

Abstract
Since the discovery of hepatitis C virus (HCV) by molecular cloning almost a quarter of a century ago, unprecedented at the time because the virus had never been grown in cell culture or detected serologically, there have been impressive strides in many facets of our understanding of the natural history of the disease, the viral life cycle, the pathogenesis, and antiviral therapy. It is apparent that the virus has developed multiple strategies to evade immune surveillance and eradication. This Review covers what we currently understand of the temporal and spatial immunological changes within the human innate and adaptive host immune responses that ultimately determine the outcomes of HCV infection.
The magnitude of the clinical problem
Hepatitis C virus (HCV) infection represents the most common blood-borne viral infection without a vaccine, and an estimated 3% of the world’s population is infected (i.e., approximately 180 million individuals) (1). Only a minority (~20%) of individuals exposed to HCV can spontaneously clear the infection, and most infected patients remain undiagnosed (2). HCV-related liver failure is a leading cause of cirrhosis and liver cancer and is a primary indication for liver transplantation (3, 4). There have been extraordinary advances in HCV treatment in the last two decades, and the current standard of care involves pegylated IFN, ribavirin, and as of May 2011, a protease inhibitor targeting genotype 1 (either boceprevir or telaprevir) (1). Complicated regimens, drug toxicities, and costs remain significant hurdles for many patients, and triple therapy may not be available for the majority of HCV-infected patients (5). Further, approximately one-third of treated patients fail to experience a sustained virologic response and therefore remain at risk for disease progression, with the proportion being even higher in prior nonresponders and others, all of whom comprise the difficult-to-treat patient groups (6). However, improved treatments are on the horizon, and in the near future, all-oral regimens not requiring IFN and given for shorter treatment durations will become a reality (7).
The virus and the innate hepatocyte response
First cloned in 1989 (8), hepatitis C is an enveloped, positive-stranded RNA hepacivirus that is approximately 9.6 kb in length. Following binding to cell surface proteins and entry by receptor-mediated endocytosis (reviewed in refs. 9, 10), HCV translation and replication begin in the cytosol. Pattern recognition receptors (PRRs) play major roles in the recognition of HCV RNA, such as retinoic-inducible gene I (RIG-I), which serves as a cytoplasmic viral sensor. In addition, the PRR toll-like receptor 3 (TLR-3) recognizes extracellular double-stranded RNAs (dsRNAs) generated from virus released from an infected cell and subsequently relocalizes to the endosome. A single-point mutation in RIG-I and a lack of TLR-3 expression in the human hepatocellular carcinoma–derived cell line Huh-7.5 and its derivatives contribute to a 50-fold greater permissiveness for HCV replication (11, 12). In 2005, the cloning of Japanese fulminant hepatitis (JFH-1) — an HCV genotype 2a isolate with exceptional efficiency at viral genome replication that does not require adaptive mutations (13–15) — combined with expression in Huh-7.5–derived cells, for the first time allowed the production of workable titers of infectious virus in culture, overcoming a major obstacle that had hitherto hindered the development of antiviral agents (9).
Innate recognition of HCV in hepatocytes occurs through dsRNA sensor protein kinase R (PKR), RIG-I, and TLR-3 (Figure 1A). PKR binds to the HCV internal ribosomal entry site (IRES) as early as 2 hours after infection and prior to the interaction with RIG-I; both pathways result in the recruitment of mitochondrial antiviral signaling (MAVS, also known as CARDIF/IPS-1/VISA) and tumor necrosis factor receptor–associated factor 3 (TRAF3) (16). PKR preferentially induces IFN-stimulated genes (ISGs) including the ubiquitin-like modifier ISG15 that negatively regulates RIG-I ubiquitylation. ISG15 induction inhibits the ability of RIG-I to recruit MAVS and TRAF3, and thereby may lead to a net proviral effect (16, 17). The latter is supported by recent data indicating that pharmacological PKR inhibition decreases HCV replication and increases IFN induction (18). RIG-I binds the polyuridine motif of the HCV genome 3′ nontranslated region, i.e., HCV pathogen–associated molecular patterns (PAMPs), leading to the recruitment of a signaling complex that activates transcription factors and the production of type I and III IFNs as well as proinflammatory cytokines (refs. 19, 20, and Figure 1A). The signals driving this response are relayed through MAVS localized within both mitochondria and peroxisomes (21). The ER contains a specialized domain, the mitochondrial-associated membrane (MAM), which physically links the ER to mitochondria and has been implicated in NLRP3 inflammasome signaling (21). Once sufficient viral proteins have accumulated in the cytosol, HCV uses its multifunctional NS3/4A protease, essential for HCV replication, to target the MAM-anchored synapse, cleaving MAVS from the MAM (but not from the mitochondria) and ablating RIG-I–mediated innate immune signaling (21). Another independent signaling pathway involves the binding of activated TLR-3 to the adaptor TRIF (Toll/interleukin-1 receptor domain–containing adapter–inducing IFN-β), which can also be cleaved by NS3/4A (19). Thus, the NS3 serine protease inhibitors that are part of the current triple-therapy regimen inhibit replication but would also be expected to restore innate responses within hepatocytes. Signaling from either MAVS or TRIF leads to the activation of various transcription factors, which in turn induce the production of type I and type III IFNs (via IFN regulatory factors [IRFs]), as well as proinflammatory cytokines and chemokines (via NF-κB and AP-1).
Figure 1. Hepatocyte innate immune responses.

(A) LDL receptors (LDLRs) on the basolateral hepatocyte surface, SR-BI, CD81, and tight-junction proteins CLDN-1 (claudin-1) and OCLN (occludin) are essential for HCV uptake (9). Intracellular HCV recognition occurs through dsRNA sensors such as RIG-I and TLR-3. MAMs function as the central scaffold that coordinates MAVS-dependent signaling of the RIG-I pathway between mitochondria and peroxisomes (21). MAVS interacts with the essential adapter protein TRAF3 to further recruit downstream kinases; activation of the kinases IKK-ε and TANK-binding kinase 1 (TBK1), which phosphorylate the transcription factors IRF-3 and IRF-7 (19), and binding to NF-κB lead to the induction of antiviral and immunomodulatory genes, including types I and III IFNs, as well as chemokines and proinflammatory cytokines that function in parallel with IFNs to mediate the response to HCV (147). (B) Binding of type I IFNs to the IFN-α/β receptors (IFN-αR1 and -2) and type III IFNs (IFN-λ) to the heterodimeric IL-28Ra/IL-10Rβ receptor (24) results in activation of the JAK/STAT pathway, conferring stable association with IRF-9. The resulting IFN-stimulated gene factor 3 (ISGF3) transcription factor complex localizes to the hepatocyte nucleus, where it binds to the ISREs within the promoter/enhancer region of hundreds of ISGs (147). Autocrine and paracrine signaling in neighboring cells results in anti-HCV amplification loops (including IRF-7, which binds to IFN promoters). HCV core protein subverts immunity by the induction of suppressors of cytokine signaling (SOCS1/SOCS3) and by impairing the binding of ISGF3 to nuclear IFN ISREs (19). See text for details.
Type III IFNs, which consist of four IFN-λs, are antiviral cytokines that display type I IFN-like (IFN-α/β–like) antiviral activity (22, 23), but are structurally and genetically closer to the members of IL-10 family of cytokines. Type III IFNs signal through a heterodimeric receptor composed of IL-28Rα (also known as IFN-λR1) and the IL-10 receptor β chain (IL-10Rβ) (ref. 24 and Figure 1). In contrast to the nearly ubiquitous expression of the IFN-α receptor (IFN-αR), IL-28Rα has been found primarily on epithelial cells and dendritic cells (DCs) (24). Following both in vitro and in vivo HCV infection, type III IFNs are upregulated at the mRNA and protein levels to an even greater extent than type I IFNs (25, 26). IFNs activate the JAK/STAT pathway, culminating in the induction of hundreds of ISGs encoding effector proteins that include ISG56, IFITM1, viperin, and 2′–5′ oligoadenylate synthase (OAS1), which restrict HCV infection within hepatocytes (27, 28), as well as PKR and IRF-7. Moreover, the promoters of early ISGs can be stimulated directly by IRF-3 (16). The relative requirement for IRF-3, IRF-7, or both vary according to cell and IFN type (29). Notably, type III IFNs, in addition to inducing well-known ISGs, activate a distinct set of genes in primary human hepatocytes from the type I IFNs (including those involved in chemotaxis and antigen presentation) with different kinetics of induction, suggesting divergent signaling pathways following receptor engagement (25).
Multiple proteins expressed by HCV have evolved important interactions with host cell proteins that benefit the viral cycle either directly or indirectly by disarming antiviral responses (reviewed in refs. 10, 30, 31). In addition to actions of the NS3/4A protease described above, for example, HCV core protein interferes with STAT signaling (via the induction of SOCS1/3) and may contribute to IFN resistance by diminished binding of ISGF3 to nuclear IFN–stimulated response elements (ISREs) (Figure 1B and ref. 19). An additional negative feedback loop is exemplified by USP18, a protease that decreases ISGylation of cellular proteins, thereby attenuating tyrosine phosphorylation of STAT1 and the expression of ISGs, decreasing the antiviral activity of IFNs (32). Paradoxically, higher hepatic ISG expression is predictive of a nonresponse to IFN-α treatment (33, 34), and a number of mechanisms have been proposed. The induction of type IIIs as the predominant antiviral pathway and driver of ISG induction (25, 29) may render hepatocytes refractory to further type I IFN action, conceptually supported by the observation that blocking type III IFN enhances the antiviral activity of exogenous IFN-α (ref. 25 and Figure 1B).
Association of genetic variation in IFN-λ genes and HCV recovery
One of the most ground-breaking discoveries in the area of HCV host response within the past five years has been that SNPs in chromosome 19 within or near the IFNL3 gene (encoding IFN-λ3, also known as IL-28B) are highly predictive of both antiviral success and spontaneous recovery in untreated patients (35, 36). As described above, in humans, four genes encode the members of the type III IFN family, i.e., IFN-λ1 (IFNL1, also known as IL29), IFN-λ2 (IFNL2 or IL28A), IFN-λ3 (IFNL3), and IFN-λ4 (IFNL4). Among these, IFN-λ3 is the most potent in inhibiting JFH-1 replication (25, 37). A genome-wide association study that examined the frequencies of about 600,000 SNPs demonstrated that patients homozygous for the C allele at the rs12979860 SNP of IFNL3 had a 2-fold greater chance of cure with pegylated IFN and ribavirin compared with those with the TT genotype (35). The advantageous allele is more frequently found in mixed–European descent and Asian populations relative to African-Americans, and much of the race-related impairment in response to antiviral therapy is explained by population differences in the incidence of IFNL3 genotypes. Subsequent reports have confirmed that the IFNL3 genotype is the most important baseline predictor of sustained virologic response (SVR) after treatment with pegylated IFN plus ribavirin in patients with genotype 1 infection (38). The effect of the IFNL3 genotype is evident within the first 48 hours following treatment initiation, indicating that IFN-λ3 somehow primes the host response to HCV, decreasing the threshold for virologic control with treatment (24). The favorable IFNL3 variant is associated with lower ISG expression in pretreatment liver biopsies (39), and exogenous IFN-α induces a rapid antiviral state (40). Interestingly, serum levels of IFN-γ–induced protein 10 (IP-10 or CXCL10), a well-characterized marker of HCV (41) that may antagonize T cell recruitment in chronic infection (42), enhance the predictive value of the IFNL3 genotype in patients receiving dual therapy (43). In addition, the dinucleotide ss469415590 variant of IFNL4 (44), a newly identified gene upstream of IFNL3, provides an even greater prediction of impaired viral kinetics among African-Americans than rs12979860 variants of IFNL3 (44). Recent data suggest that while IFN-λ4 induces weak expression of ISGs that could provide an antiviral response to lower HCV load, it uniquely induces genes involved in HCV-related liver injury (e.g., chemokine CCL5 and the proto-oncogene FOS) and also reduces the responsiveness to type I and type III IFNs that are required for efficient HCV clearance (44).
The multicellular immune response to HCV
DCs.
The orchestration of diverse, multifunctional cell types following HCV infection (Figure 2) ultimately governs the outcome of infection, and these cell types represent potential targets for pharmacologic and immunotherapeutic approaches (31). DCs play crucial roles in innate pathogen sensing as well as in the initiation of adaptive immunity (45). Several major subsets of DCs have been identified, including plasmacytoid DCs (pDCs) and myeloid DCs (mDCs). pDCs are the main producer of type I IFNs, synthesizing up to 109 IFN molecules per cell within 12 hours after activation (46). IFN production by pDCs requires cell-to-cell contact and is proportional to the number of HCV-infected hepatocytes; further, IFN production is mediated by TLR7 activation independently of HCV RNA replication within pDCs (47). Recently, HCV RNA–containing exosomes produced by infected hepatocytes have been shown to transfer their RNA to pDCs that subsequently respond by secreting IFN-α (48); moreover, HCV subgenomic replicon cells that replicate viral RNA without producing infectious virus particles can also trigger type I IFNs. Recent work from our group (49) has demonstrated that intracellular sensing of the HCV PAMP by pDCs leads to robust types I and III IFN production and is mediated by signaling through RIG-I, challenging the dogma that TLR, and not RLR, signaling within pDC predominates (50). Furthermore, conditioned medium from HCV PAMP–transfected human pDCs greatly inhibits HCV replication and induces STAT1 and IRF9 within Huh 7.5.1 cells. Although these collective data implicate sentinel roles for pDCs in HCV sensing, further work is necessary to understand the characteristics of pDCs that favor activation, which occurs in only a small subset, and the mechanisms that lead to their depletion or functional impairment in chronic infection, as described in some but not all studies (48, 51–54). In this regard, the ligation of C-type lectin immunoreceptors, blood DC antigen 2 (BDCA2), and DC immunoreceptor (DCIR) on pDCs by HCV E2 glycoprotein antagonizes the production of IFNs (55), and the relative expression of these receptors likely affects the ability of pDCs to respond to HCV within the hepatic microenvironment. Another C-type lectin, CLEC9A, is expressed by a distinct population of DCs, blood DC antigen 3 (BDCA3)+, which are enriched in the liver and produce significantly higher levels of type III (but relatively lower levels of type I) IFN compared with other DC populations following coculture with HCV (56). The latter study found that the IFNL3 allele associated with a more favorable response also correlates with greater type III IFN production than the less favorable IFNL3 genotypes, shedding some light on the puzzling lack of association between IFNL3 SNPs and hepatic expression of IFN-λs (57). Although hepatocytes comprise most of the mRNA message from liver biopsies, recent data suggest that there may be cell-specific effects on expression, and BDCA3+ DCs and pDCs are potent sources of IFN-λs (56). This concept is further supported by the recent demonstration that donor and recipient IFNL3 polymorphisms differentially affect HCV outcomes following liver transplantation (58).
Figure 2. Immune response to HCV infection within the liver.

Viral RNA is transferred to pDCs, triggering robust production of IFNs that inhibit HCV replication in hepatocytes. pDCs produce more type I (IFN-α and IFN-β) IFNs, whereas BDCA3+ DCs produce more type III IFNs with HCV infection and do not require direct cell-to-cell contact. NK and NKT cells comprise a large proportion of intrahepatic lymphocytes, mediating antiviral functions through a combination of IFN type II (IFN-γ) production and cytolytic function. IFNα-induced TRAIL is associated with the control of HCV (78, 82). Hepatic accumulation of NKp46hi NK cells is associated with lower viral replication and attenuated fibrosis (78). KCs phagocytose HCV, leading to the induction of innate immune (IFN-β) as well as inflammatory (IL-1β) responses. HCV core protein inhibits type I IFN responses (89) and also drives proinflammatory responses, augmenting processes that result in liver fibrosis (87). IFN-γ induces KC upregulation of Gal-9 and PD-L1, inhibitory ligands that promote T cell dysfunction. LSECs can pinocytose viral particles and produce a broad array of IFNs. Multispecific and polyfunctional CD4+ T (Th) cells provide “help” for clonal expansion of B cells and CTLs required for spontaneous viral control. Early expression of CD127, IL-2 production, development of neutralizing Abs, and HCV-specific CTL cells contribute to immune response (148, 149). PD-1 and TIM3 demarcate functionally impaired CTLs. Moreover, CD33+ myeloid–derived suppressor cells (150) and FOXP3+ Tregs (10, 151) attenuate T cell responses and immune-mediated liver injury.
NK cell phenotype and function associated with HCV infection outcomes.
NK cells constitute an early host defense against viral pathogens (59–61), eliminating virus-infected cells both directly via cytolytic mechanisms and indirectly by secreting cytokines such as IFN-γ (62, 63). Although NK cells have been classically viewed as innate immune cells, their effects can extend into periods of adaptive immunity, and hepatic NK cells demonstrate adaptive immunity to structurally diverse antigens (64, 65). NK cell activity is stringently controlled by activating and inhibitory NK receptors (NKRs). NKRs include the predominantly inhibitory killer Ig–like receptors (KIRs), C-type lectin-like receptors of the NKG2 family comprising inhibitory (NKG2A) and activatory (NKG2C/D) isoforms, as well as the natural cytotoxicity receptors (NCRs) such as NKp30 (also known as NCR3), NKp44 (also known as NCR2), and NKp46 (also known as NCR1) that deliver activating signals (66–68). Emerging data indicate that NK cells play central roles in every stage of HCV infection, from the protection against infection in injection drug users (IDUs) to the prediction of antiviral success or failure with IFN-based therapies.
By convention, CD56+ NK cells have been assigned to functional categories on the basis of the relative cell surface density of CD56; namely, CD56dimCD16+ NK cells, predominant in peripheral blood, display potent cytolytic activity, whereas the poorly cytolytic CD56bright NK cells enriched in tissue are responsible for cytokine production, although recent data call into question this simple classification (69). IDUs repeatedly exposed to HCV infection but who remain uninfected have proportionally higher circulating frequencies of CD56dim mature effector NK cells (70). The balance between positive and negative signals from activating and inhibitory cell surface receptors is likely to result in NK cell functional changes (71). In our IDU cohort, there was a correlation between the frequency of NK cells expressing the activating receptor NKp30 and in vitro lymphokine-activated killing. NKp30, which is induced by IL-2, is most highly expressed on the NK and NKT cells of exposed, uninfected subjects (70). The NKp46 receptor is considered the major human natural cytotoxicity receptor involved in NK cell–mediated killing (72, 73) and is more highly expressed on the NK cells of women and people of mixed European descent (i.e., populations known to demonstrate higher rates of spontaneous resolution of HCV infection) (74–76). NKp46 ligand expression is induced on hepatocytes following HCV infection (76), although whether this represents a specific HCV component or an unspecific stress response is not yet clear. TLR stimulation of purified NKp46hi is associated with increased transcription of cytotoxicity-related genes as compared with NKp46lo counterparts. Two recent studies (76, 77) with further discussion in an editorial (78) highlight that NKp46hi NK cells (or ligation with an agonist NKp46 antibody) have increased anti-HCV activity in vitro, a process mediated by IFN-γ; accordingly, intrahepatic accumulation of NKp46hi NK cells is inversely correlated with HCV RNA levels (77). An inverse correlation also exists with the stage of fibrosis and ex vivo intrahepatic NKp46hi frequencies; in vitro, blockade of NKp46 reduces NK cell–mediated killing of human hepatic stellate cells (HSCs), implicating potentially important antifibrotic roles (77) for this population of NK cells (Figure 2).
NK cells are rapidly activated by cytokine stimulation, and thus, studies have examined whether the response to anti-HCV therapy (the major component of which is IFN-α) would be predicted by the phenotype and function of NK cells. Pretreatment expansion of CD56– NK cells functionally skewed toward MIP-1β production (rather than IFN-γ) is a predictor of failure to pegylated IFN-α and ribavirin (79). Patients with impaired viral kinetics following initiation of antiviral therapy have NK cells with higher expression levels of inhibitory receptors (80), and a multivariate model based on two inhibitory receptors (NKG2A and CD158e) is highly predictive of SVR. Pegylated IFN-α transcriptionally upregulates ISGs and TNF-related apoptosis–inducing ligand (TRAIL) in NK cells. HCV also enhances the susceptibility of primary human hepatocytes to TRAIL-mediated killing via increased expression of the death receptors DR4 and DR5 (81). The ex vivo expression of TRAIL and CD107a on NK cells increases as early as hours following the initiation of antiviral therapy (82, 83), correlates with the induction of phosphorylated STAT1 levels (84), and is associated with early virologic response. Thus, on aggregate, these data identify NK cell markers associated with protection from early infection and predictors of response to IFN-based therapy. Moreover, NK cells can demonstrate regulatory and reciprocal interactions with B and T cells, DCs, macrophages, and endothelial cells (ECs), thus functioning to amplify or attenuate immune responses (60).
Liver-resident macrophages and ECs.
Kupffer cells (KCs) constitute the first macrophage population with which pathogens, bacterial endotoxins, and microbial debris derived from the gastrointestinal tract come into contact, and together with the liver sinusoidal ECs (LSECs), make up the hepatic reticuloendothelial system (ref. 85 and Figure 2). ISG upregulation within KCs is a strong positive and independent predictor of subsequent response to antiviral therapy with pegylated IFN and ribavirin, whereas hepatocyte ISG expression was associated with nonresponse (86), underscoring the importance of examining different patterns of cellular activation. HCV enters KCs (but not undifferentiated monocytes) via a phagocytic uptake that is independent of productive infection, leading to the induction of IFN-β–dependent innate immune responses through RIG-I/MAVS, as well as inflammatory IL-1β–dependent signaling through TLR7/MyD88-dependent and NLRP3 inflammasome pathways (87). The induction of proinflammatory genes may lead to the recruitment of immune cells to the liver and augment the processes that result in liver fibrosis and cirrhosis.
Tolerance is a critical mechanism that protects against excessive inflammation. However, monocytes/macrophages from patients with chronic HCV are activated and lack tolerance to proinflammatory cytokine–inducing TLR ligands (88). Moreover, the combination of HCV core protein, IFN-γ, and LPS during macrophage differentiation recapitulates the ex vivo findings in chronic HCV patients (88). Recombinant HCV core protein, which engages TLR-2, induces primary human KCs to secrete IL-1β, TNF-α, and IL-10 and inhibits TLR-3–mediated type I IFN responses (89). The HCV core further subverts host immunity by attenuating KC expression of TRAIL, a transcriptional target of IRF-3 (90) and strongly upregulates the expression of programmed death ligand 1 (PD-L1), which is known to promote T cell dysfunction and the development of viral persistence (89, 91, 92). These effects are largely mediated through phosphoinositide 3-kinase (PI3K), indicating shared regulation between MyD88- and TRIF-dependent TLR signaling pathways (89). Galectin-9 (Gal-9), another important inhibitory ligand (93), is expressed robustly by KCs and circulates at high levels in HCV-infected patients (94). Gal-9 binds to T cell Ig and mucin domain–containing molecule 3 (TIM3), inducing apoptosis of HCV-specific CD8+ T cells and expanding Tregs (94). Further, KC-derived Gal-9 mediates T cell dysfunction and predicts poor prognosis in patients with hepatitis B–related hepatocellular carcinoma (95). Gal-9 has pleiotropic roles that include the inhibition and regulation of NK cell functions that may impact viral persistence (96).
LSECs function to clear waste molecules that have entered the circulation (97) and comprise approximately 50% of nonparenchymal cells in the liver. Although long recognized as central to the induction of cytotoxic CD8+ T cell (CTL) tolerance and apoptosis (98, 99), an understanding of the roles in antiviral responses of LSECs, which express a wide array of PRRs (100), is just beginning to emerge. Their strategic anatomic position (Figure 2) places LSECs as the first cells in contact with blood flow in the sinusoids (101). Expression of the C-type lectin L-SIGN by LSECs was postulated to mediate capture of HCV particles and transcytosis of the virus across the endothelial barrier, thereby concentrating infectious particles and facilitating their direct contact with hepatocytes (102). Moreover, LSECs are highly efficient scavengers that pinocytose particles less than 0.2 μm in size, which typically encompasses virus-sized particles (103). The recent demonstration that LSECs, rather than KCs, clear the bulk of blood-borne human adenovirus underscores their importance during the viremic phase of any natural viral infection (103). Even if LSECs do not sustain active replication, viral sensing and its downstream effector responses, including exosomal transfer of ISG products (ref. 104 and Figure 2), may impact other processes within the hepatic microenvironment and warrant further investigation.
B cells and humoral immunity.
Whether the humoral response plays an important role in controlling HCV infection remains controversial (105, 106); indeed, the fact that hypogammaglobulinemic humans can spontaneously eradicate HCV (107) may suggest that antibody responses are dispensable. Studies showing a lack of association between neutralizing antibodies (NAbs) and viral clearance were confounded by poorly defined viral inoculum and heterogeneous patient populations. However, in a study of healthy young women infected with identical, single-source viral inoculum, spontaneous resolvers demonstrated an early induction of NAb responses directed against HCV envelope glycoproteins, including broader cross-neutralization of heterologous strains, whereas chronically evolving subjects had delayed and low-titered NAb responses (108). The use of retroviral pseudoparticles bearing autologous HCV glycoproteins (HCVpp) from the initial inoculum allowed the precise examination of strain-specific neutralization. The late phase of chronic infection is accompanied by the induction of NAb responses as well as an increasing rate of envelope viral sequence evolution (106), indicative of ongoing humoral selection pressure. Viral escape may occur through several additional mechanisms (108), including an impaired cross-neutralizing activity that selects the outgrowth of viral variants and the interplay of HCV glycoproteins with HDL and the scavenger receptor SR-BI (Figure 1A), which can prevent the effect of NAbs and enhance cell entry of HCV and infection (109). Only a minority of patients with resolved HCV infection exhibit NAbs 10–17 years after viral clearance (108), which may underlie the absent or limited protective immunity against reinfection (110). Moreover, human liver chimeric mice infused with HCV E2–specific human monoclonal antibodies directed against a conserved epitope are protected against heterologous HCV (111), raising the possibility that future approaches using a combination of such NAbs and blocking receptors involved in viral entry (112) might demonstrate efficacy in high-risk populations (e.g., to prevent allograft reinfection in patients who have undergone liver transplantation or in healthcare workers with needle-stick injuries).
CTLs in HCV infection.
CTLs are the primary effector cells that mediate viral clearance through the secretion of antiviral cytokines, which is about 100- to 1,000-fold more effective than cytolytic activity (10) (113). In the absence of viral clearance, the display of HCV peptides persists on the surface of hepatocytes, and the continued presence of CTLs results in significant immune-mediated liver injury (31). Further, the secretion of inflammatory and profibrotic cytokines such as TGF-β activates stellate cells, the primary source of fibrosis, ultimately leading to progressive liver damage. Chimpanzee and human studies were highly instructive in understanding the distinguishing features of CTL responses that provide permanent viral control versus viral persistence (10, 114–118), including the recognition that CD4+ T cells are critical both for limiting immune evasion and priming effector memory CTLs (119, 120). A complex interplay between the opposing forces of immune selection pressure and viral fitness costs drives mutations in HCV (Figure 3). The high rate of replication coupled with the lack of proofreading ability of its polymerase provides the HCV genome with a means to escape HLA-restricted immune responses (10, 121). The ability of HCV to mutate at an amino acid residue is constrained, however, by fitness costs, and compensatory mutations may be required to restore HCV replicative capacity (121–123).
Figure 3. Interplay of host T cell responses and the evolution of HCV epitopes.
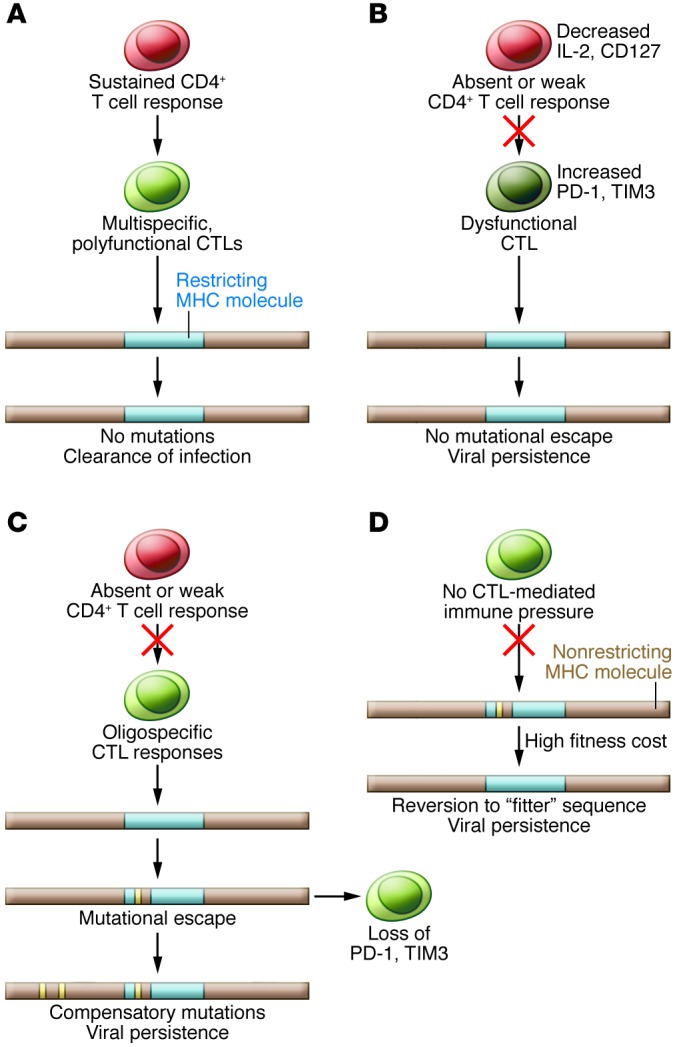
(A) A sustained CD4+ T cell response with multispecific CTLs may constrain the development of viral escape mutations, leading to viral clearance during acute infection. (B) If CTL responses are weak (manifested by high PD-1 and TIM3 expression) with impaired CD4 help and priming, escape mutations likely will not develop. High viral levels and intact HCV epitopes are associated with increased levels of inhibitory receptors. (C) Failure of the CD4+ T cell response in the presence of a narrow but vigorous CTL response favors the development of escape mutations. Additional compensatory mutations may be required for replicative fitness (120–122). Viral amino acid substitution is associated with decreased inhibitory receptor expression on CTLs, perhaps accounting for their robust proliferation to wild-type, nonmutated virus (115). (D) Without a restricting HLA allele and immune selective pressure, reversion to the wild-type sequence likely occurs because of the high fitness cost associated with an escape mutation. (Adapted with permission from The Journal of Experimental Medicine [ref. 118].)
Anergy occurs when T cells are initially primed improperly by signaling through the TCR in the absence of costimulatory or inflammatory signals (124, 125), which results in a failure to develop proper function from the outset. In contrast, exhausted T cells are primed by antigen, costimulation, and inflammation. Although these T cells initially develop effector functions, prolonged, excessive stimulation leads to the progressive loss of function over time (124). Compelling evidence exists for both anergy and exhaustion in chronic HCV infection. Programmed cell death protein 1 (PD-1) is an immunoreceptor tyrosine–based inhibition motif–containing (ITIM-containing) receptor expressed on activated T cells that mediates hyporesponsiveness. PD-1 is upregulated in chronic infection within the intrahepatic compartment (91, 126) and its ligand PD-L1 is expressed by a number of nonparenchymal cells in the liver including KCs, LSECs, and DCs, as well as IFN-exposed hepatocytes (10). Moreover, PD-1 expression on HCV-specific CTLs inversely correlates with early and sustained virologic response to combination IFN-based antiviral therapy (127) and is associated with impaired control of replication in vitro (128). In association with PD-1, the coexpression of multiple inhibitory receptors, including 2B4, CD160, KLRG1, and TIM3, identifies HCV-specific CTLs with impaired proliferation and cytokine production and correlates with the absence of sequence variation within cognate epitopes, consistent with the necessity for ongoing antigen triggering (129, 130). In this regard (131), the frequency of dual TIM3posPD-1pos HCV–specific CTLs is predictive of viral persistence in patients with acute HCV infection and is increased within the central memory subset (compared with the effector memory population) and within the hepatic compartment relative to the peripheral compartment. Increased levels of TIM3 and PD-1 expression are associated with CTL dysfunction, with TIM3hi/PD-1hi CTLs producing less IFN-γ, TNF-α, and CD107a than their TIM3lo/PD-1lo counterparts. Importantly, blockade of different coinhibitory pathways may differentially enhance CTL effector functions. CTL avidity, also termed “functional avidity” (132, 133), refers to the overall sensitivity of the T cell response to antigen density. High-avidity CTLs are those that secrete substantial amounts of IFN-γ when stimulated with relevant APCs loaded with low concentrations of peptide. Considerable data in other model systems demonstrate the immense potential of high-avidity CTLs to preferentially control certain viral infections as well as tumors (134). In infections that can rapidly evolve escape mutations, such as with HCV, another potentially important attribute of CTLs is the ability to cross-recognize mutated epitopes (135–137). From a standpoint of the design of novel immunotherapeutic approaches, the use of TCRs derived from CTLs demonstrating relatively higher avidity and cross-reactivity would seem optimal (Figure 4). HCV TCR transduction of human T cells renders them responsive to HCV peptide–loaded hepatocytes (138) and inhibits viral replication in vitro (139).
Figure 4. Paradigm for avidity and cross-reactivity pertaining to HCV-specific CTLs, their antiviral efficacy and probability of mutational escape.

(i) CTLs with low functional avidity rarely select for escape variants, particularly with broad cross-recognition of mutant virus. HCV-specific CTLs that respond effectively to low concentrations of peptide have greater potency than those requiring high peptide concentrations. (ii) CTL responses rapidly selecting for HCV escape variants require a low concentration of peptide for stimulation (high avidity), (iii) but if coupled with poor cross-recognition, they are associated with decreased antiviral efficacy and potentially impaired recognition of variant peptides as they emerge (note that there is the least amount of supportive data in this poor cross-recognition paradigm). (iv) CTLs with low functional avidity rarely select for escape variants. The most effective CTL specificities express both functional attributes (in light purple) during the earliest stages of infection and predate the resolution of viremia. Taken together, the data suggest that strong immunity will eradicate HCV infection, weak immunity will drive few mutations, and intermediate immunity will select for escape but allow for persistence, resulting in a broader viral quasispecies.
Future directions
The past five years have seen a remarkable degree of progress in the understanding of HCV pathogenesis, and we are in the midst of an extraordinary phase of development with direct-acting antiviral agents that may cure the majority of patients undergoing antiviral therapy. However, as antiviral therapy will not be accessible for most patients in resource-limited regions, the development of a safe and effective HCV vaccine is paramount. HCV poses multiple important obstacles to vaccine development (140), including a considerable primary sequence divergence that is greater than HIV (141). In vaccinated animals, despite initial control, viral persistence is associated with T cell immune escape (142), higher viral mutation rates (143, 144), and expression of inhibitory receptors, which suggest that in the absence of rapid viral clearance, an environment for selective pressure or T cell exhaustion remains. Longitudinal liver biopsies revealed correlations between higher PD-1 expression and failure to control viremia, pointing to the possibility that strong induction and activation of CTLs may be counterproductive if they target promiscuous HCV epitopes that do not impact viral fitness (145). HCV is able to suppress the early innate immune response both within hepatocytes and innate lymphocytes, such as NK cells, and can attenuate or escape T cell responses. The nature and extent of immune restoration that occurs with regimens not including IFN-α, which is known to have antiproliferative effects (146), remain to be defined. Greater understanding of the complex crosstalk between different cell types within the hepatic microenvironment may soon allow the identification of targeted approaches to address the sequelae of chronic infection, including fibrosis and the development of cancer.
Acknowledgments
I would like to thank Michael Gale and Stacy Horner (University of Washington) Emmanuel Thomas (University of Miami), Todd Allen (Massachusetts General Hospital/Broad Institute), Arash Grakoui (Emory University), Michael Nishimura (Loyola University), Martha Alexander-Miller (Wake Forest School of Medicine), Stephen Polyak (University of Washington), Young Hahn (University of Virginia), Robert Thimme (University of Freiburg), and members of my research laboratory for their helpful discussions. I would also like to acknowledge support from NIH grants RO1DK60590, RO1DK071560, U19 AI1066328, R21AI103361, R56AI100991, K24AI083742, and R01HD075549, and from VA Merit Review grants.
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(10):4121–4130. doi:10.1172/JCI67714.
References
- 1.Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364(25):2429–2438. doi: 10.1056/NEJMcp1006613. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology. 2010;51(3):729–733. doi: 10.1002/hep.23561. [DOI] [PubMed] [Google Scholar]
- 3.Davis GL, Albright JE, Cook SF, Rosenberg DM. Projecting future complications of chronic hepatitis C in the United States. Liver Transpl. 2003;9(4):331–338. doi: 10.1053/jlts.2003.50073. [DOI] [PubMed] [Google Scholar]
- 4.Biggins SW, et al. Projected future increase in aging hepatitis C virus-infected liver transplant candidates: a potential effect of hepatocellular carcinoma. Liver Transpl. 2012;18(12):1471–1478. doi: 10.1002/lt.23551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aronsohn A, Jensen D. Distributive justice and the arrival of direct-acting antivirals: who should be first in line? Hepatology. 2011;53(6):1789–1791. doi: 10.1002/hep.24374. [DOI] [PubMed] [Google Scholar]
- 6.Dabbouseh NM, Jensen DM. Future therapies for chronic hepatitis C. Nat Rev Gastroenterol Hepatol. 2013;10(5):268–276. doi: 10.1038/nrgastro.2013.17. [DOI] [PubMed] [Google Scholar]
- 7.Gane EJ, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368(1):34–44. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- 8.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 9.Rice CM. New insights into HCV replication: potential antiviral targets. Top Antivir Med. 2011;19(3):117–120. [PMC free article] [PubMed] [Google Scholar]
- 10.Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119(7):1745–1754. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartenschlager R, Pietschmann T. Efficient hepatitis C virus cell culture system: what a difference the host cell makes. Proc Natl Acad Sci U S A. 2005;102(28):9739–9740. doi: 10.1073/pnas.0504296102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li K, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102(8):2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11(7):791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102(26):9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309(5734):623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 16.Arnaud N, et al. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011;7(10):e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim MJ, Hwang SY, Imaizumi T, Yoo JY. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol. 2008;82(3):1474–1483. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnaud N, et al. Hepatitis C virus controls interferon production through PKR activation. PLoS One. 2010;5(5):e10575. doi: 10.1371/journal.pone.0010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horner SM, Gale M. Intracellular innate immune cascades and interferon defenses that control hepatitis C virus. J Interferon Cytokine Res. 2009;29(9):489–498. doi: 10.1089/jir.2009.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454(7203):523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horner SM, Liu HM, Park HS, Briley J, Gale M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108(35):14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81(14):7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choubey D, Moudgil KD. Interferons in autoimmune and inflammatory diseases: regulation and roles. J Interferon Cytokine Res. 2011;31(12):857–865. doi: 10.1089/jir.2011.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balagopal A, Thomas DL, Thio CL. IL28B and the control of hepatitis C virus infection. Gastroenterology. 2010;139(6):1865–1876. doi: 10.1053/j.gastro.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas E, et al. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology. 2012;142(4):978–988. doi: 10.1053/j.gastro.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park H, et al. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology. 2012;56(6):2060–2070. doi: 10.1002/hep.25897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helbig KJ, et al. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology. 2011;54(5):1506–1517. doi: 10.1002/hep.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raychoudhuri A, Shrivastava S, Steele R, Kim H, Ray R, Ray RB. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol. 2011;85(24):12881–12889. doi: 10.1128/JVI.05633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179(6):3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 30.Yamane D, McGivern DR, Masaki T, Lemon SM. Liver injury and disease pathogenesis in chronic hepatitis C. Curr Top Microbiol Immunol. 2013;369:263–288. doi: 10.1007/978-3-642-27340-7_11. [DOI] [PubMed] [Google Scholar]
- 31.Mengshol JA, Golden-Mason L, Rosen HR. Mechanisms of disease: HCV-induced liver injury. Nat Clin Pract Gastroenterol Hepatol. 2007;4(11):622–634. doi: 10.1038/ncpgasthep0961. [DOI] [PubMed] [Google Scholar]
- 32.Randall G, et al. Silencing of USP18 potentiates the antiviral activity of interferon against hepatitis C virus infection. Gastroenterology. 2006;131(5):1584–1591. doi: 10.1053/j.gastro.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 33.Sarasin-Filipowicz M, et al. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol. 2009;29(17):4841–4851. doi: 10.1128/MCB.00224-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Afdhal NH, et al. Hepatitis C pharmacogenetics: state of the art in 2010. Hepatology. 2011;53(1):336–345. doi: 10.1002/hep.24052. [DOI] [PubMed] [Google Scholar]
- 35.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 36.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461(7265):798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcello T, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131(6):1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 38.Thompson AJ, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139(1):120–129.e18. doi: 10.1053/j.gastro.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 39.Urban TJ, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52(6):1888–1896. doi: 10.1002/hep.23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarasin-Filipowicz M, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105(19):7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beinhardt S, et al. Serum level of IP-10 increases predictive value of IL28B polymorphisms for spontaneous clearance of acute HCV infection. Gastroenterology. 2012;142(1):78–85.e2. doi: 10.1053/j.gastro.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 42.Casrouge A, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest. 2011;121(1):308–317. doi: 10.1172/JCI40594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darling JM, et al. Quantitation of pretreatment serum interferon-gamma-inducible protein-10 improves the predictive value of an IL28B gene polymorphism for hepatitis C treatment response. Hepatology. 2011;53(1):14–22. doi: 10.1002/hep.24056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prokunina-Olsson L, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45(2):164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lande R, Gilliet M. Plasmacytoid dendritic cells: key players in the initiation and regulation of immune responses. Ann N Y Acad Sci. 2010;1183:89–103. doi: 10.1111/j.1749-6632.2009.05152.x. [DOI] [PubMed] [Google Scholar]
- 46.Hagberg N, et al. IFN-alpha production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1beta and LFA-1. J Immunol. 2011;186(9):5085–5094. doi: 10.4049/jimmunol.1003349. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi K, et al. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A. 2010;107(16):7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dreux M, et al. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe. 2012;12(4):558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stone AE, et al. Hepatitis C virus pathogen associated molecular pattern (PAMP) triggers production of lambda-interferons by human plasmacytoid dendritic cells. PLoS Pathog. 2013;9(4):e1003316. doi: 10.1371/journal.ppat.1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20(1):17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Sagan SM, Sarnow P. Plasmacytoid dendritic cells as guardians in hepatitis C virus-infected liver. Proc Natl Acad Sci U S A. 2010;107(17):7625–7626. doi: 10.1073/pnas.1002943107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wertheimer AM, Bakke A, Rosen HR. Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology. 2004;40(2):335–345. doi: 10.1002/hep.20306. [DOI] [PubMed] [Google Scholar]
- 53.Liang H, et al. Differential effects of hepatitis C virus JFH1 on human myeloid and plasmacytoid dendritic cells. J Virol. 2009;83(11):5693–5707. doi: 10.1128/JVI.02671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shiina M, Rehermann B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology. 2008;47(2):385–395. doi: 10.1002/hep.21996. [DOI] [PubMed] [Google Scholar]
- 55.Florentin J, et al. HCV glycoprotein E2 is a novel BDCA-2 ligand and acts as an inhibitor of IFN production by plasmacytoid dendritic cells. Blood. 2012;120(23):4544–4551. doi: 10.1182/blood-2012-02-413286. [DOI] [PubMed] [Google Scholar]
- 56.Yoshio S, et al. Human blood dendritic cell antigen 3 (BDCA3) dendritic cells are a potent producer of interferon-lambda in response to hepatitis C virus. Hepatology. 2013;57(5):1705–1715. doi: 10.1002/hep.26182. [DOI] [PubMed] [Google Scholar]
- 57.Honda M, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139(2):499–509. doi: 10.1053/j.gastro.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 58.Biggins SW, et al. Differential effects of donor and recipient IL28B and DDX58 SNPs on severity of HCV after liver transplantation. J Hepatol. 2013;58(5):969–976. doi: 10.1016/j.jhep.2012.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biron CA. Initial and innate responses to viral infections--pattern setting in immunity or disease. Curr Opin Microbiol. 1999;2(4):374–381. doi: 10.1016/S1369-5274(99)80066-6. [DOI] [PubMed] [Google Scholar]
- 60.Vivier E, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163–194. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 62.Biron CA, Brossay L. NK cells and NKT cells in innate defense against viral infections. Curr Opin Immunol. 2001;13(4):458–464. doi: 10.1016/S0952-7915(00)00241-7. [DOI] [PubMed] [Google Scholar]
- 63.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633–640. doi: 10.1016/S1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 64.Paust S, et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat Immunol. 2010;11(12):1127–1135. doi: 10.1038/ni.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang X, Chen Y, Peng H, Tian Z. Memory NK cells: why do they reside in the liver? Cell Mol Immunol. 2013;10(3):196–201. doi: 10.1038/cmi.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moretta A, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- 67.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 68.Moretta A, Biassoni R, Bottino C, Mingari MC, Moretta L. Natural cytotoxicity receptors that trigger human NK-cell-mediated cytolysis. Immunol Today. 2000;21(5):228–234. doi: 10.1016/S0167-5699(00)01596-6. [DOI] [PubMed] [Google Scholar]
- 69.De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-γ on activation. Proc Natl Acad Sci U S A. 2011;108(2):728–732. doi: 10.1073/pnas.1012356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Golden-Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology. 2010;52(5):1581–1589. doi: 10.1002/hep.23896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Golden-Mason L, Rosen HR. Natural killer cells: primary target for hepatitis C virus immune evasion strategies? Liver Transpl. 2006;12(3):363–372. doi: 10.1002/lt.20708. [DOI] [PubMed] [Google Scholar]
- 72.Sivori S, et al. NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol. 1999;29(5):1656–1666. doi: 10.1002/(SICI)1521-4141(199905)29:05<1656::AID-IMMU1656>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 73.Mandelboim O, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409(6823):1055–1060. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- 74.Thomas DL, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA. 2000;284(4):450–456. doi: 10.1001/jama.284.4.450. [DOI] [PubMed] [Google Scholar]
- 75.Rosen HR, et al. Selective decrease in hepatitis C virus-specific immunity among African Americans and outcome of antiviral therapy. Hepatology. 2007;46(2):350–358. doi: 10.1002/hep.21714. [DOI] [PubMed] [Google Scholar]
- 76.Golden-Mason L, Stone AE, Bambha KM, Cheng L, Rosen HR. Race- and gender-related variation in NKp46 expression associated with differential anti-HCV immunity. Hepatology. 2012;56(4):1214–1222. doi: 10.1002/hep.25771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kramer B, et al. Natural killer p46High expression defines a natural killer cell subset that is potentially involved in control of hepatitis C virus replication and modulation of liver fibrosis. Hepatology. 2012;56(4):1201–1213. doi: 10.1002/hep.25804. [DOI] [PubMed] [Google Scholar]
- 78.Heeg M, Thimme R. NK cells and hepatitis C: NKp46 expression linked to antiviral and antifibrotic activity. Hepatology. 2012;56(4):1197–1200. doi: 10.1002/hep.25858. [DOI] [PubMed] [Google Scholar]
- 79.Gonzalez VD, et al. Expansion of functionally skewed CD56-negative NK cells in chronic hepatitis C virus infection: correlation with outcome of pegylated IFN-alpha and ribavirin treatment. J Immunol. 2009;183(10):6612–6618. doi: 10.4049/jimmunol.0901437. [DOI] [PubMed] [Google Scholar]
- 80.Golden-Mason L, et al. Natural killer inhibitory receptor expression associated with treatment failure and interleukin-28B genotype in patients with chronic hepatitis C. Hepatology. 2011;54(5):1559–1569. doi: 10.1002/hep.24556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lan L, et al. Hepatitis C virus infection sensitizes human hepatocytes to TRAIL-induced apoptosis in a caspase 9-dependent manner. J Immunol. 2008;181(7):4926–4935. doi: 10.4049/jimmunol.181.7.4926. [DOI] [PubMed] [Google Scholar]
- 82.Stegmann KA, et al. Interferon-alpha-induced TRAIL on natural killer cells is associated with control of hepatitis C virus infection. Gastroenterology. 2010;138(5):1885–1897. doi: 10.1053/j.gastro.2010.01.051. [DOI] [PubMed] [Google Scholar]
- 83.Ahlenstiel G, et al. Early changes in natural killer cell function indicate virologic response to interferon therapy for hepatitis C. Gastroenterology. 2011;141(4):1231–1239. doi: 10.1053/j.gastro.2011.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Edlich B, et al. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology. 2012;55(1):39–48. doi: 10.1002/hep.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vollmar B, Menger MD. The hepatic microcirculation: mechanistic contributions and therapeutic targets in liver injury and repair. Physiol Rev. 2009;89(4):1269–1339. doi: 10.1152/physrev.00027.2008. [DOI] [PubMed] [Google Scholar]
- 86.Chen L, et al. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology. 2010;138(3):1123–1123. doi: 10.1053/j.gastro.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 87.Negash AA, et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9(4):e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dolganiuc A, et al. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology. 2007;133(5):1627–1636. doi: 10.1053/j.gastro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tu Z, Pierce RH, Kurtis J, Kuroki Y, Crispe IN, Orloff MS. Hepatitis C virus core protein subverts the antiviral activities of human Kupffer cells. Gastroenterology. 2010;138(1):305–314. doi: 10.1053/j.gastro.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 90.Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev. 2005;16(1):1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 91.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. . J Virol. 2007;81(17):9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kassel R, Cruise MW, Iezzoni JC, Taylor NA, Pruett TL, Hahn YS. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatology. 2009;50(5):1625–1637. doi: 10.1002/hep.23173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anderson AC, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318(5853):1141–1143. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 94.Mengshol JA, et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS One. 2010;5(3):e9504. doi: 10.1371/journal.pone.0009504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li H, et al. Tim-3/galectin-9 signaling pathway mediates T cell dysfunction and predicts poor prognosis in patients with HBV-associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342–1351. doi: 10.1002/hep.25777. [DOI] [PubMed] [Google Scholar]
- 96.Golden-Mason L, et al. Galectin-9 functionally impairs natural killer (NK) cells in humans and mice. J Virol. 2013;87(9):4835–4845. doi: 10.1128/JVI.01085-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seternes T, Sorensen K, Smedsrod B. Scavenger endothelial cells of vertebrates: a nonperipheral leukocyte system for high-capacity elimination of waste macromolecules. Proc Natl Acad Sci U S A. 2002;99(11):7594–7597. doi: 10.1073/pnas.102173299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Karrar A, Broome U, Uzunel M, Qureshi AR, Sumitran-Holgersson S. Human liver sinusoidal endothelial cells induce apoptosis in activated T cells: a role in tolerance induction. Gut. 2007;56(2):243–252. doi: 10.1136/gut.2006.093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tiegs G, Lohse AW. Immune tolerance: what is unique about the liver. J Autoimmun. 2010;34(1):1–6. doi: 10.1016/j.jaut.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 100.Wu J, et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. 2010;129(3):363–374. doi: 10.1111/j.1365-2567.2009.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huebert RC, et al. Immortalized liver endothelial cells: a cell culture model for studies of motility and angiogenesis. Lab Invest. 2010;90(12):1770–1781. doi: 10.1038/labinvest.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cormier EG, et al. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc Natl Acad Sci U S A. 2004;101(39):14067–14072. doi: 10.1073/pnas.0405695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ganesan LP, Mohanty S, Kim J, Clark KR, Robinson JM, Anderson CL. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PLoS Pathog. 2011;7(9):e1002281. doi: 10.1371/journal.ppat.1002281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li J, et al. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat Immunol. 2013;14(8):793–803. doi: 10.1038/ni.2647. [DOI] [PubMed] [Google Scholar]
- 105.Rehermann B. Cellular immune response to the hepatitis C virus. J Viral Hepat. 1999;6(suppl 1):31–35. doi: 10.1046/j.1365-2893.1999.00008.x. [DOI] [PubMed] [Google Scholar]
- 106.Liu L, Fisher BE, Dowd KA, Astemborski J, Cox AL, Ray SC. Acceleration of hepatitis C virus envelope evolution in humans is consistent with progressive humoral immune selection during the transition from acute to chronic infection. J Virol. 2010;84(10):5067–5077. doi: 10.1128/JVI.02265-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Christie JM, et al. Clinical outcome of hypogammaglobulinaemic patients following outbreak of acute hepatitis C: 2 year follow up. Clin Exp Immunol. 1997;110(1):4–8. doi: 10.1111/j.1365-2249.1997.508-ce1412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pestka JM, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A. 2007;104(14):6025–6030. doi: 10.1073/pnas.0607026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bartosch B, et al. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J Virol. 2005;79(13):8217–8229. doi: 10.1128/JVI.79.13.8217-8229.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lai ME, et al. Hepatitis C virus in multiple episodes of acute hepatitis in polytransfused thalassaemic children. Lancet. 1994;343(8894):388–390. doi: 10.1016/S0140-6736(94)91224-6. [DOI] [PubMed] [Google Scholar]
- 111.Bukh J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology. 2012;142(6):1279–1287.e3. doi: 10.1053/j.gastro.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 112.Lacek K, et al. Novel human SR-BI antibodies prevent infection and dissemination of HCV in vitro and in humanized mice. J Hepatol. 2012;57(1):17–23. doi: 10.1016/j.jhep.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 113.Jo J, et al. Analysis of CD8+ T-cell-mediated inhibition of hepatitis C virus replication using a novel immunological model. Gastroenterology. 2009;136(4):1391–1401. doi: 10.1053/j.gastro.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 114.Lechner F, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191(9):1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Smyk-Pearson S, Tester IA, Lezotte D, Sasaki AW, Lewinsohn DM, Rosen HR. Differential antigenic hierarchy associated with spontaneous recovery from hepatitis C virus infection: implications for vaccine design. J Infect Dis. 2006;194(4):454–463. doi: 10.1086/505714. [DOI] [PubMed] [Google Scholar]
- 116.Tester I, et al. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J Exp Med. 2005;201(11):1725–1731. doi: 10.1084/jem.20042284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436(7053):946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 118.Bowen DG, Walker CM. Mutational escape from CD8+ T cell immunity: HCV evolution, from chimpanzees to man. . J Exp Med. 2005;201(11):1709–1714. doi: 10.1084/jem.20050808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Grakoui A, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302(5645):659–662. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 120.Smyk-Pearson S, et al. Spontaneous recovery in acute human hepatitis C virus infection: functional T-cell thresholds and relative importance of CD4 help. J Virol. 2008;82(4):1827–1837. doi: 10.1128/JVI.01581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oniangue-Ndza C, et al. Compensatory mutations restore the replication defects caused by cytotoxic T lymphocyte escape mutations in hepatitis C virus polymerase. J Virol. 2011;85(22):11883–11890. doi: 10.1128/JVI.00779-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Neumann-Haefelin C, et al. Human leukocyte antigen B27 selects for rare escape mutations that significantly impair hepatitis C virus replication and require compensatory mutations. Hepatology. 2011;54(4):1157–1166. doi: 10.1002/hep.24541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fitzmaurice K, et al. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut. 2011;60(11):1563–1571. doi: 10.1136/gut.2010.228403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138(1):30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 125.Rosen HR. Transplantation immunology: what the clinician needs to know for immunotherapy. Gastroenterology. 2008;134(6):1789–1801. doi: 10.1053/j.gastro.2008.02.062. [DOI] [PubMed] [Google Scholar]
- 126.Radziewicz H, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81(6):2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Golden-Mason L, Klarquist J, Wahed AS, Rosen HR. Cutting edge: programmed death-1 expression is increased on immunocytes in chronic hepatitis C virus and predicts failure of response to antiviral therapy: race-dependent differences. J Immunol. 2008;180(6):3637–3641. doi: 10.4049/jimmunol.180.6.3637. [DOI] [PubMed] [Google Scholar]
- 128.Seigel B, Bengsch B, Lohmann V, Bartenschlager R, Blum HE, Thimme R. Factors that determine the antiviral efficacy of HCV-specific CD8(+) T cells ex vivo. . Gastroenterology. 2013;144(2):426–436. doi: 10.1053/j.gastro.2012.10.047. [DOI] [PubMed] [Google Scholar]
- 129.Bengsch B, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6(6):e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wood NA, Linn ML, Bowen DG. Exhausted or just sleeping: awakening virus-specific responses in chronic hepatitis C virus infection. Hepatology. 2011;54(5):1879–1882. doi: 10.1002/hep.24602. [DOI] [PubMed] [Google Scholar]
- 131.McMahan RH, et al. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. . J Clin Invest. 2010;120(12):4546–4557. doi: 10.1172/JCI43127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Snyder JT, Alexander-Miller MA, Berzofskyl JA, Belyakov IM. Molecular mechanisms and biological significance of CTL avidity. Curr HIV Res. 2003;1(3):287–294. doi: 10.2174/1570162033485230. [DOI] [PubMed] [Google Scholar]
- 133.Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol. 2001;2(8):711–717. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- 134.Sedlik C, et al. In vivo induction of a high-avidity, high-frequency cytotoxic T-lymphocyte response is associated with antiviral protective immunity. J Virol. 2000;74(13):5769–5775. doi: 10.1128/JVI.74.13.5769-5775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee JK, et al. T cell cross-reactivity and conformational changes during TCR engagement. J Exp Med. 2004;200(11):1455–1466. doi: 10.1084/jem.20041251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yerly D, et al. Increased cytotoxic T-lymphocyte epitope variant cross-recognition and functional avidity are associated with hepatitis C virus clearance. J Virol. 2008;82(6):3147–3153. doi: 10.1128/JVI.02252-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dazert E, et al. Loss of viral fitness and cross-recognition by CD8+ T cells limit HCV escape from a protective HLA-B27-restricted human immune response. . J Clin Invest. 2009;119(2):376–386. doi: 10.1172/JCI36587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang Y, et al. Transduction of human T cells with a novel T-cell receptor confers anti-HCV reactivity. PLoS Pathog. 2010;6(7):e1001018. doi: 10.1371/journal.ppat.1001018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pasetto A, et al. TCR-redirected human T cells inhibit hepatitis C virus replication: hepatotoxic potential is linked to antigen specificity and functional avidity. J Immunol. 2012;189(9):4510–4519. doi: 10.4049/jimmunol.1201613. [DOI] [PubMed] [Google Scholar]
- 140.Houghton M. Prospects for prophylactic and therapeutic vaccines against the hepatitis C viruses. Immunol Rev. 2011;239(1):99–108. doi: 10.1111/j.1600-065X.2010.00977.x. [DOI] [PubMed] [Google Scholar]
- 141.Gottwein JM, et al. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49(2):364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 142.Folgori A, et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat Med. 2006;12(2):190–197. doi: 10.1038/nm1353. [DOI] [PubMed] [Google Scholar]
- 143.Zubkova I, et al. T-cell vaccines that elicit effective immune responses against HCV in chimpanzees may create greater immune pressure for viral mutation. Vaccine. 2009;27(19):2594–2602. doi: 10.1016/j.vaccine.2009.02.045. [DOI] [PubMed] [Google Scholar]
- 144.Feinstone SM, Hu DJ, Major ME. Prospects for prophylactic and therapeutic vaccines against hepatitis C virus. Clin Infect Dis. 2012;55(suppl 1):S25–S32. doi: 10.1093/cid/cis362. [DOI] [PubMed] [Google Scholar]
- 145.Rollier CS, et al. Vaccine-induced early control of hepatitis C virus infection in chimpanzees fails to impact on hepatic PD-1 and chronicity. Hepatology. 2007;45(3):602–613. doi: 10.1002/hep.21573. [DOI] [PubMed] [Google Scholar]
- 146.Burton JR, et al. Prospective analysis of effector and regulatory CD4+ T cells in chronic HCV patients undergoing combination antiviral therapy. . J Hepatol. 2008;49(3):329–338. doi: 10.1016/j.jhep.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 147.Gale M, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436(7053):939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 148.Golden-Mason L, et al. Loss of IL-7 receptor α-chain (CD127) expression in acute HCV infection associated with viral persistence. Hepatology. 2006;44(5):1098–1109. doi: 10.1002/hep.21365. [DOI] [PubMed] [Google Scholar]
- 149.Shin EC, et al. The frequency of CD127(+) hepatitis C virus (HCV)-specific T cells but not the expression of exhaustion markers predicts the outcome of acute HCV infection. . J Virol. 2013;87(8):4772–4777. doi: 10.1128/JVI.03122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tacke RS, et al. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 2012;55(2):343–353. doi: 10.1002/hep.24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Radziewicz H, Dunham RM, Grakoui A. PD-1 tempers Tregs in chronic HCV infection. J Clin Invest. 2009;119(3):450–453. doi: 10.1172/JCI38661. [DOI] [PMC free article] [PubMed] [Google Scholar]