

Background: The small G protein Rap1 is phosphorylated within its carboxyl terminus by the cAMP-dependent protein kinase PKA.
Results: This phosphorylation removes Rap1 from the plasma membrane to limit Rap1 signaling.
Conclusion: Rap1 phosphorylation switches Rap1 off the membrane and terminates its activation.
Significance: Carboxyl-terminal phosphorylation may be common among small G proteins to regulate GTP/GDP cycling and downstream signaling.
Keywords: Cell Adhesion, Cell Migration, Low Molecular Weight G Proteins, Phosphorylation, Protein Kinase A (PKA), Rap1, cAMP, Electrostatic Switch, Geranylgeranylation
Abstract
The small G protein Rap1 can mediate “inside-out signaling” by recruiting effectors to the plasma membrane that signal to pathways involved in cell adhesion and cell migration. This action relies on the membrane association of Rap1, which is dictated by post-translational prenylation as well as by a stretch of basic residues within its carboxyl terminus. One feature of this stretch of acidic residues is that it lies adjacent to a functional phosphorylation site for the cAMP-dependent protein kinase PKA. This phosphorylation has two effects on Rap1 action. One, it decreases the level of Rap1 activity as measured by GTP loading and the coupling of Rap1 to RapL, a Rap1 effector that couples Rap1 GTP loading to integrin activation. Two, it destabilizes the membrane localization of Rap1, promoting its translocation into the cytoplasm. These two actions, decreased GTP loading and decreased membrane localization, are related, as the translocation of Rap1-GTP into the cytoplasm is associated with its increased GTP hydrolysis and inactivation. The consequences of this phosphorylation in Rap1-dependent cell adhesion and cell migration were also examined. Active Rap1 mutants that lack this phosphorylation site had a minimal effect on cell adhesion but strongly reduced cell migration, when compared with an active Rap1 mutant that retained the phosphorylation site. This suggests that optimal cell migration is associated with cycles of Rap1 activation, membrane egress, and inactivation, and requires the regulated phosphorylation of Rap1 by PKA.
Introduction
Rap1 is targeted to the plasma membrane via prenylation (geranylgeranylation) at its carboxyl-terminal CAAX motif, but this modification by itself is not sufficient to anchor proteins to the lipid bilayer (1, 2). For Rap1, and for other prenylated small G proteins such as K-Ras (3, 4) and RhoA (5), localization requires a second mechanism involving a stretch of positively charged basic amino acids that interact with negatively charged phospholipids within the membrane (1, 6). Interestingly, these polybasic regions are often interrupted by consensus sequences for protein phosphorylation (7–9). Phosphorylations at these sites neutralize the positive charges required for membrane association, and have the potential to destabilize membrane localization. These “electrostatic switches” occur within polybasic stretches adjacent to the prenylation sites within many prenylated proteins including the tyrosine kinase Src, via PKA (7, 8), and the small G protein K-Ras, via PKC (9). Here, we examine a potential electrostatic switch in the small G protein Rap1.
Rap1 regulates both cell adhesion and cell motility through its “inside-out” activation of integrins. Intuitively, for cell migration to occur Rap1 activation of integrins and cell adhesion must be reversible to allow for cell detachment. The mechanisms that underlie this reversible behavior are not known. We show here that PKA phosphorylation of Rap1-GTP destabilizes its association with the plasma membrane and promotes Rap1 inactivation. Using phosphorylation-deficient mutants, we demonstrate that Rap1 can potentiate cell migration only when this phosphorylation site is available for PKA-dependent phosphorylation. We propose that PKA phosphorylation provides a mechanism for cycling of Rap1 activation and inactivation during cell migration.
EXPERIMENTAL PROCEDURES
Plasmids
The cDNA of bovine Rap1b (encoding the same amino acid sequence as human Rap1b) was cloned into the vector pcDNA3 (Invitrogen) containing either an N-terminal GFP or FLAG epitope (pcDNA3-FLAG), as previously described (10, 11). Mutations encoding RapE63, and the corresponding PKA site mutations S179A/S180A (AA)3 or S180D were introduced by site-directed mutagenesis. The plasmid GFP-Rap1DK was generated by PCR amplification using a primer designed to introduce the sequence “KRKKRA” amino-terminal to the S180D mutation of GFP-Rap1D. FLAG-RapGAP1 was described previously (12). Human RapL (Nore1B, RassF3) was purchased form Origene Technologies (Rockville, MD) and inserted into a pcDNA3 vector containing the FLAG epitope (pcDNA3-FLAG). Complementary pairs of short hairpin RNA (shRNA) for Rap1a were described previously (10). Human Epac1 was gift from Johannes Bos, Utrecht University, and FLAG-Epac1 has been described previously (13).
Antibodies
Anti-Rap1 (SC65), anti-B-Raf (H145), anti-GFP (SC8334), and anti-Rap1a (SC1482) were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Anti-Rap1a/b, agarose-coupled anti-FLAG (M2), and anti-FLAG (M2) were from Sigma. Anti-GFP and anti-cadherin were from Abcam (Cambridge, MA). Anti-phospho-(Ser/Thr) PKA substrate antibody (RRXS*/T*) was from Cell Signaling Technology (Beverly, MA). Anti-mouse and anti-rabbit secondary antibodies for Western blots were from GE Healthcare. Cy3-conjugated secondary antibody for immunofluorescent microscopy was from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA).
Chemicals
Forskolin, 3-isobutyl-1-methylxanthine, 5-isoquinolinesulfonamide (H89), and PKA inhibitor (14-22 amide, cell permeable, myristoylated, or myr-PKI) were purchased from Calbiochem (Riverside, CA). 8-(4-Chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (OMe-cAMP) was purchased from Biolog Life Sciences (Bremen, Germany).
Cell Culture, Transfection, and Treatments
H1299 cells were provided by Mushui Dai (Oregon Health and Science University (OHSU)), Ovcar3 cells were provided by Grover C. Bagby (OHSU), MDM-MB-231 cells were provided by Rosalie R. Sears (OHSU), and RCC10 cells were provided by David Z. Qian (OHSU). HEK293, H1299, MDM-MB-231, and RCC10 were cultured in DMEM plus 10% fetal calf serum, penicillin-streptomycin, and l-glutamine at 37 °C and 5% CO2. SK-MEL-24 cells were cultured as previously described (11). Ovcar3 cells were cultured in RPMI 1640 plus 10% fetal calf serum supplemented with penicillin, streptomycin, and l-glutamine. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, and serum starved prior to treatment. Cells treated with 10 μm forskolin also received 100 μm 3-isobutyl-1-methylxanthine for 20 min, unless otherwise indicated. H89 was used at 10 μm and added 20 min prior to treatment. OMe-cAMP was used at 50 μm for 20 min. For the stable cell line, H1299 cells were transfected with FLAG-RapE63, RapE63AA, FLAG-Rap1WT, FLAG-RapAA, or pcDNA3 vector and selected with 0.5 mg/ml of G418 (Invitrogen) for 4 weeks.
Western Blotting
Immunoprecipitations and Western blotting were performed as previously described (11, 13). To detect a mobility shift of phosphorylated Rap1, proteins were separated on an 8% polyacrylamide gel to allow resolution of phosphorylated and non-phosphorylated bands. Rap1 activation assay was performed using GST-tagged Ras-binding domain of Ral-GDS as described previously (14). All experiments were performed at least three times and representative gels are shown.
Fractionation
HEK293, H1299, or HEK293 cells transiently transfected with the indicated cDNAs in 10-cm dishes were lysed and homogenized in hypotonic buffer (10 mm KCl, 1.5 mm MgCl2, 1 mm Na-EDTA, 1 mm DTT, 1 μm leupeptin, 10 μg/ml of soybean trypsin inhibitor, 0.1 μm aprotinin, 1 mm sodium orthovanadate, 1 mm β-glycerophosphate, and 10 mm Tris-HCl, pH 7.4). A small fraction was separated into aliquots as total cell lysate. Cell fractionation was performed as previously described (10). The endogenous cadherin proteins were examined as markers for the P (membranous) fractions, and endogenous B-Raf was examined as a marker for the S (cytosolic) fractions.
Immunofluorescent Staining
Staining was performed as previously described (11). Briefly, SK-Mel-24 cells were grown on fibronectin-coated coverslips and then serum starved overnight. After forskolin treatment, cells were fixed, permeablized, blocked, and then incubated with anti-Rap1 antibody overnight. After washing, cells were incubated with secondary antibody and wash.
Confocal Imaging and Quantification
For live cell imaging, cells were plated on to the coverslips coated with fibronectin (R&D Systems) and transfected and serum starved 4–6 h prior to imaging. All imaging experiments were performed using a confocal microscope (Zeiss LSM780) on coverslips clamped into a heated imaging chamber. The images were acquired with a ×63 oil immersion objective (N.A. 1.3), captured over the period of time indicated in the figure legends and processed with ZEN 2008 software (Carl Zeiss, Inc.). All images were analyzed using Image J software (NIH). The images are representative of at least 15 cells examined, in each of three separate experiments.
To quantify the change in intensity of GFP within the cytoplasm and the plasma membrane, we measured fluorescence intensities within two defined 0.72-μm2 areas, along the plasma membrane and at a fixed distance within the cytoplasm. Intensities within the plasma membrane and cytoplasm areas were measured at time 0. Intensities at the plasma membrane and cytoplasm at 10 and 30 min of forskolin treatment were also measured and the ratios of these intensities at each time point were calculated and normalized to the ratio at time 0. For each construct, 10–15 random cells were imaged before and after forskolin treatment in a single experiment, and experiments were independently repeated 3 times. The data were presented as the relative ratio of intensities within the cytoplasm to the plasma membrane.
Adhesion Assay
96-Well flat bottom plates were coated with fibronectin (1–5 μg/ml as indicated) followed by blocking with 3% bovine serum albumin/phosphate-buffered saline for 1 h at 37 °C. Stably transfected cells serum starved were overnight before the adhesion assay, and detached by trypsinization and resuspended in serum-free medium containing 10 mm HEPES, 0.5% BSA, rotated in suspension at 37 °C for 1 h and plated in quadruplicate at 25,000 cells/well. In studies with H89 (10 μm), cells were incubated at 37 °C for 45 min before plating wells. After incubating at 37 °C for the indicated times, non-adherent cells were removed and adherent cells were fixed and stained with 0.5% crystal violet. After extensive washing, the retained dye was eluted and the absorbance at 590 nm was read. The number of total adherent cells was determined at 2 h post-plating, and the percentage of adherent cells was plotted.
Migration Assay
Wound healing assays were performed using stably transfected cells plated onto fibronectin (1 μg/ml)-coated 24-well plates in duplicate or triplicate and serum starved overnight. After cells were grown as a monolayer, a wound was introduced by scratching with a 200-μl tip and washed, then replaced with 1% serum-containing medium. Migration across the wound was measured at 0 and 8 h.
For shRNA experiments, cells were transfected three times on consecutive days and serum starved overnight. shRNA expressing cells or stably transfected cells were grown as a monolayer, a wound was introduced. Cells were washed and incubated with 1% serum containing medium with or without treatment. Migration across the wound was monitored and measured using IncuCyte ZOOM (Essen Bioscience, Ann Arbor, MI).
Modified Boyden chamber assays were performed using stably transfected cells were serum starved overnight and then stained with Calcein AM for 1 h, then detached by trypsinization and resuspended in 5 (for Fig. 6C) or 1% (for Fig. 6D) serum-contained medium. A total of 5,000 cells were loaded into duplicate or triplicate wells of a 24-well BD FluoroBlockTM chamber (modified Boyden chamber), and allowed to migrate toward medium containing with or without treatment. After 24 h, cells migrating through inserts were imaged using an inverted fluorescence microscope and counted manually.
FIGURE 6.
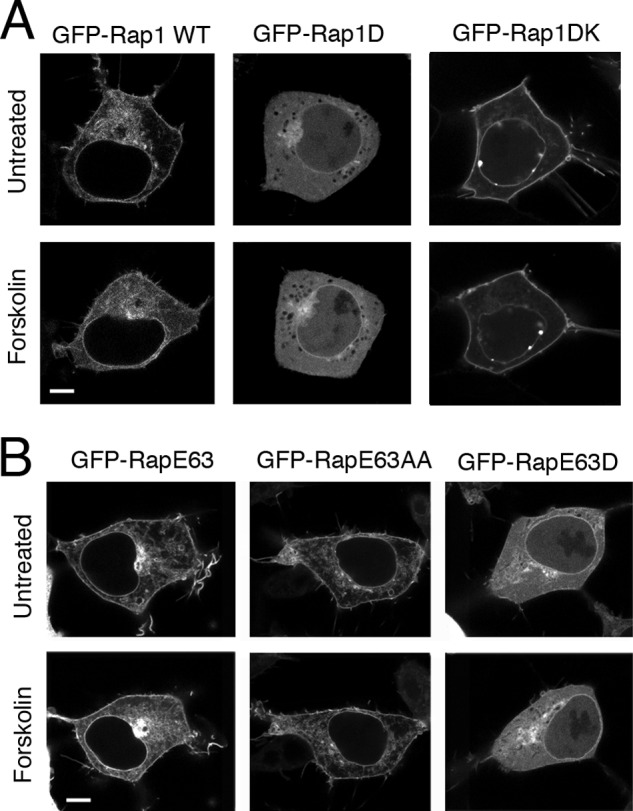
Localization of Rap1 mutants in cells. A, the introduction of basic amino acids restores plasma membrane relocalization of the Rap1D mutant. Micrographs of HEK293 cells transfected with GFP-tagged Rap1 constructs, GFP-Rap1WT, GFP-Rap1D, and GFP-Rap1DK, are shown. Images were taken before and 20 min after forskolin treatment. For each transfection, a representative cell is shown. The scale bar is 5 μm in length. B, localization of RapE63 is regulated by PKA phosphorylation. Shown here are micrographs of HEK293 cells transfected with the GFP-tagged RapE63 constructs, GFP-RapE63 WT, GFP-RapE63AA, and GFP-RapE63D. For each transfection, a representative cell is shown before and after forskolin treatment. The scale bar is 5 μm in length.
Statistics
All of the experiments were repeated at least three times. Prism version 3 (GraphPad Software, La Jolla, CA) was used for data plotting and analysis. Data were shown as mean ± S.E., and significant differences were analyzed using un-paired t test and Tukey's method.
RESULTS
Rap1 Is Phosphorylated on Serine 179/180 by PKA
The Rap family of small G proteins includes Rap1 and Rap2. Rap1 is well known for its role in cell adhesion and cell migration (15). Rap1 exists as two isoforms, Rap1a and Rap1b that are evolutionarily conserved, both being modified by a geranylgeranylation at their carboxyl-terminal cysteines. Lying adjacent to these cysteines are serine residues within consensus recognition sites for the cAMP-dependent protein kinase PKA (Fig. 1A). Rap1a has a single serine at this site, whereas Rap1b has two tandem serine residues (Fig. 1A). In 1991, phosphorylation of endogenous Rap1b by PKA was demonstrated in vitro and predicted to occur at one of two serines within the carboxyl terminus (16). Later it was shown that endogenous Rap1b was phosphorylated both in vitro and in vivo at serine 180 by PKA (17), but when this site was mutated to alanine (S180A), the adjacent Ser-179 became a PKA substrate (17). Therefore, to create a phosphorylation-deficient mutant, we used the double mutant Rap1 S179A/S180A (Rap1AA). Phosphorylation of Rap1 can be triggered by forskolin, a potent activator of adenylate cyclase and PKA. This phosphorylation was identified by an antibody recognizing phosphorylated PKA substrates (PKAS Ab), in both wild type Rap1 (Fig. 1B) and a constitutively activated mutant of Rap1, RapE63 (Fig. 1C). This phosphorylation did not occur in Rap1AA, as judged by the loss of detection by the PKAS Ab (Fig. 1B). Phosphorylation of endogenous Rap1 is complete by 20 min of forskolin treatment (Fig. 1D) and was inhibited by H89 (Fig. 1E). A similar time course of phosphorylation of wild type Rap1 was monitored by a mobility shift that is absent in the Rap1AA mutant (Fig. 1F).
FIGURE 1.

PKA phosphorylation of the carboxyl-terminal domain of Rap1. A, alignment of the carboxyl-terminal sequences of Rap1a, Rap1b (human, bovine, and mouse), and Rap1 (Drosophila melanogaster). The amino acid residue number is shown below. The conserved cysteines demarcating the sites of geranylgeranyl modification within the CAAX domains are indicated with the arrow. PKA consensus phosphorylation sites are shown in gray. B, phosphorylation of serine 179/180 in Rap1b. HEK293 cells were transfected with wild type FLAG-tagged Rap1b (Flag-Rap1WT) or FLAG-Rap1AA (mutated at the consensus PKA sites Ser-179 and Ser-180), and treated with forskolin (F) or left untreated (U). Forskolin induced the phosphorylation in FLAG-Rap1WT, but not FLAG-Rap1AA, as detected by the PKAS Ab (first panel). Levels of FLAG-Rap1 in the total cell lysate (TCL) are shown (second panel). C, phosphorylation of serine 179/180 in constitutively active Rap1E63 (RapE63). HEK293 cells were transfected with FLAG-tagged RapE63 (FLAG-RapE63) and treated with forskolin (F) or left untreated (U). Forskolin induced the phosphorylation in FLAG-RapE63, as detected by the PKAS Ab, and these actions were prevented by pretreament with H89 (FH) (first panel). Levels of FLAG-RapE63 within the immunoprecipitate (IP) and total cell lysate are shown in the second and third panels, respectively. D, the phosphorylation of endogenous Rap1 can be monitored by PKAS antibody. HEK293 cells were treated with forskolin for the times indicated. Cells were lysed and endogenous Rap1 was immunoprecipitated and assayed for PKA-dependent phosphorylation, detected using PKAS Ab (first panel). Phosphorylation can be seen at 15 min of forskolin and is maximal by 20 min. Rap1 levels within the total cell lysate are shown (second panel). E, the phosphorylation of endogenous Rap1 by forskolin is PKA-dependent. HEK293 cells were untreated (U), treated with forskolin alone (F), or in the presence of pretreatment with H89 (FH). Cells were lysed, endogenous Rap1 was immunoprecipitated and assayed for PKA-dependent phosphorylation, detected using PKAS Ab following Rap1 IP (first panel). Rap1 levels within the immunoprecipitate and total cell lysate are shown in the second and third panels, respectively. F, the phosphorylation of Rap1b by forskolin can be monitored by gel mobility shift. Cells were transfected with FLAG-Rap1b or FLAG-Rap1AA and treated with forskolin for the indicated times. The mobility of FLAG-Rap1b is shown by Western blot and can be seen as a gel shift that is complete by 20 min. This shift requires serines 179/180, as it is absent in FLAG-Rap1AA.
GTP Loading of Rap1 Is Decreased by PKA Phosphorylation
Rap1 can be activated by cAMP through the activation of the family of guanine nucleotide exchange proteins called Epacs (exchangers activated by cAMP) (18). This action of cAMP is independent of PKA. To determine how PKA phosphorylation could affect activation of Rap1 by cAMP, we examined Rap1 activation in HEK293 cells transfected with Epac1. In these cells, we stimulated Rap1 activation using either forskolin or the Epac agonist 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (OMe-cAMP) (19). To inhibit PKA, we utilized the PKA inhibitor H89. Rap1 activation was monitored biochemically using GST-Ral-GDS. We show that forskolin treatment induced a modest level of Rap1 activation that was further increased when PKA was inhibited by H89 (Figs. 2, A and B). The same high level of Rap1 activation was also achieved using OMe-cAMP, which activates Epac without activating PKA. Moreover, H89 did not further increase the activation of Rap1 induced by OMe-cAMP (Fig. 2A). Importantly, the enhancing effect of H89 was lost in the Rap1AA mutant (Fig. 2, A and B). Taken together, the data show that PKA reduces the level of activation of Rap1 achieved following Epac activation and demonstrate a direct role for PKA phosphorylation of Rap1 in this reduced activation. Consistent with this model, a phosphomimetic mutant containing an aspartate in place of Ser-180 (Rap1S180D or Rap1D) displayed very limited Rap1 activation by forskolin (Fig. 2B). This PKA-dependent inhibition of cAMP activation of endogenous Rap1 was seen in multiple cell lines (Fig. 2, C–E).
FIGURE 2.

The level of activated Rap1 is decreased by PKA phosphorylation. A, inhibition of PKA enhances Rap1 activity. HEK293 cells were transfected with wild type FLAG-Rap1 (Rap1WT) or FLAG-Rap1AA (Rap1AA), along with FLAG-Epac1, and left untreated (U) or treated with forskolin (F), OMe-cAMP (OMe), and H89, as indicated. Activity of FLAG-Rap1 (Flag-Rap1-GTP) was measured by a GST-Ral-GDS pull-down assay (Ral-GDS), followed by a FLAG Western blot (first panel). The expression levels of FLAG-Rap1 (WT or AA) and FLAG-Epac1 within the total cell lysates (TCL) are shown in the second and third panels, respectively. B, the level of Rap1 activity is decreased following by PKA-dependent phosphorylation. HEK293 cells were transfected with wild type FLAG-Rap1 (Rap1WT), FLAG-Rap1AA (Rap1AA), or FLAG-Rap1D (Rap1D), as indicated, along with FLAG-Epac1. Cells were treated with forskolin in the absence of H89 (F), in the presence of H89 pre-treatment (FH), or left untreated (U). Activity of FLAG-Rap1 (Flag-Rap1-GTP) was measured by Ral-GDS assay, followed by a FLAG Western blot (first panel). The expression levels of total FLAG-Rap1 (WT or mutants) and FLAG-Epac1 within the total cell lysates are shown in the second and third panels, respectively. The relative levels of Rap1 activation (Rap-GTP) averaged from four independent experiments are shown in the bar graph, n = 4 ± S.E. *, denotes statistical significance at <0.05. **, denotes statistical significance at <0.01. ns, denotes statistical significance at ≥0.05. C–E, inhibition of PKA enhances endogenous Rap1 activity in multiple cell lines. RCC10 (C), MDM-MB-231 (D), and Ovcar3 (E) cells were treated with H89 alone (H), forskolin in the absence of PKA inhibitors (F), in the presence of either H89 (FH) or myr-PKI (F/PKI), or left untreated (U). Cell lysates were examined for activated Rap1 (Rap-GTP) by a Ral-GDS assay followed by a Rap1 Western blot (first panel) and total Rap protein levels are shown in the second panel.
The PKA-dependent Decrease in the GTP Loading of Rap1 Reflects Increased Inactivation of Rap1
The ability of PKA phosphorylation of Rap1 to reduce Rap1 activation could be due to decreased Rap1 activation or increased Rap1 inactivation. These two possibilities were tested by comparing the time course of activation by forskolin of wild type Rap1 with Rap1AA. In Fig. 3A, we show that the time course of Rap1AA activation was more sustained than that of wild type Rap1, suggesting that PKA phosphorylation decreased the levels of Rap1 activity after it was already activated, possibly by increasing the rate of Rap1 inactivation.
FIGURE 3.
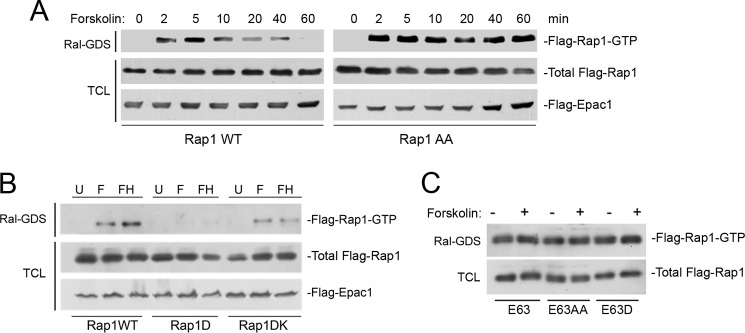
PKA enhances GAP-dependent inactivation of Rap1-GTP. A, PKA inhibits the duration of Rap1 activation by forskolin. HEK293 cells were transfected with FLAG-Epac1 and wild type FLAG-Rap1 (Rap1WT, left panels) or FLAG-Rap1AA (Rap1AA, right panels), and treated with forskolin for the times indicated. Activity of FLAG-Rap1 (Flag-Rap1-GTP) was measured by a Ral-GDS assay, followed by a FLAG Western blot (first panel). The levels of total FLAG-Rap1 (WT or AA) within the total cell lysates (TCL) are shown in the second panel. The levels of FLAG-Epac1 are also shown (third panel). B, the introduction of basic amino acids restores the activity of the phosphomimetic mutant Rap1D. HEK293 cells were transfected with wild type FLAG-Rap1 (Rap1WT), FLAG-Rap1D (Rap1D), or FLAG-Rap1DK (Rap1DK), as indicated, along with FLAG-Epac1. Cells were treated with forskolin alone (F) or following H89 pre-treatment (FH) or left untreated (U). Activity of FLAG-Rap1 (Flag-Rap1-GTP) was measured by a Ral-GDS assay, followed by a FLAG Western blot (first panel). The expression levels of FLAG-Rap1 (WT or mutants) and FLAG-Epac1 within the total cell lysates are shown in the second and third panels, respectively. C, PKA cannot inhibit the GTP loading of the GAP-insensitive mutant RapE63. HEK293 cells were transfected with FLAG-tagged Rap mutants (E63, E63AA, E63D), treated with forskolin (+) or left untreated (−), and Rap1 activity was measured by Ral-GDS assay followed by a FLAG Western blot (first panel). The levels of FLAG-Rap mutants E63, E63AA, and E63D within the total cell lysates are shown in the second panel.
The inability of Rap1D to be activated by forskolin (shown in Fig. 2B) was examined further. PKA phosphorylation of Rap1 introduces negative charges adjacent to the positive charges provided by the polybasic motif that is required for proper membrane attachment (Fig. 1A). Similarly, we reasoned that the introduction of a phosphomimetic aspartate (Rap1D) might also counteract the charges within this motif to destabilize membrane attachment. Because Rap1 activation usually occurs via membrane-targeted guanine nucleotide exchangers, destabilizing membrane attachment might also affect Rap1 activation. To test this, we introduced an additional polybasic motif between the aspartate mutation and the carboxyl cysteine residue to generate Rap1DK (described under “Experimental Procedures”). Rap1DK largely restored the deficit in activation seen in Rap1D (Fig. 3B). This suggests that the Rap1D mutant was inactive because of the electrostatic effects of the phosphomimetic mutant.
Rap1 is inactivated by the hydrolysis of GTP via Rap1-GAP (GTPase-activating protein). Mutants such as RapV12 and RapE63 are poor targets of Rap1-GAP and, therefore, are constitutively GTP-loaded (activated). To determine whether PKA inhibition of Rap1 required GTP hydrolysis, we examined the action of forskolin on RapE63. Forskolin was unable to affect the Rap1 activity of RapE63. Furthermore, two phosphorylation mutants also mutated at the Glu-63 site, RapE63AA, and RapE63D, showed similar levels of activation to RapE63, both basally and following forskolin treatment (Fig. 3C). These data suggest that the S180D mutation did not disrupt the constitutive GTP loading of Rap1 mutants. Taken together, PKA phosphorylation of Rap1 does not affect the intrinsic GTP loading of Rap1, but only lowers Rap1-GTP levels when Rap1 is a target for GTP hydrolysis.
This regulation of Rap1 activation by phosphorylation was also reflected in the binding of Rap1 to the Rap1 effector RapL (also called Nore1B or RassF3). RapL is a well characterized effector linking Rap1 to integrin activation (20) and motility (21). RapL binds Rap1 in a GTP-dependent manner where it forms a complex with the integrin α chain to alter integrin affinity and localization (22–24). Here, we show that the binding of Rap1 to RapL requires the GTP-loading state of Rap1, and is decreased by the PKA-dependent phosphorylation of Ser-180 (Fig. 4A). In Fig. 4B, we show that this binding is GTP-dependent. The GTP-loaded Rap1 constructs RapE63, RapE63AA, and RapE63D all bound RapL equally well and this binding was not regulated by treatment with forskolin.
FIGURE 4.
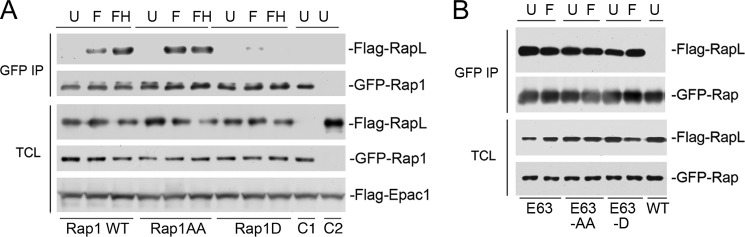
PKA effects binding of Rap1 to the Rap1 effector RapL. A, the binding of Rap1 to its effector RapL is decreased by PKA-dependent phosphorylation. HEK293 cells were transfected with FLAG-RapL and wild type GFP-Rap1 (Rap1WT), GFP-Rap1AA, or GFP-Rap1D, as indicated, along with FLAG-Epac1. Cells were treated with forskolin in the absence of H89 (F), in the presence of H89 pre-treatment (FH), or left untreated (U). RapL binding to GFP-Rap1 and mutants was shown by a FLAG Western blot following GFP immunoprecipitation (IP) (first panel). Levels of GFP-Rap WT and mutants in the immunoprecipitation are shown in the second panel. The levels of expression of FLAG-RapL (third panel), GFP-Rap1WT and mutants (fourth panel), and FLAG-Epac1 (fifth panel) in the total cell lysates (TCL) are shown. The last two lanes include cells transfected with FLAG-Epac1 and GFP-Rap1 (C1) or FLAG-Epac1 and FLAG-RapL (C2). B, the binding of constitutively activated Rap1 to its effector RapL is not affected by PKA-dependent phosphorylation. HEK293 cells were transfected with FLAG-RapL and either wild type GFP-Rap1 (WT) or mutants E63, E63AA, or E63D, as indicated. Cells were treated with forskolin (F) or left untreated (U). RapL binding to GFP-Rap1 mutants was determined by a FLAG Western blot following GFP IP (first panel). Levels of Rap1 mutants within the IP are shown in the second panel. The expression levels of FLAG-RapL (third panel) and RapE63 mutants (fourth panel) within the total cell lysates are shown. The last lane includes cells transfected with Rap1WT (WT) and left untreated (U), showing the GTP dependence of the interaction with RapL.
The Localization of Rap1 at the Plasma Membrane Is Regulated by PKA Phosphorylation
Small G proteins are generally only capable of being activated on the plasma membrane because their exchangers are activated on, or recruited to, the plasma membrane. Both FLAG-Rap1b and endogenous Rap1 are largely localized to the plasma membrane before forskolin treatment, and relocalize to the cytosol after treatment (Fig. 5, A and B, respectively). This movement of endogenous Rap1 is also seen in H1299 cells (a human non-small cell lung carcinoma cell line) (Fig. 5C). Translocation of Rap1 did not require Rap1 inactivation as forskolin-dependent translocation of the constitutively active mutant, RapE63, was also seen (Fig. 5D).
FIGURE 5.

Rap1 localization is regulated by PKA phosphorylation. A, Rap1 translocates from the membrane to the cytosol upon forskolin treatment, whereas Rap1-GAP is always cytosolic. HEK293 cells were transfected with FLAG-Rap1-GAP and FLAG-Rap1WT and were left untreated (−) or treated with forskolin (+), then fractionated into soluble (S, left) and particulate (P, right) fractions. FLAG-Rap1-GAP (first panel) and FLAG-Rap1 (second panel) were detected by a FLAG Western blot. B and C, forskolin induces the translocation of endogenous Rap1 from the plasma membrane and into the cytosol in HEK293 (B) and H1299 cells (C). Cells were treated with forskolin for 20 min, and lysates were separated into particulate (P) and soluble (S) fractions. The first and second panels show the levels of the cytoplasmic protein B-Raf and the membrane protein cadherin as markers for S and P fractions, respectively. The third panel shows the localization of endogenous Rap1 protein within the S and P fractions, as detected by a Rap1 Western blot. The bottom inset shows the levels of ERK2 (upper panel) and Rap1 (lower panel) within the total cell lysates (TCL) before and after forskolin treatment. D, forskolin-induced translocation of Rap1 from the plasma membrane to the cytosol requires Rap1 phosphorylation. Forskolin induces a relocalization of FLAG-RapE63 (E63), but not FLAG-RapE63AA (E63AA) to the soluble fraction. HEK293 cells were transfected with the indicated FLAG-tagged proteins, treated with forskolin for 20 min, and lysates were separated into soluble and particulate fractions. The first and second panels show the levels of the cytoplasmic protein B-Raf and the membrane protein Cadherin as markers for S and P fractions, respectively. The third panel shows the localization of FLAG-Rap mutants within S and P fractions, as detected by a FLAG Western blot. The bottom inset shows the levels of FLAG-Rap mutant proteins (upper panel) and ERK2 (lower panel) within the total cell lysates before and after forskolin treatment. E, immunofluorescence of SK-MEL-24 cells. Left, a representative untreated cell showing endogenous Rap1 at the plasma membrane. Right, a representative cell treated with forskolin (20 min). The upper and lower panels represent two optical sections through the same cells. The optical planes in the upper panels are 0.32 μm above those in the lower panels. The scale bar is 5 μm in length. F, time course of Rap1 relocalization by forskolin. HEK293 cells were transfected with GFP-Rap1WT (first panel) or GFP-RapAA (second panel). Cells were left untreated (Un) or treated with forskolin for 10 and 30 min, as indicated. The scale bar is 5 μm in length. G, quantification of Rap1 at the plasma membrane in real time. The fluorescence intensities of GFP-Rap1WT or GFP-RapAA at the plasma membrane and the cytosol at 0, 10, and 30 min after forskolin treatment were quantified as described under “Experimental Procedures.” The data are plotted as the ratio of cytoplasmic to plasma membrane intensities, normalized to that ratio calculated for 0 min, n = 3, ± S.E. ***, denotes statistical significance at <0.001.
Importantly, this relocalization required phosphorylation of Rap1 within its C terminus, as there was no relocation of the phosphorylation-deficient mutant RapE63AA (Fig. 5D). In contrast, Rap1 GTPase-activating protein (Rap1GAP) is localized in the cytosol and forskolin does not affect either its localization or levels (Fig. 5A). Therefore we suggest that PKA phosphorylation and subsequent translocation increases the proximity of Rap1 and Rap1GAP, thereby enhancing Rap1 inactivation.
The Removal of Rap1 from the Plasma Membrane Was Confirmed Microscopically
Forskolin treatment relocated endogenous Rap1 from the plasma membrane to the cytoplasm (Fig. 5E). Live cell images of HEK293 cells showed that wild type GFP-Rap1 and GFP-Rap1AA were predominantly at the membrane. Forskolin treatment of cells relocalized GFP-Rap1WT from the plasma membrane into the cytoplasm (Fig. 5, F and G). In contrast, forskolin did not induce the translocation of GFP-RapAA (Fig. 5F), suggesting that this relocalization required phosphorylation of Rap1 within its C terminus.
In contrast to wild type GFP-Rap1, GFP-Rap1D was largely cytoplasmic and remained cytoplasmic following forskolin treatment (Fig. 6A). However, Rap1DK restored the membrane localization (Fig. 6A). This demonstrates that the activation deficits seen in Rap1D (Fig. 3B) are due to the movement off the plasma membrane and can be reversed by neutralizing the acidic residue. The ability of PKA phosphorylation to relocate Rap1 to the cytosol can also be seen microscopically using the constitutively active mutant RapE63 (Fig. 6B). Similarly, RapE63AA and RapE63D were located on the plasma membrane and cytosol independently of forskolin treatment (Fig. 6B).
PKA Phosphorylation of Rap1 Is Not Required for Cell Adhesion
Constitutively GTP-loaded RapV12 and RapE63 both stimulate cell adhesion on a variety of extracellular matrices in a wide range of cell types (25–28). This was also true in H1299 cells stably expressing RapE63, when compared with cells expressing a control vector (Fig. 7A). Inhibition of PKA using H89 enhanced adhesion only in vector expressing cells (Fig. 7B).
FIGURE 7.
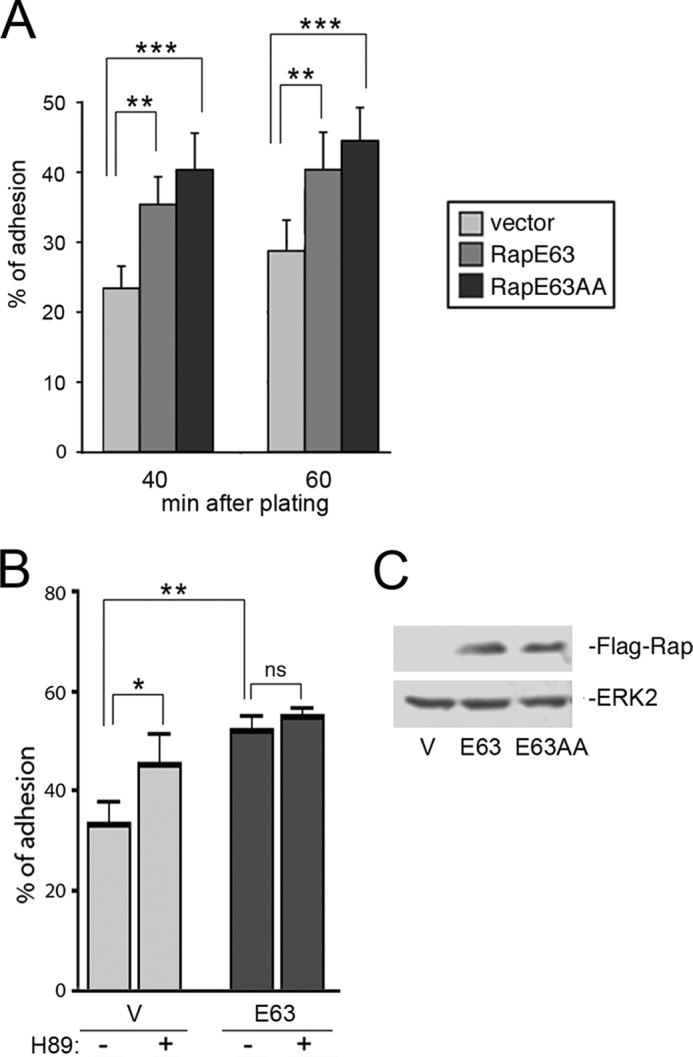
RapE63AA and RapE63 stimulate adhesion in H1299 cells. A, adhesion assay. Stably transfected H1299 cells expressing pcDNA3 vector (Vector) or FLAG-RapE63 (RapE63) or FLAG-RapE63AA (RapE63AA) were resuspended and plated in quadruplicate onto fibronectin (1 μg/ml)-coated wells and adherent cells were quantitated as described under “Experimental Procedures.” The values shown are the average of 4 independent experiments, ± S.E. **, denotes statistical significance at <0.01. ***, denotes statistical significance at <0.001. B, adhesion assay. Stably transfected H1299 cells expressing pcDNA3 vector (V) or FLAG-RapE63 (E63) were incubated with H89 40 min before being plated onto fibronectin (1.5 μg/ml)-coated wells and cells were allowed to adhere for 60 min. The percentage of adherent cells were quantitated as described under “Experimental Procedures.” The values shown are the average of 5 independent experiments, ± S.E. *, denotes statistical significance at <0.05. **, denotes statistical significance at <0.01. ns, denotes statistical significance at ≥0.05. C, stable cells lines. H1299 cells expressing pcDNA3 vector (V), FLAG-RapE63 (E63), or FLAG-RapE63AA (E63AA) were stably selected as described. The levels of transfected Rap proteins are shown in the first panel. The levels of ERK2 are shown in the second panel as a loading control.
We compared Rap1-dependent adhesion in H1299 cells stably expressing RapE63 with cells expressing equivalent levels of RapE63AA (Fig. 7C). Like RapE63, RapE63AA expressing cells showed enhanced adhesion compared with vector expressing cells (Fig. 7A), confirming that RapE63AA was functional and could activate integrin signaling. RapE63AA appeared to show enhanced adhesion compared with RapE63, although this was not statistically significant. These data suggest that PKA phosphorylation of Rap1 is not required for adhesion.
PKA Phosphorylation of Rap1 Is Required for Cell Migration
Rap1 is also required for integrin-dependent cell migration (21, 29–32). To address whether PKA phosphorylation of Rap1 would affect cell migration, we examined Rap1-dependent migration in two models: a wound healing assay (Fig. 8, A and B) and a modified Boyden chamber assay (Fig. 8, C and D). Initial experiments utilized RapE63 to eliminate the known effects of cAMP to regulate Rap1 through specific Rap1 exchangers (18, 33, 34). In both assays, transfection of RapE63 significantly increased migration compared with vector alone (Fig. 8, A and C). Unlike that seen for cell adhesion, these increases were blocked by H89 (Fig. 8B). Importantly, this increase was not seen in cells expressing RapE63AA (Fig. 8A). The expression of constitutively active mutants of Rap1 permits us to examine effects of forskolin that are independent of cAMP activation of Rap1. To examine Rap1 and mutants that retain the capacity for cAMP-dependent activation, we stably expressed wild type Rap1 and Rap1AA in H1299 cells (Fig. 8, D and E). As with the Glu-63 mutants, expression of wild type Rap1, but not Rap1AA, stimulated migration compared with vector expressing cells (Fig. 8D). However, in all cells, forskolin enhanced migration. In Fig. 8, F and G, we show that the depletion of Rap1a reduced cell migration. These data support a role for endogenous Rap1 in cell migration. The inhibition was statistically significant but modest because of the continued presence of Rap1b. Taken together, these results are consistent with a model that reversible phosphorylation of Rap1 is necessary for migration.
FIGURE 8.

Cell migration assays show Rap1-dependent migration. A, wound healing assay. Stably transfected H1299 cells expressing pcDNA3 vector (V), FLAG-RapE63 (E63), or FLAG-RapE63AA (E63AA) were examined by a wound healing assay as described under “Experimental Procedures.” The bar graph shows the percentage of wound closure across the scratch at 8 h. The values shown are the average of 5 independent experiments, ± S.E. **, denotes statistical significance at <0.01. B, wound healing assay. Stably transfected H1299 cells expressing pcDNA vector (V) or FLAG-RapE63 (E63) were prepared for a wound healing assay as described under “Experimental Procedures.” Then cells were incubated in 1% serum with (+) or without (−) H89. Cell migration across the gap was monitored and analyzed after 8 h. The bar graph shows the percentage of wound closure. n = 3, ± S.E. *, denotes statistical significance at <0.05. ***, denotes statistical significance at <0.001. C, modified Boyden chamber assay. Stably transfected H1299 cells expressing pcDNA vector (Vector), FLAG-RapE63 (E63), or FLAG-RapE63AA (E63AA) were loaded into duplicate wells of a 24-well BD FluoroBlockTM plates (modified Boyden chamber) and migrating cells were quantitated as described under “Experimental Procedures.” n = 4, ± S.E. *, denotes statistical significance at <0.05. **, denotes statistical significance at <0.01. D, modified Boyden chamber assay. Stably transfected H1299 cells expressing pcDNA vector (V), FLAG-Rap1WT (WT), or FLAG-RapAA (AA) were serum staved overnight and loaded into duplicate wells, then allowed to migrate into 1% serum contain medium with (+) or without (−) forskolin (10 μm)/3-isobutyl-1-methylxanthine (100 μm) of a 24-well BD FluoroBlockTM plates (modified Boyden chamber). Migrating cells were quantitated as described under “Experimental Procedures.” n = 3, ± S.E. **, denotes statistical significance at <0.01. ***, denotes statistical significance at <0.001. E, stable cells lines used in S180D. H1299 cells expressing pcDNA3 vector (V), FLAG-Rap1 (WT), or FLAG-RapAA (AA) were stably selected as described. The levels of transfected Rap proteins are shown in the first panel. The levels of ERK2 are shown in the second panel as a loading control. F, wound healing assay. H1299 cells expressing either pcDNA3 vector (V), scrambled (scr), or Rap1a shRNA (sh-Rap1a) were examined by a wound healing assay as described under “Experimental Procedures.” The bar graph shows the percentage of wound confluence at 6 h. n = 4, ± S.E. *, denotes statistical significance at <0.05. G, stable cells lines were as described in F. The efficiencies of knockdown of endogenous Rap1a by pcDNA3 vector (V), scrambled (scr), or Rap1a shRNA (sh-Rap1a) are shown in the first panel and total endogenous ERK is shown as a loading control in the second panel.
DISCUSSION
The ability of a cell to migrate requires the coordination of two cellular processes: cell adherence and cell motility. Cell adherence is governed by a family of transmembrane proteins called integrins, whose activation is tightly controlled by the small G protein Rap1 (15, 35, 36). Rap1 signaling to integrins requires the recruitment of one of a number of specific Rap1 effectors including RapL, RIAM (Rap1-GTP-interacting adaptor molecule), or the paralogue of RIAM, Lamellipodin (37, 38). Bringing these effectors to the plasma membrane is essential for the binding of talin to the cytoplasmic tail of integrin. This is the final step in the process of “inside-out signaling” that governs integrin activation (38, 39). However, the regulated turnover of active and inactive Rap1 between the compartments of plasma membrane and cytoplasm is not well characterized. Here we show that this dynamic process is controlled by PKA-dependent phosphorylation of Rap1.
cAMP and PKA both regulate Rap1 signaling in multiple ways. For example, PKA can inhibit Rap1-dependent angiogenesis, acting downstream of Rap1 (40). cAMP-dependent activation of Epac can regulate Rap1-dependent adhesion and migration (27, 41–44). PKA and other kinases can decrease the signaling actions of Epac by regulating the availability of cAMP through phosphodiesterases (45–48). Here we examine an additional level of PKA regulation by direct phosphorylation of Rap1 itself.
Using biochemical and microscopic methods, we show that phosphorylation of a consensus PKA site within the polybasic region at the carboxyl terminus releases Rap1 from the plasma membrane. This was supported by experiments using a phosphomimetic mutant of Rap1 (Rap1D) and a phosphorylation-deficient Rap1 mutant (Rap1AA). Rap1AA was localized on the plasma membrane, whereas Rap1D was largely localized within the cytoplasm. Forskolin had no effect on the location of either mutant. Importantly, neutralizing the acidic charge introduced by the phosphomimetic mutation restored the plasma membrane location of the protein.
Previous studies have proposed that Rap1 phosphorylation promotes Rap1 degradation (49). However, in our hands, Rap1 protein levels were not altered by phosphorylation. Other studies have proposed that phosphorylation of Rap1 regulates the level of interaction of effectors with GTP-loaded (activated) Rap1 (50, 51). However, when we examine binding of RapL to GTP-loaded Rap1, we see no additional effect of phosphorylation at Ser-180. Indeed, the levels of Rap1 activation in the wild type or phosphomimetic mutants Rap1D and Rap1AA measured in vitro correlated with their binding to the effector RapL in vivo, as measured by co-immunoprecipitation (Fig. 4, A and B). Moreover, when variants of the constitutively active mutant RapE63 were examined, all phosphorylation site mutants showed identical GTP loading and binding to RapL (Fig. 4B), demonstrating that phosphorylation of Rap1 did not affect the affinity of Rap1 for RapL independently of its effect on GTP loading. Therefore, we propose a simpler model that phosphorylation of Rap1 may induce changes in the activation of membrane-bound effectors by removing Rap1 from effector pathways normally triggered at the plasma membrane, such as RapL-dependent integrin activation.
We show that phosphorylation of Rap1 decreased the levels of Rap1-GTP. We propose that this is due to an increase in hydrolysis of GTP rather than decrease in GTP loading for the following reasons. One, phosphorylation of Rap1 occurs after Rap1 activation. Two, the consequence of Rap1 phosphorylation is a reduction in the duration of Rap1 activation rather than in its magnitude of activation. Three, the inhibitory effects of the phosphorylation of PKA on Rap1 GTP levels are not seen when using the RapE63 mutant. Because RapE63 is no longer inhibited by Rap-GAPs, this suggests that GTP hydrolysis is required for the inhibitory effects of PKA on Rap1. The only Rap-specific GAP that is expressed in HEK293 cells is Rap1GAP (52, 53), and it is entirely cytoplasmic. It is likely the release of Rap1 from the plasma membrane into the cytoplasm promotes its inactivation by increasing its chances of encountering the cytoplasmic pool of Rap1GAP. We propose that Rap1D is deficient in its activation because it is localized within the cytoplasm. This model is supported by the finding that the Rap1DK mutant restores both its activation deficit and plasma membrane localization.
Because cell migration requires cycles of Rap1 activation and inactivation, we predict that the presence of an electrostatic switch may enhance this cycling to promote integrin-dependent cell migration. We propose that both activation and inactivation of Rap1 participates in integrin-dependent cellular movements and that this occurs through cycles of PKA-dependent phosphorylation of Rap1.
Cell migration requires the reversibility of integrin-dependent contacts (54). We propose that it also depends on the reversibility of Rap1-dependent integrin activation. Here, we use phosphorylation site mutants to examine the role of phosphorylation of Rap1 in cell adhesion and cell migration. We show that eliminating the electrostatic switch with phosphorylation-deficient mutants of Rap1 blocked cell migration but did not block cell adhesion. This suggests that local cycling of Rap1 on and off the membrane is important for migration, but not for adhesion. Indeed, studies with the phosphomimetic RapV12D mutant suggest that locking this site into a permanent phosphorylated state may also not be optimal for migration (data not shown). We propose that the ability of Rap1 to be phosphorylated by PKA is essential for the reversibility of integrin-dependent adhesion that is required during cell migration. This reversibility may underscore other motile processes as well, such as axonal pathfinding, lymphocyte extravasation, and tumor cell metastasis (55). For example, preventing either the activation or inactivation of Rap1 limits its ability to promote metastasis of cancer cells into the lungs (55).
The inability of phosphorylation-deficient mutants of Rap1 to promote migration suggests that Rap1 was phosphorylated by PKA at some stage during migration. Because we examined cell migration in the absence of any extrinsic stimulators of PKA, we postulate that the stimulation of PKA activity initiated from intrinsic cues. One intrinsic trigger of PKA activation is integrin activation itself. cAMP/PKA activity is generated locally by integrin clustering and activation (56, 57) at the leading edge of the migrating cell (58). Importantly, cycles of PKA activation and PKA inactivation generation at the leading edge are required for cell migration (59–63). These cycles of PKA activation have been shown to regulate Rac1 and RhoA (60, 63). We propose that these cycles of PKA activation also regulate Rap1 signaling to provide cyclical regulation of integrins as well.
Integrin-dependent fluctuations in PKA activity are thought to act as pacemakers for RhoA inactivation during cell migration (60, 63). By regulating Rap1 as well, PKA can coordinate the actions of RhoA and Rap1 to orchestrate cell motility and cell adhesion during cell migration. This has broad significance for all processes that rely on the coupling of cell adhesion and cell migration including development, immune function, and cancer.
Acknowledgment
We acknowledge the technical and scientific assistance of Stefanie Kaech, the Advanced Light Microscopy Core, OHSU.
This work was supported, in whole or in part, by National Institutes of Health Grant DK090309.
- AA
- S179A/S180A
- HEK293
- human embryonic kidney 293
- GAP
- GTPase activating protein
- OMe
- OMe-cAMP, 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate
- PKA
- cAMP dependent protein kinase
- PKI
- protein kinase inhibitor (of PKA)
- Epac
- exchangers activated by cAMP
- GDS
- guanine nucleotide dissociation stimulator.
REFERENCES
- 1. Heo W. D., Inoue T., Park W. S., Kim M. L., Park B. O., Wandless T. J., Meyer T. (2006) PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McLaughlin S., Aderem A. (1995) The myristoyl-electrostatic switch. A modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20, 272–276 [DOI] [PubMed] [Google Scholar]
- 3. Roy M. O., Leventis R., Silvius J. R. (2000) Mutational and biochemical analysis of plasma membrane targeting mediated by the farnesylated, polybasic carboxy terminus of K-ras4B. Biochemistry 39, 8298–8307 [DOI] [PubMed] [Google Scholar]
- 4. Silvius J. R., Bhagatji P., Leventis R., Terrone D. (2006) K-ras4B and prenylated proteins lacking “second signals” associate dynamically with cellular membranes. Mol. Biol. Cell 17, 192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forget M. A., Desrosiers R. R., Gingras D., Béliveau R. (2002) Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem. J. 361, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ghomashchi F., Zhang X., Liu L., Gelb M. H. (1995) Binding of prenylated and polybasic peptides to membranes. Affinities and intervesicle exchange. Biochemistry 34, 11910–11918 [DOI] [PubMed] [Google Scholar]
- 7. Murray D., Ben-Tal N., Honig B., McLaughlin S. (1997) Electrostatic interaction of myristoylated proteins with membranes. Simple physics, complicated biology. Structure 5, 985–989 [DOI] [PubMed] [Google Scholar]
- 8. Schmitt J. M., Stork P. J. (2002) PKA phosphorylation of Src mediates cAMP's inhibition of cell growth via Rap1. Mol. Cell 9, 85–94 [DOI] [PubMed] [Google Scholar]
- 9. Bivona T. G., Quatela S. E., Bodemann B. O., Ahearn I. M., Soskis M. J., Mor A., Miura J., Wiener H. H., Wright L., Saba S. G., Yim D., Fein A., Pérez de Castro I., Li C., Thompson C. B., Cox A. D., Philips M. R. (2006) PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 21, 481–493 [DOI] [PubMed] [Google Scholar]
- 10. Liu C., Takahashi M., Li Y., Song S., Dillon T. J., Shinde U., Stork P. J. (2008) Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Mol. Cell. Biol. 28, 7109–7125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu C., Takahashi M., Li Y., Dillon T. J., Kaech S., Stork P. J. (2010) The interaction of Epac1 and Ran promotes Rap1 activation at the nuclear envelope. Mol. Cell. Biol. 30, 3956–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carey K. D., Dillon T. J., Schmitt J. M., Baird A. M., Holdorf A. D., Straus D. B., Shaw A. S., Stork P. J. (2000) CD28 and the tyrosine kinase lck stimulate mitogen-activated protein kinase activity in T cells via inhibition of the small G protein Rap1. Mol. Cell. Biol. 20, 8409–8419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z., Dillon T. J., Pokala V., Mishra S., Labudda K., Hunter B., Stork P. J. (2006) Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol. Cell. Biol. 26, 2130–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franke B., Akkerman J. W., Bos J. L. (1997) Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 16, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bos J. L. (2005) Linking Rap to cell adhesion. Curr. Opin. Cell Biol. 17, 123–128 [DOI] [PubMed] [Google Scholar]
- 16. Fischer T. H., Collins J. H., Gatling M. N., White G. C. (1991) The localization of the cAMP-dependent protein kinase phosphorylation site in the platelet rat protein, rap 1B. FEBS Lett. 283, 173–176 [DOI] [PubMed] [Google Scholar]
- 17. Altschuler D., Lapetina E. G. (1993) Mutational analysis of the cAMP-dependent protein kinase-mediated phosphorylation site of Rap1b. J. Biol. Chem. 268, 7527–7531 [PubMed] [Google Scholar]
- 18. Bos J. L. (2006) Epac proteins. Multipurpose cAMP targets. Trends Biochem. Sci. 31, 680–686 [DOI] [PubMed] [Google Scholar]
- 19. Holz G. G., Chepurny O. G., Schwede F. (2008) Epac-selective cAMP analogs. New tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell. Signal. 20, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Price L. S., Bos J. L. (2004) RAPL. Taking the Rap in immunity. Nat. Immunol. 5, 1007–1008 [DOI] [PubMed] [Google Scholar]
- 21. Raab M., Wang H., Lu Y., Smith X., Wu Z., Strebhardt K., Ladbury J. E., Rudd C. E. (2010) T cell receptor “inside-out” pathway via signaling module SKAP1-RapL regulates T cell motility and interactions in lymph nodes. Immunity 32, 541–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dustin M. L., Bivona T. G., Philips M. R. (2004) Membranes as messengers in T cell adhesion signaling. Nat. Immunol. 5, 363–372 [DOI] [PubMed] [Google Scholar]
- 23. Fujita H., Fukuhara S., Sakurai A., Yamagishi A., Kamioka Y., Nakaoka Y., Masuda M., Mochizuki N. (2005) Local activation of Rap1 contributes to directional vascular endothelial cell migration accompanied by extension of microtubules on which RAPL, a Rap1-associating molecule, localizes. J. Biol. Chem. 280, 5022–5031 [DOI] [PubMed] [Google Scholar]
- 24. Raab M., Smith X., Matthess Y., Strebhardt K., Rudd C. E. (2011) SKAP1 protein PH domain determines RapL membrane localization and Rap1 protein complex formation for T cell receptor (TCR) activation of LFA-1. J. Biol. Chem. 286, 29663–29670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reedquist K. A., Ross E., Koop E. A., Wolthuis R. M., Zwartkruis F. J., van Kooyk Y., Salmon M., Buckley C. D., Bos J. L. (2000) The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J. Cell Biol. 148, 1151–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Z., Rehmann H., Price L. S., Riedl J., Bos J. L. (2005) AF6 negatively regulates Rap1-induced cell adhesion. J. Biol. Chem. 280, 33200–33205 [DOI] [PubMed] [Google Scholar]
- 27. Enserink J. M., Price L. S., Methi T., Mahic M., Sonnenberg A., Bos J. L., Taskén K. (2004) The cAMP-Epac-Rap1 pathway regulates cell spreading and cell adhesion to laminin-5 through the α3β1 integrin but not the α6β4 integrin. J. Biol. Chem. 279, 44889–44896 [DOI] [PubMed] [Google Scholar]
- 28. Smolen G. A., Schott B. J., Stewart R. A., Diederichs S., Muir B., Provencher H. L., Look A. T., Sgroi D. C., Peterson R. T., Haber D. A. (2007) A Rap GTPase interactor, RADIL, mediates migration of neural crest precursors. Genes Dev. 21, 2131–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kinashi T., Katagiri K. (2004) Regulation of lymphocyte adhesion and migration by the small GTPase Rap1 and its effector molecule, RAPL. Immunol. Lett. 93, 1–5 [DOI] [PubMed] [Google Scholar]
- 30. Almahariq M., Tsalkova T., Mei F. C., Chen H., Zhou J., Sastry S. K., Schwede F., Cheng X. (2013) A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 83, 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hernández-Varas P., Coló G. P., Bartolomé R. A., Paterson A., Medraño-Fernández I., Arellano-Sánchez N., Cabañas C., Sánchez-Mateos P., Lafuente E. M., Boussiotis V. A., Strömblad S., Teixidó J. (2011) Rap1-GTP-interacting adaptor molecule (RIAM) protein controls invasion and growth of melanoma cells. J. Biol. Chem. 286, 18492–18504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He Y., Kapoor A., Cook S., Liu S., Xiang Y., Rao C. V., Kenis P. J., Wang F. (2011) The non-receptor tyrosine kinase Lyn controls neutrophil adhesion by recruiting the CrkL-C3G complex and activating Rap1 at the leading edge. J. Cell Sci. 124, 2153–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emery A. C., Eiden M. V., Mustafa T., Eiden L. E. (2013) Rapgef2 connects GPCR-mediated cAMP signals to ERK activation in neuronal and endocrine cells. Sci. Signal. 6, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vossler M. R., Yao H., York R. D., Pan M. G., Rim C. S., Stork P. J. (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89, 73–82 [DOI] [PubMed] [Google Scholar]
- 35. Bos J. L., de Rooij J., Reedquist K. A. (2001) Rap1 signalling. Adhering to new models. Nat. Rev. Mol. Cell Biol. 2, 369–377 [DOI] [PubMed] [Google Scholar]
- 36. Bos J. L., de Bruyn K., Enserink J., Kuiperij B., Rangarajan S., Rehmann H., Riedl J., de Rooij J., van Mansfeld F., Zwartkruis F. (2003) The role of Rap1 in integrin-mediated cell adhesion. Biochem. Soc. Trans. 31, 83–86 [DOI] [PubMed] [Google Scholar]
- 37. Lafuente E. M., van Puijenbroek A. A., Krause M., Carman C. V., Freeman G. J., Berezovskaya A., Constantine E., Springer T. A., Gertler F. B., Boussiotis V. A. (2004) RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev. Cell 7, 585–595 [DOI] [PubMed] [Google Scholar]
- 38. Mor A., Dustin M. L., Philips M. R. (2007) Small GTPases and LFA-1 reciprocally modulate adhesion and signaling. Immunol. Rev. 218, 114–125 [DOI] [PubMed] [Google Scholar]
- 39. Boettner B., Van Aelst L. (2009) Control of cell adhesion dynamics by Rap1 signaling. Curr. Opin. Cell Biol. 21, 684–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Menon J., Doebele R. C., Gomes S., Bevilacqua E., Reindl K. M., Rosner M. R. (2012) A novel interplay between Rap1 and PKA regulates induction of angiogenesis in prostate cancer. PLoS One 7, e49893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Breckler M., Berthouze M., Laurent A. C., Crozatier B., Morel E., Lezoualc'h F. (2011) Rap-linked cAMP signaling Epac proteins. Compartmentation, functioning and disease implications. Cell Signal 23, 1257–1266 [DOI] [PubMed] [Google Scholar]
- 42. Grandoch M., Rose A., ter Braak M., Jendrossek V., Rübben H., Fischer J. W., Schmidt M., Weber A. A. (2009) Epac inhibits migration and proliferation of human prostate carcinoma cells. Br. J. Cancer 101, 2038–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rangarajan S., Enserink J. M., Kuiperij H. B., de Rooij J., Price L. S., Schwede F., Bos J. L. (2003) Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the β2-adrenergic receptor. J. Cell Biol. 160, 487–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ross S. H., Post A., Raaijmakers J. H., Verlaan I., Gloerich M., Bos J. L. (2011) Ezrin is required for efficient Rap1-induced cell spreading. J. Cell Sci. 124, 1808–1818 [DOI] [PubMed] [Google Scholar]
- 45. Baillie G. S., Scott J. D., Houslay M. D. (2005) Compartmentalisation of phosphodiesterases and protein kinase A. Opposites attract. FEBS Lett. 579, 3264–3270 [DOI] [PubMed] [Google Scholar]
- 46. Houslay M. D. (2010) Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 35, 91–100 [DOI] [PubMed] [Google Scholar]
- 47. Raymond D. R., Wilson L. S., Carter R. L., Maurice D. H. (2007) Numerous distinct PKA-, or EPAC-based, signalling complexes allow selective phosphodiesterase 3 and phosphodiesterase 4 coordination of cell adhesion. Cell. Signal. 19, 2507–2518 [DOI] [PubMed] [Google Scholar]
- 48. Serrels B., Sandilands E., Frame M. C. (2011) Signaling of the direction-sensing FAK/RACK1/PDE4D5 complex to the small GTPase Rap1. Small GTPases 2, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rundell C. J., Repellin C. E., Yarwood S. J. (2004) Protease inhibitors prevent the protein kinase A-dependent loss of Rap1 GTPase from the particulate fraction of COS1 cells. Biochem. Biophys. Res. Commun. 315, 1077–1081 [DOI] [PubMed] [Google Scholar]
- 50. Edreira M. M., Li S., Hochbaum D., Wong S., Gorfe A. A., Ribeiro-Neto F., Woods V. L., Jr., Altschuler D. L. (2009) Phosphorylation-induced conformational changes in Rap1b. Allosteric effects on switch domains and effector loop. J. Biol. Chem. 284, 27480–27486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu C. D., Kariya K., Okada T., Qi X., Song C., Kataoka T. (1999) Effect of phosphorylation on activities of Rap1A to interact with Raf-1 and to suppress Ras-dependent Raf-1 activation. J. Biol. Chem. 274, 48–51 [DOI] [PubMed] [Google Scholar]
- 52. Mochizuki N., Ohba Y., Kiyokawa E., Kurata T., Murakami T., Ozaki T., Kitabatake A., Nagashima K., Matsuda M. (1999) Activation of the ERK/MAPK pathway by an isoform of Rap1GAP associated with Gαi. Nature 400, 891–894 [DOI] [PubMed] [Google Scholar]
- 53. Polakis P. G., Rubinfeld B., Evans T., McCormick F. (1991) Purification of a plasma membrane-associated GTPase-activating protein specific for rap1/Krev-1 from HL60 cells. Proc. Natl. Acad. Sci. U.S.A. 88, 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Broussard J. A., Webb D. J., Kaverina I. (2008) Asymmetric focal adhesion disassembly in motile cells. Curr. Opin. Cell Biol. 20, 85–90 [DOI] [PubMed] [Google Scholar]
- 55. Freeman S. A., McLeod S. J., Dukowski J., Austin P., Lee C. C., Millen-Martin B., Kubes P., McCafferty D. M., Gold M. R., Roskelley C. D. (2010) Preventing the activation or cycling of the Rap1 GTPase alters adhesion and cytoskeletal dynamics and blocks metastatic melanoma cell extravasation into the lungs. Cancer Res. 70, 4590–4601 [DOI] [PubMed] [Google Scholar]
- 56. Lim C. J., Kain K. H., Tkachenko E., Goldfinger L. E., Gutierrez E., Allen M. D., Groisman A., Zhang J., Ginsberg M. H. (2008) Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol. Biol. Cell 19, 4930–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. O'Connor K. L., Shaw L. M., Mercurio A. M. (1998) Release of cAMP gating by the α6β4 integrin stimulates lamellae formation and the chemotactic migration of invasive carcinoma cells. J. Cell Biol. 143, 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Paulucci-Holthauzen A. A., Vergara L. A., Bellot L. J., Canton D., Scott J. D., O'Connor K. L. (2009) Spatial distribution of protein kinase A activity during cell migration is mediated by A-kinase anchoring protein AKAP Lbc. J. Biol. Chem. 284, 5956–5967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McKenzie A. J., Campbell S. L., Howe A. K. (2011) Protein kinase A activity and anchoring are required for ovarian cancer cell migration and invasion. PLoS One 6, e26552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O'Connor K. L., Mercurio A. M. (2001) Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J. Biol. Chem. 276, 47895–47900 [DOI] [PubMed] [Google Scholar]
- 61. Whittard J. D., Akiyama S. K. (2001) Positive regulation of cell-cell and cell-substrate adhesion by protein kinase A. J. Cell Sci. 114, 3265–3272 [DOI] [PubMed] [Google Scholar]
- 62. Meyer C. J., Alenghat F. J., Rim P., Fong J. H., Fabry B., Ingber D. E. (2000) Mechanical control of cyclic AMP signalling and gene transcription through integrins. Nat Cell Biol. 2, 666–668 [DOI] [PubMed] [Google Scholar]
- 63. Tkachenko E., Sabouri-Ghomi M., Pertz O., Kim C., Gutierrez E., Machacek M., Groisman A., Danuser G., Ginsberg M. H. (2011) Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat Cell Biol. 13, 660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]