

Abstract
The activating receptor NK cell group 2 member D (NKG2D) mediates antitumor immunity in experimental animal models. However, whether NKG2D ligands contribute to tumor suppression or progression clinically remains controversial. Here, we have described 2 novel lines of “humanized” bi-transgenic (bi-Tg) mice in which native human NKG2D ligand MHC class I polypeptide-related sequence B (MICB) or the engineered membrane-restricted MICB (MICB.A2) was expressed in the prostate of the transgenic adenocarcinoma of the mouse prostate (TRAMP) model of spontaneous carcinogenesis. Bi-Tg TRAMP/MICB mice exhibited a markedly increased incidence of progressed carcinomas and metastasis, whereas TRAMP/MICB.A2 mice enjoyed long-term tumor-free survival conferred by sustained NKG2D-mediated antitumor immunity. Mechanistically, we found that cancer progression in TRAMP/MICB mice was associated with loss of the peripheral NK cell pool owing to high serum levels of tumor-derived soluble MICB (sMICB). Prostate cancer patients also displayed reduction of peripheral NK cells and high sMIC levels. Our study has not only provided direct evidence in “humanized” mouse models that soluble and membrane-restricted NKG2D ligands pose opposite impacts on cancer progression, but also uncovered a mechanism of sMIC-induced impairment of NK cell antitumor immunity. Our findings suggest that the impact of soluble NKG2D ligands should be considered in NK cell–based cancer immunotherapy and that our unique mouse models should be valuable for therapy optimization.
Introduction
NK cell group 2 member D (NKG2D) and its ligands are important in antitumor immunity, as evidenced in experimental animal models. NKG2D is an activating receptor expressed by all NK cells, most NKT cells, subsets of γδ T cells, all human CD8 T cells, and activated mouse CD8 T cells (1, 2). Engagement of NKG2D can activate NK cells and costimulate CD8 and γδ T cells in vitro (3–5). Enforced expression of NKG2D ligand causes tumor cells to be rejected in syngeneic mice in a manner that is dependent on NK cells and, in some cases, primed CD8 T cells (6, 7). Neutralizing NKG2D in vivo with a specific antibody enhances host sensitivity to carcinogen-induced spontaneous tumor initiation (8). NKG2D-deficient transgenic adenocarcinoma of the mouse prostate (TRAMP) mice are 3 times more likely to develop aggressive poorly differentiated (PD) prostate carcinoma than NKG2DWT TRAMP counterparts (9). Moreover, in NKG2DWT TRAMP mice, progression to PD prostate carcinoma was mostly associated with downregulation of NKG2D ligand expression by tumor cells (9).
Curiously, most human tumors, in particular those of epithelial origins, express abundant NKG2D ligands yet progress to advanced diseases (10, 11), suggesting that NKG2D function is compromised in cancer patents. Various mechanisms have been proposed to explain how tumor cells evade NKG2D immunity. One prevailing concept is that exhaustion of NKG2D function by chronic exposure to its ligands contributes significantly to tumor immune evasion and progression (12–19). However, the prognostic value of NKG2D ligand expression in cancer patients is inconsistent at best (20–24). Levels of NKG2D ligand expression were reported to correlate with better clinical prognosis in colorectal cancer and early stages of breast cancer (22–24), but with poor survival in ovarian and invasive breast cancer (20, 21). Moreover, the underlying mechanisms for NKG2D exhaustion in cancer patients are also under debate. One hypothesis proposes that soluble NKG2D ligands, as a result of tumor-associated shedding, are the negative regulator for NKG2D function and confer the mechanisms of immune evasion. Indeed, clinical data demonstrated that elevated serum levels of soluble NKG2D ligands correlated with advanced epithelial malignancies (13, 14, 25–29). An alternative hypothesis proposes that chronic exposure to membrane-bound NKG2D ligands on tumor cell surface also impairs NKG2D immunity, based on observations that constitutive and ectopic expression of NKG2D ligands in normal mouse downmodulates NKG2D function (12, 15, 19). These inconsistent, to some extent confusing, findings have caused puzzlement in stratifying NKG2D-based immunotherapy for human malignancies (30). Thus, more studies are necessary to clarify the impact of NKG2D ligands on cancer progression.
Currently, there are no suitable mouse models for studying the impact of human NKG2D ligands on cancer progression and host immunity due to divergence of NKG2D ligands between humans and mice. Although NKG2D function is conserved between humans and rodents, the nature and expression pattern of its ligands are highly dissimilar between the 2 species (10, 31). In humans, known ligands for NKG2D include the MHC class I chain-related molecules (MIC) family members MHC class I polypeptide-related sequence A (MICA) and MICB and a family of UL-16–binding proteins (ULBPs) 1–5 (10, 31). In murine systems, the known ligands for NKG2D include the retinoic acid early inducible family of proteins RAE-1, the minor histocompatibility antigen H60 and its variants, and the murine ULBP-like transcript 1 (MULT1) (10, 31). No homolog of human MIC has yet been described in mice. In general, human or mouse NKG2D does not recognize the ligands of their counterparts, except that mouse NKG2D can recognize human MICB and selective alleles of human MICA (32–34). Most importantly, tumor shedding of NKG2D ligands to downregulate NKG2D function has been described and proposed as one of the immune evasion mechanisms in human cancer, but has not been described in mice. In mice, NKG2D-mediated suppression of ligand expression on tumor cells was postulated to be the main mechanism of tumor immune evasion (9). These differences create a barrier to studying the impact of human NKG2D ligands on cancer progression.
To clarify the impact of human NKG2D ligands on cancer, we exploited the knowledge that the human NKG2D ligand MICB can stimulate mouse NKG2D immunity (33, 34) and generated 2 unique lines of “humanized” bi-transgenic (bi-Tg) mouse models, TRAMP/MICB and TRAMP/MICB.A2. The former expresses the native human MICB, which can be shed by tumor cells, while the latter expresses an engineered membrane-restricted MICB (designated as MICB.A2) in the prostate of the autochthonous TRAMP mouse (33, 35, 36). Using these models, for what we believe is the first time, we conclusively demonstrate the distinct roles of membrane-bound NKG2D ligands and tumor-derived soluble NKG2D ligands on cancer progression. Moreover, we have also shown that prostate cancer metastatic progression is associated with profound loss of peripheral NK cells and elevated levels of circulating soluble NKG2D ligands in both men and mice.
Results
Bi-Tg TRAMP/MICB mice exhibit accelerated progression to PD prostate carcinoma and metastasis.
To define the role of NKG2D ligand in tumor progression, we used the minimal rat probasin (rPb) promoter (35) to direct the expression of the native human NKG2D ligand MICB and the engineered membrane-restricted MICB.A2 (36) encoding transgenes to the prostate epithelium in independent lines of C57BL/6 mice (designated as MICB/B6 and MICB.A2/B6 respectively, Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI69369DS1). Single copy integration and prostate-specific expression of the transgenes were confirmed, respectively, by genomic PCR against a limited template dilution standard (data not shown) and RT-PCR (Supplemental Figure 1). These transgenic mice exhibit normal prostate physiology and immune constitution as wild-type B6 animals (Supplemental Figures 1 and 2).
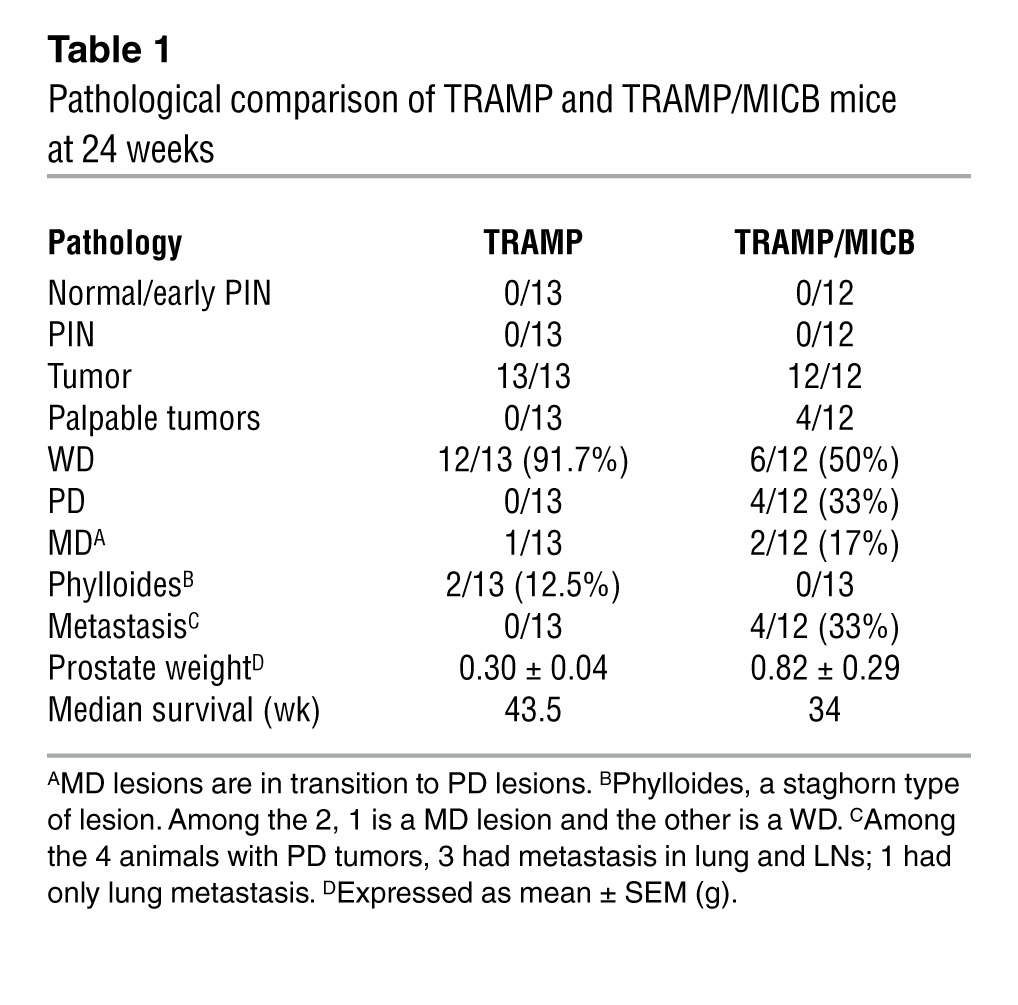
To investigate the role of native human NKG2D ligand on tumor immunity, male MICB/B6 mice were crossed with TRAMP female mice on a B6 background to generate the bi-Tg TRAMP/MICB mice. Normally, male TRAMP mice of B6 background develop hyperplasia and adenocarcinomas by 18 weeks of age and metastatic disease by 30 weeks of age (35, 37), with rare incidence of PD carcinomas. Remarkably, TRAMP/MICB mice elicited much more aggressive tumor growth and progression than TRAMP littermates, with significant reduction in overall survival (Figure 1A and Table 1). When specifically examined at 24 weeks of age, 33% (4/12) of the TRAMP/MICB mice had palpable prostate tumors and had significantly elevated prostate weight at sacrifice in comparison with age-matched TRAMP mice (n = 13) (Figure 1B). The palpable tumors were grossly large in volume and uniformly found to be PD carcinomas in histology (Figure 1, C and D). Among the nonpalpable tumors from TRAMP/MICB mice, 75% (6/8) exhibited well-differentiated (WD) carcinomas characterized by intact glandular architecture (Table 1). The remaining tumors (2/8) were mildly differentiated (MD) carcinomas, a transitional lesion in progression that was characterized by nearly anaplastic sheets of cells with remnants of glandular architecture (Figure 1D and ref. 37). In age-matched TRAMP male littermates (n = 13), only 8% (1/13) had palpable prostate tumor which, however, did not exhibit the pathological characteristics of PD lesions, but phylloides-like (PHY) lesions characterized by staghorn luminal patterns (Table 1 and Figure 1D), similar to a rare lesion in human prostate cancer with unclear prognosis (37, 38). Furthermore, all the 4 TRAMP/MICB mice that developed palpable PD carcinomas had metastatic deposits in the pelvic draining LNs, lung, and/or liver, as confirmed by immunostaining for the SV40T oncoprotein (Figure 1, E and F, and data not shown). Conversely, none of the 13 age-matched TRAMP littermates had evidence of metastatic lesions in the LNs, lung, liver, or bone (Table 1). These observations demonstrate that expression of native NKG2D ligand MICB accelerated carcinoma progression in TRAMP mice.
Figure 1. Prostate-specific expression of native MICB promotes the development of PD prostate carcinoma and metastasis.

(A) Kaplan-Meier plot showing significantly reduced survival of TRAMP/MICB mice in comparison with TRAMP mice (P < 0.001). Only tumor-associated incidence of death was considered in the analyses. Numbers indicate animals were surveyed during the indicated survival time. (B) Significant increased prostate weight in cohorts of 24-week-old TRAMP/MICB mice (n = 12) in comparison with TRAMP littermates (n = 13). (C) Representative images of gross appearance of tumors and associated LNs from TRAMP/MICB mice (representative of 4 out of 12 mice) and TRAMP littermates (representative of 12 out of 13 mice). (D) Representative images of H&E staining of prostate carcinomas from TRAMP/MICB and TRAMP mice. Note that WD tumors from TRAMP and TRAMP/MICB mice share the same pathological characteristics; thus, data only representatively shows WD tumors from TRAMP mice. (E) Representative images of H&E staining of LNs from TRAMP/MICB and TRAMP mice. Inserts are the magnified view of the boxed area. Arrowhead indicates tumor metastatic deposits. (F) SV40 Tag immunostaining (brown) of LNs from TRAMP and TRAMP/MICB mice to confirm tumor metastatic lesions. Scale bars: 50 μm (D–F).
Table 1.
Pathological comparison of TRAMP and TRAMP/MICB mice at 24 weeks
Rapid progression of prostate carcinoma in TRAMP/MICB mice is associated with elevated serum levels of soluble MICB.
To understand the underlying mechanism under which expression of NKG2D ligand MICB expedites the progression of prostate carcinoma in TRAMP mice, we first examined MICB expression in the prostate of TRAMP/MICB mice by immunohistochemistry with the mAb 6D4.6, which is specific for the α1α2 ectodomain of MICA and MICB (11). Interestingly, premalignant prostate intraepithelia neoplasia-like (PIN-like) lesions and WD carcinoma displayed intensive MIC immunostaining, predominantly located on the epithelial cell surface of the prostate gland, whereas little or no MICB expression was detected on the surface of PD tumor cells (Figure 2, A and B). Instead, intense MIC immunostaining in the interstitial spaces of the PD lesions was seen (Figure 2A, arrow). This pattern of MICB immunoreactivity in WD and PD carcinomas is similar to what was found in our previous observation of MIC expression in high-grade and low-grade prostate carcinomas in cancer patients (28). Furthermore, quantitative RT-PCR revealed that MICB was expressed equally in WD and PD lesions at the mRNA level (Figure 2C), suggesting that the loss of tumor cell surface MICB expression in the PD lesions was due to modifications at the posttranscriptional level. In prostate cancer patients, loss of tumor cell surface MIC was shown to be a result of shedding (28). We thus tracked serum levels of soluble MICB (sMICB) in these mice during carcinoma development. sMICB was detected in all TRAMP/MICB mice at all ages at varying levels (Figure 2D). However, mice that developed PD carcinomas (n = 4) by 24 weeks of age had remarkably elevated levels of serum sMICB compared with those that only developed WD carcinomas (n = 6) (Figure 2D, P < 0.01). Furthermore, there is a significant correlation between serum sMIC and final tumor volume as determined at the age of 24 weeks (Figure 2E, R2 = 0.88, P < 0.001). Together, these data strongly suggest that the progression to PD carcinomas in TRAMP/MICB mice was associated with loss of tumor cell surface membrane-bound MICB and elevated serum levels of sMICB.
Figure 2. Accelerated progression to PD carcinomas in TRAMP/MICB mice is associated with elevated serum sMICB and loss of surface MICB.

(A) Representative IHC staining demonstrating the pattern of MICB expression (brown) in TRAMP/MICB mice. Scale bars: 100 μm. (B) Summary IHC score of surface MICB expression on tumor cells. (C) Quantitative RT-PCR showing comparable levels of MICB expression in PD and WD tumors at the mRNA level. (D) ELISA measurement of serum levels of sMICB in the cohorts of 24-week-old TRAMP/MICB mice at various ages during carcinoma development. *P < 0.01; **P < 0.001. Note that development of PD carcinomas is associated with marked elevation of serum levels of sMICB. (E) Correlation of serum sMICB with tumor volume in TRAMP/MICB mice at the age of 24 weeks. (F) Quantitative RT-PCR showing comparable levels of endogenous NKG2D ligand RAE-1 expression in PD and WD tumors at the mRNA level. (G) Representative micrograph and added summary scores of IHC staining demonstrating pattern of endogenous NKG2D ligand RAE-1 expression in TRAMP/MICB carcinomas. Scale bars: 100 μm. Data represent results from 4 independent analyses.
Tumor shedding of NKG2D ligands to impair the NKG2D receptor function has been demonstrated in many types of human cancers and proposed to be one of the mechanisms of immune evasion. In TRAMP mice, however, tumors have been shown to evade NKG2D immune surveillance via an alternative mechanism. Guerra et al. have demonstrated that expression of the endogenous NKG2D ligand, e.g., RAE-1, was downregulated by the receptor NKG2D during tumor progression (9). To address whether this alternative mechanism contributes, at least in part, to the development of PD tumors in TRAMP/MICB mice, we examined the expression of RAE-1, the most abundantly expressed endogenous mouse NKG2D ligand in TRAMP tumors (9). Comparable mRNA and protein levels of RAE-1 expression were found in PD and WD tumors from TRAMP/MICB mice (Figure 2, F and G). These data suggest that development of PD carcinoma in TRAMP/MICB mice is not due to immune editing of endogenous NKG2D ligand expression. We thus conclude that tumor-derived sMICB is the major cause of tumor progression.
sMICB perturbs NK cell peripheral maintenance and facilitates tumor metastasis.
To understand the mechanisms by which sMICB facilitates tumor progression, we examined splenic CD8 T and NK cell populations and their NKG2D expression in TRAMP/MICB mice versus their TRAMP littermates. All the TRAMP/MICB and TRAMP mice had comparable frequency and absolute numbers of splenic CD8 T cells (Figure 3A and Supplemental Figure 3A). Only a small fraction of the CD8 T cells expressed NKG2D, and the expression levels were not significantly different between TRAMP/MICB and TRAMP mice. Given that NKG2D is only expressed by activated mouse CD8 T cells and that antigen-specific CD8 T cell tolerance is well documented in TRAMP mice (39, 40), this observation is expected. Intriguingly, the frequency and absolute number of NK cells in the spleen were significantly reduced in TRAMP/MICB mice that developed PD carcinomas and metastasis (Figure 3B and Supplemental Figure 3B). There was a significant inverse correlation between serum levels of sMICB and numbers of residual splenic NK cells in the cohort of TRAMP/MICB mice (Figure 3C, P = 0.002, R2 = –0.94). No major difference in the splenic NK cell population was observed among the TRAMP littermates.
Figure 3. Prostate carcinoma progression is associated with sMICB-induced impairment in NK cell peripheral maintenance.

(A and B) Comparisons of splenic CD8 (A) and NK (B) cells and NKG2D+ population in cohorts of 24-week-old TRAMP/MICB, TRAMP, and wild-type B6 mice. Left panel, representative dot plots of flow cytometry analyses. Right panel, pooled statistical data of flow cytometry analyses of each cohort. **P < 0.001 in comparison with TRAMP mice or with TRAMP/MICB mice that developed WD tumors. (C) Significant inverse correlation of serum sMICB with splenic NK cell population as analyzed from the cohort of 24-week-old TRAMP/MICB mice. (D) Representative dot plots of flow cytometry analyses and statistics of pooled data demonstrating systemic (LN, BM, and blood) depletion of NK cells in TRAMP/MICB mice that developed PD carcinoma. (E) Representative dot plots of flow cytometry analyses and statistical data demonstrating significant reduction of NK cells in tumor infiltrated lymphocytes (TILs) in PD tumors from TRAMP/MICB mice. All data show represents results from 3 independent assays of at least 5 animals.
We further addressed whether sMICB-associated depletion of splenic NK cells in TRAMP/MICB mice is a systemic effect or splenic-specific effect due to impaired trafficking. Despite marked reduction of splenic NK cells in association with high serum levels of sMICB, no accumulation of CD3–NK1.1+ cell population in the LNs or bone marrow was seen. Rather, a significant reduction of NK cell frequency in the LNs was also observed (Figure 3D). Only a modest but not significant reduction of NK cells was seen in the bone marrow (Figure 3D). Consistent with the findings in the peripheral lymphoid organs, NK cell infiltration in the tumor parenchyma of PD carcinomas was also significantly reduced (Figure 3E), whereas CD8 T cell infiltration was not affected (Supplemental Figure 3C). Nevertheless, NKG2D+ CD8 T cells were significantly reduced among CD8 T cells infiltrated in the PD carcinomas (Supplemental Figure 3C). These observations strongly suggest that the loss of NK cells in the secondary lymphoid organs is not due to development block or increased egress of NK cells into the tumor parenchyma.
To confirm that tumor progression and metastasis is facilitated by sMICB through impairing NK cell homeostasis, we determined the growth characteristics of TRAMP carcinoma-derived syngeneic metastatic cell line TRAMP-C2 (TC2) (41, 42) with and without sMICB expression after intravenous inoculation (Figure 4A). Three weeks after implantation, animals were treated with the sMICB-neutralizing antibody B10G5 or control IgG for a 2-week duration (Supplemental Methods and Supplemental Figure 4). As expected, micrometastatic deposits in the lung were significantly higher in mice implanted with TC2 cells expressing sMICB (TC2-sMICB) than in those implanted with TC2 cells (Figure 4B). Concomitantly, a significant reduction of NK cells was found in the spleen, LNs, and lung of mice implanted with TC2-sMICB cells in comparison with those implanted with TC2 cells (Figure 4C and Supplemental Figure 5). Moreover, lung micrometastatic deposits of TC2-sMICB were significantly reduced by sMICB-neutralizing antibody (Figure 4B), with the concurrent restoration of a peripheral pool of NK cells (Figure 4C). By BrdU pulsing, we found that highly proliferating NK cells failed to accumulate in the spleen of TC2-sMICB–bearing mice, but restored with the treatment of sMICB-neutralizing antibody (Figure 4D). Taken together, these data clearly show that sMICB facilitates metastasis of tumor cells and that such an activity is associated strongly with disruption of NK cell peripheral maintenance.
Figure 4. sMICB impairs NK cell peripheral homeostasis and facilitates lung metastasis in experimental TRAMP-C2 lung metastatic model.

(A) Experiment design. (B) Representative images of H&E staining (left panel) and quantification of lung deposits of micrometastasis (arrowhead on micrograph, right arrowhead). (C) Representative dot plots (left panel) of flow cytometry analyses and pooled statistical data (right panel) demonstrating NK1.1 frequency in bone marrow, LNs, and spleens. *P < 0.05 in comparison with anti-sMICB treatment group and all animals injected with TRAMP-C2 cells. (D) Representative flow cytometry analyses and quantification of splenic BrdU+ NK cell accumulation over 5 days. Data are representative of 5 animals per subgroup and 3 independent experiments.
To further address whether the maintenance of established NK cell populations is negatively affected by sMICB and to confirm the potential underlying mechanisms, we adoptively transferred cell trace dye V450-labeled congenic CD45.1+ splenocytes into mice 4 weeks after implantation with TC2 or TC2-sMICB cells as described above. These mice were pretreated with sMICB-neutralizing antibody B10G5 or control IgG (twice weekly) 1 week prior to the transfer. Five days after transfer, we examined the frequency of congenic NK cells in the spleen and LNs. In comparison with mice implanted with TC2 cells, mice that were implanted with TC2-sMICB cells had significantly reduced frequency of CD45.1+ NK cells in both spleens and LNs (Figure 5, A and B, and Supplemental Figure 6A). However, treatment with sMICB-neutralizing antibody led to a significant increase of congenic CD45.1+ NK cells in mice inoculated with TC2-sMICB cells (Figure 5, A and B, and Supplemental Figure 6A). The sMICB-neutralizing antibody did not have a significant effect on the frequency of CD45.1+ NK cells in mice implanted with TC2 cells (Figure 5, A and B, and Supplemental Figure 6A). V450 dilution analyses of the transferred congenic CD45.1+ cells revealed a significant increase in proliferation of CD45.1+ NK cells in both spleen and LNs with the treatment of sMICB-neutralizing antibody in mice implanted with TC2-sMICB cells (Figure 5, C and D, and Supplemental Figure 6B). We did not detect a significant difference of the expression of NK cell homing receptor CD62L in the spleen or LNs among all groups of animals (Supplemental Figure 6C), suggesting that sMIC-associated loss of NK cells in the peripheral was not likely due to impaired NK trafficking. This adoptive transfer experiment has clearly and conclusively demonstrated that sMICB perturbs NK cell peripheral maintenance, in part through impairing NK cell proliferation.
Figure 5. sMICB perturbs NK cell proliferation in metastatic TRAMP-C2-sMICB mice.

1 × 107 V450-labeled CD45.1+ congenic splenocytes were transferred into CD45.2+ mice that were implanted with TRAMP-C2-sMICB or into TRAMP-C2 mice. These mice were treated with sMICB-neutralizing antibody or control IgG before and after receiving CD45.1+ congenic splenocytes. Mice were euthanized 5 days after congenic transfer. (A and B) Percentage of CD45.1+ NK cells in the total CD45.1+ population in the spleen (A) and LNs (B). (C and D) summary of V450 dilution assay indicating that sMICB perturbs congenic CD45.1+ NK cell proliferation in the spleen (C) and LNs (B). 5–8 animals were used in each group. Data represent 3 independent experiments.
Stable expression of the nonshedding membrane-bound NKG2D ligand by tumor cells prevents tumorigenic progression.
To further determine that tumor progression in TRAMP/MICB mice is driven by sMICB rather than tumor surface MICB and to define the role of membrane-bound NKG2D ligands in tumor immunity, we generated bi-Tg MICB.A2/TRAMP mice by crossing male MICB.A2/B6 mice to female TRAMP (B6 background) mice. Of note, the engineered MICB.A2 was generated by replacing a shedding regulatory region of MICB with the corresponding sequence of HLA-A2. MICB.A2 could not be shed from tumor cell surface and is membrane-restricted (36). Overall, more than 90% of the TRAMP/MICB.A2 mice showed long-term, tumor-free survival (Figure 6A). When cohorts of TRAMP/MICB.A2 mice was examined at 24 weeks of age, prostate weights (0.20 ± 0.01 g) were only slightly higher than those of wild-type B6 littermates (0.17 ± 0.01 g; P = 0.16) but significantly lower than those of TRAMP littermates (0.30 ± 0.04 g; P < 0.01, Figure 6B). Remarkably, no single TRAMP/MICB.A2 mouse developed prostate carcinoma. The normal architecture of prostate gland was mostly preserved, as demonstrated by p63 immunostaining (Figure 6C). Among the 21 TRAMP/MICB.A2 mice, prostate glands from 8 animals exhibited normal histology, with intact basal cell layers. Prostates from the remaining 13 of these animals exhibited varying grades of PIN-like lesions represented by reduced p63 positivity in the interior of the multilayered lesions and an increased ratio of luminal cell to p63-positive basal cells (Figure 6C and ref. 37). Furthermore, the SV40T oncogenic protein that drives carcinoma development in TRAMP mice remained present in the epithelium of prostates from all TRAMP/MICB.A2 mice (35). Importantly, no soluble form of sMICB was detected in the sera of these animals (Figure 6D). These data clearly demonstrate that the membrane-bound form of MICB suppresses prostate cancer oncogenesis and progression.
Figure 6. Expression of membrane-restricted NKG2D ligand MICB.A2 in the TRAMP prostate prevents tumor progression.

(A) Kaplan-Meier plot demonstrating tumor-free survival of TRAMP/MICB.A2 mice. Numbers indicate animals being surveyed. (B) Significantly reduced prostate weight in TRAMP/MICB.A2 bi-Tg mice (n = 21) in comparison with age-matched TRAMP (n = 13) littermates. Age-matched wild-type B6 are shown as normal controls. Numbers of animals are shown in parentheses. (C) Representative images of H&E and p63 IHC staining demonstrate benign prostate histology in TRAMP/MICB.A2 mice. The expression of SV40 Tag in the benign prostate epithelium was also shown. Scale bars: 50 μm. (D) No sMICB was detected in the sera of TRAMP/MICB.A2 mice by ELISA. Asterisk indicates sera from TRAMP/MICB mice as the assay positive control. (E) Blocking NKG2D function with an antibody in TRAMP/MICB.A2 mice resulted in significantly elevated prostate weight. Data in E represent 2 independent experiments.
To ascertain that tumor suppression in TRAMP/MICB.A2 mice is mediated by NKG2D immunity as a consequence of prostate-specific expression of MICB.A2, cohorts of TRAMP/MICB.A2 mice (n = 6/cohort) of 16 to 18 week of age were treated with the NKG2D-blocking antibody C7 or control rat IgG twice weekly for an 8-week duration. Without exception, all the mice treated with C7 had elevated prostate weight and developed prostate carcinoma (Figure 6E and data not shown).
Remarkably, unlike TRAMP/MICB mice, which exhibited a significant reduction of NK cells in the spleen (Figure 3), the frequency of both NK and CD8 T cell populations was comparable to that of TRAMP or wild-type B6 littermates (Figure 7). The level of NKG2D expression on NK cells in TRAMP/MICB.A2 mice was also unperturbed (Figure 7A). Moreover, when stimulated with the NKG2D ligand RAE-1–expressing cell line ex vivo, splenic NK cells from TRAMP/MICB.A2 and wild-type mice showed comparable levels of IFN-γ production and NKG2D-dependent cytotoxicity (Figure 7, B and C). Moreover, the splenic NKG2D+ CD8 T cells increased in frequency in TRAMP/MICB.A2 mice in comparison with TRAMP littermates, although total CD8 T cell frequency remained similar (Figure 7D). These data support the notion that expression of membrane-bound NKG2D ligands on tumor cells promotes CD8 T cell response because NKG2D expression correlates with the activation status of mouse CD8 T cells (6). Collectively, these data provided direct in vivo evidence that sustained expression of the membrane-bound form of NKG2D ligand does not impair, but rather stimulates host, immunity to suppress tumorigenic progression.
Figure 7. Membrane-restricted MICB.A2 sustains NKG2D antitumor immunity in both NK and CD8 T cells.

(A) Flow cytometry analyses of splenic NK cells from cohorts of 24-week-old TRAMP/MICB.A2, TRAMP, and wild-type B6 mice and relative levels of NKG2D expression. Left panel, representative dot plots. Right panel, quantification of pooled data. (B and C) Splenic NK cells form TRAMP/MICB.A2 mice and B6 mice have comparable levels of IFN-γ production (B) and lytic ability (C) when stimulated with RMA–RAE-1 cells for 4 hours. Cytotoxicity assay was performed using the standard 51Cr release assay. Anti-mouse NKG2D antibody C7 was used as the blocking antibody. (D) Representative dot plots of flow cytometry analyses and statistics of splenic CD8 T cell population and their NKG2D expression. All data represent 3 independent analyses of a minimum of 5 animals.
Metastatic prostate cancer in men is associated with reduced peripheral NK cells and elevated serum sMIC.
To understand the clinical significance of our findings, we analyzed CD3–CD56+ NK cell populations in the peripheral blood of 36 newly diagnosed treatment-naive prostate cancer patients with varying Gleason scores, of whom 11 had distant metastatic diseases and 25 had only localized diseases. We found that the frequency of circulating NK cells was profoundly reduced in prostate cancer patients (n = 36) in comparison with age-matched healthy men (n = 10). Strikingly, men who developed metastatic diseases had a significantly lower frequency of circulating NK cells than those who were only diagnosed with localized diseases (Figure 8A). No significant difference was observed in circulating T cells (CD3+CD56–) among all the analyzed subjects (Figure 8B). Moreover, serum levels of sMIC were significantly higher in men with metastatic disease than those with localized diseases (Figure 8C). Furthermore, there was a significant inverse correlation between the serum levels of sMIC and NK cell frequency in the circulation (Figure 8D). No correlation of T cell frequency with sMIC was found (data not shown). These data revealed a clear association of prostate cancer metastasis with loss of NK cells in the peripheral lymphocyte pool and high serum levels of sMIC in patients and confirmed the close immunological and pathological similitude of our TRAMP/MICB mouse model to prostate cancer patients.
Figure 8. Development of metastatic prostate cancer in men is associated with reduced NK cell frequency in the blood and high levels of serum sMIC.

Data show NK cell (CD3–CD56+) frequency (A), T cell (CD3+CD56–) frequency (B), and serum levels of sMIC (C) from 36 treatment-naive prostate cancer patients, of whom 25 were diagnosed with localized disease and 11 were diagnosed with distant metastasis. Samples from age-matched healthy men (n = 10) were used as controls. Data represent results from 3 independent assays. (D) Significant inverse correlation between serum sMIC and the frequency of circulating NK cells from all patients.
Discussion
Ample evidence from experimental animal models supports the significance of NKG2D function in tumor immunity. However, to date, studies have not been able to clarify whether sustained expression of NKG2D ligands on tumor cells or chronic exposure of NKG2D to its ligands expressed on tumor cells is most beneficial for tumor immunity (12, 15, 19). To this end, we established “humanized” bi-Tg TRAMP/MICB and TRAMP/MICB.A2 mouse models in which a human NKG2D ligand was specifically expressed on a spontaneous tumor with concurrent expression of the oncogenic protein SV40T, closely emulating human cancers. Moreover, we differentially expressed 2 forms of NKG2D ligands to distinguish their effects on tumor progression: the native NKG2D ligand MICB, which can be shed by tumors to generate the soluble form as naturally occurred in cancer patients, and the mutant membrane-restricted NKG2D ligand MICB.A2, which could not be shed from tumors. With these 2 lines of mouse models, we have for what we believe is the first time clearly demonstrated that membrane-restricted and soluble NKG2D ligands pose opposite impacts on tumor progression and metastasis. We conclusively demonstrate that membrane-restricted NKG2D ligand MICB.A2 could sustain NKG2D protective immunity and prevent spontaneous tumorigenesis and that native NKG2D ligand MICB facilitates tumor progression through soluble ligand-mediated impairment of NK cell peripheral maintenance.
Mechanisms such as downmodulation of NKG2D expression by tumor-derived soluble NKG2D ligands (13, 14, 25, 28, 43) and TGF-β (44) and obstruction of MIC-NKG2D interaction by core2 O-glycans (45) have been described as tumor-immune evasions of NKG2D-mediated immunity. An observation was made in our current study regarding the profound impairment of NK cell homeostasis in both mice and men by tumor-derived soluble NKG2D ligands. We found that peripheral NK cells were profoundly depleted as a result of high levels of circulating sMIC in bi-Tg TRAMP/MICB mice. This tumor evasion mechanism was further confirmed in an experimental lung TRAMP-C2-sMICB model and also indicated to be of significance in prostate cancer patients. As shown in the animal models, depletion of NK cells was most profound in the peripheral lymphoid organs and less so in the bone marrow, suggesting that homeostatic maintenance of the NK cell pool in the peripheral was affected. This point was further strengthened by our adoptive transfer experiment in which we observed the depletion of proliferating NK cells by sMICB that can be reversed by treatment of neutralizing antibody. Our findings are intriguing and could provide explanations, at least in part, for many clinical observations. For instance, decreased intratumoral NK cell numbers have been found to predict a poor clinical outcome in certain types of cancer patient (46, 47). Also, retrospective studies suggest that serum sMIC has a predictive value for epithelial cancer progression to metastasis (13, 14, 29). The failure of clinical trials of adoptive NK cell transfer to treat human malignancy (48, 49) may also be explained in part by soluble NKG2D ligand-mediated NK cell depletion. One of the challenges for improving the therapeutic outcome is to sustain infused NK cells in cancer patients (48, 49). Our current study suggests that elimination of soluble NKG2D ligands may be one of the viable avenues for exploring way to improve NK cell–based cancer immunotherapy. Further studies are necessary to uncover the cellular and molecular mechanism by which sMIC impairs NK cell peripheral homeostasis.
Our finding that membrane-bound NKG2D ligand protects antitumor immunity contradicts the conclusion from other reports that chronic exposure to membrane-bound ligands impairs NKG2D function and thus increases susceptibility to tumorigenesis (15, 19). We reason that the discrepancy lies in the differences among various experimental models. In 2 studies, the ligand of NKG2D was constitutively and broadly expressed in non–tumor prone wild-type mice (15, 19), not in a tissue-specific fashion in the context of a spontaneous tumor, which is highly nonphysiological. For instance, 1 transgenic mouse model that was created by expressing human MICA under the constitutive and ubiquitous mouse MHC class I H-2Kb promoter on a C57BL/6 background showed impaired ability of NK cells to reject MICA-transfected RMA tumors in comparison with the wild-type counterparts (19). In other models (15), NKG2D ligand RAE-1ε was expressed in normal mice under the constitutive involucrin promoter (inducing squamous epithelium expression) or the ubiquitous chicken β-actin promoter; local and systemic NKG2D downregulation was noted in these mice in comparison with the wild-type counterparts. Notably, in these transgenic mouse models, NKG2D ligand expression was “ectopic” and under the direction of a constitutive or ubiquitous promoter in somatic cells. Given the magnitude of ligand-induced NKG2D signaling on activating NK cells, downmodulation of NKG2D function is expected in these transgenic mice in comparison with an otherwise wild-type counterpart as a self-regulatory mechanism to allow normal embryonic development. Thus, although the findings from these mouse models are interesting, they do not represent the real situation in cancer patients. On the contrary, we created unique transgenic mice that express NKG2D ligands in the prostate that is prone to develop cancer spontaneously due to concurrent expression of SV40 T antigen. The organ-specific and temporal expression of NKG2D ligand during cancer progression closely resembles that in human cancers. The results from our studies may reflect more closely what is happening in human patients, which is validated by our consistent clinical observations: prostate cancer progression correlated profoundly with increased soluble MIC and concurrent depletion of NK cells pools in the circulation.
Giving the opposing roles of membrane-bound and soluble NKG2D ligands in cancer progression, it is not surprising that the prognostic significance of NKG2D ligands in human cancer remains controversial (20–24). Most of the studies did not specify whether NKG2D ligands present on tumor tissues were membrane bound or soluble. This ambiguity may account for the contradiction in prognostic value of NKG2D ligands in patients, even with the same type of cancer at different disease stages. For example, high expression of NKG2D ligand MIC in tumors was reported to predict a good prognosis for early breast cancer but a poor prognosis for invasive breast carcinoma (21, 24). Tumor shedding of sMIC is mediated by members of MMP families (50, 51). One of the members, MT1-MMP, that is shown to mediate MIC shedding (50) is upregulated in invasive breast carcinomas, and its expression suggests a poor clinical prognosis in patients with invasive breast cancer (52). Thus, it is possible that different forms of NKG2D ligands were detected in early and invasive breast cancer tissues. Because soluble NKG2D ligands negatively regulate the function of multiple antitumor effector cells within tumor tissue and systemically, we believe that a combination of vigilant scoring of NKG2D ligand expression in tumor tissues with serum levels of sMIC is needed to provide an accurate prognosis of a malignancy.
In summary, we have generated a mouse model of spontaneous carcinoma that recapitulates the specific interactions between NKG2D and its cognate ligands during tumor progression. We further demonstrate that metastatic progression of prostate cancer is associated with loss of NK cells in the circulating and high levels of soluble MIC in both mice and men. Our study not only clearly defines the impact of NKG2D ligand expression on tumor progression, but also presents a valuable preclinical autochthonous mouse model that can be exploited to develop and optimize cancer immunotherapy based on the biology of NKG2D and its ligands.
Methods
Human blood sample collection.
Blood samples of treatment-naive and newly diagnosed prostate cancer patients (n = 36, age 52–79) were obtained from the outpatient Urology Clinics at the Veterans Affair Puget Sound Health Care System, the Seattle Cancer Care Alliance, and the Hollings Cancer Center. All subjects enrolled in this study had normal white blood cell counts (4,000–11,000 cells/μl). Twenty-five of these men were diagnosed with localized disease only. Eleven of these men were diagnosed with distant metastasis (bone or LNs). Blood samples from healthy control age-matched men (n = 10, age 50–68) were obtained from human subjects recruited at the University of Washington. Healthy subject was defined as no history of cancer and no immunological disorder. Whole blood was collected in heparinized tubes. Serum was collected and PBMCs were separated by Ficoll-Hypaque density.
Transgenic mice.
Mice were bred and housed under specific pathogen–free conditions in the animal facility of the University of Washington and the Medical University of South Carolina in accordance with institutional guidelines with approved IACUC protocols. All mice used in this study were on the B6 background. The rPB-MICB and rPB-MICB.A2 expression cassettes were constructed by replacing the SV40T fragment from rPB-SV40T expression cassettes (35) with the cDNA fragments encoding MICB or MICB.A2, respectively. The cDNA encoding for MICB.A2 was similarly described (33). Briefly, a small region of MICB α3 domain (aa 238–252) spanning the motif that regulates MICB shedding (36) was replaced with a corresponding sequence from HLA.A2. The molecule MICB.A2 has the same function as MICB in recognition of NKG2D (33, 36). The entire rPB-MICB or rPB-MICB.A2 expression cassette was gel purified following digestion with HindIII and was microinjected into fertilized B6 embryos, respectively (performed at University of Washington Comparative Medicine Transgenic Core Facility). Transgenic progeny were identified by PCR analysis of DNA extracted from tail biopsies using the forward primer specific for rPB (5′-ACAAGTGCATTTAGCCTCTCCAGTA-3′) and the reverse primer specific for MICB (5′-TGTGTCTTGGTCTTCATGGC-3′) or MICB.A2 (5′-CAGAGACAGCGTGGTGAGTCATATG-3′).
The male rPB-MICB and rPB-MICB.A2 animals were bred with female TRAMP mice to generate the experimental male TRAMP-MICB and TRAMP-MICB.A2 animals. Presence of double transgenes was identified by PCR analysis of DNA extracted from tail biopsies using the primers specific for SV40 Tag (forward, 5′-GATATGGCTGATCATGAACAGACT-3′; reverse 5′-TTTGAGGATGTAAAGGGCACTG-3′) and MICB or MICB.A2 as described above. All experimental mice were randomly assigned to cohorts and sacrificed at the defined age for evaluation. In some cohorts, animals were administrated i.p. with anti-NKG2D blocking antibody C7 (100 μg/ mouse, eBiosciences) or control IgG twice weekly for an 8-week duration. Prostates, LNs, liver, lung, and femur were collected for histological evaluation. In some experiments, LN and bone marrow were also collected along with spleens for single-cell suspension and immunological evaluation. Blood were collected for assaying serum levels of sMICB by ELISA.
Experimental lung metastatic model.
TRAMP-C2 (TC2) or TC2 cells expressing sMICB (TC2-sMICB) were i.v. transplanted into 2 groups of syngenic MICB/B6 transgenic mice (n = 6) (0.5 × 106 cells/mouse) at the age of 8 to 10 weeks. As mice do not naturally express homologs to human MIC, we chose to use the transgenic MICB/B6 mice instead of B6 mice in this experiment to avoid the effect of autoantibody against the human molecule sMICB during the experiment. Six weeks after transplantation, mice in each group were further randomized into 2 subgroups: with anti-MICB mAb B10G5 (Supplemental Methods) treatment or control mouse IgG. Each mouse in the treatment group received i.p. the dose of 100 μg mAb B10G5 or mouse IgG twice weekly for 2 weeks before sacrifice. For BrdU feeding, mice were supplied with 0.8 mg/ml of BrdU (Sigma-Aldrich) in drinking water made fresh daily. For congenic experiments, 1 × 107 congenic CD45.1+ splenocytes were labeled with Cell Trace Dye V450 before being transferred i.v. into each mouse. After euthanization, spleens, bone marrows, and LNs were harvested for single-cell suspension analyses. Lung were harvested, fixed in 10% neutral fixation buffer, paraffin embedded, and sectioned for histology evaluation.
Histological and immunohistochemical examination.
The mouse prostate and other soft tissues were fixed in 10% formaldehyde and embedded in paraffin wax. The bone tissues were decalcificated for 24 hours before they were embedded. Five-micrometer sections were cut and stained with H&E for pathological evaluation. Sections were also stained with the following: (a) anti-MICB (and MICB.A2) mAb 6D4.6 (mouse IgG; 1:500; Biolegend); (b) anti–panRAE-1 antibody (rat mAb IgG2a; 1:500; R&D); (c) anti-SV40T antigen (rabbit polyclonal IgG, 1:200; Santa Cruz Biotechnology Inc.); (d) anti-mouse p63 antibody (mouse mAb IgG2a; 1:200; Thermo Scientific). Sections were deparaffinized and incubated for 10 minutes in 10 mM citrate buffer (pH 6.0) at 95°C for antigen retrieval. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide. After quenching endogenous peroxidase activity and blocking nonspecific binding, slides were incubated with specific primary antibody overnight at 4°C followed by subsequent incubation with the appropriate biotinylated secondary antibody: goat anti-rabbit IgG (Vector); rabbit anti-rat IgG (Vector); and goat anti-mouse IgG (Vector) at a 1:1000 dilution for 20 minutes at 37°C. Immunoreactive antigens were detected using the Vectastain Elite ABC Immunoperoxidase Kit and DAB. All slides were counterstained with hematoxylin (Vector) and mounted with Permount (Fisher Scientific).
IHC evaluation.
Scoring of IHC staining was similar to that described previously (53). Briefly, the intensity of specific staining was double-blind graded on a scale of 1 to 3 (1 = weak staining, 2 = moderate staining, and 3 = strong staining). A semiquantitative score on a 10% increment scale ranging from 0% to 100% was used to assess the percentage of stained cells of the intensity. Approximately 500 cells were analyzed for each case. The final composite staining score for immunostaining was based on the intensity of staining (1, 2, or 3) multiplied by the percentage of immunopositive cells (0%–100%). The maximum composite score was 300.
Pathological evaluation.
Ten randomly selected fields of H&E-stained sections of the prostate from individual mice of each cohort were independently scored by 2 scientists for incidence and percentage of area corresponding to each pathologic stage. Pathologic grading of the prostate was performed according to the recommendations of published studies (35, 38).
Flow cytometry analysis.
For analyses of NK cell frequency in human PBMCs, single-cell suspensions were stained with FITC-conjugated anti-human CD3 and PE-conjugated anti-human CD56 antibodies (eBiosciences). Samples were analyzed with BD FACScan. NK cells were defined as CD3–CD56+. For analyzing mouse samples, single-cell suspensions of splenocytes, LN, and bone marrow were incubated on ice for 30 minutes with a combination of FITC-conjugated mNK-specific mAb NK1.1 or DX5 or mCD8-specific antibody (eBioscience) in combination with PE-conjugated anti-mouse NKG2D mAb CX5 (eBioscience) and PerCP-conjugated anti-mouse CD3 antibody (eBioscience). Cells were analyzed using a BD FACscan or Fortessa. Data were analyzed using the BD FlowJo software (Tree Star).
Cytotoxicity assay.
Mouse NK cells were isolated as previously described from pooled splenocytes from 2 to 3 representative mice with similar prostate disease progression within the same experimental group and were used as effectors (33). 51Cr-labeled TRAMP-MICB.A2 cells were used as target cells. Cyototoxicity assay was performed using a standard 4-hour 51Cr release assay as described previously (33). 30 μg/ml of anti-NKG2D antibody C7 (eBioscience) was for blocking NKG2D function.
ELISA for sMIC.
Serum was diluted 1:2 with PBS. Amount of soluble MIC in human serum was measured using MICA DuoSet Sandwich ELISA kit (R&D Systems) following the manufacture’s protocol. Amount of sMICB in mouse serum was measured using the human MICB DuoSet Sandwich ELISA kit (R&D Systems) as previously described (33).
Statistics.
All statistical data were expressed as mean ± SEM. Difference between means of populations was compared by standard Student’s t test for unpaired, 1-tailed samples. Survival was determined via Kaplan-Meier analysis with comparison of curves using the Mantel-Haenszel log-rank test. A P value of 0.05 or less was considered significant. GraphPad Prism software was used for all analyses.
Study approval.
The collection and the use of human peripheral blood samples and relevant clinical information were approved by the University of Washington Institutional Review Board and the Medical University of South Carolina. All subjects gave written informed consent for the study. All animal studies were approved by the IACUC of the University of Washington and the Medical University of South Carolina.
Supplementary Material
Acknowledgments
Supported by DOD-USMRC IDEA Development Award W81XWH-06-1-0014, NIH Temin Award 1K01CA116002, NIH grant 1R01CA149405, and A. David Mazzone — PCF Challenge Award to J.D. Wu. We thank Veterans Affair Research Service (S.R. Plymate) for help with recruiting prostate cancer patients. We thank X. Yu and C. Paulos for helpful discussions and critical reading of the manuscript.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(10):4410–4422. doi:10.1172/JCI69369.
Norman M. Greenberg’s present address is: MedImmune LLC, Gaithersburg, Maryland, USA.
References
- 1.Long EO. Versatile signaling through NKG2D. Nat Immunol. 2002;3(12):1119–1120. doi: 10.1038/ni1202-1119. [DOI] [PubMed] [Google Scholar]
- 2.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3(10):781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 3.Bauer S, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285(5428):727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 4.Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol. 2001;2(3):255–260. doi: 10.1038/85321. [DOI] [PubMed] [Google Scholar]
- 5.Wu J, Groh V, Spies T. T cell antigen receptor engagement and specificity in the recognition of stress-inducible MHC class I-related chains by human epithelial gamma delta T cells. J Immunol. 2002;169(3):1236–1240. doi: 10.4049/jimmunol.169.3.1236. [DOI] [PubMed] [Google Scholar]
- 6.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413(6852):165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A. 2001;98(20):11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med. 2005;202(5):583–588. doi: 10.1084/jem.20050994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guerra N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28(4):571–580. doi: 10.1016/j.immuni.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene. 2008;27(45):5944–5958. doi: 10.1038/onc.2008.272. [DOI] [PubMed] [Google Scholar]
- 11.Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A. 1999;96(12):6879–6884. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coudert JD, et al. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood. 2005;106(5):1711–1717. doi: 10.1182/blood-2005-03-0918. [DOI] [PubMed] [Google Scholar]
- 13.Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICB in malignant diseases: analysis of diagnostic significance and correlation with soluble MICA. Cancer Immunol Immunother. 2006;55(12):1584–1589. doi: 10.1007/s00262-006-0167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer. 2006;118(3):684–687. doi: 10.1002/ijc.21382. [DOI] [PubMed] [Google Scholar]
- 15.Oppenheim DE, et al. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol. 2005;6(9):928–937. doi: 10.1038/ni1239. [DOI] [PubMed] [Google Scholar]
- 16.Raffaghello L, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. 2004;6(5):558–568. doi: 10.1593/neo.04316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salih HR, et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 2003;102(4):1389–1396. doi: 10.1182/blood-2003-01-0019. [DOI] [PubMed] [Google Scholar]
- 18.Salih HR, Goehlsdorf D, Steinle A. Release of MICB molecules by tumor cells: mechanism and soluble MICB in sera of cancer patients. Hum Immunol. 2006;67(3):188–195. doi: 10.1016/j.humimm.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Wiemann K, et al. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J Immunol. 2005;175(2):720–729. doi: 10.4049/jimmunol.175.2.720. [DOI] [PubMed] [Google Scholar]
- 20.McGilvray RW, Eagle RA, Rolland P, Jafferji I, Trowsdale J, Durrant LG. ULBP2 and RAET1E NKG2D ligands are independent predictors of poor prognosis in ovarian cancer patients. Int J Cancer. 2010;127(6):1412–1420. doi: 10.1002/ijc.25156. [DOI] [PubMed] [Google Scholar]
- 21.Madjd Z, et al. Upregulation of MICA on high-grade invasive operable breast carcinoma. Cancer Immun. 2007;7:17. [PMC free article] [PubMed] [Google Scholar]
- 22.Watson NF, et al. Expression of the stress-related MHC class I chain-related protein MICA is an indicator of good prognosis in colorectal cancer patients. Int J Cancer. 2006;118(6):1445–1452. doi: 10.1002/ijc.21510. [DOI] [PubMed] [Google Scholar]
- 23.McGilvray RW, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res. 2009;15(22):6993–7002. doi: 10.1158/1078-0432.CCR-09-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Kruijf EM, et al. NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: an observational study. BMC Cancer. 2012;12:24. doi: 10.1186/1471-2407-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419(6908):734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 26.Jinushi M, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A. 2008;105(4):1285–1290. doi: 10.1073/pnas.0711293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamaki S, et al. Soluble MICB serum levels correlate with disease stage and survival rate in patients with oral squamous cell carcinoma. Anticancer Res. 2010;30(10):4097–4101. [PubMed] [Google Scholar]
- 28.Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest. 2004;114(4):560–568. doi: 10.1172/JCI22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rebmann V, et al. Soluble MICA as an independent prognostic factor for the overall survival and progression-free survival of multiple myeloma patients. Clin Immunol. 2007;123(1):114–120. doi: 10.1016/j.clim.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Hayakawa Y. Targeting NKG2D in tumor surveillance. Expert Opin Ther Targets. 2012;16(6):587–599. doi: 10.1517/14728222.2012.681378. [DOI] [PubMed] [Google Scholar]
- 31.Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev. 2010;235(1):267–285. doi: 10.1111/j.0105-2896.2010.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friese MA, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003;63(24):8996–9006. [PubMed] [Google Scholar]
- 33.Wu JD, Atteridge CL, Wang X, Seya T, Plymate SR. Obstructing shedding of the immunostimulatory MHC class I chain-related gene B prevents tumor formation. Clin Cancer Res. 2009;15(2):632–640. doi: 10.1158/1078-0432.CCR-08-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nature Immunol. 2000;1(2):119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 35.Greenberg NM, et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92(8):3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Lundgren AD, Singh P, Goodlett DR, Plymate SR, Wu JD. An six-amino acid motif in the alpha3 domain of MICA is the cancer therapeutic target to inhibit shedding. Biochem Biophys Res Commun. 2009;387(3):476–481. doi: 10.1016/j.bbrc.2009.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gingrich JR, Barrios RJ, Foster BA, Greenberg NM. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate Cancer Prostatic Dis. 1999;2(2):70–75. doi: 10.1038/sj.pcan.4500296. [DOI] [PubMed] [Google Scholar]
- 38.Kaplan-Lefko PJ, et al. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate. 2003;55(3):219–237. doi: 10.1002/pros.10215. [DOI] [PubMed] [Google Scholar]
- 39.Anderson MJ, Shafer-Weaver K, Greenberg NM, Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. J Immunol. 2007;178(3):1268–1276. doi: 10.4049/jimmunol.178.3.1268. [DOI] [PubMed] [Google Scholar]
- 40.Bai A, Higham E, Eisen HN, Wittrup KD, Chen J. Rapid tolerization of virus-activated tumor-specific CD8+ T cells in prostate tumors of TRAMP mice. Proc Natl Acad Sci U S A. 2008;105(35):13003–13008. doi: 10.1073/pnas.0805599105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren C, et al. Cancer gene therapy using mesenchymal stem cells expressing interferon-beta in a mouse prostate cancer lung metastasis model. Gene Ther. 2008;15(21):1446–1453. doi: 10.1038/gt.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varghese S, Rabkin SD, Liu R, Nielsen PG, Ipe T, Martuza RL. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006;13(3):253–265. doi: 10.1038/sj.cgt.7700900. [DOI] [PubMed] [Google Scholar]
- 43.Holdenrieder S, et al. Soluble NKG2D ligands in hepatic autoimmune diseases and in benign diseases involved in marker metabolism. Anticancer Res. 2007;27(4A):2041–2045. [PubMed] [Google Scholar]
- 44.Doubrovina ES, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol. 2003;171(12):6891–6899. doi: 10.4049/jimmunol.171.12.6891. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki Y, et al. MUC1 carrying core 2 O-glycans functions as a molecular shield against NK cell attack, promoting bladder tumor metastasis. Int J Oncol. 2012;40(6):1831–1838. doi: 10.3892/ijo.2012.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 47.Gulubova M, Manolova I, Kyurkchiev D, Julianov A, Altunkova I. Decrease in intrahepatic CD56+ lymphocytes in gastric and colorectal cancer patients with liver metastases. APMIS. 2009;117(12):870–879. doi: 10.1111/j.1600-0463.2009.02547.x. [DOI] [PubMed] [Google Scholar]
- 48.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salagianni M, Baxevanis CN, Papamichail M, Perez SA. New insights into the role of NK cells in cancer immunotherapy. Oncoimmunology. 2012;1(2):205–207. doi: 10.4161/onci.1.2.18398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu G, Atteridge CL, Wang X, Lundgren AD, Wu JD. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J Immunol. 2010;184(7):3346–3350. doi: 10.4049/jimmunol.0903789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waldhauer I, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008;68(15):6368–6376. doi: 10.1158/0008-5472.CAN-07-6768. [DOI] [PubMed] [Google Scholar]
- 52.Perentes JY, et al. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res. 2011;71(13):4527–4538. doi: 10.1158/0008-5472.CAN-10-4376. [DOI] [PubMed] [Google Scholar]
- 53.Wu JD, Lin DW, Page ST, Lundgren AD, True LD, Plymate SR. Oxidative DNA damage in the prostate may predispose men to a higher risk of prostate cancer. Transl Oncol. 2009;2(1):39–45. doi: 10.1593/tlo.08217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.