resin (Pierce) at 4°C for 1 hour, 1 mg cell lysate was incubated with pERK or total ERK antibodies at 4°C overnight. After further incubation with protein A/G–conjugated resin at 4°C for 2 hours, IPs were analyzed by immunoblotting.
Cell fractionation.
3 × 105 primary lung fibroblasts were plated in 100 mm plates, infected with virus, harvested at the times indicated, and fractionated using Qproteome Cell Compartment kit (Qiagen) according to the manufacturer’s instructions. Nuclear and cytoplasmic fractions were then analyzed by immunoblotting as described above, using antibodies against pERK, lamin B1 (Abcam), α-tubulin (Applied Biological Materials), and β-actin.
Immunohistochemistry.
Mice were euthanized at the indicated times. Lung tissues were perfused and fixed with 10% neutral buffered formalin. Paraffin-embedded sections were prepared using standard techniques and stained with H&E for histological examination. Human lung cancer samples were obtained at the time of surgery from the surgical pathology lab at the Hospital of the University of Pennsylvania. Formalin-fixed, paraffin-embedded tissue blocks were screened and made into a tissue microarray (TMA). The array contained 68 tumor samples (arrayed in triplicate) and corresponding normal lung tissue.
Immunostaining was performed as previously described (69) using antibodies against CD31 (BD Pharmingen), TSP-1 (NeoMarkers), Ki67 (NeoMarkers), pan-RasAsp12 (Calbiochem), pERK (Cell Signaling Technology), and CD68 (AbD Serotec) with secondary antibodies from Molecular Probes. Immunostaining of p21cip1 was performed as previously described (30) using antibody to p21cip1 (Santa Cruz) and DAB Peroxidase Substrate kit (Vector Labs).
Duolink staining was performed on lung tumors harvested from mice 21 weeks after intranasal inhalation of Ad-Cre, using antibodies against TSP-1 and pERK and the Duolink kit (Olink Biosciences) according to the manufacturer’s instructions.
Immunofluorescence.
104 primary lung epithelial cells were plated on coverslips coated with 0.1% gelatin for 24 hours, fixed with 4% formaldehyde in PBS for 15 minutes, and permeabilized with 0.1% Triton X-100 for 15 minutes. After blocking with 5% BSA in PBS/0.1% Triton X-100 for 1 hour at RT, the cells were incubated with antibodies against SP-C or PML (Milipore) diluted in blocking solution for 1 hour at RT. Cells were further incubated with secondary antibodies for 30 minutes at RT, then counterstained with Hoechst dye to visualize cell nuclei.
Microvessel density.
Lung tissues harboring similar-sized tumor nodules were harvested and paraffin embedded. After staining with CD31 to visualize tumor vasculature, microvessel density was determined as previously described (69).
Statistics.
Overall survival of mice after intranasal Ad-Cre administration was evaluated by a Kaplan-Meier survival analysis, and the log-rank test was used to determine statistical significance of survival differences between KrasG12D×Tsp-1+/+ and KrasG12D×Tsp-1–/– mice. For other studies, the difference between 2 groups was analyzed using Student’s 2-tailed unpaired t test. A P value less than 0.05 was considered significant.
Study approval.
Animal studies were approved by the University of Pennsylvania Medical School IACUC as set forth in the NIH Guide for the Care and Use of Laboratory Animals.
Supplementary Material
Acknowledgments
The authors thank members of the Ryeom lab, Lars Zender, and David Feldser for helpful discussions and advice. This work was supported by Samsung Biomedical Research Institute grant SBRI B-B0-008-3 (to K.H. Baek), the V Foundation for Cancer Research (to C.F. Kim), an American Cancer Society Research Scholar Grant (to C.F. Kim), NIH grant P01CA66726 (to S.M. Albelda), NCI/NIH grant CA100193 (to G.I. Evan), NIH grant R01CA118375 (to S. Ryeom), NIH grant P01CA45548 (to S. Ryeom), The Garrett B. Smith Foundation (to S. Ryeom), and the Ted Driven Foundation (to S. Ryeom).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(10):4375–4389. doi:10.1172/JCI67465.
References
- 1.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10(1):51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. . Cell. 1997;88(5):593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 3.Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436(7051):660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 4.Sarkisian CJ, et al. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9(5):493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 5.Dankort D, et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. . Genes Dev. 2007;21(4):379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwanaga K, et al. Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. . Cancer Res. 2008;68(4):1119–1127. doi: 10.1158/0008-5472.CAN-07-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michaloglou C, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. . Nature. 2005;436(7051):720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 9.Courtois-Cox S, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10(6):459–472. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27(20):2801–2809. doi: 10.1038/sj.onc.1210950. [DOI] [PubMed] [Google Scholar]
- 11.Guerra C, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. . Cancer Cell. 2003;4(2):111–120. doi: 10.1016/S1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 12.Tuveson DA, et al. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. . Cancer Cell. 2004;5(4):375–387. doi: 10.1016/S1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 13.Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20(2):150–155. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Collado M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 15.Kamijo T, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. . Cell. 1997;91(5):649–659. doi: 10.1016/S0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 16.Sharpless NE, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. . Nature. 2001;413(6851):86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 17.Kang TW, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 18.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9(2):81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 19.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acosta JC, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 21.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 22.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. . Genes Dev. 2001;15(24):3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6(1):1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malkinson AM. The genetic basis of susceptibility to lung tumors in mice. Toxicology. 1989;54(3):241–271. doi: 10.1016/0300-483X(89)90062-0. [DOI] [PubMed] [Google Scholar]
- 26.Jackson EL, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65(22):10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 27.Lin AW, et al. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12(19):3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferbeyre G, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. . Mol Cell Biol. 2002;22(10):3497–3508. doi: 10.1128/MCB.22.10.3497-3508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junttila MR, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468(7323):567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feldser DM, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468(7323):572–575. doi: 10.1038/nature09535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaumont-Leclerc MF, Mukhopadhyay UK, Goumard S, Ferbeyre G. PEA-15 is inhibited by adenovirus E1A and plays a role in ERK nuclear export and Ras-induced senescence. J Biol Chem. 2004;279(45):46802–46807. doi: 10.1074/jbc.M403893200. [DOI] [PubMed] [Google Scholar]
- 32.Fontanini G, et al. Thrombospondins I and II messenger RNA expression in lung carcinoma: relationship with p53 alterations, angiogenic growth factors, and vascular density. Clin Cancer Res. 1999;5(1):155–161. [PubMed] [Google Scholar]
- 33.Ferbeyre G, et al. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14(16):2015–2027. [PMC free article] [PubMed] [Google Scholar]
- 34.Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265(5178):1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 35.Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. . Science. 1997;277(5327):831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 36.Pantoja C, Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. 1999;18(35):4974–4982. doi: 10.1038/sj.onc.1202880. [DOI] [PubMed] [Google Scholar]
- 37.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 38.Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. . Proc Natl Acad Sci U S A. 2001;98(9):5025–5030. doi: 10.1073/pnas.091100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chuderland D, Seger R. Protein-Protein interactions in the regulation of the extracellular signal-regulated kinase. Mol Biotechnol. 2005;29(1):57–74. doi: 10.1385/MB:29:1:57. [DOI] [PubMed] [Google Scholar]
- 40.Xu J, Xiao N, Xia J. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci. 2010;13(1):22–24. doi: 10.1038/nn.2459. [DOI] [PubMed] [Google Scholar]
- 41.Söderberg O, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. . Nat Methods. 2006;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 42.Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40(12):2707–2719. doi: 10.1016/j.biocel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 43.Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12(3):915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brunet A, et al. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18(3):664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doyen V, et al. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J Exp Med. 2003;198(8):1277–1283. doi: 10.1084/jem.20030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Favata MF, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273(29):18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 47.Azzarone B, Suarez H, Mingari MC, Moretta L, Fauci AS. 4F2 monoclonal antibody recognizes a surface antigen on spread human fibroblasts of embryonic but not of adult origin. J Cell Biol. 1984;98(3):1133–1137. doi: 10.1083/jcb.98.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen WY, Grant ME, Schor AM, Schor SL. Differences between adult and foetal fibroblasts in the regulation of hyaluronate synthesis: correlation with migratory activity. J Cell Sci. 1989;94(pt 3):577–584. doi: 10.1242/jcs.94.3.577. [DOI] [PubMed] [Google Scholar]
- 49.Ellis I, Banyard J, Schor SL. Differential response of fetal and adult fibroblasts to cytokines: cell migration and hyaluronansynthesis. Development. 1997;124(8):1593–1600. doi: 10.1242/dev.124.8.1593. [DOI] [PubMed] [Google Scholar]
- 50.Hernandez L, et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling inprogeria. Dev Cell. 2010;19(3):413–425. doi: 10.1016/j.devcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sha W, Thompson K, South J, Baron M, Leask A. Fibrogenesis Tissue Repair. Loss of PPARγ expression by fibroblasts enhances dermal wound closure [published online ahead of print April 13, 2012]. doi: 10.1186/1755-1536-5-5 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malumbres M, et al. Cellular response to oncogenic ras involves induction of the Cdk4 and Cdk6 inhibitor p15INK4b. . Mol Cell Biol. 2000;20(8):2915–2925. doi: 10.1128/MCB.20.8.2915-2925.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 55.Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. . Mol Cell Biol. 1997;17(9):5588–5597. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woods D, et al. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. . Mol Cell Biol. 1997;17(9):5598–5611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Formstecher E, et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell. 2001;1(2):239–250. doi: 10.1016/S1534-5807(01)00035-1. [DOI] [PubMed] [Google Scholar]
- 58.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4(8):556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 59.Ebisuya M, Kondoh K, Nishida E. The duration, magnitude and compartmentalization of ERK MAP kinase activity: mechanisms for providing signaling specificity. J Cell Sci. 2005;118(pt 14):2997–3002. doi: 10.1242/jcs.02505. [DOI] [PubMed] [Google Scholar]
- 60.Yamaguchi M, Sugio K, Ondo K, Yano T, Sugimachi K. Reduced expression of thrombospondin-1 correlates with a poor prognosis in patients with non-small cell lung cancer. Lung Cancer. 2002;36(2):143–150. doi: 10.1016/S0169-5002(01)00470-6. [DOI] [PubMed] [Google Scholar]
- 61.Papadaki C, et al. Tumoral expression of TXR1 and TSP1 predicts overall survival of patients with lung adenocarcinoma treated with first-line Docetaxel-Gemcitabine regimen. Clin Cancer Res. 2009;15(11):3827–3833. doi: 10.1158/1078-0432.CCR-08-3027. [DOI] [PubMed] [Google Scholar]
- 62.Hamburg B, Bauer E, Isenberg J, Lee J. Chronic cigarette smoke exposure leads to increased thrombospondin-1 concentrations, apoptosis, and parenchymal cell loss in the lungs. Am J Respir Crit Care Med. 2010;181:A2923. [Google Scholar]
- 63.Rodella LF, et al. Nicotine-induced morphological changes in rat aorta: the protective role of melatonin. Cells Tissues Organs. 2012;195(3):252–259. doi: 10.1159/000324919. [DOI] [PubMed] [Google Scholar]
- 64.Johannessen CM, et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102(24):8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fasbender A, et al. Incorporation of adenovirus in calcium phosphate precipitates enhances gene transfer to airway epithelia in vitro and in vivo. J Clin Invest. 1998;102(1):184–193. doi: 10.1172/JCI2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghosh M, et al. Role of cytosolic phospholipase A2 in prostaglandin E2 production by lung fibroblasts. . Am J Respir Cell Mol Biol. 2004;30(1):91–100. doi: 10.1165/rcmb.2003-0005OC. [DOI] [PubMed] [Google Scholar]
- 67.Antunes MB, et al. Murine nasal septa for respiratory epithelial air-liquid interface cultures. Biotechniques. 2007;43(2):195–204. doi: 10.2144/000112531. [DOI] [PubMed] [Google Scholar]
- 68.Woodworth BA, Antunes MB, Bhargave G, Palmer JN, Cohen NA. Murine tracheal and nasal septal epithelium for air-liquid interface cultures: a comparative study. Am J Rhinol. 2007;21(5):533–537. doi: 10.2500/ajr.2007.21.3068. [DOI] [PubMed] [Google Scholar]
- 69.Ryeom S, et al. Targeted deletion of the calcineurin inhibitor DSCR1 suppresses tumor growth. Cancer Cell. 2008;13(5):420–431. doi: 10.1016/j.ccr.2008.02.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.