

Abstract
Sphingolipids are components of the plasma membrane whose metabolic manipulation is of interest as a potential therapeutic approach in a number of diseases. Sphingosine kinase 1 (SphK1), the major kinase that phosporylates sphingosine to sphingosine-1-phosphate (S1P), was previously shown by our group and others to modulate inflammation in murine models of inflammatory arthritis, inflammatory bowel disease and asthma. Sphingosine kinase 2’s (SphK2) impact on inflammation is less well known, as variable results were reported depending on the disease model. A specific SphK2 inhibitor inhibited inflammatory arthritis in one model, while siRNA knockdown of SphK2 worsened arthritis in another. We previously demonstrated that SphK1 deficient mice are protected against development of hTNF-α induced arthritis. To investigate the role of SphK2 in TNF-α induced arthritis, we developed SphK2 deficient hTNF-α overexpressing mice and separately treated hTNF-α mice with ABC294640, a SphK2 specific inhibitor. Our data show that genetic inhibition of SphK2 did not significantly impact the severity or progression of inflammatory arthritis, while pharmacologic inhibition of SphK2 led to significantly more severe arthritis. Compared to vehicle-treated mice, ABC294640 treated mice also had less S1P in whole blood and inflamed joint tissue, although the differences were not significant. ABC294640 treatment did not affect SphK1 activity in the inflamed joint while little SphK2 activity was detected in the joint. We conclude that the differences in the inflammatory phenotype in genetic inhibition vs. pharmacologic inhibition of SphK2 can be attributed to the amount of ABC294640 used in the experiments versus the impact of acute inhibition of SphK2 with ABC294640 vs. genetically-induced life-long SphK2 deficiency. Thus, inhibition of SphK2 appears to be proinflammatory in contrast to the clear anti-inflammatory effects of blocking SphK1. Therapies directed at this sphingosine kinase pathways will need to be specific in their targeting of sphingosine kinases.
Keywords: Sphigosine kinase 2, inflammatory arthritis, sphingolipids, TNF
Introduction
The etiology of many diseases results from the dysregulation of inflammation. Sphingolipids are bioactive molecules present in the plasma membrane and are being investigated for their effects on inflammation. There are 2 isoenzymes responsible for phosphorylation of sphingosine to sphingosine 1 phosphate (S1P), sphingosine kinases 1 and 2 (SphK1 and 2). SphK1 is the major enzyme responsible for S1P formation [1], however distinct differences exist in the other functions of SphK1 and SphK2. SphK2 also phosphorylates dihydrosphingosine, phytosphingosine [2, 3] and FTY720 [5, 6], a synthetic receptor agonist of S1P that leads to lymphopenia and downregulation of S1P1, one of the S1P receptors [7–10]. It also is found later in embryonic development compared to SphK1 [3]. Additionally, only SphK1 affects degranulation, migration, and CCL2 production of IgE/antigen stimulated mast cells [4].
SphK2’s effects on inflammation were studied in arthritis models. Reduction of SphK2 expression by siRNA in collagen-induced arthritic (CIA) mice led to an increased incidence, mean articular index, number of affected paws and serum levels of pro-inflammatory cytokines (i.e. IL-6, IFN-γ and TNF-α) compared to similar mice treated with SphK1-specific or scrambled siRNA. However, less IL-10, an anti-inflammatory cytokine, was present [11], suggesting that inhibition of SphK2 induces a more severe inflammatory arthritis in this model. Treating with a SphK2 specific inhibitor, however, had the opposite effect of improving arthritis in murine models of disease.
Reduction of SphK2 was also studied in a cancer model using a xenograft of MCF-7 cells in nude mice. Twenty-five days post-injection of SphK2-deficient MCF-7cells, demonstrated a slower tumor volume progression and a lower average tumor volume vs. injection of SphK2-competent MCF-7 cells. Tumor associated macrophages (TAMs) were isolated from SphK2-deficient MCF-7 xenografts had significantly higher levels of TNF-α, CCL2 and IL-12 pre- and post stimulation with LPS/IFN, a pro-inflammatory profile. Significantly less IL-10 was noted prestimulation with LPS and IFN-γ [12], concluding the anti-tumor effects resulted from a proinflammatory environment.
Using an adoptive transfer model of IBD, SphK2-competent or -deficient, CD4+, CD25−, CD45RB high T-cells were transferred into C.B-17-scid mice. Transfer of SphK2-deficient T-cells into scid mice resulted in a more severe and rapid progression of inflammation compared to transfer of SphK2 competent T-cells. Isolated mesenteric lymph nodes from SphK2-deficient T-cell recipients produced higher levels of IL-2, IFN-γ and IL-17 [13].
Treatment of mice with dextran sodium sulfate (DSS), azoxymethane (AOM) and ABC294640, a specific inhibitor of SphK2 [14], demonstrated decreased colon cancer incidence and tumor burden (macroscopic and microscopic) [15]. However, mice given DSS only and ABC294640 had lower colonic levels of pro-inflammatory cytokines (i.e. TNF-α, IL-6, IL-1β, and IFN-γ), with longer colonic lengths and lower histological scores than control [16]. Therefore, in DSS only-induced IBD, pharmacologic inhibition of SphK2 has contrasting effects on pro-inflammatory cytokine levels.
Based on the dichotomy of results in the literature, we studied the differences in pharmacologic and genetic inhibition of SphK2 on the development of TNF-α induced arthritis to determine if manipulation of SphK2 impacts arthritis when studied from both a genetic and pharmacologic strategy. In the hTNF-α model of inflammatory arthritis, pharmacologic inhibition led to a more severe arthritis while genetic inhibition had minimal to no effect on arthritis development.
Methods
Mice
SphK2−/− mice [6] were obtained and bred with hTNF-α transgenic mice (Taconic Farms, Germantown, NY). No obvious phenotypes were noted in SphK2−/− mice. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at MUSC. Each mouse was screened for the presence or absence of the hTNF-α transgene and the presence or absence of modified SphK2, using PCR amplification of tail DNA. The primer sequences to detect the genotypes of interest and PCR parameters are previously published [17, 6].
Arthritis Scoring
Mice were observed by a blinded observer and assigned an arthritis score based on the degree of swelling and deviation present as previously described [17].
Gavage Study
After obtaining 10 hTNF/SphK+/+ mice at 3 months of age, the mice were divided into 2 groups of 5 and the mice were assigned as the “vehicle” group or the “treatment” group. The “treatment” group was gavaged with 100mL of 50mg/kg of ABC294640 in suspension with the vehicle composed of 0.375% polysorbate (tween)-80. Mice in the “vehicle” group received 100mL of 0.375% polysorbate (tween)-80 only. Both groups were gavaged twice daily for 12 weeks. Arthritis scores were measured by a blinded observer as previously published [17].
Tissue and Whole Blood sphingolipid levels
Ankle joints and whole blood were isolated from the mice in the gavage study at the time of sacrifice. Whole blood was collected in heparin-containing tubes to prevent clotting while the joint tissue was homogenized in SphK lysis buffer. After whole blood and tissue collection, samples (100 ml serum and 1 mg protein) were sent to the Medical University of South Carolina’s Lipidomics Core Facility, where advanced analyses were performed of sphingosine and ceramide species as previously described [18].
Sphingosine Kinase Activity Assay
Sphingosine kinase assay was followed as previously described [19–21]. Briefly, joint tissue of gavaged mice were collected and homogenized in SphK lysis buffer. The protein concentration of each homgineate was determined using BCA protein Assay kit (Pierce, Rockford, IL) for normalization of protein amount for each animal. Equal amounts of protein were added to labeled tubes corresponding to each mouse. D-erytho sphingosine was reconstituted with 100% ethanol and combined with fatty acid free BSA (Sigma-Aldrich St. Louis, MO) to make 1mM BSA-sphingosine solution. Radiolabeled ATP and sphingosine-BSA solution were added to each tube with joint the homogenates, incubated at 37°C and placed on TLC plates. The S1P bands were scraped and radioactivity was measured by the scintillation counter.
Results
We previously demonstrated a key role of SphK1 in hTNF-α induced arthritis [17], and extended these studies to investigate the effect of SphK2 in this model of inflammatory arthritis. All mice expressing hTNFα developed inflammatory arthritis regardless of SphK2 genotype. Mice that did not overexpress hTNF-α and were SphK2 deficient (SphK2−/−) did not develop inflammatory arthritis. As expected, mice that overexpress hTNFα and are SphK2-competent (hTNF/SphK2+/+) developed inflammatory arthritis as demonstrated by a significant increase of the arthritis score (Figure 1). The absence of SphK2 had no significant affect on the time of development of arthritis, overall arthritis progression or the percentage of mice developing arthritis. When comparing the arthritis scores of hTNF/SphK2+/+ vs. hTNF/SphK2−/−, a significantly lower, but clinically insignificant, arthritis score was noted only when the mice were 7 months old, while arthritis was not impacted at all at other time points.
Figure 1.
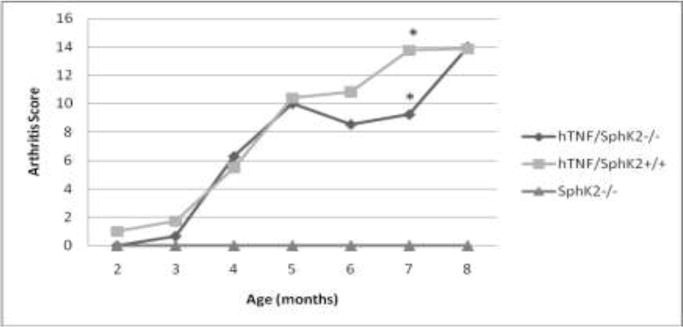
Arthritis Scores of mice varying in hTNFα and SphK2 genotypes. Significantly higher arthritis scores were noted in both hTNF/SphK2+/+ and hTNF/SphK2−/− mice vs. SphK2−/− mice. hTNF/SphK2−/− mice had similar arthritis scores in all months compared to hTNF/SphK2+/+ mice. A significant decrease in overall score was noted at 7 months in hTNF/SphK2−/− mice. *p< 0.001, by two-way ANOVA analysis with Bonferroni’s Multiple Comparison Test
To determine if pharmacologic SphK2 inhibition had a similar impact on inflammatory arthritis as genetic deficiency, we utilized a SphK2-pharmacologic inhibitor, ABC294840. Beginning at week one post initiation of treatment, mice that were receiving ABC294840 developed more severe arthritis compared to vehicle treated group, a trend that continued throughout the 12 weeks of treatment (Figure 2). Therefore, in contrast to genetic inhibition of SphK2, pharmacologic inhibition of SphK2 in hTNF-α transgenic mice led to a significantly more severe inflammatory arthritis phenotype. It is notable that the scores for the ABC294840 mice are comparable to the scores of the mice in the genetic intervention study in Figure 1. The vehicle treated mice had lower overall joint scores than any of the other groups. These studies were not done concurrently and the mice in the two experiments were derived independently. We cannot explain the lower scores in the vehicle group other than variation in disease in litters.
Figure 2. Arthritis score of mice treated with ABC294640 or vehicle.
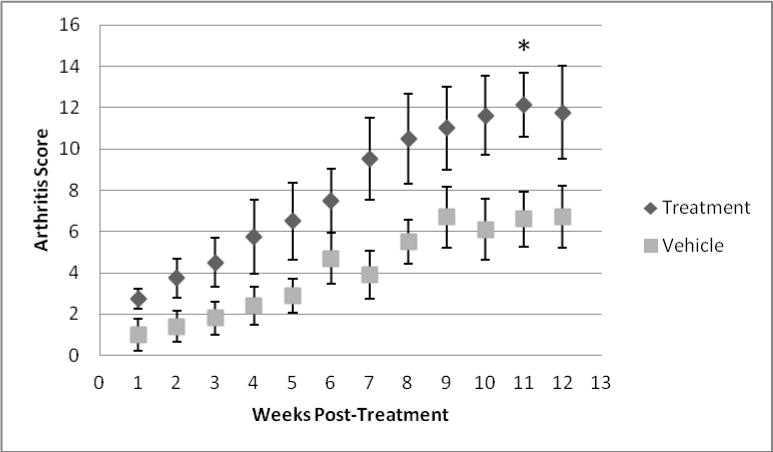
Vehicle treated mice had a significantly lower arthritis score compared to ABC294640 treated mice. * p < 0.05, by non-parametric t-Test
Since SphK2 produces S1P, we wanted to determine the impact of ABC29464 on SphK2’s functional capacity to form S1P after pharmacologic inhibition. The hTNF/SphK2+/+, ABC294640 treated mice had lower mean systemic levels of S1P as compared to hTNFα-vehicle treated mice (Figure 3A), however, the difference did not reach statistical significance due to wide variation in values within each group. Similarly, mean tissue levels of S1P were decreased in the treatment group compared to the control group (Figure 3B), yet the difference did not approach statistical significance. There was no correlation between measures of neither whole blood S1P nor tissue S1P and clinical measures of disease severity.
Figure 3a. S1P levels in whole blood samples from gavaged mice.
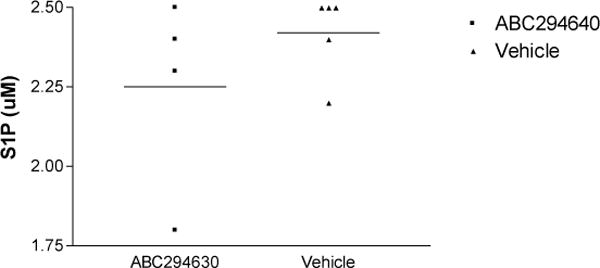
S1P was measured in mice treated with ABC294640 compared to vehicle-treated mice. p= 0.4127, by non-parametric t-Test.
Figure 3b. S1P levels in tissue samples.
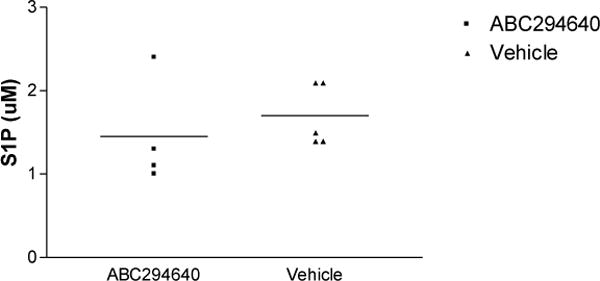
S1P was assessed in knee tissue in ABC294640-treated mice compared to vehicle treated mice. p= 0.4851, by non-parametric t-Test.
Finally, we directly determined the enzymatic activity of SphK1 and 2 in joint tissue specifically. As expected, no difference was noted in the enzymatic activity of SphK1 in the treatment group vs. the control group. SphK2 enzymatic activity could not be detected in either group and therefore could not be quantified (data not shown).
Discussion
Inhibition of SphK2 yields different outcomes in disease progression depending on the disease model in question. Our results revealed differences in severity of TNFα-induced inflammatory arthritis depending on genetic vs. pharmacologic inhibition SphK2. The overall lack of differences observed in genetic inhibition of SphK2 may be due to compensatory mechanisms that develop in the absence of SphK2 from birth. Genetic compensation was shown in SphK2 null mice that were able to activate S1P1 agonists, like FTY720, a known function of SphK2, in the absence of SphK2 [6]. These findings demonstrate that genetically-deficient SphK2 mice can compensate for a deficiency in SphK2. In experiments where SphK2 was acutely inhibited using siRNA or the introduction of SphK2-deficient cells into a cancer model, the mice developed a pro-inflammatory phenotype similar to our results in the ABC294640 treated mice.
Exceptions to the pro-inflammatory effect of SphK2 inhibition were shown when ABC294640 was used in models of IBD [15, 16] where decreased inflammation was seen. Similarly, in a CIA murine model and a rat model of inflammatory arthritis, ABC294640 was also shown to decrease disease expression, especially in conjunction with methotrexate (MTX), a disease modifying agent used in RA patients [22]. In our data, the dose used was doubled, based on additional PK testing in normal mice, which differs from previous experiments [16, 15]. However, we recognize that Fitzgerald et. al used similar doses in the rat arthritis model with positive results [22]. Yet, the addition of MTX may account for differences in arthritis severity noted. Another possibility is treatment with ABC294640 preferentially protects the colon from inflammatory assault despite little SphK2 mRNA is detected there [3].
Higher levels of S1P were found in whole blood vs. tissue samples as expected; whole blood acts as an S1P reservoir due to the absence of S1P degrading enzymes in RBCs [Rev. [2]]. Additionally, we expected no statistical difference in S1P levels in ABC294640 vs. vehicle-treated mice because SphK2 is not the major enzyme involved in S1P production [1, 6]. Finally, stable SphK1 enzymatic activity is logical since ABC294640 specific for SphK2. But, inability to detect SphK2 may be attributed to the decreased expression in joints. Most SphK2 RNA was detected in liver and kidney [3, 6] with little detection in skeletal muscle [3]. Even though joint tissue has not been directly tested, we can infer that the lack of detection in our assay is due to a lack of SphK2 in intrinsic joint cells.
SphK2 may affect other cellular targets due to its minimal affect on S1P production (approximately 25%) [6, 1]. While genetic inhibition of SphK1 affected the development of hTNF-α induced arthritis due to decreased S1P [17], inhibition of SphK2 affected cytokine production, producing a pro-inflammatory phenotype [4, 13, 12]. Even though we did not measure systemic cytokine levels in this model due to the localization of inflammation to the joints [17], we infer that inhibition of SphK2 with ABC294640 lead to a more potent arthritis due to increased inflammatory mediator production. Therefore, we hypothesize that inhibition of SphK2 may be more influential in its effects on the cytokine profile in vivo than S1P production.
SphK2 is interesting in that its modulation can affect disease processes depending on the etiology of the disease. Decreasing levels of SphK2 can be potentially therapeutic in situations where pro-inflammatory states are warranted (i.e. cancers that thrive in anti-inflammatory states). However, decreasing SphK2 may not be helpful in condition in which inflammation is exaggerated (auto-immune diseases). Thus, further investigation is needed to find the proper balance of SphK2 to best utilize its therapeutic potential. These findings are of further importance in emphasizing the need for use of specific SphK inhibitors rather than more promiscuous inhibitors as, at least in the hTNF model of inflammatory arthritis, the two SphKs have marked differences in their in vivo effects on disease. In summary, based on these and prior experiments, SphK1 plays an important role in the development of inflammatory arthritis in murine models of disease and may be a useful target for therapy. In contrast, SphK2, in most published results, appears to be anti-inflammatory or neutral in its effects, and thus, is a candidate for cancer therapy or enhancing immune responses, but its blockade should be avoided in autoimmune/autoinflammatory diseases.
References
- 1.Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. Molecular cloning and functional characterization of murine sphingosine kinase. J Biol Chem. 1998;273(37):23722–23728. doi: 10.1074/jbc.273.37.23722. [DOI] [PubMed] [Google Scholar]
- 2.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60(2):181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem. 2000;275(26):19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 4.Oskeritzian CA, Alvarez SE, Hait NC, Price MM, Milstien S, Spiegel S. Distinct roles of sphingosine kinases 1 and 2 in human mast-cell functions. Blood. 2008;111(8):4193–4200. doi: 10.1182/blood-2007-09-115451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zemann B, Kinzel B, Muller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood. 2006;107(4):1454–1458. doi: 10.1182/blood-2005-07-2628. [DOI] [PubMed] [Google Scholar]
- 6.Kharel Y, Lee S, Snyder AH, Sheasley-O’neill SL, Morris MA, Setiady Y, Zhu R, Zigler MA, Burcin TL, Ley K, Tung KS, Engelhard VH, Macdonald TL, Pearson-White S, Lynch KR. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005;280(44):36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- 7.Lan YY, De Creus A, Colvin BL, Abe M, Brinkmann V, Coates PT, Thomson AW. The sphingosine-1-phosphate receptor agonist FTY720 modulates dendritic cell trafficking in vivo. Am J Transplant. 2005;5(11):2649–2659. doi: 10.1111/j.1600-6143.2005.01085.x. [DOI] [PubMed] [Google Scholar]
- 8.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 9.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 10.Graeler M, Goetzl EJ. Activation-regulated expression and chemotactic function of sphingosine 1-phosphate receptors in mouse splenic T cells. Faseb J. 2002;16(14):1874–1878. doi: 10.1096/fj.02-0548com. [DOI] [PubMed] [Google Scholar]
- 11.Lai WQ, Irwan AW, Goh HH, Melendez AJ, McInnes IB, Leung BP. Distinct roles of sphingosine kinase 1 and 2 in murine collagen-induced arthritis. J Immunol. 2009;183(3):2097–2103. doi: 10.4049/jimmunol.0804376. [DOI] [PubMed] [Google Scholar]
- 12.Weigert A, Schiffmann S, Sekar D, Ley S, Menrad H, Werno C, Grosch S, Geisslinger G, Brune B. Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards an anti-inflammatory phenotype. Int J Cancer. 2009;125(9):2114–2121. doi: 10.1002/ijc.24594. [DOI] [PubMed] [Google Scholar]
- 13.Samy ET, Meyer CA, Caplazi P, Langrish CL, Lora JM, Bluethmann H, Peng SL. Cutting edge: Modulation of intestinal autoimmunity and IL-2 signaling by sphingosine kinase 2 independent of sphingosine 1-phosphate. J Immunol. 2007;179(9):5644–5648. doi: 10.4049/jimmunol.179.9.5644. [DOI] [PubMed] [Google Scholar]
- 14.French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 333(1):129–139. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chumanevich AA, Poudyal D, Cui X, Davis T, Wood PA, Smith CD, Hofseth LJ. Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis. 1787;31(10):1787–1793. doi: 10.1093/carcin/bgq158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maines LW, Fitzpatrick LR, French KJ, Zhuang Y, Xia Z, Keller SN, Upson JJ, Smith CD. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53(4):997–1012. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker DA, Barth J, Chang R, Obeid LM, Gilkeson GS. Genetic sphingosine kinase 1 deficiency significantly decreases synovial inflammation and joint erosions in murine TNF-alpha-induced arthritis. J Immunol. 2010;185(4):2570–2579. doi: 10.4049/jimmunol.1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39(2):82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, Obeid LM. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. Faseb J. 2009;23(1):143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson KR, Becker KP, Facchinetti MM, Hannun YA, Obeid LM. PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA) J Biol Chem. 2002;277(38):35257–35262. doi: 10.1074/jbc.M203033200. [DOI] [PubMed] [Google Scholar]
- 21.Olivera A, Kohama T, Tu Z, Milstien S, Spiegel S. Purification and characterization of rat kidney sphingosine kinase. J Biol Chem. 1998;273(20):12576–12583. doi: 10.1074/jbc.273.20.12576. [DOI] [PubMed] [Google Scholar]
- 22.Fitzpatrick LR, Green C, Frauenhoffer EE, French KJ, Zhuang Y, Maines LW, Upson JJ, Paul E, Donahue H, Mosher TJ, Smith CD. Attenuation of arthritis in rodents by a novel orally-available inhibitor of sphingosine kinase. Inflammopharmacology. 19(2):75–87. doi: 10.1007/s10787-010-0060-6. [DOI] [PubMed] [Google Scholar]