

Background: The sumoylation pathway is conserved in Plasmodium falciparum.
Results: The small ubiquitin-related modifier (SUMO) E1 and E2 enzymes are not functionally interchangable between humans and the malaria parasite, P. falciparum.
Conclusion: P. falciparum E1 and E2 interactions have significantly diverged from humans.
Significance: Divergent E1 and E2 interaction could be exploited for the design of parasite specific inhibitors.
Keywords: Crystal Structure, Malaria, Plasmodium, Sumo, Sumoylation, Ubiquitin-conjugating Enzyme (Ubc)
Abstract
Small ubiquitin-related modifiers (SUMOs) are post-translationally conjugated to other proteins and are thereby essential regulators of a wide range of cellular processes. Sumoylation, and enzymes of the sumoylation pathway, are conserved in the malaria causing parasite, Plasmodium falciparum. However, the specific functions of sumoylation in P. falciparum, and the degree of functional conservation between enzymes of the human and P. falciparum sumoylation pathways, have not been characterized. Here, we demonstrate that sumoylation levels peak during midstages of the intra-erythrocyte developmental cycle, concomitant with hemoglobin consumption and elevated oxidative stress. In vitro studies revealed that P. falciparum E1- and E2-conjugating enzymes interact effectively to recognize and modify RanGAP1, a model mammalian SUMO substrate. However, in heterologous reactions, P. falciparum E1 and E2 enzymes failed to interact with cognate human E2 and E1 partners, respectively, to modify RanGAP1. Structural analysis, binding studies, and functional assays revealed divergent amino acid residues within the E1-E2 binding interface that define organism-specific enzyme interactions. Our studies identify sumoylation as a potentially important regulator of oxidative stress response during the P. falciparum intra-erythrocyte developmental cycle, and define E1 and E2 interactions as a promising target for development of parasite-specific inhibitors of sumoylation and parasite replication.
Introduction
Malaria is a mosquito-borne infectious disease caused by protists of the genus Plasmodium, with Plasmodium falciparum being the most virulent species infecting humans. It is estimated that malaria affects more than 300 million people each year, with nearly one million or more of those affected dying of complications caused by the disease (1). Despite ongoing efforts, an effective vaccine to prevent malaria still remains an elusive goal. Current treatment and preventative measures therefore rely on artemisinin-based combination drug treatments, long-lasting insecticide-treated bed nets and insecticide spraying. Despite the current effectiveness of these strategies, resistance to artemisinin-based treatments is an increasingly serious concern (2). Therefore, there is an urgent need to identify and develop next generation drugs that can be used to effectively treat malaria.
The malaria parasite life cycle is complex, involving morphological, biochemical, and physiological transformations that enable growth and replication in mosquitoes and humans, as well as transmission between these two hosts. In humans, the intra-erythrocyte developmental cycle (IDC)3 represents one of the best studied phases of the parasite life cycle. The IDC consists of three distinct stages: a morphologically defined ring stage, a trophozoite stage in which the parasite breaks down hemoglobin into amino acids and toxic heme, and a schizont stage in which the parasite undergoes 3–5 rounds of asexual replication (3). Comprehensive transcriptome and proteomic studies have revealed that a continuous cascade of gene and protein expression, in which most genes and proteins exhibit a single peak of expression, is associated with progression through the IDC (4–6). Given this rigid mode of regulation at the level of gene and protein expression, it is anticipated that post-translational protein modifications play particularly important regulatory roles during the IDC. Consistent with this, more than half of all P. falciparum proteins detected during the IDC are present as two or more post-translationally modified isoforms (5, 7). Functionally important post-translational protein modifications identified and characterized in P. falciparum during the IDC include phosphorylation, acetylation, methylation, lipidation, N- and O-linked glycosylation, ubiquitylation, and sumoylation (8).
Sumoylation involves the covalent attachment of SUMOs (small ubiquitin-related modifiers), ∼100 amino acid proteins, to lysine residues in target proteins (Fig. 1a). Sumoylation regulates the functions of a wide range of proteins involved in nearly all aspects of cell function, including proteins involved in transcription, DNA replication and repair, chromosome segregation, mitochondrial fission, ion transport, and signal transduction (9). Sumoylation is therefore essential in organisms ranging from yeast to humans. Sumoylation also has vital roles in promoting cell survival under a variety of stress conditions, including hypoxia, heat, and oxidative stress (10). Oxidative stress is of particular significance during the middle and late stages of the malaria parasite IDC, when the degradation of hemoglobin results in production of toxic-free heme and the generation of reactive oxygen species (11). A preliminary list of ∼20 sumoylated proteins has been identified in P. falciparum during the IDC (12). However, the functional significance of the modification of these proteins, and the importance of sumoylation in progression through the IDC and protection from oxidative stress, remains to be fully characterized. Notably, an inhibitor of desumoylation blocks P. falciparum replication in human red blood cell cultures, demonstrating that sumoylation is essential during the IDC and that inhibitors of the sumoylation pathway have potential as effective anti-malarial drugs (13).
FIGURE 1.

The sumoylation pathway is conserved in P. falciparum. a, SUMO is conjugated to proteins through an enzymatic cascade involving E1-activating and E2-conjugating enzymes and E3 ligases. Deconjugation is catalyzed by isopeptidases. b, enzymes of the P. falciparum sumoylation pathway identified by sequence similarity searches (% identity to human proteins is indicated). Gene ID and predicted molecular weights (MW) were obtained from PlasmoDB. c, amino acid sequence alignment of SP-RING domains from human (Hs), S. cerevisiae (Sc), Schizosaccharomyces pombe (Sp), and Plasmodium falciparum (Pf) SUMO E3 ligases, as well as RING domains from c-Cbl and BRCA1. Sequence conservation is highlighted in gray and black. The SP-RING domain is indicated by the top bar and Zn2+ coordinating cysteine and histidine residues are indicated by black dots. Cysteine and histidine residues unique to SP-RING domains are indicated with asterisks.
The development of anti-malarial drugs targeting the sumoylation pathway of P. falciparum requires a detailed molecular understanding of the enzymes involved in conjugation and deconjugation, and most importantly, an understanding of how these enzymes differ from their human counterparts. SUMO-specific proteases (SENPs) are involved in processing SUMO precursors to generate mature SUMO, and in cleaving the isopeptide bond between the C-terminal glycine of SUMO and the lysine side chain of target proteins to mediate deconjugation (Fig. 1a). The P. falciparum genome encodes for two putative SENPs (PfSENP1 and PfSENP2), of which to date only PfSENP1 has been shown to be functionally active (13) (Fig. 1b). SUMO conjugation is dependent on an essential heterodimeric E1-activating enzyme that activates SUMO through the ATP-dependent formation of a high-energy thioester bond between the C-terminal glycine of SUMO and an active site cysteine in the E1. Activated SUMO is subsequently transferred to the active site cysteine residue of an E2-conjugating enzyme, known as Ubc9. Ubc9 subsequently binds target proteins, either directly or assisted by one of several E3 ligases, and catalyzes the formation of an isopeptide bond between a lysine residue in the target protein and the C-terminal glycine of SUMO (14). Bioinformatic analysis has identified predicted SUMO E1 (PfAos1/PfUba2)-activating and E2 (PfUbc9)-conjugating enzymes in the P. falciparum genome, however, no biochemical analysis of the encoded proteins has been reported (12) (Fig. 1b). Detailed biochemical and structural studies of the E1 and E2 enzymes from human and yeast have been reported, and are available for guiding comparative studies (15–19).
To begin to understand the sumoylation pathway of P. falciparum at the molecular level, we reconstituted SUMO conjugation in vitro using purified recombinant proteins and solved a crystal structure of PfUbc9. Our studies, revealed specific features of P. falciparum E1 and E2 interactions that are distinct from human E1 and E2 interactions. We also developed a monoclonal antibody specific for PfSUMO and found that sumoylation peaks during midstages of the IDC cycle, consistent with sumoylation having an important role in the oxidative stress response. Our findings indicate the E1- and E2-conjugating enzymes of P. falciparum may be viable drug targets, and that inhibitors of SUMO conjugation might prove effective in combination therapies with drugs that enhance oxidative stress, including artemisinin.
EXPERIMENTAL PROCEDURES
P. falciparum Cell Culture, Harvesting, and Lysate Preparation
Pf biotin carboxyl carrier protein (BCCP)-GFP dd2attB (a gift from Dr. Sean Prigge, Johns Hopkins University) and 3D7 strain parasites were cultured using a modified standard procedure (20). The mRPL2 promoter that drives BCCP-GFP expression is activated consistently across IDC stages (6) and was therefore used as a protein loading control for immunoblot analysis. Parasites were grown at 2% hematocrit in human O+ erythrocytes in malaria culture media, RPMI 1640 media (Invitrogen), supplemented with 25 mm HEPES, 0.2% sodium bicarbonate (Invitrogen), 10% human O+ serum (Interstate Blood Bank, Inc., Memphis TN), 12.5 μg/ml of hypoxanthine (Sigma) in 3% CO2, 3% O2, 96% N2 atmosphere at 37 °C. RBCs were pelleted at 600 × g, washed with 37 °C RPMI supplemented with 5 mm N-ethylmaleimide (Sigma) to inhibit SUMO isopeptidases, and treated with 0.2% saponin/PBS (w/v) for 3 min at room temperature, followed by three rounds of washing with ice-cold PBS. Parasite pellets were collected by centrifugation at 20,800 × g for 10 min at 4 °C and stored at −80 °C. Pellets were lysed in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.05% sodium deoxycholate, 0.1% SDS) supplemented with 5 mm N-ethylmaleimide for 30 min on ice with occasional vortexing. Lysates were centrifuged at 20,800 × g for 10 min at 4 °C and supernatants were quantified by BCA assay (Thermo Scientific, Rockford, IL) before the addition of reducing SDS-sample buffer.
Parasite Synchronization
PfBCCP-GFP dd2attB strain parasites were grown as described above, and synchronized with three cycles of sorbitol treatment at the ring stage (21). Cultures were pelleted at 600 × g and infected red blood cell pellets were incubated with 5% sorbitol (Sigma) for 10 min at room temperature, with occasional gentle shaking. RBCs were pelleted at 600 × g and washed three times in malaria culture media, and subcultured. Parasites were harvested at respective stages after 48 h following the third sorbitol treatment and lysates were prepared as described above.
Immunofluorescence Microscopy
3D7-infected red blood cells were cultured as indicated and harvested at 500 × g for 3 min. Cells were fixed for 30 min at room temperature with 4% paraformaldehyde, 0.0075% glutaraldehyde in 1× PBS. Cells were then washed with PBS and permeabilized for 10 min at room temperature with 0.1% Triton X-100 in PBS. After washing, aldehydes were further reduced for 10 min at room temperature with 0.1 mg/ml of NaBH4 in PBS. Following subsequent washing, cells were blocked in 3% BSA in PBS for 1 h at room temperature and incubated overnight at 4 °C with primary antibody (1:20 mAb 7E11; 1:500 anti-acyl carrier protein) in 3% BSA in PBS. After washing with PBS, cells were incubated overnight at 4 °C in the dark with secondary, Alexa Fluor-conjugated antibodies (Invitrogen) were diluted 1:3000 in 3% BSA in PBS. Cells were washed with PBS and allowed to dry on glass slides. Dried slides were treated with mounting solution containing 100 mm Tris, pH 8.8, 50% glycerol, 2.5% Dabco (Sigma), 0.2 μg/ml of 4,6-diamidino-2-phenylindole (DAPI), and images were taken using a Zeiss Observer Z1 microscope. SUMO images were taken at identical exposure settings.
Immunoblot Analysis, Antibodies
Parasite lysates were heated to 100 °C for 5 min, resolved by SDS-PAGE, and transferred to PVDF membrane. Membranes were probed with mAb 7E11, 21C7 (22), 8A2 (23), or anti-GFP (Clontech). HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) were detected by SuperSignal West Femto chemiluminescent substrate (Thermo Scientific).
mAb 7E11 was produced by immunizing Balb/c mice with full-length recombinant PfSUMO. Hybridoma cells were prepared by fusing spleen cells with the mouse myeloma cell line, Sp2/0, and screened using standard procedures.
cDNA Cloning and Plasmid Construction
Human RanGAP1, SUMO-1, Ubc9, and E1 bacterial expression vectors were as previously described (24). The coding regions for mature PfSUMO and full-length PfUbc9 were PCR-amplified from a 3D7 strain cDNA library kindly provided by Dr. Sean Prigge (Johns Hopkins University) using primers designed to introduce BamHI and XhoI sites at 5′ and 3′ ends, respectively. PCR-amplified products were inserted into the expression vectors pGEX-4T-1 or pGEX-6P-1 (GE Healthcare). PfUba2 and PfAos1 cDNAs were codon optimized for expression in Escherichia coli using the Codon Juggle module of Gene Design (25). The codon-optimized cDNAs were chemically synthesized (Genewiz, South Plainfield, NJ) and used as templates for PCR amplification and cloning. PfUba2 was cloned into NcoI/BamHI sites of pET32a (EMD Millipore, Billerica, MA), re-amplified with the N-terminal His6-S tag and cloned into the BamHI/XhoI sites of pFastBac1 (Invitrogen). PfAos1 was amplified without a tag and also cloned into the BamHI/XhoI sites of pFastBac1. Human and P. falciparum Uba2Ufd domains (HsUba2Ufd, residues 444–561; PfUba2Ufd, residues 508–625) were subcloned into pGEX-6P-1 expression vectors (GE Healthcare) using BamHI and XhoI sites.
PfUbc9 N-terminal domain swap mutants were generated by the fusion PCR technique (26). Additional amino acid substitution mutants were produced using standard site-directed mutagenesis. All constructs were verified by DNA sequence analysis.
Recombinant Protein Expression and Purification
Human RanGAP1, SUMO-1, Ubc9, and E1 were expressed and purified as previously described (24). In general, all other bacterial expression plasmids were transformed into E. coli host strain Rosetta 2(DE3) and LB cultures were grown at 37 °C to an A600 of 0.6. Cultures were then induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside and continued incubation at 20 °C overnight. Cells were harvested by centrifugation at 4,500 × g and E. coli pellets were frozen at −80 °C. Frozen pellets were resuspended in 4 °C lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm DTT, 0.1% Triton X-100, 1 μg/ml of leupeptin, 1 μg/ml of pepstatin, 20 μg/ml of aprotinin, 1 mm PMSF, 10 units/ml of benzonase). After sonication, insoluble material was removed by centrifugation at 28,000 × g and supernatants containing GST-tagged proteins were run over glutathione-agarose chromatography columns (GE Healthcare). On-column cleavage was performed using purified thrombin or PreScission Proteases (GE Healthcare). Eluted proteins were further purified using ion exchange on Mono S or Mono Q columns or size exclusion on a SuperdexTM 200 column (GE Healthcare). For isothermal titration calorimetry (ITC) analysis, concentrated Ubc9 and Uba2UFD domains were dialyzed together into 50 mm glycine, pH 8.5, 150 mm NaCl, and 0.5 mm Tris(2-carboxyethyl)phosphine. Protein concentrations were determined by UV absorption at 280 nm using a Nanodrop spectrophotometer (Thermo Scientific), or by Bradford colorimetric assays (Bio-Rad).
P. falciparum E1 enzyme was purified from baculovirus-infected insect cells. pFastBac1 vectors containing PfUba2 or PfAos1 were used to make P4 viral stocks according to the manufacturer's protocol (Invitrogen). For expression of recombinant proteins, 0.1% (v/v) P4 PfUba2 or PfAos1 viral stocks were added individually to 1 × 106 Sf9 cells/ml in SFMII media (Invitrogen), and shaken at 110 rpm, for 72 h at 26 °C. Infected cells were combined, lysed in buffer, and purified on nickel-nitrilotriacetic acid beads according to the manufacturer's instructions (Qiagen).
Isothermal Titration Calorimetry
Binding affinities and thermodynamic binding parameters for interactions between Ubc9 and Uba2Ufd domains were determined using a MicroCal VP-ITC system (GE Healthcare). The ITC cell (1.4 ml) contained either HsUbc9 (18.5–23.9 μm) or PfUbc9 (14.5–20 μm), and the syringe (300 μl) contained either HsUba2Ufd (216.4–470.9 μm) or PfUba2Ufd (210.7–371.5 μm). Each run was performed at 15 °C with a 310 rpm rotating syringe, and consisted of an initial 3-μl injection of 5.1 s duration with 150 s spacing, followed by 25 additional 10-μl injections of 7.3 s duration with 350 s spacing between injections. Data were obtained with a 2-s filter period and raw heat signals were normalized to the dilution heat of the titrant. Binding isotherms were fitted to a single site model using Origin 7 software (GE Healthcare). Experiments were performed in triplicate.
In Vitro Sumoylation Assays
For assays using in vitro expressed RanGAP1, protein was transcribed and translated in rabbit reticulocyte lysate in the presence of [35S]methionine as recommended by the manufacturer (Invitrogen). Modification assays were performed in 20-μl reactions containing 15 nm E1, 45 nm Ubc9, 500 nm SUMO, 2 μl of in vitro translated RanGAP1, 1 mm ATP, 0.6 units/ml of creatine phosphokinase, 10 mm phosphocreatine, 0.6 units/ml of inorganic pyrophosphatase, 20 mm HEPES-KOH, pH 7.3, 110 mm KAc, 2 mm Mg(Ac)2, and 1 mm DTT. Reactions were incubated at 37 °C for 1 h and analyzed by SDS-PAGE and autoradiography. Modification assays using recombinant RanGAP1 were performed in 20-μl reactions containing 1 μm GST-RanGAP1 (residues 419–589), 5 μm HsSUMO-1, 50 nm HsE1, 80 or 800 nm E2, 0.6 units/ml of creatine phosphokinase, 10 mm phosphocreatine, 0.6 units/ml of inorganic pyrophosphatase, 20 mm HEPES, pH 7.4, 2 mm Mg(Ac)2, 110 mm KAc, and 5 mm ATP. Reactions were incubated at 30 °C and aliquots were combined with sample buffer for analysis by SDS-PAGE, followed by immunoblot analysis. Membranes were probed with anti-GST antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA). HRP-conjugated secondary antibody (1:15,000 GαM, Jackson) was detected by ECL Prime (GE Healthcare).
PfUbc9 Structure Determination
PfUbc9 was concentrated to 7.5 mg/ml and crystallization screening was performed using a Mosquito Crystallization Robot (TTP Labtech, Cambridge, MA). Diffraction quality crystals were grown at 20 °C using the sitting drop method, and plated as 3 drops per well, with each drop consisting of a mixture of 200 nl of protein (7.5 mg/ml), 20 nl of seed crystals from PEGs Suite (Qiagen, 130704) in a buffer of 0.2 m NaF, 20% PEG3350, and 200 nl of buffer (0.2 m NaF, 19% PEG3350) from the 60-μl buffer well. Crystals were transferred into cryogenic buffer consisting of 30% PEG3350, 10% glycerol, 0.2 m NaF, and 100 mm glycine, pH 8.5, then mounted on nylon loops and flash frozen in liquid nitrogen. Crystals were shipped to the Brookhaven National Laboratory Synchrotron Lightsource and datasets were collected under cryogenic conditions at beamline X25.
Although the XDS indexing routine (27) suggested a higher symmetry space group and the program POINTLESS (28) determined P21212 as the most likely space group, refinement in any of the eight possible P2x2x2x point groups resulted in Rwork/Rfree combinations of 40/45%. The best case scenarios did not decrease below an Rwork −38% despite exhibiting a well defined electron density map with clear side chain densities. Only when using a lower symmetry space group, in combination with twin refinement, could a successful refinement be carried out yielding a final Rwork = 13.7% and Rfree = 17.3%. The twin fraction was refined to 49%, indicating an almost perfect twin, explaining the apparent higher symmetry space group during the indexing steps.
The PfUbc9 crystals belonged to the space group P21 with cell dimensions of a = 28.9 Å, b = 94.4 Å, c = 124.9 Å, and β = 90.02°, and contained four molecules in the asymmetric unit. Data reduction and scaling was performed with XDS/XSCALE (27). A molecular replacement solution was obtained with BALBES, using a predicted homology model based on the structure of mammalian Ubc9 (PDB code 1U9A), with an initial Rfactor of 0.35 (29). Model building and refinement was performed with Coot (30) and REFMAC 5 (31) in the CCP4 and PHENIX suites (32). The final structure was analyzed with validation tools in Coot as well as MolProbity (33), indicating zero Ramachandran outliers. Although PfUbc9 crystallized as a dimer, a buried surface area of 1870 Å2 for the 4 chain assembly and its predicted size based on gel filtration chromatography are consistent with a monomer in solution.
A second crystal form was obtained at a protein concentration of 12.35 mg/ml using the Morpheus screen (Molecular Dimensions) condition of 0.12 m alcohols (1,6-hexanediol, 1-butanol, 1,2-propanediol (racemic), 2-propanol, 1,4-butanediol, and 1,3-propanediol), 0.1 m buffer (imidazole, MES, pH 6.5), and 30% precipitant (PEGMME 550; PEG 20K) yielding well diffracting crystals to 1.25 Å. These crystals belonged to space group C2 2 21 with three molecules in the asymmetric unit. Refinement was carried out using 1.5 Å as suggested high-resolution cutoff based on the recently introduced CC1/2 criteria for significant data (34). Details of the refinement statistics are shown in Table 1.
TABLE 1.
Data collection, phasing, and refinement statistics
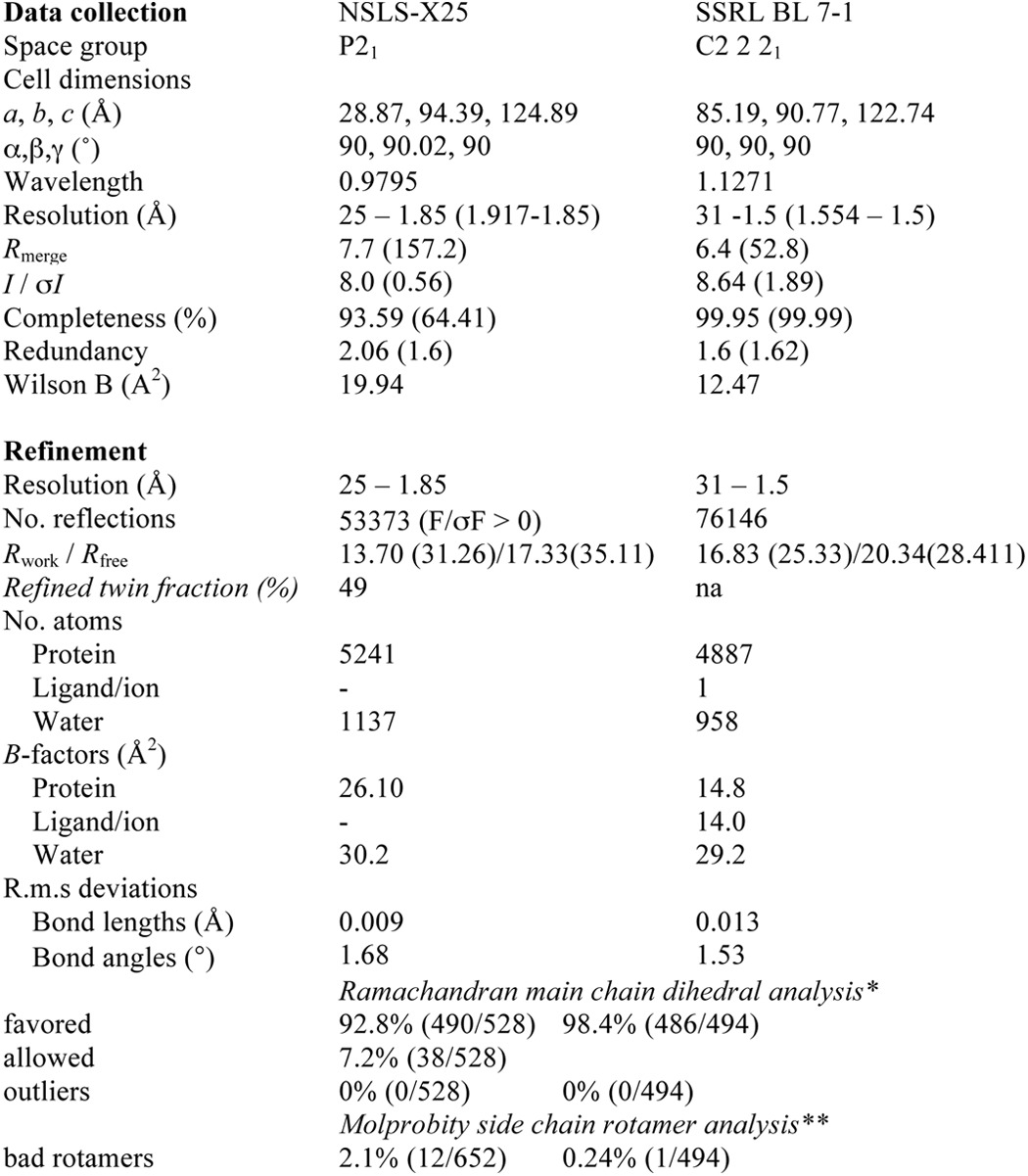
* Highest resolution shell is shown in parentheses.
** According to Molprobity (53).
Sequence Alignment and Structure Prediction
Saccharomyces cerevisiae, human, and P. falciparum Uba2Ufd and E2 sequences were aligned using the T-Coffee Expresso server (35) and determined or predicted structures. The PfUba2Ufd structure was predicted using the I-Tasser server (36).
RESULTS
Sumoylation Peaks during Late Stages of the P. falciparum IDC
Genes coding for SUMO, and enzymes of the sumoylation pathway, have been identified through bioinformatic analysis of the P. falciparum genome (12, 13) (Fig. 1b). This includes a gene coding for a protein with an SP-RING domain and belonging to the family of PIAS E3 ligases (Fig. 1c). To aid in characterizing the functional roles of sumoylation in parasites, we cloned and expressed PfSUMO and generated a mouse monoclonal antibody, mAb 7E11. By immunoblot analysis, mAb 7E11 reacted with recombinant PfSUMO, but did not recognize recombinant human SUMO-1, SUMO-2, or SUMO conjugates in uninfected human red blood cells (Fig. 2a). Analysis of P. falciparum cell lysates, prepared from parasites purified from an asynchronous culture of infected red blood cells, revealed a smear of high molecular weight proteins consistent with previous detection of P. falciparum SUMO conjugates (12). To gain insights into possible stage-specific roles for sumoylation during the IDC, we synchronized P. falciparum-infected red blood cell cultures and performed immunoblot analysis of lysates prepared from purified ring, trophozoite, and schizont stage parasites. To control for protein loading, parasites expressing GFP-tagged BCCP were used and lysates were normalized so that roughly equivalent BCCP-GFP signals were obtained using an antibody recognizing GFP (Fig. 2b). As in lysates of parasites from asynchronous cultures, a smear of high molecular weight conjugates was readily detectable at all three stages, with little if any detectable free SUMO. Notably, however, the levels of high molecular weight SUMO conjugates varied greatly between stages, with levels being lowest in ring-stage parasites and peaking in trophozoites (Fig. 2b).
FIGURE 2.

Sumoylation is differentially regulated during P. falciparum red blood cell stages. a, uninfected red blood cell lysate (RBC), 3D7 parasite lysate, and recombinant PfSUMO, HsSUMO-1, and HsSUMO-2, were analyzed using mAb 7E11 (α-PfSUMO). To demonstrate antibody specificity, lower portions of the blot were also probed with mAbs 21C7 (α-HsSUMO-1) and 8A2 (α-HsSUMO-2). b, immunoblot analysis of stage-specific P. falciparum parasite lysates using mAb 7E11 (α-PfSUMO). Detection of GFP-BCCP and Coomassie Blue staining of the immunoblot membrane are included as loading controls. c, indirect immunofluorescence microscopy of synchronized 3D7 parasites probed with anti-PfSUMO mAb 7E11 and anti-acyl carrier protein (ACP) antibodies. DNA was detected using DAPI. Bar indicates 2 μm.
To further evaluate sumoylation during the P. falciparum IDC, we performed indirect immunofluorescence microscopy on infected red blood cells. Cells were fixed and stained with mAb 7E11 together with an antibody to acyl carrier protein, an apicoplast marker protein (37). SUMO was concentrated in the nucleus of parasites at all three stages, with no discernible association with Maurer's clefts, as previously reported (12). Consistent with immunoblot analysis, levels of detectable SUMO signal were greatest in trophozoite- and schizont-stage parasites (Fig. 2c).
P. falciparum E1- and E2-conjugating Enzymes Modify RanGAP1 at the ΨKXE Consensus Site
To characterize the P. falciparum sumoylation pathway at the molecular level, we next cloned, expressed, and purified the predicted heterodimeric E1-activating enzyme, consisting of proteins PfAos1 and PfUba2, as well as the E2-conjugating enzyme, PfUbc9. Enzyme activities were preliminarily evaluated using in vitro sumoylation assays containing in vitro transcribed and translated mammalian RanGAP1. RanGAP1 is a well characterized and efficient sumoylation substrate, modified at a single consensus sumoylation motif, ΨKXE (where Ψ is a hydrophobic amino acid, K is the modified lysine residue, X is any amino acid, and E is glutamic acid) that is recognized through direct interactions with Ubc9 (15, 38). In the absence of exogenously added factors, expression of wild type RanGAP1 in rabbit reticulocyte lysate produces predominantly unmodified protein migrating at ∼70 kDa, and also a minor fraction of sumoylated protein migrating at ∼85 kDa. In reactions containing purified human E1 and E2 enzymes, wild type RanGAP1 is sumoylated quantitatively and migrates at ∼85 kDa, consistent with modification at the single consensus site (38) (Fig. 3a). As predicted, modification of mutant forms of RanGAP1, in which the consensus site glutamic acid or lysine residues were substituted with alanine, was significantly reduced or undetectable (Fig. 3a). In reactions containing purified P. falciparum E1 and E2 enzymes, wild type RanGAP1 also shifted from ∼70 to 85 kDa, again consistent with modification at the single consensus site. Consensus site mutations also reduced or inhibited modification by the P. falciparum enzymes (Fig. 3a). These findings validate the orthologous identity of the predicted P. falciparum SUMO-activating and -conjugating enzymes and demonstrate a conservation of consensus site recognition by PfUbc9.
FIGURE 3.

Human and P. falciparum SUMO E1 and E2 enzyme interactions are distinct. a, P. falciparum enzymes modify RanGAP1 at the consensus site lysine. In vitro transcribed and translated wild type RanGAP1 (WT) or consensus sumoylation site mutants (E528A and K526A), were incubated with recombinant human (Hs) or P. falciparum (Pf)-conjugating enzymes. Conjugation was detected by autoradiography. b, human and P. falciparum E1-conjugating enzymes activate both human and P. falciparum SUMOs. Increasing concentrations of E1-activating enzymes were incubated with human or P. falciparum GFP-SUMO in the presence of ATP and reaction products were separated by non-reducing SDS-PAGE and analyzed by immunoblot analysis. The asterisk indicates a nonspecific GFP-SUMO-2 band. c, P. falciparum and human SUMOs are interchangeable. SUMO conjugation assays were performed in the presence of the indicated recombinant human or P. falciparum proteins. Reaction products were analyzed by immunoblot analysis. d, human and P. falciparum SUMO E2 enzymes are not interchangeable. SUMO conjugation assays were performed in the presence of the indicated recombinant human or P. falciparum proteins. Reaction products were analyzed by immunoblot analysis. e, E2∼SUMO thioesters are not formed in reactions containing heterologous E1 enzymes. The indicated recombinant proteins were incubated in the presence of ATP and reaction products were separated by non-reducing SDS-PAGE and analyzed by immunoblot analysis.
Interactions between Human and P. falciparum E1- and E2-conjugating Enzymes Are Functionally Unique
To identify potentially distinct properties of the P. falciparum E1 and E2 enzymes, we next performed a series of in vitro assays using heterologous conditions in which purified recombinant human and P. falciparum proteins were interchanged. First, we investigated the ability of human and P. falciparum E1 enzymes to activate human and P. falciparum SUMOs. E1 enzymes were incubated with SUMO in the presence of ATP, and E1∼SUMO thioester intermediates were identified by non-reducing SDS-PAGE and immunoblot analysis (Fig. 3b). This analysis revealed that both human and P. falciparum E1-activating enzymes have the ability to recognize and activate both human and P. falciparum SUMOs. As exemplified in the assays shown, the human E1-activating enzyme consistently produced higher levels of E1∼SUMO intermediates, suggesting either more efficient SUMO activation or formation of a more stable thioester intermediate.
We next performed RanGAP1-conjugation assays in which human and P. falciparum SUMOs were interchanged (Fig. 3c). In reactions containing human E1 and E2 enzymes, RanGAP1 was modified by both human and P. falciparum SUMOs, indicating conservation of SUMO features required for both E1 activation, E2 loading, and substrate modification. Similarly, both human and P. falciparum SUMOs were conjugated to RanGAP1 in reactions containing P. falciparum E1 and E2 enzymes. In another series of conjugation assays, human and P. falciparum E1 and E2 enzymes were interchanged (Fig. 3d). In contrast to reactions containing all human components, modification of RanGAP1 was not observed in reactions containing human E1, human SUMO-2, and P. falciparum E2. Similarly, and in contrast to reactions containing all P. falciparum components, modification of RanGAP1 was undetectable in reactions containing P. falciparum E1, P. falciparum SUMO, and human E2. Thus, although human and P. falciparum SUMOs are functionally interchangeable, human and P. falciparum E1- and E2-conjugating enzymes are not.
The finding that RanGAP1 sumoylation did not occur in reactions containing a heterologous mixture of human and P. falciparum E1 and E2 enzymes, suggested a possible defect in the transfer of activated SUMO from the E1 enzyme to Ubc9 under these assay conditions. We therefore assayed for E2 thioester formation in reactions performed in the absence of RanGAP1. Reaction products were separated by non-reducing SDS-PAGE and analyzed by immunoblot analysis (Fig. 3e). In reactions containing all human or all P. falciparum components, E2∼SUMO thioester intermediates were detected in an E2 concentration-dependent manner. In contrast, E2∼SUMO thioester intermediates were not detected in reactions containing heterologous mixtures of human and P. falciparum E1 and E2 enzymes. These findings indicate a possible defect in functional interactions between human and P. falciparum E1 and E2 enzymes.
To investigate the molecular basis for the observed defects in human and P. falciparum E1 and E2 functional interactions, we performed ITC to evaluate the relative binding affinities of the enzymes. For this analysis, we expressed and purified the C-terminal ubiquitin-fold domains of human and P. falciparum Uba2 (Uba2ufd), which are predicted to mediate interactions with Ubc9 (16, 19). Consistent with this prediction, we observed endothermic binding between human Ubc9 and human Uba2ufd with the following characteristics: stoichiometry (N) = 1.3 ± 0.13, Kd = 1.26 ± 0.4 μm, ΔH = 2700 ± 67 kcal mol−1, and ΔS = 36 ± 0.9 cal mol−1 K−1 (mean ± S.D. from three independent trials) (Fig. 4a). Similarly, endothermic binding between P. falciparum Ubc9 and P. falciparum Uba2ufd was also observed: stoichiometry (N) = 0.92 ± 0.06, Kd = 0.5 ± 0.1 μm, ΔH = 8867 ± 500 kcal mol−1, and ΔS = 60 ± 1.4 cal mol−1 K−1 (Fig. 4b). In contrast, no measurable interactions between human Ubc9 and P. falciparum Uba2ufd or between P. falciparum Ubc9 and human Uba2ufd were detected by ITC (Fig. 4, c and d). Collectively, these findings reveal evolutionarily divergent modes of interaction between human and P. falciparum E1-activating and E2-conjugating enzymes.
FIGURE 4.
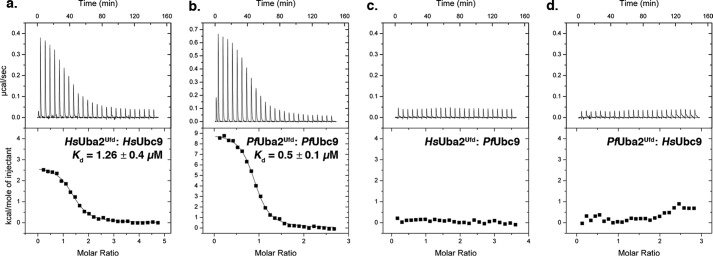
HsUbc9 and PfUbc9 interact specifically with human and P. falciparum E1-activating enzymes, respectively. Representative isotherms of ITC binding data for (a) HsUba2Ufd and HsUbc9, (b) PfUba2Ufd and PfUbc9, (c) HsUba2Ufd and PfUbc9, and (d) PfUba2Ufd and HsUbc9.
Divergent Interfaces Mediate Human and P. falciparum E1 and E2 Interactions
To further define the defects in binding between human and P. falciparum E1 and E2 enzymes at the molecular level, we determined a crystal structure of PfUbc9 at 1.85-Å resolution (Fig. 5a, Table 1). PfUbc9 and HsUbc9 share 63% sequence identity and a high degree of backbone conservation, as reflected by a small root mean square deviation value of 0.72 Å over all Cα atoms. Comparison of the overall electrostatic potential on the surface of PfUbc9 and HsUbc9 revealed potentially significant charge differences due to divergent amino acid residues in the α1 helix and the β1-β2 loop (Fig. 5, b–d). Based on crystallographic analysis of S. cerevisiae E1 and E2 interactions (19), the α1 helix and the β1-β2 loop are predicted to form the E1 interaction interface. Of particular interest, several residues within this region of PfUbc9, including Lys-5 and Lys-6, Ala-13 and Glu-14, and Pro-29 and Lys-34 have diverged from residues in HsUbc9 (Fig. 6b). Residues at several of these positions are conserved between human and yeast Ubc9, and in the case of yeast Ubc9, are known to be directly involved in E1 binding (19) (Fig. 6b). Residues within the α4 helix of HsUbc9 are also important for low affinity interactions with the catalytic cysteine domain of HsUba2 (39), however, these residues are conserved in PfUbc9.
FIGURE 5.
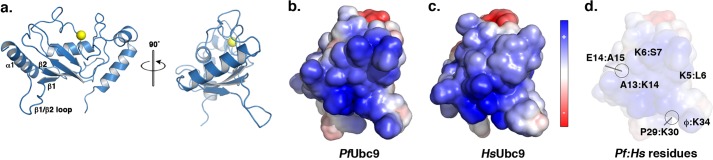
Crystallographic structure of PfUbc9. a, ribbon diagram of the determined structure of PfUbc9 with secondary structures comprising the predicted E1 interface numbered. The active site cysteine is highlighted in yellow. b and c, electrostatic surface representations of the α1 helix and β1-β2 loop regions of PfUbc9 and HsUbc9. Electrostatic potentials were calculated using APBS (52) and colored from positive (+5kT/e; blue) to negative (−5kT/e; red). d, specific divergent residues in the α1 helix and β1-β2 loop region of HsUbc9 and PfUbc9 are indicated.
FIGURE 6.

Human and P. falciparum Uba2Ufd and Ubc9 interfaces are divergent. a, sequence alignment of S. cerevisiae (Sc) Uba2Ufd sequence, with corresponding regions from human (Hs) and P. falciparum (Pf) Uba2. Secondary structure is indicated with arrows for β sheets, and dashed closed lines for α-helices. Residues shown to be important for interactions between S. cerevisiae Uba2Ufd and Ubc9 are indicated in blue (19). Conserved residues are highlighted in green, and similarity is highlighted in yellow, with shading corresponding to degree of shared identity between the sequences. b, sequence alignment of S. cerevisiae, human and P. falciparum Ubc9. Residues involved in the S. cerevisiae Uba2Ufd and Ubc9 interaction, but which have diverged between human and P. falciparum Ubc9, are highlighted as black asterisks. Conservation is indicated as above. c–e, comparison of determined interactions between S. cerevisiae Uba2Ufd and Ubc9, and predicted interactions between the human and P. falciparum E1 and E2 enzyme interfaces.
Because known structures of E1 and E2 enzyme complexes for human or P. falciparum proteins are not available, we used in silico approaches to superimpose known or predicted structures of individual Uba2ufd domains and Ubc9. The solved structure of a complex between S. cerevisiae Uba2ufd (ScUba2ufd) and ScUbc9 was used to guide protein alignment and build homology models (19). Results from this analysis indicated that the E1 and E2 enzymes have co-evolved to maintain surface residues compatible for interaction. As one example, Leu-6 of ScUbc9 makes hydrophobic contacts with Leu-511 and Leu-478 in ScUba2ufd (Fig. 6c). In humans, Leu-6 of HsUbc9 is conserved and predicted to be surrounded by a loop in HsUba2ufd also containing hydrophobic residues, including Ile-489, Gly-485, Gly-487, and Ile-482. In contrast, Lys-5 replaces Leu-6 in PfUbc9. To accommodate this basic amino acid substitution, the predicted interaction surface of the PfUba2ufd has co-evolved to contain a cluster of acidic residues, including Asp-544, Asp-546, and Asp-547 (Fig. 6c). As a second example, Lys-14 of ScUbc9, also in the α1 helix, forms a salt bridge with Asp-474 of ScUba2ufd (Fig. 6d). Again, this lysine residue is conserved in HsUbc9, as is the acidic nature of the interacting surface on HsUba2ufd, including conservation of a complimentary aspartic acid at position 479. In PfUbc9, Lys-14 is replaced by Ala-13. To accommodate this small, non-polar amino acid substitution, the predicted interaction surface of PfUba2ufd, formed in part by Phe-587 and Phe-542, has co-evolved to become bulkier and more hydrophobic in nature (Fig. 6d).
Several other notable substitutions occur within the β1-β2 loop regions of HsUbc9 and PfUbc9. HsUbc9 Lys-30, for example, is positioned to potentially interact with Glu-497 of HsUba2ufd. This interaction is conserved in S. cerevisiae (Fig. 6e). However, Lys-30 is replaced by Pro-29 in PfUbc9 and is predicted to make no direct contacts with PfUba2ufd (Fig. 6e). Another key difference between the human and P. falciparum β1-β2 loops is the insertion of Lys-34 in PfUbc9, creating a longer loop. This lysine residue clashes with Trp-574 in the predicted PfUba2Ufd structure, suggesting that binding leads to changes in the topology of the loop (Fig. 6e). Lys-34 of PfUbc9 may also, however, interfere with binding to HsUba2Ufd, as the predicted E1 interface presents potential clashes with Lys-486, Asn-501, and Lys-504 (not shown). Collectively, these findings reveal that co-evolution of substitutions in the E1 and E2 enzyme interfaces can account for the observed specificity in interactions between the human and P. falciparum proteins.
We next took an experimental approach to identify residues in PfUbc9 that are critical for functional E1 enzyme interactions. We designed and expressed PfUbc9 mutants in which domains or specific residues within the predicted E1 interface were substituted with their human counterparts (Fig. 7a). These mutants included a substitution of the first 81 amino acids of PfUbc9 with residues from HsUbc9(1–81), as well as more limited swaps encompassing the α1 helix and β1-β2 loop together (helix + loop), or individually (helix and loop). Mutants HP2 and HP3 contained loop or helix swaps in combination with other point mutations, respectively. The HP1 mutant contained substitutions at 5 residues predicted to make E1 contacts based on ScUbc9 and ScUba2ufd interactions (including residues Lys-5, Ala-13, and Pro-29 highlighted in Fig. 6, c–e) (19).
FIGURE 7.

Ubc9 chimeras restore E1 and E2 interaction. a, sequence of N-terminal residues of PfUbc9, HsUbc9, and humanized PfUbc9 chimeras. HsUbc9-specific residues are highlighted in dark red. Residues predicted to be critical for E1-E2 interactions are indicated in blue. Secondary structure is illustrated, as described above. b, GST-RanGAP1 conjugation assays comparing low and high PfUbc9 chimera concentrations. Reactions were performed in the presence of human E1-activating enzyme and human SUMO-1. Products were analyzed by immunoblot analysis with anti-GST antibodies.
RanGAP1 conjugation assays were performed to test whether the humanized PfUbc9 mutants could restore functional interactions with human E1. High and low E2 concentrations (80 and 800 nm) were used to detect a potentially broad range of E1 interactions (Fig. 7b). The 1–81 mutant exhibited RanGAP1 conjugation activity equivalent to that detected with HsUbc9 at low and high E2 concentrations, consistent with E1 interactions being mediated almost exclusively through the N-terminal domain. The helix + loop mutant also displayed conjugation activity at low E2 concentrations, but unexpectedly the observed activity was lower than that of the 1–81 mutant. When individual helix or loop substitution mutants were assayed at low E2 concentrations, no detectable activity was observed. RanGAP1 sumoylation was, however, detected with the individual α1 helix mutant when assayed at high E2 concentrations, indicating partial restoration of E1 interactions. In assays containing P. falciparum E1, the α1 helix substitution mutant also displayed partial activity, whereas the helix + loop mutant exhibited no detectable activity (Fig. 8). These findings indicate that amino acid substitutions in both the α1 helix and β1-β2 loop of PfUbc9 contribute to the observed specificity in E1 binding. Consistent with this, the HP2 mutant, combining the β1-β2 loop swap with two amino acid substitutions in the α1 helix, also showed partial activity at high E2 concentrations. Notably, the HP1 mutant showed no sumoylation activity at low or high E2 concentrations, indicating an important role for other residues in addition to those predicted to make direct E1 contacts based on yeast E1 and E2 interactions (Fig. 7b).
FIGURE 8.
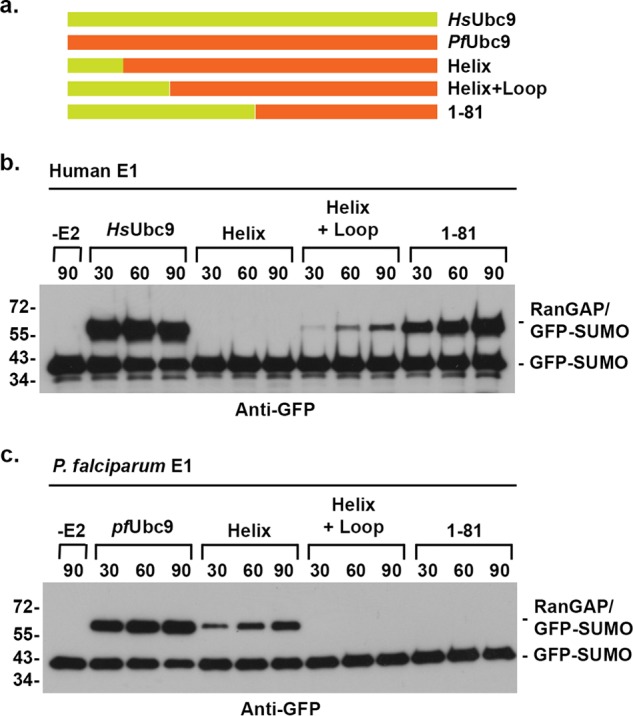
Ubc9 chimeras affect human and P. falciparum E1 enzyme interactions. a, schematic of wild type and chimeric Ubc9 proteins. b, assays were performed in the presence of human GFP-tagged SUMO-1, human E1-activating enzyme, and the indicted Ubc9 protein. c, assays were performed in the presence of human GFP-tagged SUMO-1, P. falciparum E1-activating enzyme, and the indicated Ubc9 protein. Reaction products were detected by immunoblot analysis with anti-GFP antibodies.
DISCUSSION
Sumoylation is essential for cell viability. This, in combination with a dependence on multiple distinct enzymatic steps, makes the sumoylation pathway an attractive target for pathogen drug design. Although expected to be conserved and essential in protozoans, relatively few studies have investigated the roles of sumoylation in this important class of organisms. Previous studies have identified genes encoding for putative components of the sumoylation pathway in P. falciparum (12, 13), however, biochemical characterization of these components has not been extensively investigated. Biochemical analysis of factors involved in SUMO conjugation is of particular importance, given the high degree of primary sequence identity between the E1 and E2 enzymes involved in ubiquitylation, sumoylation, and neddylation. Using in vitro biochemical assays, we have demonstrated that genes coding for predicted SUMO E1 and E2 enzymes are true SUMO pathway orthologs. Importantly, we have also provided evidence that functional interactions between P. falciparum and human E1 and E2 enzymes are molecularly distinct due to a divergence of residues in the E1 and E2 interface.
SUMO Functions
Sumoylation is an essential regulator of gene expression, DNA repair, and chromosome segregation, and has also emerged as a vital component of cellular stress tolerance pathways (10, 40–42). In response to a variety of stresses, including heat shock, hypoxia, and oxidative stress, sumoylation of a wide variety of proteins is dramatically up-regulated (43, 44). The enhanced sumoylation that occurs in response to stress promotes cell survival, although the underlying mechanisms are still not fully understood. Our analysis of sumoylation levels in malaria parasites revealed a notable increase in high molecular weight conjugates in trophozoites, consistent with up-regulation of SUMO gene expression during mid to late stages of the IDC (45). It is during this stage of parasite development that hemoglobin is taken up from the host erythrocyte and digested into amino acids and free heme. Although most free heme is sequestered into non-toxic crystalline hemozoin, heme that escapes sequestration is oxidized, leading to elevated levels of reactive oxygen species including superoxide and hydrogen peroxide (11). P. falciparum is known to utilize thioredoxin- and glutathione-based systems to maintain redox equilibrium and limit oxidative stress (46), and our findings reveal that sumoylation may also be an important part of the oxidative stress response during the IDC. Of particular interest, the toxicity of a number of antimalarial drugs, including chloroquine-related drugs and artemisinin, is related at least in part to induction of heme-mediated oxidative stress (47). Thus, inhibitors of the natural oxidative stress response pathways in the parasite, including sumoylation, could prove effective in combination therapies with these drugs. Although it remains to be formally demonstrated that sumoylation itself is essential for P. falciparum development and replication, inhibitors of PfSENP1 block parasite replication during late stages of the IDC (13).
P. falciparum SUMO Substrate Recognition
As in other organisms, the P. falciparum sumoylation pathway is predicted to contain a single heterodimeric E1-activating enzyme and a single E2-conjugating enzyme (Fig. 1b). We also identified a single PIAS-like E3 ligase encoded by the P. falciparum genome, which is predicted to regulate the modification of specific SUMO substrates (14). In addition to being E3-mediated, SUMO substrate recognition may also occur through direct binding of the E2-conjugating enzyme to ΨKXE consensus motifs in protein targets (15, 38, 48). Our in vitro biochemical studies revealed that PfUbc9 recognizes the ΨKXE consensus motif in RanGAP1, and is therefore capable of also playing a direct role in substrate recognition (15). Although direct evidence for conjugation to lysines within this same motif remains to be obtained in P. falciparum, at least a subset of candidate SUMO substrates identified in red blood cell-cultured parasites contains ΨKXE motifs (12).
The first enzymatic step in sumoylation involves ATP-dependent activation of the C-terminal residue of SUMO and formation of a thioester intermediate with the active site cysteine of the E1-activating enzyme. SUMO is subsequently transferred from the E1 enzyme to the E2-conjugating enzyme, a step that also involves formation of a thioester intermediate (49). The transfer of SUMO from E1 to E2 involves direct binding between the two enzymes, as revealed by both biochemical and structural studies of yeast and human enzymes (16, 19, 39). Our analysis of the P. falciparum E1 and E2 enzymes, PfAos1/PfUba2 and PfUbc9, respectively, provided several lines of evidence to indicate that the interactions between these enzymes are molecularly distinct from interactions between their human counterparts. First, we found that SUMO was effectively conjugated to RanGAP1 in reactions containing P. falciparum E1 and E2 enzymes, but not in reactions containing combinations of human and P. falciparum enzymes. Second, ITC analysis revealed robust interactions between PfUbc9 and PfUba2ufd as well as between HsUbc9 and HsUba2ufd, but failed to detect interactions between the P. falciparum and human enzymes. Last, we solved a crystal structure of PfUbc9 and used in silico protein-protein docking to reveal interactions with the predicted structure of the PfUba2ufd domain. In comparison with known or predicted structures of yeast and human E1 and E2 interactions, this analysis revealed amino acid substitutions in PfUbc9 that explain its specific interactions with the P. falciparum E1 enzyme.
In particular, our analysis revealed divergent residues in the α1 helix and the β1-β2 loop of PfUbc9 that enable interactions with P. falciparum E1 but prevent interactions with the human E1. Analysis of chimeric Ubc9 proteins indicated that residues in both the α1 helix and the β1-β2 loop contribute to specific binding, as substituting one region or the other of PfUbc9 with residues from HsUbc9 was insufficient to promote full RanGAP1 conjugation activity in reactions with human E1. Similarly, substitution of the α1 helix alone in PfUbc9 only partially reduced activity in reactions with P. falciparum E1. Substituting both the α1 helix and the β1-β2 loop regions of PfUbc9 with residues from HsUbc9 was sufficient to enable interactions with human E1, as indicated by RanGAP1 conjugation activity. Surprisingly, however, full activity was only seen when residues 1–81 of PfUbc9 were substituted with residues from HsUbc9. It is not clear how the additional residues C-terminal to the β1-β2 loop region affect RanGAP1 conjugation, as these residues are not predicted to affect E1 binding or RanGAP1 interactions based on available structures (15, 19). It is possible that these additional residues may allow for more optimal folding of the chimeric protein. Notably, however, the majority of residues divergent between human and P. falciparum are surface exposed, whereas the hydrophobic core is highly conserved. Within the α1 helix and β1-β2 loop region, for example, all divergent residues are surface exposed. Thus, it is not expected that the domain swaps made to generate chimeric proteins should have major effects on overall structural integrity. Similar heterologous conjugation studies have been performed with HsUbc9 and with ScE1 (50). Unlike human and P. falciparum enzymes, human and yeast enzymes formed functional interactions, although at low efficiency.
PfUbc9 Conservation
Ubc9 is highly conserved across species, sharing >60% identity between human and P. falciparum and >95% identity among Plasmodium species. This high degree of conservation likely reflects selective pressure to maintain functional interactions with multiple factors, including SUMO, the E1-activating enzyme, E3 ligases, as well as substrates containing consensus modification sites. Our findings that PfUbc9 effectively conjugates human SUMOs to mammalian RanGAP1 are consistent with the high degree of conservation of residues that form the PfUbc9 active site and that are required for both substrate recognition and SUMO thioester formation. The less stringent conservation of residues required for E1 interaction is consistent with observations regarding related ubiquitin E2-conjugating enzymes, which in general share a highly conserved catalytic core and a more divergent outer “shell” important for selective E1 binding (51). Importantly, amino acid substitutions in PfUbc9 believed to be critical for SUMO E1 interaction, as exemplified by K5 and A13, are accompanied by compensatory amino acid substitutions within the PfUba2ufd interface, indicative of co-evolution of the two proteins to support species-specific functionality.
In summary, we have definitively identified the P. falciparum SUMO E1-activating and E2-conjugating enzymes and demonstrated that the molecular basis for their functional interactions is distinct from that of their human counterparts. These findings provide the basis for identification of small molecule inhibitors that could be used to selectively block parasite sumoylation. In addition to their clinical value, these inhibitors could also be used to investigate the essential functions of sumoylation during P. falciparum development that, as indicated by our findings, are likely to include an important role in oxidative stress response.
Acknowledgments
We thank members of the Matunis and Bosch laboratories, Dr. David Sullivan, and Dr. Sean Prigge for helpful discussions and assistance during the course of the studies. We also thank the beamline scientists at NSLS beamline X25 for their help and support during data collection.
This work was supported, in whole or in part, by National Institutes of Health Grant GM060980 (to M. J. M.), the Sommer Scholar's Program (to K. R.), and the Bloomberg Philanthropies and Johns Hopkins Malaria Research Institute (to J. B. and M. J. M.).
The atomic coordinates and structure factors (codes 4JUE and 4M1N) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- IDC
- intra-erythrocyte developmental cycle
- SUMO
- small ubiquitin-related modifiers
- SENP
- SUMO-specific protease
- ITC
- isothermal titration calorimetry
- BCCP
- biotin carboxyl carrier protein.
REFERENCES
- 1. Murray C. J., Rosenfeld L. C., Lim S. S., Andrews K. G., Foreman K. J., Haring D., Fullman N., Naghavi M., Lozano R., Lopez A. D. (2012) Global malaria mortality between 1980 and 2010. A systematic analysis. Lancet 379, 413–431 [DOI] [PubMed] [Google Scholar]
- 2. O'Brien C., Henrich P. P., Passi N., Fidock D. A. (2011) Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr. Opin. Infect. Dis. 24, 570–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tilley L., Dixon M. W., Kirk K. (2011) The Plasmodium falciparum-infected red blood cell. Int. J. Biochem. Cell Biol. 43, 839–842 [DOI] [PubMed] [Google Scholar]
- 4. Bozdech Z., Llinás M., Pulliam B. L., Wong E. D., Zhu J., DeRisi J. L. (2003) The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 1, E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Foth B. J., Zhang N., Chaal B. K., Sze S. K., Preiser P. R., Bozdech Z. (2011) Quantitative time-course profiling of parasite and host cell proteins in the human malaria parasite Plasmodium falciparum. Mol. Cell. Proteomics 10, M110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le Roch K. G., Zhou Y., Blair P. L., Grainger M., Moch J. K., Haynes J. D., De La Vega P., Holder A. A., Batalov S., Carucci D. J., Winzeler E. A. (2003) Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301, 1503–1508 [DOI] [PubMed] [Google Scholar]
- 7. Foth B. J., Zhang N., Mok S., Preiser P. R., Bozdech Z. (2008) Quantitative protein expression profiling reveals extensive post-transcriptional regulation and post-translational modifications in schizont-stage malaria parasites. Genome Biol. 9, R177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung D. W., Ponts N., Cervantes S., Le Roch K. G. (2009) Post-translational modifications in Plasmodium. More than you think!. Mol. Biochem. Parasitol. 168, 123–134 [DOI] [PubMed] [Google Scholar]
- 9. Geiss-Friedlander R., Melchior F. (2007) Concepts in sumoylation. A decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 10. Tempé D., Piechaczyk M., Bossis G. (2008) SUMO under stress. Biochem. Soc. Trans. 36, 874–878 [DOI] [PubMed] [Google Scholar]
- 11. Becker K., Tilley L., Vennerstrom J. L., Roberts D., Rogerson S., Ginsburg H. (2004) Oxidative stress in malaria parasite-infected erythrocytes. Host-parasite interactions. Int. J. Parasitol. 34, 163–189 [DOI] [PubMed] [Google Scholar]
- 12. Issar N., Roux E., Mattei D., Scherf A. (2008) Identification of a novel post-translational modification in Plasmodium falciparum. Protein sumoylation in different cellular compartments. Cell. Microbiol. 10, 1999–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ponder E. L., Albrow V. E., Leader B. A., Békés M., Mikolajczyk J., Fonović U. P., Shen A., Drag M., Xiao J., Deu E., Campbell A. J., Powers J. C., Salvesen G. S., Bogyo M. (2011) Functional characterization of a SUMO deconjugating protease of Plasmodium falciparum using newly identified small molecule inhibitors. Chem. Biol. 18, 711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gareau J. R., Lima C. D. (2010) The SUMO pathway. Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bernier-Villamor V., Sampson D. A., Matunis M. J., Lima C. D. (2002) Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell 108, 345–356 [DOI] [PubMed] [Google Scholar]
- 16. Lois L. M., Lima C. D. (2005) Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 24, 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reverter D., Lima C. D. (2005) Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature 435, 687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bencsath K. P., Podgorski M. S., Pagala V. R., Slaughter C. A., Schulman B. A. (2002) Identification of a multifunctional binding site on Ubc9p required for Smt3p conjugation. J. Biol. Chem. 277, 47938–47945 [DOI] [PubMed] [Google Scholar]
- 19. Wang J., Taherbhoy A. M., Hunt H. W., Seyedin S. N., Miller D. W., Miller D. J., Huang D. T., Schulman B. A. (2010) Crystal structure of UBA2(ufd)-Ubc9. Insights into E1-E2 interactions in Sumo pathways. PloS One 5, e15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trager W., Jensen J. B. (2005) Human malaria parasites in continuous culture. 1976. J. Parasitol. 91, 484–486 [DOI] [PubMed] [Google Scholar]
- 21. Lambros C., Vanderberg J. P. (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 [PubMed] [Google Scholar]
- 22. Matunis M. J., Coutavas E., Blobel G. (1996) A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 135, 1457–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang X. D., Goeres J., Zhang H., Yen T. J., Porter A. C., Matunis M. J. (2008) SUMO-2/3 modification and binding regulate the association of CENP-E with kinetochores and progression through mitosis. Mol. Cell 29, 729–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu J., Zhu S., Guzzo C. M., Ellis N. A., Sung K. S., Choi C. Y., Matunis M. J. (2008) Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. J. Biol. Chem. 283, 29405–29415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Richardson S. M., Wheelan S. J., Yarrington R. M., Boeke J. D. (2006) GeneDesign. Rapid, automated design of multikilobase synthetic genes. Genome Res. 16, 550–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Szewczyk E., Nayak T., Oakley C. E., Edgerton H., Xiong Y., Taheri-Talesh N., Osmani S. A., Oakley B. R. (2006) Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 1, 3111–3120 [DOI] [PubMed] [Google Scholar]
- 27. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans P. R. (2011) An introduction to data reduction. Space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Long F., Vagin A. A., Young P., Murshudov G. N. (2008) BALBES. A molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) REFMAC5 dictionary. Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee. A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 36. Roy A., Xu D., Poisson J., Zhang Y. (2011) A protocol for computer-based protein structure and function prediction. J. Vis. Exp. 57, e3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gallagher J. R., Matthews K. A., Prigge S. T. (2011) Plasmodium falciparum apicoplast transit peptides are unstructured in vitro and during apicoplast import. Traffic 12, 1124–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sampson D. A., Wang M., Matunis M. J. (2001) The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 276, 21664–21669 [DOI] [PubMed] [Google Scholar]
- 39. Wang J., Hu W., Cai S., Lee B., Song J., Chen Y. (2007) The intrinsic affinity between E2 and the Cys domain of E1 in ubiquitin-like modifications. Mol. cell 27, 228–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cubeñas-Potts C., Matunis M. J. (2013) SUMO. A multifaceted modifier of chromatin structure and function. Dev. Cell 24, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wan J., Subramonian D., Zhang X. D. (2012) SUMOylation in control of accurate chromosome segregation during mitosis. Curr. Protein Pept. Sci. 13, 467–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jackson S. P., Durocher D. (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807 [DOI] [PubMed] [Google Scholar]
- 43. Golebiowski F., Matic I., Tatham M. H., Cole C., Yin Y., Nakamura A., Cox J., Barton G. J., Mann M., Hay R. T. (2009) System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2, ra24. [DOI] [PubMed] [Google Scholar]
- 44. Lee Y. J., Miyake S., Wakita H., McMullen D. C., Azuma Y., Auh S., Hallenbeck J. M. (2007) Protein SUMOylation is massively increased in hibernation torpor and is critical for the cytoprotection provided by ischemic preconditioning and hypothermia in SHSY5Y cells. J. Cereb. Blood Flow Metab. 27, 950–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Le Roch K. G., Johnson J. R., Florens L., Zhou Y., Santrosyan A., Grainger M., Yan S. F., Williamson K. C., Holder A. A., Carucci D. J., Yates J. R., 3rd, Winzeler E. A. (2004) Global analysis of transcript and protein levels across the Plasmodium falciparum life cycle. Genome Res. 14, 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jortzik E., Becker K. (2012) Thioredoxin and glutathione systems in Plasmodium falciparum. Int. J. Med. Microbiol. 302, 187–194 [DOI] [PubMed] [Google Scholar]
- 47. Müller I. B., Hyde J. E. (2010) Antimalarial drugs. Modes of action and mechanisms of parasite resistance. Future Microbiol. 5, 1857–1873 [DOI] [PubMed] [Google Scholar]
- 48. Rodriguez M. S., Dargemont C., Hay R. T. (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659 [DOI] [PubMed] [Google Scholar]
- 49. Johnson E. S. (2004) Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382 [DOI] [PubMed] [Google Scholar]
- 50. van Waardenburg R. C., Duda D. M., Lancaster C. S., Schulman B. A., Bjornsti M. A. (2006) Distinct functional domains of Ubc9 dictate cell survival and resistance to genotoxic stress. Mol. Cell. Biol. 26, 4958–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Wijk S. J., Timmers H. T. (2010) The family of ubiquitin-conjugating enzymes (E2s). Deciding between life and death of proteins. FASEB J. 24, 981–993 [DOI] [PubMed] [Google Scholar]
- 52. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Structure validation by Cα geometry. φ,ψ and C β deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]