

Background: Prefoldin consisting of six subunits prevents protein misfolding.
Results: Prefoldin prevented proteasome inhibitor- and endoplasmic reticulum stress-induced protein aggregation, and mutation of a prefoldin subunit resulted in loss of its activity.
Conclusion: In addition to its protein folding activity, prefoldin protects cells from aggregated protein-induced cell death.
Significance: Prefoldin is a quality control protein against protein aggregation.
Keywords: Cell Death, Chaperone Chaperonin, Neurodegeneration, Proteasome, Protein Aggregation, Prefoldin
Abstract
Prefoldin is a molecular chaperone composed of six subunits, PFD1–6, and prevents misfolding of newly synthesized nascent polypeptides. Although it is predicted that prefoldin, like other chaperones, modulates protein aggregation, the precise function of prefoldin against protein aggregation under physiological conditions has never been elucidated. In this study, we first established an anti-prefoldin monoclonal antibody that recognizes the prefoldin complex but not its subunits. Using this antibody, it was found that prefoldin was localized in the cytoplasm with dots in co-localization with polyubiquitinated proteins and that the number and strength of dots were increased in cells that had been treated with lactacystin, a proteasome inhibitor, and thapsigargin, an inducer of endoplasmic reticulum stress. Knockdown of prefoldin increased the level of SDS-insoluble ubiquitinated protein and reduced cell viability in lactacystin and thapsigargin-treated cells. Opposite results were obtained in prefoldin-overexpressed cells. It has been reported that mice harboring a missense mutation L110R of MM-1α/PFD5 exhibit neurodegeneration in the cerebellum. Although the prefoldin complex containing L110R MM-1α was properly formed in vitro and in cells derived from L110R MM-1α mice, the levels of ubiquitinated proteins and cytotoxicity were higher in L110R MM-1α cells than in wild-type cells under normal conditions and were increased by lactacystin and thapsigargin treatment, and growth of L110R MM-1α cells was attenuated. Furthermore, the polyubiquitinated protein aggregation level was increased in the brains of L110R MM-1α mice. These results suggest that prefoldin plays a role in quality control against protein aggregation and that dysfunction of prefoldin is one of the causes of neurodegenerative diseases.
Introduction
Prefoldin is a molecular chaperone found in eukarya and archaea domains and assists the folding of newly synthesized polypeptide chains such as actin and tubulin (1, 2). Prefoldin binds to proteins that had been synthesized in ribosomes and transports them to chaperonin Tric/CCT in cooperation with HSP70 and HSP40 (1–4). Prefoldin thus plays a role in maintaining homeostasis in cells through stimulation of proper folding of proteins. Prefoldin is composed of six subunits containing two α-subunits (PFD3 and PFD5) and four β-subunits (PFD1, PFD2, PFD4, and PFD6). The coiled-coiled regions present in both N- and C-terminal regions in prefoldin form a “jellyfish-like” structure and bind to substrates with their tentacle-like structures (5, 6). This unique structure of prefoldin is thought to determine specificity of substrate recognition by prefoldin. Furthermore, it has been reported that archaeal prefoldin stimulates formation of soluble amyloid β oligomers to inhibit their fibril formation in vitro (7) and that human prefoldin inhibits Aβ fibrillation to form nontoxic Aβ aggregates (8). We have reported that prefoldin inhibited aggregate and inclusion formation of exogenously added pathogenic huntingtin, a cause of Huntington disease (9). These findings suggest that prefoldin plays a modifier role against the toxicity of misfolded proteins, including proteins that cause neurodegenerative diseases. Little is known, however, about the physiological role of prefoldin in neurodegenerative diseases.
In addition to functioning as a subunit of prefoldin, each prefoldin subunit has its respective function. MM-1α is PFD5 and has been identified by us as a c-MYC-binding protein (10). MM-1α/PFD5 is a tumor suppressor protein and inhibits c-Myc function in various ways, resulting in suppression of cell cycle movement and cell transformation (11–14). PFD3 is a pVHL-binding protein (VBP1) and stimulates protein degradation through the ubiquitin-proteasome system (15, 16). PFD2 makes a large protein complex to regulate the expression of nutrition-related genes (17). Mice harboring a missense mutation of the MM-1α/PFD5 gene that causes an amino acid substitution from leucine to arginine at amino acid number 110 (L100R) have recently been established after exposure of mice to N-ethyl-N-nitrosourea (18). L100R MM-1α mice exhibit various phenotypes, including cerebellar atrophy with death of Purkinje cells and male infertility (18). PFD1-knock-out mice also exhibit cerebellar atrophy (19). It is not known whether phenotypes of these mice are due to dysfunction of the prefoldin complex or of its subunits. Because an antibody that recognizes the prefoldin complex but not its subunits is not available and because antibodies that recognize prefoldin subunits have been used to detect the prefoldin complex, it is not possible to distinguish functions of the prefoldin complex and its subunits by using these antibodies.
In this study, we first established a monoclonal antibody that recognizes the prefoldin complex but not its subunits and then examined the effect of prefoldin on lactacystin-induced polyubiquitination of proteins and cell toxicity. The results showed that prefoldin prevented polyubiquitination of proteins and subsequent cell toxicity and that polyubiquitinated proteins were increased in L100R MM-1α mice irrespective of formation of the prefoldin complex, suggesting that recognition activity of prefoldin in L100R MM-1α mice is decreased. We discuss prefoldin-dependent protection mechanisms of neuronal cells against the toxicity.
EXPERIMENTAL PROCEDURES
Cells and Mice
Human HeLa and mouse myeloma X63/Ag8-653 cells were purchased from American Tissue Culture Collection (ATCC) and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% calf serum and in RPMI 1640 medium with 10% fetal calf serum, respectively. ATG5(+/+) and ATG(−/−) cells were kindly provided by Dr. Noboru Mizushima. Cell lines from wild-type and L100R MM-1α mice were established as follows. Whole cells from newborn mice were cut out, digested with trypsin, and seeded on a 10-cm dish in DMEM with 10% calf serum. Cells were then transfected with an expression vector for T antigen of simian virus 40 (SV40), pMTI (20). About 2 weeks after transfection, immortalized cells appeared and were cloned.
Homozygous C57BL/6-Pfdn5nmf5a/Pfdn5nmf5a mice harboring L110R mutation in MM-1α/PFD5 (18) were kindly provided by Dr. Patsy M. Nishina and are referred to here as MM-1α L110R mice. C57BL/6 and MM-1α L110R mice were fed a normal diet (D12337, Research Diets, Inc., New Brunswick, NJ).
Establishment of an Anti-prefoldin Monoclonal Antibody
Four prefoldin subunits (PFD1, PFD2, PFD5, and PFD6) and two prefoldin subunits (PFD3 and PFD4) were fused with glutathione S-transferase (GST) and maltose-binding protein, respectively. The proteins were expressed in and purified from Escherichia coli BL21 and digested with PreScission protease (GE Healthcare) to cleave off GST and maltose-binding protein as described previously (21). Ten μm each of the six GST or maltose-binding protein-free prefoldin subunits in PBS(−) were mixed and incubated with urea at a final concentration of 2 m for 30 min on ice. After dialysis of prefoldin subunits against PBS(−), NaCl was added to the prefoldin solution at a final concentration of 200 mm, and the solution was concentrated by using Vivaspin 20 (GE Healthcare) and applied onto a gel filtration column (HiLoad 16/600 Superdex 200, GE Healthcare) in an HPLC system (AKTA explore 10s/100 UNICORN 5.0, GE Healthcare). After elution of samples with PBS(−), fractions containing the prefoldin complex were obtained. The prefoldin complex obtained was then treated with 3.5 mm disuccinimidyl suberate for 2 h on ice, dialyzed against PBS(−), and injected into footpads of rats. Two weeks after immunization, lymphocytes were extracted from rats, fused with mouse myeloma X63/Ag8-653 cells, and cultured. Anti-prefoldin antibodies present in culture media were screened by using the enzyme-linked immunosorbent assay (ELISA). In the first screening by ELISA, the prefoldin complex was used as an immunogen on plates, and positive clones were then screened by ELISA with a mixture of the six prefoldin subunits on plates (second screening).
Knockdown and Overexpression of Prefoldin
Nucleotide sequences of the upper and lower strands for siRNA were as follows: 5′-GCCUAGUGAUCGAUACACUGA-3′ and 5′-AGUGUAUCGAUCACUAGGCUG-3′ for PFD2; 5′-GUCUGAACGUGCUGAACAAGA-3′ and 5′-UUGUUCAGCACGUUCAGACAG-3′ for PFD5, and 5′-CGUACGCGGAAUACUUCGATT-3′ and 5′-UCGAAGUAUUCCGCGUACGTT-3′ for luciferase. HeLa cells were first transfected with 5 pm siRNA targeting PFD2, PFD5, and luciferase using Lipofectamine 2000 reagent (Invitrogen). Forty eight h after transfection, 10 μm lactacystin was added to the medium, and the cells were cultured for another 24 h. HeLa cells in a 10-cm dish were transfected with 100 ng each of CMV promoter-based expression vectors for PFD1–PFD6 using Lipofectamine 2000. Twenty four h after transfection, proteins extracted from cells were subjected to Western blotting, filter trap, glycerol density gradient centrifugation, and MTT2 and LDL assays as described below.
Western Blotting
Proteins were extracted from cells after incubation of cells with a buffer containing 50 mm Tris-HCl (pH 7.4), 120 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, and protease inhibitors (20 mg/ml leupeptin, 10 mg/ml pepstatin A, 10 mg/ml aprotinin, 2 mm phenylmethylsulfonyl fluoride) for 15 min at 4 °C. Proteins were then dissolved in the buffer containing 30 mm Tris-HCl (pH 6.8), 3% SDS, 6% β-mercaptoethanol, and 30% glycerol, boiled for 5 min, and subjected to Western blot analysis with anti-PFD1 (PA006499, Sigma), anti-PFD2 and anti-PFD3 (K-13, Santa Cruz Biotechnology), anti-PFD4 and anti-PFD5 (S-20, Santa Cruz Biotechnology), anti-PFD6 (AP2836a, Abgent, San Diego), anti-LC3 (MBL, Nagoya, Japan), anti-p62 (Progen Biotechnik GmbH, Heidelberg, Germany), and anti-actin (Chemicon) antibodies. Proteins were then reacted with an IRDye800- or Alexa Fluor 680-conjugated secondary antibody and visualized by using an infrared imaging system (Odyssey, LI-COR). Anti-PFD2 and anti-PFD4 antibodies were established by injection of GST-PFD2 and GST-PFD4, respectively, into rabbits. The anti-PFD2 antibody was purified from rabbit serum through an affinity column with GST-PFD2, and serum from GST-PFD4-injected rabbits was used as the anti-PFD4 antibody. To separate SDS-soluble and SDS-insoluble proteins, proteins were extracted from cells after incubation of cells with the buffer containing 50 mm Tris-HCl (pH 6.8), 0.1% SDS, 2% β-mercaptoethanol, and 8.7% glycerol for 15 min at 4 °C and centrifuged at 12,000 rpm for 10 min at 4 °C. Supernatant and pellet fractions were assigned to SDS-soluble and SDS-insoluble proteins, respectively. Pellet fractions were dissolved in the buffer containing 30 mm Tris-HCl (pH 6.8), 1% SDS, 2% 2-mercaptoethanol, and 10% glycerol and sonicated using a water bath-type sonicator at 4 °C twice for 1 min. Both SDS-soluble and SDS-insoluble proteins were then subjected to Western blotting as described above.
Glycerol Density Gradient Centrifugation to Separate the Prefoldin Complex
Proteins were extracted from HeLa cells as described above and separated by glycerol density gradient centrifugation at 40,000 rpm for 16 h at 4 °C using an SW41 rotor (Beckman Instruments). After samples had been fractionated into 21 fractions, proteins in fractions were precipitated with acetone at −80 °C for 1 h, dissolved in an SDS buffer containing 50 mm Tris-HCl (pH 6.8), 1% SDS, 2% β-mercaptoethanol, and 8.7% glycerol, and subjected to Western blotting or Coomassie Brilliant Blue staining on 15% SDS-containing polyacrylamide gels. Aldolase (160 kDa), bovine serum albumin (BSA) (56 kDa), and RNase A (13 kDa) were used as molecular weight markers.
Immunofluorescence Microscopy
Cells were cultured on coverslips in a 6-cm dish and treated with 10 μm lactacystin for 24 h or 2.5 and 5.0 μm thapsigargin for 8 h. The cells were then fixed with 4% paraformaldehyde in PBS at 37 °C for 20 min. After treatment of cells with 50 μg/ml digitonin for 5 min at room temperature, the cells were incubated with a blocking buffer containing 5% calf serum, 20% glycerol, and 0.02% Triton X-100 at room temperature for 16 h. The cells were stained with anti-prefoldin (1:100), anti-PFD2 (1:100), anti-PFD5 (1:50), and anti-polyubiquitin (1:100, FK2, MBL) antibodies and then reacted with Alexa Fluor 488- or Alexa Fluor 594-conjugated anti-rabbit, anti-mouse, or anti-rat IgG antibody (1:100, Invitrogen). Images were observed using an immunofluorescence microscope (BZ-8000, Keyence, Osaka, Japan). An anti-PFD5 antibody for immunofluorescence detection was developed by us after immunization of rabbits with recombinant GST-PFD5 and purified from serum through an affinity column with GST-PFD5.
Filter Trap Assay of Cell Lysates
Cells were suspended in PBS containing protease inhibitors and sonicated using a water bath-type sonicator at 4 °C for 5 min. Lysates were adjusted to a final concentration of 1% SDS/PBS, boiled for 10 min, and filtered through a cellulose acetate membrane (0.2 μm) using a dot blotter (Bio-dot SF, Bio-Rad). The membrane was incubated with 5% skim milk in PBS overnight and reacted with a mouse anti-polyubiquitin antibody (FK2, MBL). The membrane was then reacted with an Alexa Fluor 680-conjugated secondary antibody, and proteins on the membrane were visualized by an Odyssey system.
Tissue Preparation and Immunohistochemistry
The brains of male mice at 17 weeks of age were frozen and embedded with O.T.C. compound. The brains were cut into 10-μm-thick slices using a cryostat, fixed for 20 min with 4% paraformaldehyde in PBS, and then washed with PBS. The brain slices were treated with 0.3% H2O2 in methanol for 30 min at room temperature and reacted with an anti-ubiquitin antibody (1:100, Cell Signaling Technology) for 1 h at room temperature. After several washes, sections were subjected to DAB staining using a VECTASTAIN ABC kit (Vector Laboratories). Stained images were then observed under a fluorescent microscope (Biorevo BZ-9000).
Statistical Analyses
Data are expressed as means ± S.D. Statistical analyses were performed using the Tukey-Kramer test.
Ethics Statement
All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the protocols were approved by the Committee for Animal Research at Hokkaido University (permit numbers 06-0467 and 08-0467).
RESULTS
Establishment of a Monoclonal Antibody That Recognizes the Prefoldin Complex but Not Prefoldin Subunits
Because each prefoldin subunit contains activity independent of the prefoldin complex and because anti-prefoldin subunit antibodies have been used to detect both the prefoldin complex and its subunits, it has not been possible to distinguish characteristics of the prefoldin complex and its subunits. We therefore tried to establish an antibody that recognizes the prefoldin complex but not prefoldin subunits. Six recombinant subunits (PFD1–PFD6) were expressed in and purified from E. coli. PFD1–PFD6 were denatured by 2 m urea and then renatured by dialysis against PBS(−) to assemble the prefoldin complex. The prefoldin complex was then applied to a gel filtration column in parallel with marker proteins containing aldolase (160 kDa), bovine serum albumin (BSA) (67 kDa), and RNase A (13.7 kDa). The elution profile showed three major peaks, and the second peak of ∼90 kDa in molecular mass was found to correspond to the prefoldin complex (Fig. 1A). The prefoldin complex obtained was cross-linked with disuccinimidyl suberate to stabilize the complex, and rats were immunized with the prefoldin complex. Lymphocytes in immunized rats were then fused with mouse myeloma X63/Ag8-653 cells, and antibodies secreted into culture media were screened by ELISA. Positive clones obtained on ELISA plates with the prefoldin complex were further screened on those with a mixture of six prefoldin subunits. Finally, a monoclonal antibody, clone 1E9, that recognizes the prefoldin complex but not its subunits was obtained, and hereafter this antibody is called anti-prefoldin antibody. When HeLa cells were immunostained with the anti-prefoldin antibody, cytoplasmic staining with dots was observed (Fig. 1B). This staining image disappeared or was weakened after the anti-prefoldin antibody was first reacted with cross-linked prefoldin complex but not with the mixture of six prefoldin subunits (Fig. 1C). Furthermore, the prefoldin complex composed of six recombinant subunits were separated on a blue-native gel and stained with Coomassie Brilliant Blue. An ∼90-kDa band corresponding to the prefoldin complex was obtained (Fig. 1D-a, left panel). The prefoldin complex was reacted with anti-prefoldin, anti-PFD5, and anti-PFD2 antibodies by Western blot analysis (Fig. 1D-a, 3 right panels). This band of the prefoldin complex disappeared after the anti-prefoldin antibody was first reacted with cross-linked prefoldin complex but not with the mixture of six prefoldin subunits (Fig. 1D-b). These results indicate the specificity of the anti-prefoldin antibody.
FIGURE 1.

Localization of prefoldin. A, six purified prefoldin subunits were denatured and then renatured as described under “Experimental Procedures.” The protein mixture was then applied onto a gel filtration column (HiLoad 16/600 Superdex 200). mAU, measurable absorbance unit. B, HeLa cells were stained with an anti-prefoldin antibody (clone 1E9), and nuclei were stained with DAPI (left and right panels, respectively). Cells were then reacted with a secondary antibody, and cell images were observed using an immunofluorescence microscope. C, HeLa cells were stained with an anti-prefoldin antibody (clone 1E9) (panels a and b). The anti-prefoldin antibody was first incubated with the cross-linked prefoldin complex (panel c) or with a mixture of six prefoldin subunits (panel d) for 1 h at 4 °C and used for immunostaining of HeLa cells. D-a, reconstituted prefoldin complex composed of 25 μg each of six recombinant prefoldin subunits was separated on a blue native gel and stained with Coomassie Brilliant Blue (CBB) (left panel). The prefoldin complex (150 μg) and 25 μg each of six recombinant prefoldin subunits were separated on blue native gels and analyzed by Western blotting with anti-prefoldin complex, anti-PFD5, and anti-PFD2 antibodies (right 3 panels). D-b, anti-prefoldin complex antibody was first reacted with the reconstituted prefoldin complex or with six prefoldin subunits for 2 h at 4 °C and used for Western blot analysis of filters, in which the prefoldin complex and six prefoldin subunits were loaded.
Effect of a Proteasome Inhibitor on Prefoldin Localization
HeLa cells were treated with 10 μm lactacystin, a proteasome inhibitor, for 24 h and immunostained with anti-prefoldin, anti-PFD2, and anti-PFD5 antibodies. Without lactacystin treatment, similar patterns of cytoplasmic staining with faint dots were observed with all three antibodies, but nuclear staining patterns were also observed with anti-PFD2 and anti-PFD5 antibodies (Fig. 2A). After cells had been treated with lactacystin, the number and strength of dots were remarkably increased (Fig. 2A). Because lactacystin is a proteasome inhibitor, ubiquitinated proteins in cells should be increased, and proteins in dots are likely to be aggregated proteins. To confirm this, HeLa cells were treated with lactacystin and stained with anti-prefoldin and anti-ubiquitin antibodies. Cytoplasmic staining patterns with dots were clearly observed with both antibodies from 16 h after lactacystin treatment and became more evident at 24 h (Fig. 2B). Dots observed with anti-prefoldin and anti-ubiquitin antibodies completely overlapped (Fig. 2C), suggesting that prefoldin is localized on ubiquitinated protein aggregates. To confirm whether proteins observed as dots are polyubiquitinated protein aggregates, proteins were extracted from lactacystin-treated cells by an SDS-containing buffer, and SDS-soluble and SDS-insoluble proteins were analyzed by Western blotting with an anti-polyubiquitin antibody (Fig. 2D). The results showed that the level of polyubiquitinated proteins in SDS-insoluble fractions was increased in a time-dependent manner, indicating that insoluble aggregated proteins with polyubiquitination were increased upon treatment of cells with lactacystin. The levels of autophagy-related proteins LC3-1 and LC3-2 were increased both in SDS-soluble and SDS-insoluble fractions, and p62 levels were increased and decreased in SDS-soluble and SDS-insoluble fractions, respectively, suggesting induction of the proteasome-induced autophagy reaction upon lactacystin treatment of cells. To examine whether localization of prefoldin in aggregated proteins is affected by autophagy induction, wild-type and knock-out cells of Atg5, a protein involved in the early phase of autophagy, were treated with lactacystin and stained with the anti-prefoldin antibody (Fig. 2E). Even without lactacystin treatment, prefoldin was localized in protein dots in Atg5(−/−) cells, indicating that clearance activity against damaged proteins in cells is decreased due to dysfunction of the autophagy pathway as described previously (22). After treatment of cells with lactacystin, the localization levels of prefoldin in dots were increased both in Atg5(+/+) and Atg5(−/−) cells. These results indicate that lactacystin-induced prefoldin localization in dots occurs independently of the autophagy pathway but that prefoldin is localized in protein aggregates in response to autophagy dysfunction.
FIGURE 2.

Localization of prefoldin on polyubiquitinated protein aggregates in lactacystin-treated HeLa cells. A, HeLa cells were cultured with or without 10 μm lactacystin (Lac-cys) for 24 h and stained with anti-prefoldin, anti-PFD2, and anti-PFD5 antibodies. Cell images were then obtained after cells had been reacted with the respective secondary antibodies. Control indicates cells without lactacystin treatment. B, HeLa cells were cultured with 10 μm lactacystin for 0, 8, 16, and 24 h and stained with anti-prefoldin and anti-polyubiquitin antibodies. Cell images were then obtained after cells had been reacted with the respective secondary antibodies. C, HeLa cells were cultured with or without 10 μm lactacystin for 24 h. Cells were then stained with rat anti-prefoldin and rabbit anti-ubiquitin antibodies and reacted with Alexa Fluor 488-conjugated anti-rat IgG and Alexa Fluor 594-conjugated anti-rabbit IgG antibodies, respectively. Both cell images were merged. Control indicates cells without lactacystin treatment. D, HeLa cells were treated with 10 μm lactacystin for 0, 8, 16, and 24 h. Proteins extracted from cells were treated with 1% SDS and centrifuged. SDS-soluble proteins (supernatant after centrifugation) and SDS-insoluble proteins (pellet after centrifugation) were analyzed by Western blotting with anti-polyubiquitin, anti-LC3, anti-p62, and anti-actin antibodies. E, Atg5(+/+) and Atg5(−/−) cells were cultured with or without 10 μm lactacystin for 24 h and stained with an anti-prefoldin antibody. Cell images were then obtained after cells had been reacted with an Alexa Fluor 488-conjugated anti-rat IgG antibody. Control indicates cells without lactacystin treatment.
Effect of Prefoldin Expression on Aggregation of Ubiquitinated Protein
To examine the physiological role of prefoldin in protein quality control, HeLa cells were transfected with siRNAs targeting PFD2, PFD5, and luciferase as a negative control, and the effect of the reduced level of prefoldin on protein aggregation was examined. Because the prefoldin complex contains two α-type subunits (PFD3 and PFD5) and four β-type subunits (PFD1, PFD2, PFD4, and PFD6), siRNAs targeting PFD5 (α-type subunit) and PFD2 (β-type subunit) were chosen for knockdown. First, expression levels of prefoldin subunits in siRNA-transfected cells were examined by Western blotting. Because the prefoldin complex is more stable than its subunits and because excess prefoldin subunits that do not form the prefoldin complex are degraded by the ubiquitin-proteasome system, expression levels of all of the prefoldin subunits were decreased by siRNA as described previously (23) (Fig. 3A). To examine whether knockdown of a prefoldin subunit affects formation of the prefoldin complex, proteins extracted from siRNA-transfected cells were separated by glycerol density gradient centrifugation followed by Western blotting with anti-PFD2 and anti-PFD5 antibodies. The prefoldin complex was observed in fractions 8–10 by estimating fractions compared with those of protein markers, aldolase (160 kDa), BSA (56 kDa), and RNase A (13 kDa), in the gradient. As shown in Fig. 3B, amounts of the prefoldin complex in cells that had been transfected with PFD2 and PFD5 siRNAs were reduced to 12 and 15%, respectively, of that in luciferase siRNA-transfected cells, indicating that RNAi-mediated knockdown of PFD2 and PFD5 effectively reduced formation of the prefoldin complex.
FIGURE 3.

Effect of prefoldin knockdown on lactacystin-induced cell toxicity. A, HeLa cells were transfected with siRNAs targeting PFD2, PFD5, and luciferase (Luc). Twenty four h after transfection, proteins extracted from transfected cells were analyzed by Western blotting with anti-prefoldin subunits and anti-actin antibodies. B, proteins extracted from HeLa cells as described in the legend of A were separated by glycerol density gradients. Proteins in each fraction were then analyzed by Western blotting with anti-PFD2 (B-a) and anti-PFD5 (B-b) antibodies as described under “Experimental Procedures.” Total indicates proteins extracted from HeLa cells. C, HeLa cells were transfected with siRNAs targeting PFD2, PFD5, and luciferase. Twenty four h after transfection, cells were treated with 10 μm lactacystin for 24 h, and proteins extracted from the cells were treated with 1% SDS, analyzed by filter trap assays, and detected with an anti-ubiquitin antibody as described under “Experimental Procedures.” IB, immunoblot. D, intensities of bands in C were quantified. Values are means ± S.D. n = 3 experiments. Significance: **, p < 0.01. E, HeLa cells were transfected with siRNAs targeting PFD2, PFD5, and luciferase. Twenty four h after transfection, cells were treated with 10 μm lactacystin for 24 h, and their viability was examined by MTT assays. Values are means ± S.D. n = 4 experiments. Significance: **, p < 0.01.
Next, HeLa cells were transfected with PFD2, PFD5, and luciferase siRNAs. Forty eight h after transfection, cells were treated with lactacystin for 24 h. Proteins were then extracted with an SDS-containing buffer and subjected to filter trap assays, and proteins on filters were reacted with an anti-ubiquitin antibody. The results showed that knockdown of PFD5 and PFD2 significantly increased the amount of ubiquitinated SDS-insoluble aggregates in lactacystin-treated cells compared with that in control cells (Fig. 3, C and D). Furthermore, the effect of knockdown of prefoldin on cell viability was examined by MTT assays. As shown in Fig. 3E, knockdown of PFD5 and PFD2 significantly decreased the viability of lactacystin-treated cells.
To examine the effect of prefoldin on protein aggregation in lactacystin-treated cells, HeLa cells were first transfected with expression vectors for the six prefoldin subunits or a control vector. Forty eight h after transfection, the expression level of prefoldin and formation of the prefoldin complex in cells were examined by Western blotting and by glycerol density gradient centrifugation followed by Western blotting, respectively, with anti-PFD2 and anti-PFD5 antibodies. As shown in Fig. 4, A and B, the levels of expression and formation of prefoldin were increased. Next, HeLa cells were transfected with the six prefoldin subunits or a control vector. Twenty four h after transfection, cells were treated with lactacystin for 24 h, and proteins extracted by an SDS-containing buffer were subjected to filter trap and MTT assays. The results showed that overexpressed prefoldin reduced the level of ubiquitinated protein aggregates and increased cell viability in lactacystin-treated cells (Fig. 4, C–E). These results suggest that prefoldin participates in quality control against protein aggregation and resultant cell viability.
FIGURE 4.

Effect of overexpression of prefoldin on lactacystin-induced cell toxicity. A, HeLa cells were transfected with expression vectors for six prefoldin subunits. Forty eight h after transfection, proteins extracted from transfected cells were analyzed by Western blotting with anti-prefoldin subunits and anti-actin antibodies. B, proteins extracted from HeLa cells as described in the legend of A were separated by glycerol density gradients. Proteins in each fraction were then analyzed by Western blotting with anti-PFD2 (B-a) and anti-PFD5 (B-b) antibodies as described under “Experimental Procedures.” Total indicates proteins extracted from HeLa cells. C, HeLa cells were transfected with expression vectors for six prefoldin subunits. Forty eight h after transfection, cells were treated with 10 μm lactacystin for 24 h, and proteins extracted from the cells were treated with 1% SDS, analyzed by filter trap assays, and detected with an anti-ubiquitin antibody as described under “Experimental Procedures.” IB, immunoblot. D, intensities of bands in C were quantified. Values are means ± S.D. n = 3 experiments. Significance: **, p < 0.01. E, HeLa cells were transfected with expression vectors for six prefoldin subunits. Forty eight h after transfection, cells were treated with 10 μm lactacystin for 24 h, and their viability was examined by MTT assays. Values are means ± S.D. n = 4 experiments. Significance: *, p < 0.05.
Effect of a Point Mutation Located in the Coiled-coil Region in MM-1α/PFD5 on Protein Aggregation and Cell Viability
Mice harboring a missense mutation of the MM-1α/PFD5 gene that causes an amino acid substitution from leucine to arginine at amino acid 110 (L100R) have recently been established (18). These mice, L100R MM-1α mice, exhibit various phenotypes, including cerebellar atrophy with death of Purkinje cells and male infertility (18). It is not known whether the phenotype of L100R MM-1α mice is due to dysfunction of prefoldin or dysfunction of MM-1α/PFD5. To examine this issue, the prefoldin complex was assembled in vitro by using recombinant prefoldin subunits that had been expressed in and purified from E. coli. Wild-type MM-1α/PFD5 or L110R MM-1α/PFD5 was mixed with the other five prefoldin subunits as described above and applied onto a gel filtration column. As shown in Fig. 5A-a, similar elusion profiles of proteins between two samples containing wild-type MM-1α and L110R MM-1α were obtained. Two prefoldin complexes were then analyzed by Western blotting with anti-prefoldin subunit antibodies, and similar amounts of the six prefoldin subunits were observed in the two prefoldin complexes (Fig. 5A-b).
FIGURE 5.

Effect of L110R mutation of MM-1α/PFD5 on prefoldin formation. A, five prefoldin subunits (PFD1, PFD2, PFD3, PFD4, and PFD) and wild-type or L110R MM-1α/PFD5 were expressed in and purified from E. coli. The prefoldin complex was reconstituted using these six subunits as described under “Experimental Procedures” and applied onto a gel filtration column (a). mAU, measurable absorbance unit. Proteins in fractions corresponding to the prefoldin complex were analyzed by Western blotting with anti-prefoldin subunits antibodies (b). B, proteins extracted from wild-type and L110R MM-1α cells were separated by glycerol density gradients. Proteins in each fraction were then analyzed by Western blotting with anti-PFD2 and anti-PFD5 antibodies as described under “Experimental Procedures.” Total indicates proteins extracted from HeLa cells (B-a). Intensities of bands in B-a were quantified (B-b). C, proteins in fractions corresponding to the prefoldin complex shown in B-a were analyzed by Western blotting with anti-prefoldin subunits and anti-actin antibodies. IB, immunoblot.
A cell line derived from L100R MM-1α mice was then established by introduction of simian virus 40 (SV40) T-antigen into primary cells derived from wild-type and L100R MM-1α newborn mice. To examine whether L100R mutation of MM-1α/PFD5 affects formation of the prefoldin complex, proteins extracted from wild-type and L100R MM-1α cells were separated by glycerol density gradient centrifugation followed by Western blotting with anti-PFD2 and anti-PFD5 antibodies, and intensities of PFD2 and MM-1α/PFD5 were quantified. As shown in Fig. 5B, similar distribution patterns of PFD2 and MM-1α/PFD5 were obtained in both cell lines. Furthermore, proteins in prefoldin fractions were analyzed by Western blotting with anti-prefoldin subunits antibodies, and the results showed that all six subunits were present in these prefoldin fractions albeit with a slightly reduced expression level of subunits in L100R MM-1α cells (Fig. 5C). These results suggest that there is no difference or little difference of the prefoldin status in wild-type and L100R MM-1α cells.
Wild-type and L100R MM-1α cells were treated with various amounts of lactacystin for 24 h, and filter trap assays using SDS-insoluble proteins from cell extracts were carried out. Proteins on filters were then reacted with an anti-ubiquitin antibody, and their levels were quantified. As shown in Fig. 6, A-a and A-b, SDS-insoluble proteins from wild-type and L100R MM-1α cells were increased in a dose-dependent manner, and the amount of SDS-insoluble proteins in L100R MM-1α cells was larger than that in wild-type MM-1α cells. Furthermore, L100R MM-1α cells were found to be more susceptible than wild-type MM-1α cells to cytotoxicity of lactacystin as assessed by LDL assays (Fig. 6A-c). The effect of L110R mutation on cell growth was examined by trypan blue exclusion and MTT assays (Fig. 6, A-d and A-e, respectively). Both assays showed that growth of L100R MM-1α cells was attenuated compared with that of wild-type MM-1α cells. These results indicate that L100R MM-1α cells contain more SDS-insoluble ubiquitinated proteins than do wild-type MM-1α cells under normal culture conditions and that L100R MM-1α cells are more susceptible than wild-type MM-1α to lactacystin-induced stress, resulting in attenuation of cell growth and viability.
FIGURE 6.

Effect of L110R mutation of MM-1α/PFD5 on lactacystin- and thapsigargin-induced cell toxicity and cell growth. A-a, wild-type and L110R MM-1α cells were treated with various amounts of lactacystin for 24 h, and proteins extracted from the cells were treated with 1% SDS, analyzed by filter trap assays, and detected with an anti-ubiquitin antibody as described under “Experimental Procedures.” A-b, intensities of bands in A-a were quantified. Values are means ± S.D. n = 3 experiments. Significance: **, p < 0.01. A-c, wild-type and L110R MM-1α cells were treated with various amounts of lactacystin for 24 h, and cell toxicity was examined by LDL assays. n = 4 experiments. A-d and A-e, wild-type and L110R MM-1α cells were cultured and harvested for various times. Growth and viability of wild-type and L110R MM-1α cells were then examined by trypan blue exclusion (D) and MTT (E) assays. n = 4 experiments. B-a, wild-type and L110R MM-1α cells were treated with various amounts of thapsigargin for 8 h, and proteins extracted from the cells were treated with 1% SDS, analyzed by filter trap assays, and detected with an anti-ubiquitin antibody as described under “Experimental Procedures.” B-b, intensities of bands in B-a were quantified. B-c, wild-type and L110R MM-1α cells were treated with various amounts of thapsigargin for 8 h, and cell viability was measured by MTT assays. Values are means ± S.D. n = 3 experiments. Significance: *, p < 0.05; **, p < 0.01. IB, immunoblot. C, wild-type and L110R MM-1α cells were treated with 10 μm lactacystin for 24 h or with 1 μm thapsigargin for 8 h. Cells were then stained with a rat anti-prefoldin antibody and reacted with Alexa Fluor 488-conjugated anti-rat IgG antibody. Nuclei were stained with DAPI. Both cell images were merged. D, HeLa cells were treated with 2.5 and 5 μm thapsigargin for 8 h. Cells were stained as described in C.
To examine whether prefoldin is reacted with stress other than proteasomal inhibition, wild-type and L100R MM-1α cells were treated with various amounts of thapsigargin for 8 h, and filter trap and MTT assays were carried out. Thapsigargin is an inhibitor of endoplasmic reticulum Ca2+-ATPase and induces the endoplasmic reticulum stress. Although the amount of SDS-insoluble ubiquitinated proteins in L100R MM-1α cells tended to be larger than that in wild-type MM-1α cells by filter trap assays, the results were not statistically significant (Fig. 6, B-a and B-b). Cell viability of L100R MM-1α cells was, however, reduced by thapsigargin in a dose-dependent manner, and viability of L100R MM-1α cells was significantly lower than that of wild-type MM-1α cells. Furthermore, wild-type and L100R MM-1α cells were treated with lactacystin or with thapsigargin and stained with an anti-prefoldin antibody. As shown in Fig. 6C, the level of prefoldin with dots in L100R MM-1α cells was higher than that in wild-type MM-1α cells. Moreover, HeLa cells were treated with 2.5 and 5.0 μm thapsigargin for 8 h and stained with the anti-prefoldin antibody. The level of prefoldin with dots was increased in thapsigargin-treated HeLa cells (Fig. 6D). These results suggest that prefoldin is localized on protein aggregates that are induced both by lactacystin and thapsigargin.
Accumulation of Polyubiquitinated Protein in L100R MM-1α Mice
The results using the cell line from L100R MM-1α mice showed that L100R mutation in MM-1α causes accumulation of SDS-insoluble ubiquitinated proteins. To examine whether this occurs in L100R MM-1α mice, proteins extracted from the cerebellum of wild-type and L100R MM-1α mice were analyzed by Western blotting with anti-polyubiquitin and anti-MM-1α/PFD5 antibodies. Although the expression levels of MM-1α/PFD5 were not different in wild-type and L100R MM-1α mice, accumulation of polyubiquitinated proteins in L100R MM-1α mice was greater than that in wild-type MM-1α mice (Fig. 7A). Tissue sections of the brain were prepared and stained with an anti-ubiquitin antibody. As shown in Fig. 7B, stronger signals of ubiquitinated protein were observed in L100R MM-1α mice than in wild-type MM-1α mice. These results indicate that, like L100R MM-1α cells, L100R mutation in MM-1α/PFD5 stimulates polyubiquitination of proteins in mice.
FIGURE 7.
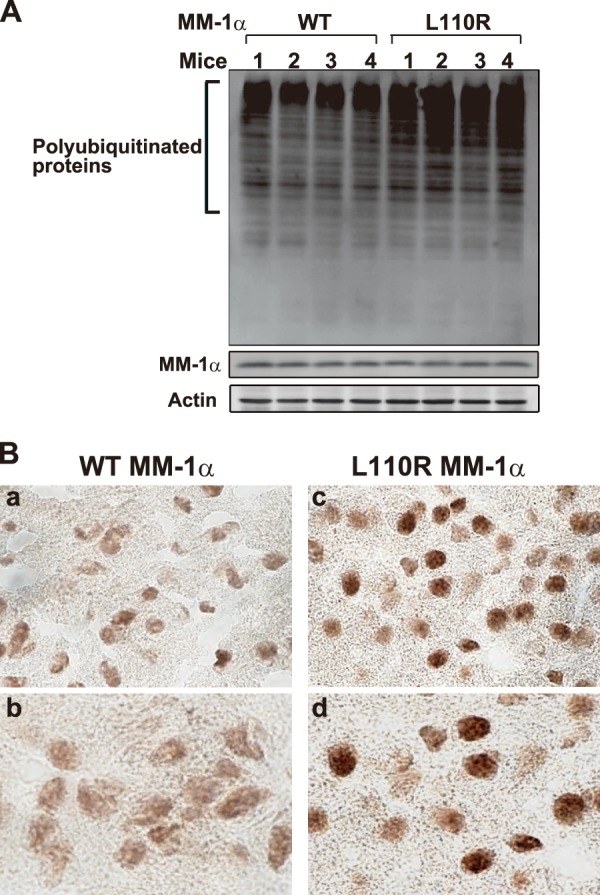
Accumulation of polyubiquitinated proteins in the brains of L110R MM-1α mice. A, proteins extracted from the cerebellum of wild-type and L100R MM-1α mice at 17 weeks of age were analyzed by Western blotting with anti-polyubiquitin, anti-MM-1α/PFD5, and anti-actin antibodies. B, brain slices from wild-type and L100R MM-1α mice at 17 weeks of age were reacted with an anti-polyubiquitin antibody, and images were visualized using an ABC kit as described under “Experimental Procedures.” Magnification of lenses used was 20 (panels a and c) and 60 (panels b and d).
DISCUSSION
Other than facilitation of folding of newly synthesized polypeptide chains such as actin and tubulin, the function of prefoldin is totally unknown. In this study, we first established an anti-prefoldin monoclonal antibody that recognizes the prefoldin complex but not its subunits. Using this antibody, prefoldin was found to be localized in the cytoplasm with dots/aggregates in which polyubiquitinated proteins were co-localized under normal conditions. Treatment of cells with lactacystin increased the number and strength of prefoldin/ubiquitin-positive dots and the amount of polyubiquitinated proteins. Knockdown of prefoldin increased the amount of insoluble ubiquitinated proteins and reduced cell viability in lactacystin-treated cells. In addition to lactacystin toxicity, prefoldin prevented cells from various stresses, including endoplasmic reticulum stress (Fig. 6B) and oxidative stress (data not shown). Although the level of SDS-insoluble proteins in thapsigargin-treated L110R MM-1α cells tended to be larger than that in wild-type MM-1α cells by filter trap assays, the results were not statistically significant (Fig. 6B-b). Cell viability against thapsigargin in L110R MM-1α cells was, however, significantly lower than that in wild-type MM-1α cells (Fig. 6B-c). Because filter trap assays detect relatively large aggregates (9), the size of aggregates formed in thapsigargin-treated cells might be not enough to be detected by filter trap assays. The levels of insoluble ubiquitinated protein and cell toxicity in cells harboring L100R MM-1α/PFD5-containing prefoldin were higher than those in wild-type cells under normal conditions, and these were increased upon lactacystin treatment. Because preventive activity of prefoldin against stresses in L110R MM-1α cells is reduced and because cells are exposed to various stresses even under a usual culture condition, it is thought that L110R MM-1α cells are more sensitive with higher levels of cytotoxicity than wild-type cells. Furthermore, polyubiquitinated protein aggregates were accumulated in the brains of L100R MM-1α mice. These results suggest that prefoldin plays a role in quality control against protein aggregation and that dysfunction of prefoldin is one of the causes of neurodegenerative disease. This is the first report showing a function of prefoldin against proteasome inhibition under physiological conditions.
Although both cytoplasmic localization and nuclear localization of proteins were observed using anti-PFD2 and anti-PFD5 antibodies, the anti-prefoldin antibody showed cytoplasmic localization of prefoldin (Figs. 1 and 2), indicating that PFD2 and PFD5 localized in the nucleus are free forms of proteins. Because polyubiquitination of proteins started at 8 h after the addition of lactacystin and because the localization of prefoldin on dots appeared from 16 h after addition of lactacystin, it is thought that prefoldin first senses polyubiquitination of proteins and then re-localizes on them. The results obtained using cultured cells and L100R MM-1α mice suggest that prefoldin plays a protective role against protein aggregation that occurs constitutively in cells even under normal conditions and that prefoldin becomes more active under pathological conditions. Although mechanisms of sensing and reduction against protein aggregation by prefoldin are not known, because prefoldin facilitates folding of newly synthesized proteins in cooperation with HSP70 and HSP40 (1–4), it is possible that prefoldin forms a complex with other chaperones such as HSP70 and HSP40 to inhibit protein aggregation through various protein degradation pathways.
In this study, we used the stresses of proteasome inhibition- and endoplasmic reticulum stress-induced protein aggregations to examine the function of prefoldin. Is a role of prefoldin specific to these stresses? It is well known that protein aggregation is induced by various stresses, resulting in the onset of various diseases, including neurodegenerative diseases. We have a preliminary result. When cells were treated with H2O2 or rotenone, both of which induce oxidative stress in cells, prefoldin was localized in aggresomes. An aggresome is a proteinaceous inclusion body that forms when the cellular degradation machinery is impaired or overwhelmed. We have reported that prefoldin inhibited aggregate and inclusion formation of exogenously added pathogenic huntingtin, a cause of Huntington disease (9). It has also been reported that archaeal prefoldin stimulates formation of soluble Aβ oligomers to inhibit their fibril formation in vitro (7) and that human prefoldin inhibits Aβ fibrillation to form nontoxic Aβ aggregates (8). These results and the in vitro finding suggest that prefoldin plays a role in preventing protein aggregations that are induced by various stresses.
It has been reported that L110R MM-1α/PFD5 mice develop a syndrome characterized by photoreceptor degeneration, central nervous system abnormalities, and male infertility (18). Although prefoldin containing L110R MM-1α/PFD5 was properly formed in vitro and in a cell line from L110R MM-1α/PFD5 mice (Fig. 5), the effects of lactacystin and thapsigargin on protein aggregation were augmented, and growth of L110R MM-1α/PFD5 cells was attenuated (Fig. 6), and ubiquitinated protein aggregates were accumulated in the brains of L110R MM-1α/PFD5 mice (Fig. 7). Leu-110 is located in the coiled-coil region. It is therefore thought that conversion from the hydrophobic amino acid leucine to the polar amino acid proline may induce conformational change of the coiled-coil region, although it is not able to be detected by methods such as gel filtration and glycerol density gradient centrifugation, resulting in reduction of recognition activity of prefoldin toward prefoldin substrates. As in L110R MM-1α/PFD5 mice, degeneration of the cerebellum, but not photoreceptor degeneration or male infertility, has been reported in PFD1-knock-out mice (19). Degeneration of the nervous systems with accumulation of ubiquitinated protein aggregates, however, has been reported in mice with knock-out of th autophagy-related genes Atg5 and Atg7 (22, 24). Because autophagy is a quality control system that operates through degradation of damaged organelle and protein aggregates, dysfunction of autophagy results in accumulation of protein aggregates, leading to degeneration of the nervous system (22, 24). Indeed, prefoldin-positive aggregates were observed in Atg5(−/−) cells under normal culture conditions, and their number and intensity were increased upon lactacystin treatment (Fig. 2E). These results also suggest that localization of prefoldin on protein aggregates occurs independently of induction of autophagy. It is therefore thought that dysfunction of prefoldin regarding quality control against protein aggregates leads to degeneration of the nervous system.
L110R MM-1α/PFD5 mice exhibit abnormality in the testis, including testicular atrophy, sloughing of spermatocytes and spermatids, and infertility (18). There are several reports regarding the relationship between a chaperone and abnormality in the testis. In mice with knock-out of DnaJA1, a co-chaperone of HSP70, excess activation of the androgen receptor in Sertoli cells causes abnormal spermatogenesis, resulting in infertility (25). In mice with knock-out of the testis-specific chaperone HSP70-2, testicular atrophy with infertility was observed (26). Prefoldin, like these chaperones, might have an important function in the testis.
Conclusion
This is the first report showing that prefoldin functions as a quality control system against protein aggregation under normal and pathogenic conditions.
Acknowledgments
We thank Patsy M. Nishina and Noboru Mizushima for C57BL/6- Pfdn5nmf5a/Pfdn5nmf5a mice and for ATG5(+/+) and ATG(−/−) cells, respectively. We also thank Tsutomu Osanai and Takae Koshiyama for their technical assistance with the mice.
This work was supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology.
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Aβ
- amyloid β.
REFERENCES
- 1. Siegers K., Waldmann T., Leroux M. R., Grein K., Shevchenko A., Schiebel E., Hartl F. U. (1999) Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin-GimC system. EMBO J. 18, 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vainberg I. E., Lewis S. A., Rommelaere H., Ampe C., Vandekerckhove J., Klein H. L., Cowan N. J. (1998) Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93, 863–873 [DOI] [PubMed] [Google Scholar]
- 3. Geissler S., Siegers K., Schiebel E. (1998) A novel protein complex promoting formation of functional α- and γ-tubulin. EMBO J. 17, 952–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hartl F. U., Hayer-Hartl M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858 [DOI] [PubMed] [Google Scholar]
- 5. Siegert R., Leroux M. R., Scheufler C., Hartl F. U., Moarefi I. (2000) Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 103, 621–632 [DOI] [PubMed] [Google Scholar]
- 6. Martín-Benito J., Gómez-Reino J., Stirling P. C., Lundin V. F., Gómez-Puertas P., Boskovic J., Chacón P., Fernández J. J., Berenguer J., Leroux M. R., Valpuesta J. M. (2007) Divergent substrate-binding mechanisms reveal an evolutionary specialization of eukaryotic prefoldin compared with its archaeal counterpart. Structure 15, 101–110 [DOI] [PubMed] [Google Scholar]
- 7. Sakono M., Zako T., Ueda H., Yohda M., Maeda M. (2008) Formation of highly toxic soluble amyloid β oligomers by the molecular chaperone prefoldin. FEBS J. 275, 5982–5993 [DOI] [PubMed] [Google Scholar]
- 8. Sörgjerd K. M., Zako T., Sakono M., Stirling P. C., Leroux M. R., Saito T., Nilsson P., Sekimoto M., Saido T. C., Maeda M. (2013) Human prefoldin inhibits amyloid-β (Aβ) fibrillation and contributes to formation of nontoxic Aβ aggregates. Biochemistry 52, 3532–3542 [DOI] [PubMed] [Google Scholar]
- 9. Tashiro E., Zako T., Muto H., Itoo Y., Sörgjerd K., Terada N., Abe A., Miyazawa M., Kitamura A., Kitaura H., Kubota H., Maeda M., Momoi T., Iguchi-Ariga S. M., Kinjo M., Ariga H. (2013) Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J. Biol. Chem. 288, 19958–19972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mori K., Maeda Y., Kitaura H., Taira T., Iguchi-Ariga S. M., Ariga H. (1998) MM-1, a novel c-Myc-associating protein that represses transcriptional activity of c-Myc. J. Biol. Chem. 273, 29794–29800 [DOI] [PubMed] [Google Scholar]
- 11. Fujioka Y., Taira T., Maeda Y., Tanaka S., Nishihara H., Iguchi-Ariga S. M., Nagashima K., Ariga H. (2001) MM-1, a c-Myc-binding protein, is a candidate for a tumor suppressor in leukemia/lymphoma and tongue cancer. J. Biol. Chem. 276, 45137–45144 [DOI] [PubMed] [Google Scholar]
- 12. Satou A., Taira T., Iguchi-Ariga S. M., Ariga H. (2001) A novel transrepression pathway of c-Myc. Recruitment of a transcriptional corepressor complex to c-Myc by MM-1, a c-Myc-binding protein. J. Biol. Chem. 276, 46562–46567 [DOI] [PubMed] [Google Scholar]
- 13. Satou A., Hagio Y., Taira T., Iguchi-Ariga S. M., Ariga H. (2004) Repression of the c-fms gene in fibroblast cells by c-Myc-MM-1-TIF1β complex. FEBS Lett. 572, 211–215 [DOI] [PubMed] [Google Scholar]
- 14. Yoshida T., Kitaura H., Hagio Y., Sato T., Iguchi-Ariga S. M., Ariga H. (2008) Negative regulation of the Wnt signal by MM-1 through inhibiting expression of the wnt4 gene. Exp. Cell Res. 314, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 15. Tsuchiya H., Iseda T., Hino O. (1996) Identification of a novel protein (VBP-1) binding to the von Hippel-Lindau (VHL) tumor suppressor gene product. Cancer Res. 56, 2881–2885 [PubMed] [Google Scholar]
- 16. Mousnier A., Kubat N., Massias-Simon A., Ségéral E., Rain J. C., Benarous R., Emiliani S., Dargemont C. (2007) von Hippel-Lindau binding protein 1-mediated degradation of integrase affects HIV-1 gene expression at a postintegration step. Proc. Natl. Acad. Sci. U.S.A. 104, 13615–13620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gstaiger M., Luke B., Hess D., Oakeley E. J., Wirbelauer C., Blondel M., Vigneron M., Peter M., Krek W. (2003) Control of nutrient-sensitive transcription programs by the unconventional prefoldin URI. Science 302, 1208–1212 [DOI] [PubMed] [Google Scholar]
- 18. Lee Y., Smith R. S., Jordan W., King B. L., Won J., Valpuesta J. M., Naggert J. K., Nishina P. M. (2011) Prefoldin 5 is required for normal sensory and neuronal development in a murine model. J. Biol. Chem. 286, 726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao S., Carlesso G., Osipovich A. B., Llanes J., Lin Q., Hoek K. L., Khan W. N., Ruley H. E. (2008) Subunit 1 of the prefoldin chaperone complex is required for lymphocyte development and function. J. Immunol. 181, 476–484 [DOI] [PubMed] [Google Scholar]
- 20. Ariga H., Sugano S. (1983) Initiation of simian virus 40 DNA replication in vitro. J. Virol. 48, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simons C. T., Staes A., Rommelaere H., Ampe C., Lewis S. A., Cowan N. J. (2004) Selective contribution of eukaryotic prefoldin subunits to actin and tubulin binding. J. Biol. Chem. 279, 4196–4203 [DOI] [PubMed] [Google Scholar]
- 22. Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 [DOI] [PubMed] [Google Scholar]
- 23. Miyazawa M., Tashiro E., Kitaura H., Maita H., Suto H., Iguchi-Ariga S. M., Ariga H. (2011) Prefoldin subunits are protected from ubiquitin-proteasome system-mediated degradation by forming complex with other constituent subunits. J. Biol. Chem. 286, 19191–19203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 [DOI] [PubMed] [Google Scholar]
- 25. Terada K., Yomogida K., Imai T., Kiyonari H., Takeda N., Kadomatsu T., Yano M., Aizawa S., Mori M. (2005) A type I DnaJ homolog, DjA1, regulates androgen receptor signaling and spermatogenesis. EMBO J. 24, 611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dix D. J., Allen J. W., Collins B. W., Mori C., Nakamura N., Poorman-Allen P., Goulding E. H., Eddy E. M. (1996) Targeted gene disruption of Hsp70–2 results in failed meiosis, germ cell apoptosis, and male infertility. Proc. Natl. Acad. Sci. U.S.A. 93, 3264–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]