

Background: It remains unclear why vasopressin induces greater antidiuresis through V2R than does oxytocin.
Results: Vasopressin sustains cAMP signaling during V2R internalization, a process promoted by β-arrestins, and is halted by the retromer complex.
Conclusion: This new noncanonical model of GPCR signaling differentiates the actions of vasopressin and oxytocin.
Significance: This emerging model may explain the physiological bias between ligands.
Keywords: Arrestin, G Protein-coupled Receptors (GPCR), Kidney Metabolism, Membrane Trafficking, Signaling, V2R
Abstract
The vasopressin type 2 receptor (V2R) is a critical G protein-coupled receptor (GPCR) for vertebrate physiology, including the balance of water and sodium ions. It is unclear how its two native hormones, vasopressin (VP) and oxytocin (OT), both stimulate the same cAMP/PKA pathway yet produce divergent antinatriuretic and antidiuretic effects that are either strong (VP) or weak (OT). Here, we present a new mechanism that differentiates the action of VP and OT on V2R signaling. We found that vasopressin, as opposed to OT, continued to generate cAMP and promote PKA activation for prolonged periods after ligand washout and receptor internalization in endosomes. Contrary to the classical model of arrestin-mediated GPCR desensitization, arrestins bind the VP-V2R complex yet extend rather than shorten the generation of cAMP. Signaling is instead turned off by the endosomal retromer complex. We propose that this mechanism explains how VP sustains water and Na+ transport in renal collecting duct cells. Together with recent work on the parathyroid hormone receptor, these data support the existence of a novel “noncanonical” regulatory pathway for GPCR activation and response termination, via the sequential action of β-arrestin and the retromer complex.
Introduction
Arrestin is thought to desensitize heterotrimeric G protein signaling at the plasma membrane by physically interacting with activated GPCR,3 thus preventing receptor-G protein coupling (1), promoting receptor internalization (2), and promoting the recruitment of enzymes such as cAMP-specific phosphodiesterase type 4D or diacylglycerol kinases to enhance cAMP or diacylglycerol degradation, respectively (3, 4). This model of arrestin-mediated desensitization has been supported with extensive work on several GPCRs such as rhodopsin and the β2-adrenergic receptor, among others, and is considered a universal and “canonical” mechanism for GPCR desensitization (5). This model has been challenged by recent studies revealing that β-arrestins prolong rather than attenuate cAMP stimulation triggered by the parathyroid hormone type 1 receptor (6). This new finding coupled with the recent observations that the PTHR along with other GPCRs can sustain heterotrimeric G protein signaling after internalization to endosomes (6–10) point to an emerging noncanonical signaling model of GPCR where β-arrestins prolong rather than attenuate ligand actions when a receptor internalizes in endosomes (11).
We do not know how many GPCRs exhibit this noncanonical form of signaling. Several factors led us to investigate the vasopressin type 2 receptor (V2R), a GS-coupled receptor that controls water and sodium ion homeostasis by regulating their reuptake in the collecting duct of the kidney (12). These include the following: 1) a high affinity complex between V2R and its native ligand, vasopressin (VP), which remains stable and active (cAMP production) under acidic conditions, pH 5.5, characteristic of early endosomes (13); 2) the redistribution of VP-V2R-arrestin complexes to early endosomes (14); 3) distinct physiological responses to its two native and structurally similar ligands, vasopressin (VP, strong antidiuretic and antinatriuretic effects) versus oxytocin (OT, weak to no effect) (15–17); and 4) evidence that a mutant of V2R associated with nephrogenic diabetes insipidus remains in the endoplasmic reticulum yet generates cAMP when challenged with a membrane permeant agonist (18). These observations raised the hypothesis that internalized VP-V2R complexes could access the machinery for signaling from membranes inside the cell to continue to stimulate the cAMP/PKA pathway via a noncanonical signaling model of GPCR.
To address this hypothesis, we used a series of biochemical, optical, and physiological assays to study V2R signaling in response to its two native ligands: vasopressin and oxytocin (13, 14). These experiments employed cultured human embryonic kidney (HEK293) cells stably expressing recombinant V2R and a line derived from renal principal cells of the mouse collecting duct, mpkCCDC14, which expresses V2R and the epithelial sodium channel (ENaC) (19, 20). We found that vasopressin induces a V2R-active state that promotes cAMP generation not only at the plasma membrane but also after β-arrestin1/2-mediated receptor internalization to endosomes. Oxytocin, however, did not induce significant β-arrestins binding or V2R internalization, and cAMP generation ended rapidly after ligand washout. Sustained cAMP generation triggered by VP was inhibited by the endosomal retromer complex. As in HEK293 cells, VP induced greater cAMP generation than OT in mpkCCDC14 cells and caused a sustained activation of ENaC on the apical plasma membrane, whereas OT did not. In both HEK293 and mpkCCDC14 cells, increased expression of β-arrestins augmented rather than attenuated the duration of cAMP production. These results indicate that noncanonical actions of β-arrestins and retromer can be extended to the regulation of V2R signaling in kidney cells, which may account for the biological differences between VP and OT on water and electrolyte homeostasis.
EXPERIMENTAL PROCEDURES
cDNA Constructs
Vasopressin type 2 receptor (V2R) and hemagglutinin (HA)-tagged V2R (HA-V2R) cDNAs were purchased from Missouri S&T cDNA Resource Center (Rolla, MO). V2R was amplified using AccuPrime Taq polymerase (Invitrogen) to introduce flanking 5′-BglII and 3′-AgeI restriction enzyme sites. The digested product was ligated into pECFP-N1 (Clontech) vector to generate V2R C-terminally tagged with enhanced CFP (V2RCFP).
Cell Culture
Cell culture reagents were obtained from Invitrogen. Human embryonic kidney cells (HEK293) (ATCC, Manassas, VA) were cultured in DMEM supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere containing 5% CO2. HEK293 cells stably expressing HA-V2R were grown in selection medium (DMEM, 10% FBS, 500 μg/ml neomycin). Culturing of mpkCCD-C14 collecting duct-derived cells was done as described previously (21, 22). AQP2-MDCK cells were cultured in DMEM supplemented with 10% fetal bovine serum and G418 (500 μg/ml) (23). For transient expression, cells were transfected with the appropriate cDNAs using FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions unless otherwise mentioned. We have optimized expression conditions to ensure moderate expression levels of fluorescent arrestins. We performed Western blots to verify the levels of fluorescent β-arrestin1 or β-arrestin2 expression relative to native β-arrestin1/2 in our experiments. We also ensured that the expression of fluorescent-labeled β-arrestin1 or β-arrestin2 was similar in the examined cells by performing experiments in cells displaying comparable fluorescence levels.
RNAi and Plasmid Transfections
We depleted the retromer subunit Vps35 from HEK293 cells using a pre-validated quartet of siRNA duplex nucleotides (SmartPool, Dharmacon). Control transfections were done with a scrambled siRNA (OriGene). Briefly, 24 h after plating, cells were transferred to serum- and antibiotic-free medium and transfected with X-TremeGene reagent (Roche Applied Science) according to the manufacturer's directions. Medium was supplemented with serum and antibiotics 24 h after transfection, and experiments were performed between 72 and 96 h after transfection. Plasmid transfections were done using FuGENE (Roche Applied Science) according to the manufacturer's directions.
Total Internal Reflection Fluorescence (TIRF) Microscopy
Cells plated on poly-d-lysine-coated glass coverslips and maintained in FRET buffer were imaged using a Nikon Ti-E inverted microscope equipped with an oil immersion ×60 NA 1.49 plan-apo TIRF objective and a TIRF excitation arm with motorized critical angle control (Nikon). Critical angles for CFP and YFP excitation were determined in advance, and identical angles were used in every experiment. In-line neutral density filters were used to minimize bleaching. Excitation light came from a 442-nm solid-state laser (CFP, Melles Griot) and the 514-nm line of an argon gas laser (YFP, Melles Griot) using appropriate dichroic filters. YFP was detected using 500 ± 20 nm (excitation) and 535 ± 30 nm (emission) filters; CFP was detected using 436 ± 20 nm (excitation) and 480 ± 40 nm (emission); and FRET was detected using a 436 ± 20 mm (excitation) and a 535 ± 30 nm (emission).
In Vitro Activation of GαS
The experiments were performed from membrane preparations of HEK293 cells stably expressing HA-V2R. Briefly, confluent cells were washed with ice-cold PBS and incubated with hypotonic buffer (10 mm Hepes, 0.5 mm EDTA, pH 7.4) for 15 min on ice. Swollen cells were harvested, collected by centrifugation (100 × g for 10 min), and resuspended in 5 volumes of 10 mm Tris, 1 mm EDTA, pH 7.4, with a mixture of proteinase inhibitors. Cells were disrupted with 30–40 strokes with a tissue grinder on ice. Lysates were centrifuged at 1000 × g for 10 min to remove unbroken cells and large cell debris. The supernatant was further centrifuged at 30,000 × g for 20 min at 4 °C. The membrane pellet was resuspended in ice-cold buffer (10 mm Hepes, 0.1 mm EDTA, pH 7.4) and rapidly frozen in liquid nitrogen. Membranes were stored at −80 °C until used. A small volume of membrane preparation was kept to determine protein concentration.
Frozen membrane aliquots (50 μg/tube) were incubated with 100 μl of assay buffer (10 mm Hepes, 100 mm NaCl, 5 mm MgCl2, pH 7.4) containing 5 μm GDP, with or without 100 nm purified β-arrestin2 (see below) for 45 min at room temperature. Then, either 100 nm vasopressin (AVP) and 10 nm [35S]GTPγS were added for different periods of time at 30 °C. Incubations were terminated by addition of 800 μl of ice-cold assay buffer and 100 μm GTPγS and transferred on ice. Cell membranes were recovered from the reaction mixture by centrifugation at 20,000 × g for 10 min. The resulting supernatant was removed. Membrane pellets were solubilized, and 2 μl of anti-GαS antibody (Millipore) was added to immunoprecipitate [35S]GTPγS-bound GαS. Samples were under agitation for 1 h at 4 °C. Then 20 μl of protein G-Sepharose (Santa Cruz Biotechnology) was added to each sample and incubated overnight at 4 °C. The beads were washed three times, suspended with 200 μl of 0.5% SDS, and incubated at 90 °C for 2–3 min. The entire contents of each tube were transferred to a vial containing 5 ml of scintillation mixture, and radioactivity was counted by β-emission spectrometry. Note that purified β-arrestin2 used in these experiments was prepared as described previously (24).
Transepithelial Isc Measurements
mpkCCDc14 cells grown on Transwell filter supports (Costar) (21) were mounted in modified Costar Ussing chambers, and the cultures were continuously short circuited with an automatic voltage clamp (Dept. of Bioengineering, University of Iowa, Iowa City). Transepithelial resistance was measured by periodically applying a 2.5-mV bipolar pulse and calculated by Ohm's law. The bathing Ringer's solution composition, gassing, and washing techniques have been described previously (25).
Aquaporin2 Phosphorylation
AQP2 stably transfected MDCK cells (AQP2-MDCK) were plated on 6-well filt ers (32 × 104 cells/well). Cells were grown on filters for 5 days to be fully polarized and treated with 50 mm indomethacin overnight to lower endogenous cAMP levels (26). Culture medium was replaced with serum- and antibiotic-free DMEM for 2 h before stimulation. Then cells were treated with or without VP or OT for 10 min. For washout experiments, cells were washed and cultured in serum- and antibiotic-free DMEM for 20 min. Filters were cut from plastic supports and incubated in lysis buffer (150 mm NaCl, 20 mm Tris-HCl, 5 mm EDTA, 1% Triton X-100 containing 10 mm NaF and 1 mm Na3VO4) for 20 min. Cell were scraped, and cell lysate was then transferred into Eppendorf tubes, rotated for 30 min at 4 °C, centrifuged for 10 min at 10,000 × g, and 12 μl of reduced samples (6 μg of protein) per lane were run on a NuPAGE 1.0-mm 15-well 4–12% BisTris gel and then transferred onto a PVDF membrane (Invitrogen). Transferred PVDF membranes were incubated in 5% skimmed milk in PBS/Tween 0.05% buffer for blocking, then incubated with primary antibody, diluted 1:5000–1:10,000 in PBS/Tween 0.05% buffer overnight (Ser(P)-256 and Ser(P)-269 AQP2 antibody) for 1 h (AQP2 polyclonal antibody). Then PVDF membranes were washed four times for 15 min in PBS/Tween 0.05% buffer (PBST), incubated with HRP-conjugated secondary antibody diluted 1:10,000 for 30 min, and washed in 0.05% PBST four times for 15 min. Signals were visualized using Western Lightning ECL and Biomax XAR film. The same PVDF membranes were stripped in stripping buffer (0.2 m glycine, 0.05% Tween 20, pH was adjusted to 2.5 by adding HCl) for 1 h, and then re-probed with the next antibody. Complete removal of the first primary antibody was confirmed by incubating the stripped membrane with secondary antibody (donkey anti rabbit IgG HRP conjugated). The order of detection was Ser(P)-269, Ser(P)-256, and then total AQP2.
Time-lapse Confocal Microscopy
HEK293 cells stably expressing HA-V2R were studied by a confocal microscope using methods described in previous studies (6, 8). In brief, cell imaging was performed at room temperature in Hepes buffer containing 0.1% BSA using a Nikon A1s confocal microscope attached to a Ti-E inverted base using a ×60 1.45 NA plan-apo objective. Live cell time-lapse confocal imaging of transfected cells treated with tetramethylrhodamine (TMR)-labeled vasopressin (VPTMR) (14) was performed at 37 °C using a Nikon A1-Rs confocal system attached to a Ti-E inverted base and an apo ×60 1.49 NA TIRF objective. The microscope was encased in a microscope cage incubator to maintain the temperature at 37 °C. HEK293 cells expressing HA-V2R were grown in a Multiwell 12-well plate (BD Biosciences). Upon reaching 70% confluency, cells were transfected using 3.75 μl of Lipofectamine 2000 and 1 μg of cDNA of green fluorescent protein (GFP)-tagged β-arrestin 2 (βarr2-GFP), GαS (GαS-GFP), or retromer (Vps29-YFP). 24 h later, cells were trypsinized and seeded in glass bottom FluoroDishes (World Precision Instruments, Sarasota, FL). 24–48 h later, the cells were washed and imaged in Hanks' Buffer (Invitrogen) with 10 mm Hepes, 25 mm sodium bicarbonate, and 10 mm glucose added to the solution. After the control picture was taken, 1 μm VPTMR was incubated with cells for 10 min, and then ligand was washed out, and images were taken at the designated time intervals.
FRET Measurements
FRET analysis was performed as described previously (8). Cells plated on poly-d-lysine-coated glass coverslips and maintained in FRET buffer were placed on a Nikon Ti-E inverted microscope equipped with an oil immersion ×60 NA 1.49 plan-apo objective and a dichroic beam splitter to allow simultaneous imaging of CFP and YFP fluorescence channels (DualView2, Photometrics, Tucson, AZ).
The emission fluorescence intensities were determined at 535 ± 15 nm (YFP) and 480 ± 20 nm (CFP) with a beam splitter DCLP of 505 nm. The FRET ratio for single experiments was corrected according to Equation 1,
 |
where FYFPex436/em535 and FCFPex436/em480 represent, respectively, the emission (em) intensities of YFP (recorded at 535 nm) and CFP (recorded at 480 nm) upon excitation (ex) at 436 nm; a and b represent correction factors for the bleed through of CFP into the 535-nm channel (a = 0.35) and the cross-talk due to the direct YFP excitation by light at 436 nm (b = 0.06). FYFPex500/em535 represents the emission intensity of YFP (recorded at 535 nm) upon direct excitation at 500 nm, and was recorded at the beginning of each experiment. Note that bleed through of YFP into the 480-nm channel was negligible. For each measurement, changes in fluorescence emissions due to photobleaching were subtracted. To ensure that CFP- and YFP-labeled molecule expression were similar in examined cells, we performed experiments in cells displaying comparable fluorescence levels.
The means of intermolecular FRET experiments were calculated according to Equation 2, which normalizes for different expression levels of CFP and YFP molecules,
 |
Receptor Degradation Assays
LLC-PK1 cells expressing V2R C-terminally tagged with enhanced GFP (V2R-GFP) (28) were grown in 6-cm dishes and transfected with 4 μg of both c-Myc-tagged Vps29 and Vps26 using 15 μl of Lipofectamine 2000. After 48 h, cells were incubated with 1 μm VP for different times of incubation (30, 60, 120, or 240 min). Cells were then lysed for 20 min at 4 °C in lysis buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS supplemented with protease inhibitors (Roche Applied Science). Before Western blot analysis, protein concentrations were measured with the bicinchoninic acid protein assay as recommended by the manufacturer (Invitrogen). Reduced samples from cell lysates (20 μg) were loaded onto each lane of a standard NUPAGE 4–12% BisTris gel and then electroblotted on Immobilon transfer membranes (Millipore, Billerica, MA). The filters were blocked with Li-Cor blocking buffer by shaking for 1 h at room temperature before the incubation (1 h) of the polyclonal anti-GFP (0.4 μg/ml, Molecular Probes) and monoclonal anti-actin antibody in Li-Cor blocking buffer, Tween 0.1%. After being washed three times in PBS/Tween 0.1% buffer, the membranes were incubated with goat anti-mouse IRdye 800CW and goat anti-rabbit IRDye 680 diluted in Li-Cor blocking buffer. After 1 h of incubation, membranes were washed three times with PBS/Tween 0.1% buffer. After two rinses in distilled water, fluorescence in the membranes was recorded by using a Li-Cor infrared imaging system (Li-Cor Biosciences). Density of protein bands was quantified using IP Lab Spectrum software (Scanalytics, Vienna, VA).
Western Blot and Immunoprecipitation
Western blot analyses were performed according to standard procedures. Briefly, HEK293 cells were transfected and harvested 48 h after transfection. The total protein extract was run on an SDS-PAGE and blotted onto 0.45-mm PVDF membranes (Bio-Rad). Immunodetection of HA-tagged V2R and retromer subunit Vps29YFP was carried out using monoclonal anti-HA antibody (Covance Inc, Denver, PA) and a polyclonal anti-GFP antibody (Invitrogen), respectively. Horseradish peroxidase-conjugated donkey anti-mouse and donkey anti-rabbit antibodies (Jackson ImmunoResearch, West Grove, PA) were used for the detection of primary antibody. Antibody binding was visualized with Immobilon ECL reagent (Millipore). Immunoprecipitations were performed as described previously (6) with the following changes. HEK293 cells were co-transfected with HA-V2R and either Vps29YFP or an empty vector control challenged with 100 nm vasopressin as indicated. At designated time points after a brief ligand challenge, 2.5 mm dithiobis[succinimidyl] propionate cross-linker (Sigma) was added and incubated at room temperature for 20 min. Medium was then washed once with PBS and cells were transferred to ice-cold RIPA buffer and incubated for 20 min. Cells were lysed, and a post-nuclear supernatant was prepared by a brief centrifugation (5 min at 5000 × g). A 50-ml aliquot was collected and frozen in Laemmli sample buffer (Bio-Rad; “2% loaded”), and then lysates were incubated overnight at 4 °C with 20 μl of anti-HA beads (Covance). Beads were washed four times in cold RIPA buffer. Dried beads were boiled for 5 min in 40 ml of Laemmli sample buffer and then loaded onto a 12% SDS-polyacrylamide gel for analysis.
Competition Binding Assay
HEK293 cells expressing HA-V2R were transfected with both cDNA plasmids of c-Myc-tagged Vps29 and Vps26 as described above. Cells were grown in 60-mm dishes until confluency (48 h after transfection). Cells were scraped and homogenized in hypo-osmotic medium (5 mm Tris-HCl, pH 7.3, 3 mm MgCl2, 1 mm EDTA, and protease inhibitors). After 10 min of centrifugation at 13,000 × g, pellets were resuspended in binding buffer (25 mm Hepes, pH 7.4, 2 mm MgCl2, 1 mm tyrosine, 1 mm phenylalanine, 0.1% bovine serum albumin) to obtain ∼35 μg of protein/ml. The [3H]VP binding competition assay was performed in a final volume of 250 μl containing ∼0.4 μg of protein, [3H]VP (3 nm, PerkinElmer Life Sciences), in the presence of an increasing concentration of either VP or OT. After 2 h at 4 °C, the incubation was terminated by adding 5.0 ml of ice-cold 25 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm MgCl2, followed immediately by two washes by filtration under vacuum through Gelman argon E glass fiber filters that had been presoaked in binding medium. Each assay was performed in triplicate. Maximum [3H]VP binding was evaluated in the absence of competitor peptide, whereas the nonspecific binding value was defined in the presence of VP (1 μm). Each filter was transferred to a vial containing 5 ml of scintillation fluid (Optic-Fluor, Packard, Groningen, The Netherlands). The radioactivity signal was determined using a liquid scintillation analyzer TriCarb 2200 CA from Packard Instrument Co. Receptors expressed strongly in HEK293 cells and bind VP with similar affinity as follows: Bmax = 7.1 ± 1.3 fmol/μg of protein and Ki = 1.4 ± 0.2 for HA-V2R; and Bmax = 6.1 ± 1.4 fmol/μg of protein and Ki = 1.6 ± 0.3 nm for V2RCFP (mean ± S.E.; n = 4).
Immunocytochemistry
AQP2-MDCK cells (8 × 104 cells/well) were plated on Transwell filters (Costar 3460) in 12 wells and incubated for 5 days to be fully polarized. Cells were treated with 50 μm indomethacin overnight, and then serum containing DMEM was replaced with serum- and antibiotic-free DMEM for 2 h. Cells were treated with or without a saturating concentration of VP or OT for 10 min. For washout experiments, cells were washed and incubated in serum- and antibiotic-free DMEM for 20 min after VP or OT incubation. Cells were fixed with 4% paraformaldehyde for 10 min, washed in PBS three times, permeabilized with 0.1% Triton X-100/PBS for 10 min, washed in PBS, blocked in 1% BSA/PBS for 30 min, and incubated with AQP2 antibody (1:1000) overnight at 4 °C. Cells were washed in PBS (10 min, three times), incubated with 1:1000 diluted donkey anti-goat IgG, FITC-conjugated antibody for 3 h at room temperature, and washed in PBS (10 min, three times). Finally, cells were mounted on glass slides with Vectashield mounting medium (Vector Laboratories, Burlingame, CA).
Statistical Analysis
Data are expressed as means ± S.E. Statistical analyses were performed using the unpaired Student's t test when applicable. Differences were considered significant at p < 0.05.
RESULTS
Sustained cAMP by Internalized V2R
We studied the V2R/VP/OT signaling systems in HEK293 cells transiently expressing recombinant HA-V2R. In the first series of experiments, we measured cAMP generation and PKA activity in response to challenge with VP or OT using FRET-based sensors expressed in the cytosol (epac1-CFP/YFP for cAMP and AKAR3 for PKA) (29, 30). We found that a brief pulse of VP followed by ligand washout resulted in a cAMP increase that remained elevated for about 20 min (Fig. 1A, black line). Oxytocin triggered a similar increase in cAMP, but cAMP returned to its basal level soon after ligand washout (Fig. 1B). In each case, the time course of PKA activity mirrored the time course of cAMP generation (Fig. 1, A and B, red lines). These results show that VP and OT induce distinct time courses of cAMP/PKA signaling: sustained after ligand washout in the case of VP, and short and sensitive to ligand washout in the case of OT.
FIGURE 1.
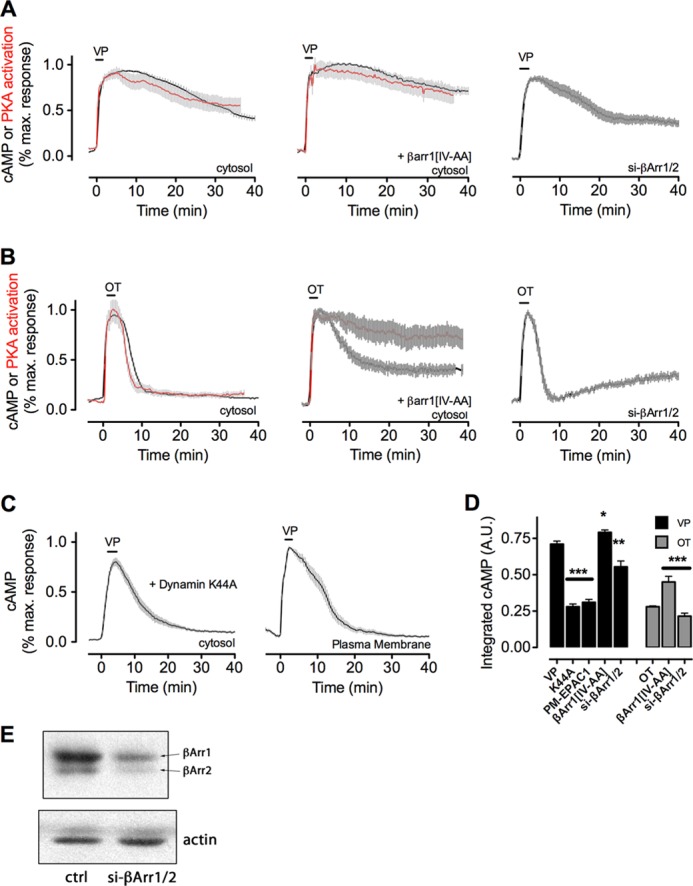
Internalization and arrestin binding regulate the time course of cAMP signaling mediated by V2R. A and B, averaged time course of cAMP generation (black) and PKA activity (red) measured by FRET-based biosensors in HEK293 cells expressing the human V2R and a cytoplasmic sensor for cAMP (Epac-CFP/YFP) or a sensor for PKA activity (AKAR3). Cells were perfused with buffer or with ligand (horizontal bars representing 30 s of a saturating concentration of either VP (100 nm) (A) or OT (10 μm). Data represent the mean ± S.E., of N ≥ 3 experiments and n ≥ 28 cells. The expression level of β-arrestins was titrated by either overexpression of a dominant-active mutant (βarr1(IV-AA), middle panel) or depletion of both β-arrestins by siRNA (right panel). C, averaged time course of cAMP generation in HEK293 cells expressing V2R and a dominant-negative mutant of dynamin 1 that prevents receptor internalization (K44A, right panel) or in which cAMP detection was limited to the plasma membrane by expression of a plasma membrane-limited mutant of the biosensor epac1CFP/YFP (right panel). Note that FRET was detected using TIRF microscopy. D, total cAMP production of data shown in A–C as measured by the integrated area under the curve. All manipulations are significant at least to p ≤ 0.05 by one-way analysis of variance and pairwise comparison with controls challenged with VP (black) or OT (gray); in all cases N ≥ 3. A.U., arbitrary unit. *, p < 0.05; **, p < 0.01; ***, p < 0.001. E, native β-arrestins were depleted from HEK293 cells as described under “Experimental Procedures,” and the reduction of native protein was shown by Western blotting. ctrl, control.
Consistent with our recent study done with the identical HEK293/HA-V2R cell model, cAMP generation in response to one “pulse” of VP is maintained at time points after internalization of V2R, which begins as soon as 5 min after challenge with a saturating concentration of VP (14). These data raised the hypothesis that sustained cAMP signaling mediated by VP is associated with internalization of V2R in early endosomes. Although residual V2R is often observed on the cell surface after challenge and ligand washout, most to all of VPTMR internalizes (14), indicating that the majority of VP-V2R complexes are within the cytoplasmic compartment and may continue to signal.
To test this hypothesis, we measured the time course of cAMP generation mediated by VP when V2R internalization was inhibited by co-expressing a dominant-negative mutant of dynamin 1, DynK44A, which blocks internalization by clathrin- and caveolin-dependent pathways (Fig. 1C, left panel) (31), or using a biosensor limited to the plasma membrane (epac1-CFP/YFPPM) (Fig. 1C, right panel) (32). In both experiments, we measured much shorter time courses of cAMP generation compared with the cytosolic cAMP sensor and V2R that is competent to internalize (Fig. 1A, left panel). These differences in cAMP generation were significant, as measured by the integrated area under the curve (Fig. 1D). These data thus support the hypothesis that internalization is a necessary component of persistent cAMP generation by V2R.
Control of V2R Signaling by Arrestin
VP is known to induce an interaction between V2R and β-arrestins that persists during receptor internalization (33). This fact coupled with our evidence for a persistent cAMP signaling by internalized V2R conflict with the traditional understanding of arrestin-mediated receptor desensitization. To investigate the influence of arrestin on V2R signaling, we first verified that β-arrestin2 (βarr2) binds and internalizes with VP-V2R. To this end, we measured the association of βarr2 and V2R mediated by VP at the plasma membrane by measuring FRET between V2R tagged with the cyan fluorescent protein (V2RCFP) and βarr2 tagged with the yellow fluorescent protein at its N terminus (YFPβarr2) (Fig. 2A). Using TIRF microscopy to limit measurements to within ∼100 nm of the plasma membrane, we found that a strong and durable association of YFPβarr2-V2R occurred within minutes, whereas a short challenge with OT induced a small and transient βarr2 association (Fig. 2B, green and blue lines). The FRET signal at the plasma membrane reached its peak within 5 min and then declined, coinciding with internalization of the V2R-βarr2 complex (as measured by the disappearance of both CFP and YFP fluorescence). We then measured FRET using wide field epifluorescence to measure the βarr2 and V2R interaction within the cell rather than at the plasma membrane. In this case, V2R-βarr2 association was detectable for at least 30 min after ligand washout, indicating that the V2R-βarr2 complex persisted during V2R endocytosis (Fig. 2B, black line). The duration of V2R-βarr2 association in this case corresponded well with the prolonged generation of cAMP observed when βarr2 was overexpressed (Fig. 1A) (6).
FIGURE 2.

Arrestin control of V2R signaling. A, Western blot showing expression of both recombinant β-arrestin1(IV-AA) (left panel) and β-arrestin2YFP (right panel). B, FRET between V2R-CFP and βarr2-YFP, as measured by TIRF microscopy (green, blue) or by epifluorescence (black). Horizontal bar indicates that cells were challenged for 30 s with either 100 nm VP or 10 μm OT and then washed with buffer. Data represent the mean ± S.E. of N ≥ 3 experiments and n ≥ 28 cells. C and D, time course (C) and basal activity (D) of [35S]GTPγS binding to GαS was measured in plasma membrane extracts of HEK293 cells expressing V2R with or without purified β-arrestin2 (red line). ANOVA, analysis of variance. Data represent mean ± S.E. of n = 4 independent experiments. ctrl, control. **, p < 0.01.
We confirmed that βarr2 does not prevent GS activation in response to VP by measuring the binding of [35S]GTPγS (a nonhydrolysable analog of GTP) to GαS by in vitro assays using purified βarr2 and plasma membrane extracts from HEK293 cells expressing V2R. Membranes were incubated at different time points with VP in the absence or presence of purified βarr2. We found that βarr2 did not inhibit the capacity of VP to activate GS but rather increased it (Fig. 2C). Importantly, our previous studies showed that purified βarr2 reduced GTPγS binding to GαS in response to isoproterenol (24). These data underline a key difference between receptors that are desensitized by β-arrestins such as the β2-AR and those that are not such as the V2R and the PTHR (11).
Retromer and the Termination of VP-V2R Signaling
If β-arrestins do not terminate V2R signaling in response to VP, what other factor could be involved? A likely candidate is the retromer complex, a soluble heterotrimer complex that binds cargo on early endosomes and sorts it to various destinations, such as the Golgi network (34–36), and that desensitizes sustained cAMP signaling after internalization of another GPCR, the PTHR (6). It is notable that one subunit of retromer, Vps26, has a remarkable structural similarity to β-arrestins (37), although the mechanistic relevance of this apparent homology remains to be shown.
We found significant co-localization between V2RCFP and the Vps29 subunit of retromer tagged at its C terminus with YFP (Vps29YFP) (Fig. 3A). This increase in co-localization was significant (Fig. 3A, lower panel) and coincided with time points when VP-stimulated cAMP generation begins to decrease (15–20 min) (Fig. 1A). Using immunoprecipitation assays, we further confirmed that Vps29 interacts with V2R at 5 min after VP challenge, and we observed a significant increase in binding by 20 min (Fig. 3B), again consistent with the timing of cAMP in these cells (Figs. 1A and 3C).
FIGURE 3.
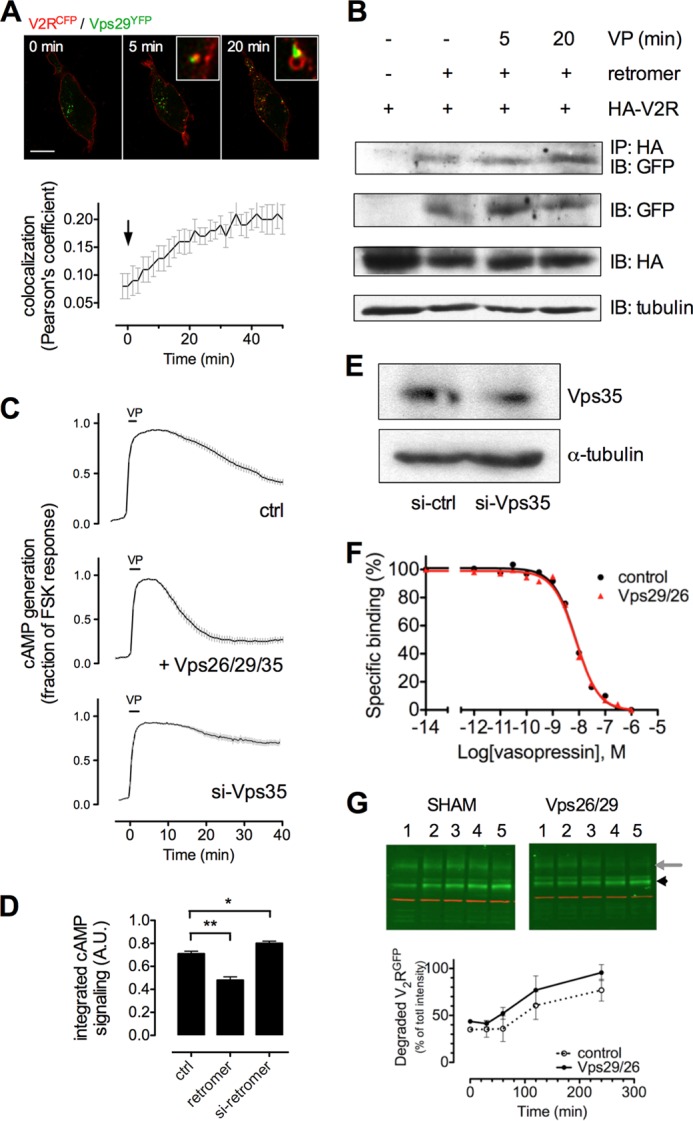
Retromer binds V2R and desensitizes VP-mediated cAMP generation. A, co-localization of V2RCFP (red) and Vps29YFP (green) in HEK293 cells challenged with 100 nm VP (arrow) and imaged using time-lapse confocal microscopy. Co-localization between V2R and retromer is shown (insets) and was quantified using Pearson's coefficient. Bars represent mean ± S.E. of N = 3 experiments and n = 14 cells. The bar represents 10 μm. B, binding of V2R and retromer was measured in HEK293 cells transfected with HA-V2R along with either Vps29YFP, Vps26, and Vps35 after a challenge with VP (100 nm) or carrier followed by immunoprecipitation (IP) and Western blot analysis. IB, immunoblot. Image is representative of n = 4 independent experiments. C, time course of cAMP generation in HEK293 cells transfected with cDNAs encoding V2R and Epac-CFP/YFP along with either siRNA against the retromer subunit Vps35 (si-Vps35) or plasmids encoding the retromer subunits Vps26, Vps29, and Vps35 (retromer). The horizontal bar indicates a 30-s challenge with 100 nm VP followed by washout with buffer. Vertical bars represent mean ± S.E. of N ≥ 3 experiments and n ≥ 28 cells. ctrl, control. D, total cAMP signaling of cAMP curves shown in A and B, as measured by the integrated area under the curve. *, p < 0.05; **, p < 0.01. E, measurement of Vps35 depletion by Western blot. F, affinity of V2R cells in the presence of overexpressed retromer was measured by competition binding assay in HEK293 cells. G, degradation of VP-challenged GFPV2R in HEK293 cells (left panel) and HEK293 cells overexpressing Vps26/29 (right panel) was measured by Western blot.
To test whether retromer influences the generation of cAMP by VP-V2R, we titrated expression of the retromer subunits Vps26/29/35 and measured the extent and time course of cAMP production using by FRET. Overexpression of the soluble Vps26/29/35 heterotrimer shortened the time course of cAMP signaling, whereas depletion of the subunit Vps35 by siRNA (Fig. 3E) (6) resulted in prolonged generation of cAMP relative to control cells (Fig. 3C). The effect of retromer depletion and overexpression on cAMP was statistically significant, as measured by quantifying the integrated area under the curve (Fig. 3D). Changing the expression level of retromer did not alter the number or affinity of V2R at the plasma membrane, as measured by radioligand binding (Fig. 3F). These data indicate that retromer inhibits cAMP signaling of VP-bound V2R on intracellular membranes.
To determine whether retromer influences degradative trafficking of V2R to the lysosome, we measured the abundance of V2R degradation by-products (28) in HEK293 cells expressing V2R along with either Vps26/29/35 or a control plasmid. Western blot analysis showed no significant increase of V2R degradation in the presence of overexpressed retromer (Fig. 3G), indicating that its influence on the degradation of V2R is negligible. It is therefore unlikely that retromer influences V2R signaling solely through influencing degradative traffic, but rather it inhibits cAMP generation by direct binding to V2R on endosomes.
Internalization of a V2R Signaling Complex
Next, we asked whether β-arrestins enhance or reduce VP-mediated cAMP generation in living cells. We used a mutant of β-arrestin 1, I386A/V387A (hereafter noted βarr1(IV-AA)) that has increased binding affinity for activated GPCR and for the AP-2 clathrin adapter (6, 38, 39). A version of this mutant tagged at its N terminus with tdTomato (tomβarr1(IV-AA)) rapidly associated with V2RCFP on the plasma membrane in response to VP and formed complexes that persisted on intracellular vesicles for up to 50 min after ligand challenge. Wild-type β-arr1 dissociated from internalized V2R and returned to the cytosol 20–40 min after ligand challenge (Fig. 4, A and B). Overexpression of tomβarr1(IV-AA) enhanced cAMP generation and PKA activity after challenge with either VP (Fig. 1A) or OT (Fig. 1B), whereas depletion of βarr1 and βarr2 by siRNA (40) reduced cAMP generation in response to VP (Fig. 1A, quantified in Fig. 1D). Depletion of β-arr1/2 had less effect on cAMP generation after OT challenge (Fig. 1B), possibly consistent with the small amount of βarr2 that binds V2R in response to OT (Fig. 2A). These data thus support a model in which β-arrestins promote rather than attenuate cAMP signaling mediated by VP. However, brief perfusion with OT did not induce detectable V2RCFP internalization or co-localization between V2RCFP and endosomes labeled with Vps29YFP (Fig. 4C), indicating that the brief generation of cAMP by OT-V2R (Fig. 1B) occurs at the plasma membrane.
FIGURE 4.
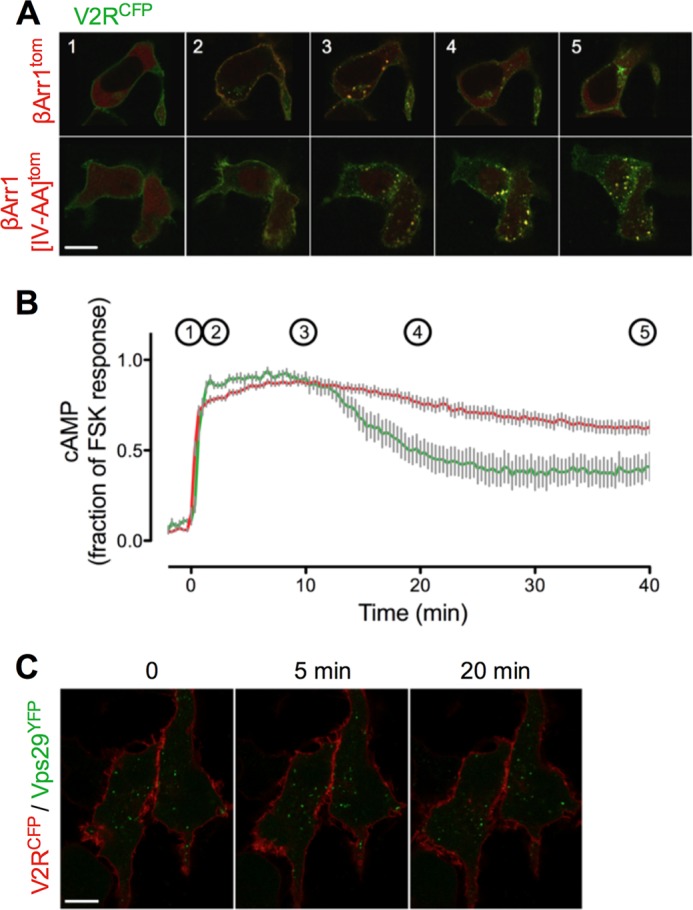
Significance of internalization for V2R signaling. A, co-localization of β-arr1tom (red, top row) or β-arr1(IV-AA)tom (red, bottom row) and V2RCFP (green) in HEK293 cells after a challenge with vasopressin (100 nm). The bar represents 10 μm. B, time course of cAMP generation in HEK293 transfected with β-arr1(IV-AA)tom (red) or a control plasmid (green). Gray bars represent the mean ± S.E., of N ≥ 3 experiments and n ≥ 28 cells. Time points indicated in B correspond to panel numbers in A. C, time-lapse confocal imaging of V2RCFP (red) and the retromer subunit Vps29YFP (green) at time points (min) shows no internalization of V2R after challenge with a saturating concentration of OT. Bar, 10 μm.
In light of reports that PTHR internalizes in a functional complex that includes βarr1 (or βarr2) and GαS (24), we sought to test whether V2R also internalizes in a complex that is competent for signaling. We found that tomβarr1(IV-AA), which significantly enhances cAMP generation by V2R, does co-internalize with GαSGFP after VP challenge of V2R (Fig. 5A). We next used TMR-labeled VP (VPTMR) and either GFP-labeled GαS (GαSGFP) or fluorescent forskolin (bodipyFSK) to show that VP-challenged V2R also internalizes with both GαS and adenylate cyclases, the key proteins necessary for cAMP generation (Fig. 5, B and C).
FIGURE 5.
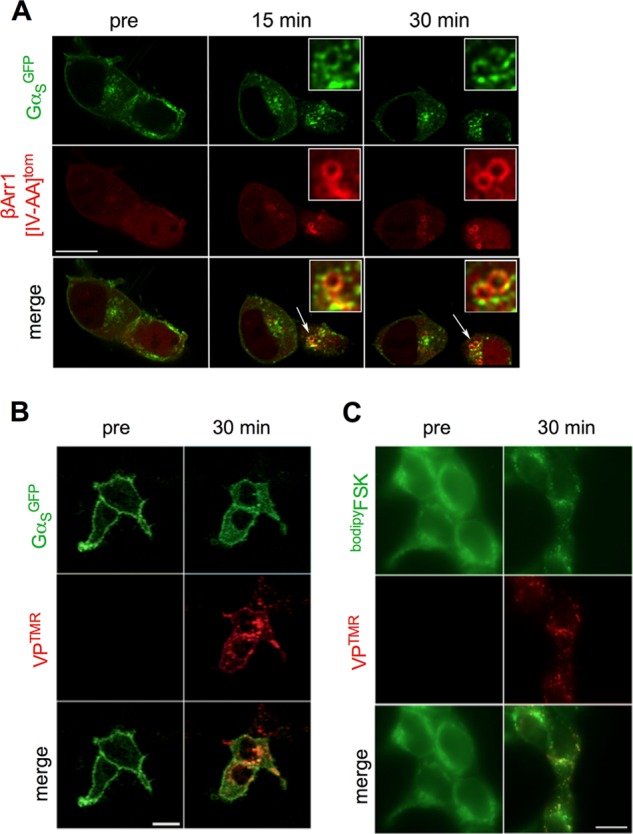
Internalization of a functional V2R-β-arrestin signaling complex. A, co-internalization of β-arrestin and GαS was demonstrated by challenging HEK293 cells expressing V2R along with GαS tagged with GFP (GαSGFP) and β-arrestin1(IV-AA) labeled at its N terminus with dTomato (dTomβarr1(IV-AA)). B and C, HEK293 cells expressing HA-V2R and GαSGFP (B) or stained with fluorescent forskolin (bodipyFSK) and then fixed (C) were challenged with VPTMR and visualized at the designated time points using confocal microscopy. Bar, 10 μm.
V2 Signaling in Cells of the Renal Collecting Duct
In principal, cells of the renal collecting duct epithelium and activation of PKA by VP lead to the accumulation of two passive channels into the apical plasma membrane as follows: the water channel aquaporin 2 (AQP2) and the ENaC (41, 42). When localized to the plasma membrane, these channels promote re-uptake of water and sodium ions from the urine by permitting their rapid transport into the cells down favorable osmotic and electrochemical gradients.
Both VP and OT stimulated the generation of cAMP in mpkCCDC14 cells, as shown by a rapid increase in the FRET ratio (FCFP/FYFP) (Fig. 6A). As the native oxytocin receptor couples with Gi or Gq but not GS (43), this shows that both OT and VP can induce V2R-mediated signaling in relevant cells of the kidney. Cyclic AMP generation by OT returned back to initial levels upon ligand washout, whereas VP induced a stronger and prolonged generation of cAMP (Fig. 6A). As in HEK293 cells, expression of β-arr1(IV-AA) prolonged significantly the cAMP elevation in response to VP (Fig. 6A), supporting the hypothesis that noncanonical signaling of V2R is also relevant in cells derived from the kidney. These results were significant, as measured by the integrated area under the curve (Fig. 6A, right panel).
FIGURE 6.
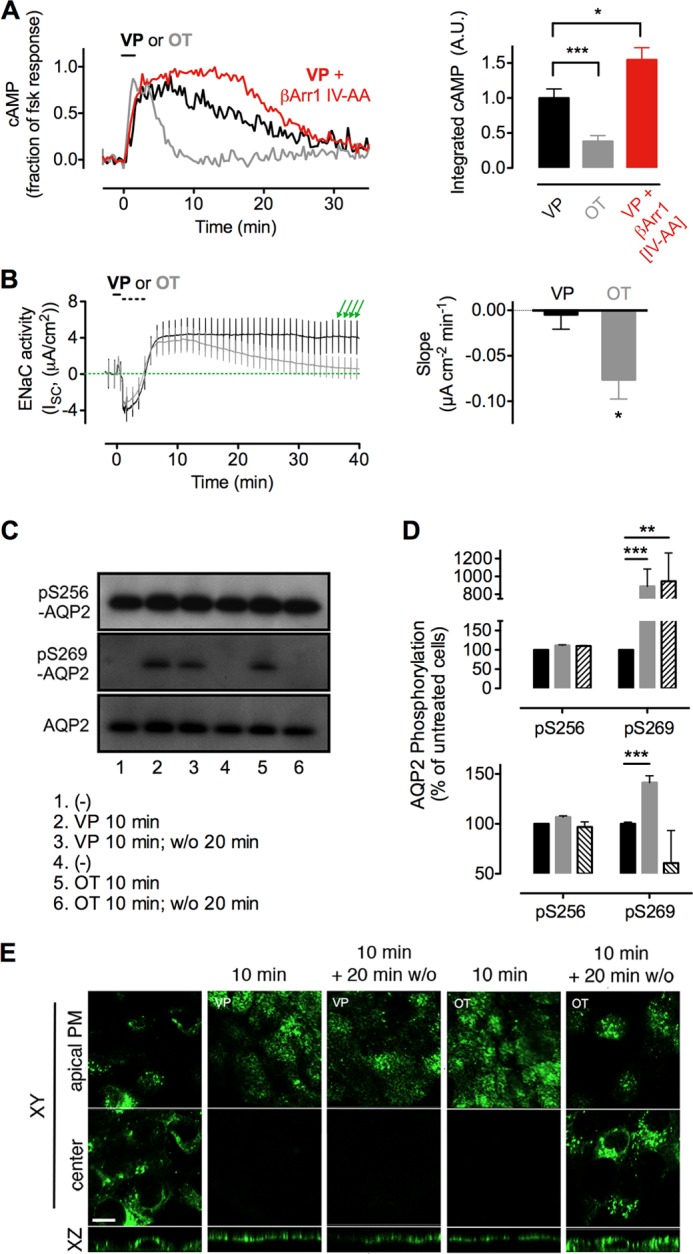
Differential actions of oxytocin and vasopressin on kidney cells. A, cAMP was measured in mpkCCDC14 cells using the epac1-CFP/YFP biosensor, as described above. Means ± S.E. of integrated cAMP generation (right panel), N ≥ 13. A.U., arbitrary unit. *, p < 0.05; **, p < 0.001. B, transepithelial current of polarized mpkCCDC14 cells was measured electrophysiologically by mounting permeable supports in Ussing chambers. Addition of VP or OT to the basolateral chamber (black bar) was followed by washout of the basolateral chamber with 5× volume of fresh medium (broken bar). The decrease in conductance at this time is an artifact attributable to washing. Vertical deviations (green arrows) represent ±2-mV voltage clamp pulses induced once each minute to verify the continued epithelial integrity. Amiloride was added at the end of each experiment to verify that >95% of the measured current was attributable to ENaC (right panel). Means ± S.E. of the slope of transepithelial current versus time after peak current at ∼6 min, N ≥ 6. *, p < 0.05. C, phosphorylation of AQP2 residues Ser-256 and Ser-269 after challenge with VP or OT for 10 min and measured without ligand washout (10 min) or after a 20-min ligand washout (10 min, w/o 20 min). D, corresponding band intensities from A were quantified by densitometry. Data represent the mean ± S.E., of n = 3 separate experiments. **, p < 0.01; ***, p < 0.001. E, translocation of AQP2 from the cytosol (center of cell) to the apical plasma membrane (apical PM) of a polarized monolayer of MDCK cells after challenge with VP or OT for 10 min and measured without ligand washout (10 min), or after a 20 min ligand washout (10 min, w/o 20 min). Bar, 10 μm.
In principal, the cells of the collecting duct, the immediate consequence of cAMP generation by V2R, is a PKA-dependent increase in the permeability of the apical membrane to water and sodium (44, 45). Sodium transport involves ENaC, an ion channel that is regulated in part by its expression level at the apical plasma membrane. In resting cells, ENaC undergoes a constitutive cycle of rapid ubiquitin-dependent internalization from the plasma membrane to specialized endosomes (46). When activated, PKA phosphorylates and inhibits the E3 ubiquitin ligase Nedd4-2, preventing the internalization of ENaC and causing it to accumulate on the apical plasma membrane (44). To evaluate the relative influence of VP and OT on ENaC activity, we measured the amiloride-sensitive transepithelial current (i.e. ENaC activity (47)) in polarized mpkCCDC14 cells following agonist challenge (Fig. 6B, black bar). A short challenge with OT or VP induced a rapid increase in apical sodium permeability, but only in the case of VP did permeability persist at its maximum level for more than 40 min after ligand washout. Sodium permeability decreased after washout of OT (Fig. 6B), as shown by measuring the slope of transepithelial current over time after its maximum at ∼6 min after ligand challenge (Fig. 6B, right panel). These results show that the apical membrane transport of ENaC correlates with the sustained cAMP response mediated by VP.
Predicting Aquaporin-2 Phosphorylation and Its Apical Membrane Translocation
As with sodium ions, the regulated reuptake of water from the urine depends on a passive channel, AQP2, which accumulates on the apical plasma membrane in response to V2R activation. Collecting duct epithelium is water-impermeable to an unusual degree (48, 49), and thus water can only pass down a favorable osmotic gradient from the urine to the blood when AQP2 channels are expressed at the apical membrane. Phosphorylation of C-terminal residues of AQP2 by PKA blocks its constitutive internalization (42, 50, 51), resulting in increased water absorption from the renal collecting duct and more concentrated urine. Our model predicts that a short challenge with VP, but not OT, should cause sustained AQP2 expression on the apical membrane of kidney cells. This would lead to greater water permeability and explain the strong antidiuretic effect of VP over other ligands of V2R. We tested this prediction in MDCK cells that stably express c-Myc-tagged AQP2.
We examined the effect of VP and OT on AQP2 phosphorylation at two key sites required for its retention at the plasma membrane, Ser-256 and Ser-269 (42). A 10-min challenge with VP caused a strong increase in Ser-269 phosphorylation (p269) by 9-fold over basal conditions. This increase persisted well after ligand washout, whereas challenge with oxytocin caused a phosphorylation increase that was both small in magnitude (≈3.3-fold over basal conditions) and brief, in that it did not persist after washout of ligand (Fig. 6, C and D). Phosphorylation at Ser-256 was not affected during these incubations.
We next challenged these cells with VP or OT and visualized localization of AQP2 by confocal microscopy using anti-AQP2 antibody (52). AQP2 localized to the cytoplasm in resting cells, but translocation to the apical plasma membrane was observed after challenge with either VP or OT (Fig. 6E). Twenty minutes after ligand washout, AQP2 localized to the apical plasma membrane in cells challenged with VP but had returned to the cytoplasm in cells challenged with OT (Fig. 6E). This further demonstrates that even a short pulse of VP induces a prolonged apical localization of AQP2, whereas OT has an effect that is much more transient.
DISCUSSION
By studying the effects of brief pulses of vasopressin on cAMP responses and V2R internalization, we have provided strong evidence indicating that prolonged generation of cAMP by V2R requires both internalization of the receptor and arrestin binding as follows: 1) a plasma membrane-restricted cAMP sensor detected much shorter generation of cAMP relative to a sensor located in the cytosol; 2) inhibition of receptor internalization with a dynamin 1 dominant-negative mutant led to a shorter generation of cAMP; 3) a high level of cAMP was observed with near-complete redistribution of VP-V2R complexes to endosomes that also contain β-arrestins, GS, and adenylate cyclases; and 4) a β-arrestin1 mutant with increased affinity for active receptors prolongs cAMP generation to VP, whereas arrestin depletion reduces the duration of the cAMP response.
Regarding desensitization of V2R, our work shows that signaling by internalized V2R is halted by the retromer sorting complex, which is most well characterized as a mediator of endosome-to-Golgi retrograde transport (34). Retromer co-localized with V2R after internalization and formed a complex with it; titrating retromer expression by transgene overexpression or by siRNA knockdown showed that retromer has an antagonistic effect on cAMP generation by V2R. Together with recent work on β-arrestins and the PTHR (6, 9, 10, 53), this work supports a new noncanonical model of GPCR signaling, wherein β-arrestins augment rather than attenuate hormone actions and in which desensitization is instead mediated by the retromer sorting complex on early endosomes.
The physiological significance of this new model is best appreciated when comparing the actions of vasopressin and oxytocin, two structurally similar V2R ligands, which stimulate the cAMP/PKA pathway resulting in water and sodium channels translocation to the apical membrane of renal collecting duct cells. This process increases the transport of water and sodium from urine in medullary and cortical collecting ducts of the kidney. Both hormones can be used for treatment of diabetes insipidus, but VP has strong antidiuretic and antinatriuretic effects as opposed to oxytocin, which has weak effects only measurable in the absence of natural vasopressin (54, 55). The mechanism that differentiates the two hormones is unclear, but this study provides a molecular and cellular basis for a better understanding of how VP and OT could induce differing effects on water and electrolyte homeostasis, whereas VP initiates a persistent mode of cAMP generation that is dependent on V2R internalization and is enhanced by arrestin, OT initiates a signaling pathway that largely does not involve arrestin and is limited to the plasma membrane. Given that V2R signaling via the cAMP/PKA pathway controls the transport of water and sodium channels to the apical membrane of collecting duct epithelia (Fig. 7), it is reasonable to link the strong antidiuretic effect of VP to its capacity to sustain a cAMP signal when the VP-V2R complex internalizes. In contrast, OT by limiting its action to V2R at the plasma membrane, transiently producing cAMP, has a lower capacity to stabilize AQP2 and ENaC expression at the apical surface, and it is thus less antidiuretic than VP. It is notable even in the case of OT that overexpression of an arrestin mutant that stabilizes the receptor-arrestin complex caused a strong increase in cAMP generation and PKA activity. This suggests that noncanonical signaling is a property of the receptor rather than unique to an agonist ligand such as VP, and that the main difference between OT and VP lies in the ability to promote a stable interaction with arrestin.
FIGURE 7.
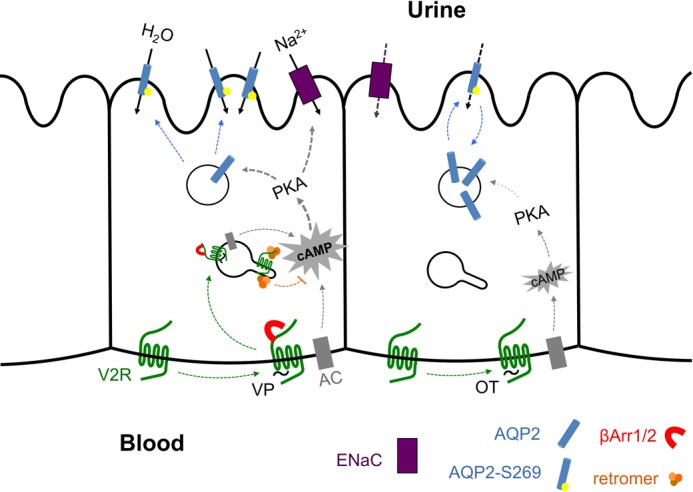
Model of regulation of water transport in epithelial cells of the kidney by V2R. VP binding to the V2R induces β-arrestin1/2 binding (red) and internalization of a VP-V2R-arrestin complex, which continues to generate cAMP via adenylyl cyclase (AC, gray) from endosomes until bound by retromer (orange). cAMP-activated protein kinase A (PKA) stimulates phosphorylation of the water channel AQP2 (blue) on serine 269, stabilizing its localization to the apical plasma membrane. OT also activates PKA via generation of cAMP from the basolateral plasma membrane, but OT does not induce a sustained cAMP response upon β-arrestin1/2 mobilization or internalization of active receptor. This causes a transient phosphorylation of AQP2 and transient localization of AQP2 in the apical plasma membrane.
Much remains unknown about how VP-V2R induces persistent signaling and OT-V2R does not. It remains to be shown, for example, how β-arrestin1/2 binding can stabilize active VP-V2R, whereas retromer destabilizes or inhibits the complex. One possibility is that arrestin stabilizes an active state of V2R that can activate GS for as long as the complex persists. Retromer, however, could bind endosomal V2R and stabilize an inactive state that is not competent to activate G proteins. If binding of retromer and β-arrestins is mutually exclusive, this would set up a competitive relationship that is consistent with previous observations (6).
One can also ask how VP-bound V2R can bind arrestin and yet continue to interact with and activate GS proteins. No evidence exists that β-arrestins can bind directly to GαS, although a modified n-formyl peptide receptor with Gαi fused to its C terminus does bind β-arrestin1 and internalize, indicating that it is possible for βarr1 to associate with a G protein-bound GPCR (56). A recent study indicates that in the case of PTHR, in fact β-arrestin1 or -2 stabilizes a ternary signaling receptor-arrestin-Gβγ complex that augments rather than attenuates PTHR signaling by increasing the steady-state level of activated GS (24). Whether or not a VP could establish a ternary signaling complex with V2R, Gβγ, and βarr1/2 that sustains GS activation remains to be determined.
If a GPCR binds its ligand with higher affinity than it binds β-arrestins, then β-arrestins could periodically leave the activated receptor exposed for further cycles of G protein activation. This is supported by FRAP analysis of βarr1-PTHR complexes on early endosomes, which showed a significant turnover of βarr1 molecules. However, a mutant βarr1 that binds activated PTHR with much higher affinity also enhances cAMP production, which would not be possible under the exchange model (6). Thus, sustained β-arrestin interaction, mediated by strong receptor-ligand binding and permitting a more stable GPCR-Gβγ binding, likely promotes the sustained signaling that characterizes a noncanonical signaling.
It is possible that V2R dimerizes (27). If so, then one receptor could bind to a β-arrestin isoform and mediate internalization, whereas the other activates GS. Finally, it is possible that the rather large third intracellular loop of V2R (42 residues) allows interactions with multiple effector proteins at one time. In this case, high affinity β-arrestin mutants, and ligands that induce stable β-arrestin binding, could prolong cAMP generation by blocking access to accessory proteins that do decouple G protein activation proteins, such as retromer, that mediate receptor desensitization.
In summary, we found that vasopressin can induce a mode of V2R signaling that is characterized by internalization of active receptor and positive feedback from β-arrestin1/2, whereas oxytocin induces a briefer signal generation that is limited to the plasma membrane. These different kinetics of cAMP signaling can explain the stark difference that we observed in the translocation of key proteins involved in water and sodium homeostasis in polarized epithelia of the kidney. Our results indicate that persistent signaling and internalization of activated V2R can at least partly explain the significant physiological differences between VP and OT when used as a therapy for disorders of water and electrolyte transport.
Acknowledgments
We thank Dr. Jin Zhang for the generous gift of the plasmid encoding AKAR3, Dr. Mark Knepper for P-AQP2 antibody, Dr. Bin Wang for optimizing the in vitro activation assay of GS, and Drs. Sei Sasaki and Shinichi Uchida for AQP2-MDCK cells. We also are grateful to Billy Andriopoulos for the preparation of the V2RCFP construct.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK087688 (to J.-P. V.), R01 DK38452, and R01 DK96586 (to D. B.), R01 DK075048 (to K. R. H.), and P30 DK079307 (to the Pittsburgh Kidney Research Center). This work was also supported by Boston Area Diabetes and Endocrinology Research Center Grant DK57521, Center for the Study of Inflammatory Bowel Disease Grant DK43341, and Cystic Fibrosis Foundation Grant KING10F0 (to D. K.).
- GPCR
- G protein-coupled receptor
- V2R
- vasopressin type 2 receptor
- VP
- vasopressin
- OT
- oxytocin
- PTHR
- parathyroid hormone type 1 receptor
- ENaC
- epithelial sodium channel
- AQP2
- aquaporin 2
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- TIRF
- total internal reflection fluorescence
- CFP
- cyan fluorescence protein
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- TMR
- tetramethylrhodamine
- MDCK
- Madin-Darby canine kidney cell.
REFERENCES
- 1. Lohse M. J., Benovic J. L., Codina J., Caron M. G., Lefkowitz R. J. (1990) β-Arrestin: a protein that regulates β-adrenergic receptor function. Science 248, 1547–1550 [DOI] [PubMed] [Google Scholar]
- 2. Laporte S. A., Miller W. E., Kim K. M., Caron M. G. (2002) β-Arrestin/AP-2 interaction in G protein-coupled receptor internalization: identification of a β-arrestin binding site in β2-adaptin. J. Biol. Chem. 277, 9247–9254 [DOI] [PubMed] [Google Scholar]
- 3. Nelson C. D., Perry S. J., Regier D. S., Prescott S. M., Topham M. K., Lefkowitz R. J. (2007) Targeting of diacylglycerol degradation to M1 muscarinic receptors by β-arrestins. Science 315, 663–666 [DOI] [PubMed] [Google Scholar]
- 4. Perry S. J., Baillie G. S., Kohout T. A., McPhee I., Magiera M. M., Ang K. L., Miller W. E., McLean A. J., Conti M., Houslay M. D., Lefkowitz R. J. (2002) Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science 298, 834–836 [DOI] [PubMed] [Google Scholar]
- 5. Luttrell L. M., Lefkowitz R. J. (2002) The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 6. Feinstein T. N., Wehbi V. L., Ardura J. A., Wheeler D. S., Ferrandon S., Gardella T. J., Vilardaga J. P. (2011) Retromer terminates the generation of cAMP by internalized PTH receptors. Nat. Chem. Biol. 7, 278–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calebiro D., Nikolaev V. O., Gagliani M. C., de Filippis T., Dees C., Tacchetti C., Persani L., Lohse M. J. (2009) Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 7, e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ferrandon S., Feinstein T. N., Castro M., Wang B., Bouley R., Potts J. T., Gardella T. J., Vilardaga J.-P. (2009) Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 5, 734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kotowski S. J., Hopf F. W., Seif T., Bonci A., von Zastrow M. (2011) Endocytosis promotes rapid dopaminergic signaling. Neuron 71, 278–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mullershausen F., Zecri F., Cetin C., Billich A., Guerini D., Seuwen K. (2009) Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat. Chem. Biol. 5, 428–434 [DOI] [PubMed] [Google Scholar]
- 11. Vilardaga J. P., Gardella T. J., Wehbi V. L., Feinstein T. N. (2012) Non-canonical signaling of the PTH receptor. Trends Pharmacol. Sci. 33, 423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown D., Breton S., Ausiello D. A., Marshansky V. (2009) Sensing, signaling and sorting events in kidney epithelial cell physiology. Traffic 10, 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zalyapin E. A., Bouley R., Hasler U., Vilardaga J. P., Lin H. Y., Brown D., Ausiello D. A. (2008) Effects of the renal medullary pH and ionic environment on vasopressin binding and signaling. Kidney Int. 74, 1557–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen S., Webber M. J., Vilardaga J. P., Khatri A., Brown D., Ausiello D. A., Lin H. Y., Bouley R. (2011) Visualizing microtubule-dependent vasopressin type 2 receptor trafficking using a new high-affinity fluorescent vasopressin ligand. Endocrinology 152, 3893–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Balment R. J., Brimble M. J., Forsling M. L., Kelly L. P., Musabayane C. T. (1986) A synergistic effect of oxytocin and vasopressin on sodium excretion in the neurohypophysectomized rat. J. Physiol. 381, 453–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou C. L., DiGiovanni S. R., Luther A., Lolait S. J., Knepper M. A. (1995) Oxytocin as an antidiuretic hormone. II. Role of V2 vasopressin receptor. Am. J. Physiol. 269, F78–F85 [DOI] [PubMed] [Google Scholar]
- 17. Chou C. L., DiGiovanni S. R., Mejia R., Nielsen S., Knepper M. A. (1995) Oxytocin as an antidiuretic hormone. I. Concentration dependence of action. Am. J. Physiol. 269, F70–F77 [DOI] [PubMed] [Google Scholar]
- 18. Robben J. H., Kortenoeven M. L., Sze M., Yae C., Milligan G., Oorschot V. M., Klumperman J., Knoers N. V., Deen P. M. (2009) Intracellular activation of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus by nonpeptide agonists. Proc. Natl. Acad. Sci. U.S.A. 106, 12195–12200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gumz M. L., Cheng K. Y., Lynch I. J., Stow L. R., Greenlee M. M., Cain B. D., Wingo C. S. (2010) Regulation of αENaC expression by the circadian clock protein Period 1 in mpkCCD(c14) cells. Biochim. Biophys. Acta 1799, 622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robert-Nicoud M., Flahaut M., Elalouf J. M., Nicod M., Salinas M., Bens M., Doucet A., Wincker P., Artiguenave F., Horisberger J. D., Vandewalle A., Rossier B. C., Firsov D. (2001) Transcriptome of a mouse kidney cortical collecting duct cell line: effects of aldosterone and vasopressin. Proc. Natl. Acad. Sci. U.S.A. 98, 2712–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., Vandewalle A. (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 22. Carattino M. D., Edinger R. S., Grieser H. J., Wise R., Neumann D., Schlattner U., Johnson J. P., Kleyman T. R., Hallows K. R. (2005) Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 280, 17608–17616 [DOI] [PubMed] [Google Scholar]
- 23. Yui N., Lu H. A., Chen Y., Nomura N., Bouley R., Brown D. (2012) Basolateral targeting and microtubule-dependent transcytosis of the aquaporin-2 water channel. Am. J. Physiol. Cell Physiol. 304, C38–C48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wehbi V. L., Stevenson H. P., Feinstein T. N., Calero G., Romero G., Vilardaga J. P. (2013) Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gβγ complex. Proc. Natl. Acad. Sci. U.S.A. 110, 1530–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Butterworth M. B., Edinger R. S., Johnson J. P., Frizzell R. A. (2005) Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J. Gen. Physiol. 125, 81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Balkom B. W., Graat M. P., van Raak M., Hofman E., van der Sluijs P., Deen P. M. (2004) Role of cytoplasmic termini in sorting and shuttling of the aquaporin-2 water channel. Am. J. Physiol. Cell Physiol. 286, C372–C379 [DOI] [PubMed] [Google Scholar]
- 27. Pioszak A. A., Harikumar K. G., Parker N. R., Miller L. J., Xu H. E. (2010) Dimeric arrangement of the parathyroid hormone receptor and a structural mechanism for ligand-induced dissociation. J. Biol. Chem. 285, 12435–12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bouley R., Lin H. Y., Raychowdhury M. K., Marshansky V., Brown D., Ausiello D. A. (2005) Downregulation of the vasopressin type 2 receptor after vasopressin-induced internalization: involvement of a lysosomal degradation pathway. Am. J. Physiol. Cell Physiol. 288, C1390–C1401 [DOI] [PubMed] [Google Scholar]
- 29. Allen M. D., Zhang J. (2006) Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem. Biophys. Res. Commun. 348, 716–721 [DOI] [PubMed] [Google Scholar]
- 30. Nikolaev V. O., Bünemann M., Hein L., Hannawacker A., Lohse M. J. (2004) Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 279, 37215–37218 [DOI] [PubMed] [Google Scholar]
- 31. Damke H., Baba T., Warnock D. E., Schmid S. L. (1994) Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127, 915–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DiPilato L. M., Zhang J. (2009) The role of membrane microdomains in shaping β2-adrenergic receptor-mediated cAMP dynamics. Mol. Biosyst. 5, 832–837 [DOI] [PubMed] [Google Scholar]
- 33. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., Caron M. G. (1999) Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem. 274, 32248–32257 [DOI] [PubMed] [Google Scholar]
- 34. Collins B. M. (2008) The structure and function of the retromer protein complex. Traffic 9, 1811–1822 [DOI] [PubMed] [Google Scholar]
- 35. Gullapalli A., Wolfe B. L., Griffin C. T., Magnuson T., Trejo J. (2006) An essential role for SNX1 in lysosomal sorting of protease-activated receptor-1: evidence for retromer-, Hrs-, and Tsg101-independent functions of sorting nexins. Mol. Biol. Cell 17, 1228–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Temkin P., Lauffer B., Jäger S., Cimermancic P., Krogan N. J., von Zastrow M. (2011) SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 13, 715–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi H., Rojas R., Bonifacino J. S., Hurley J. H. (2006) The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nat. Struct. Mol. Biol. 13, 540–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burtey A., Schmid E. M., Ford M. G., Rappoport J. Z., Scott M. G., Marullo S., Simon S. M., McMahon H. T., Benmerah A. (2007) The conserved isoleucine-valine-phenylalanine motif couples activation state and endocytic functions of β-arrestins. Traffic 8, 914–931 [DOI] [PubMed] [Google Scholar]
- 39. Keyel P. A., Thieman J. R., Roth R., Erkan E., Everett E. T., Watkins S. C., Heuser J. E., Traub L. M. (2008) The AP-2 adaptor β2 appendage scaffolds alternate cargo endocytosis. Mol. Biol. Cell 19, 5309–5326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gesty-Palmer D., Chen M., Reiter E., Ahn S., Nelson C. D., Wang S., Eckhardt A. E., Cowan C. L., Spurney R. F., Luttrell L. M., Lefkowitz R. J. (2006) Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 281, 10856–10864 [DOI] [PubMed] [Google Scholar]
- 41. Bhalla V., Hallows K. R. (2008) Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 19, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 42. Hoffert J. D., Fenton R. A., Moeller H. B., Simons B., Tchapyjnikov D., McDill B. W., Yu M. J., Pisitkun T., Chen F., Knepper M. A. (2008) Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J. Biol. Chem. 283, 24617–24627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoare S., Copland J. A., Strakova Z., Ives K., Jeng Y. J., Hellmich M. R., Soloff M. S. (1999) The proximal portion of the COOH terminus of the oxytocin receptor is required for coupling to Gq, but not Gi. Independent mechanisms for elevating intracellular calcium concentrations from intracellular stores. J. Biol. Chem. 274, 28682–28689 [DOI] [PubMed] [Google Scholar]
- 44. Hallows K. R., Wang H., Edinger R. S., Butterworth M. B., Oyster N. M., Li H., Buck J., Levin L. R., Johnson J. P., Pastor-Soler N. M. (2009) Regulation of epithelial Na+ transport by soluble adenylyl cyclase in kidney collecting duct cells. J. Biol. Chem. 284, 5774–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Katsura T., Gustafson C. E., Ausiello D. A., Brown D. (1997) Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am. J. Physiol. 272, F817–F822 [PubMed] [Google Scholar]
- 46. Edinger R. S., Bertrand C. A., Rondandino C., Apodaca G. A., Johnson J. P., Butterworth M. B. (2012) The epithelial sodium channel (ENaC) establishes a trafficking vesicle pool responsible for its regulation. PLoS ONE 7, e46593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mall M., Bleich M., Greger R., Schreiber R., Kunzelmann K. (1998) The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Invest. 102, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morgan T., Berliner R. W. (1968) Permeability of the loop of Henle, vasa recta, and collecting duct to water, urea, and sodium. Am. J. Physiol. 215, 108–115 [DOI] [PubMed] [Google Scholar]
- 49. Morgan T., Sakai F., Berliner R. W. (1968) In vitro permeability of medullary collecting ducts to water and urea. Am. J. Physiol. 214, 574–581 [DOI] [PubMed] [Google Scholar]
- 50. Brown D., Bouley R., Păunescu T. G., Breton S., Lu H. A. (2012) New insights into the dynamic regulation of water and acid-base balance by renal epithelial cells. Am. J. Physiol. Cell Physiol. 302, C1421–C1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu H. A., Sun T. X., Matsuzaki T., Yi X. H., Eswara J., Bouley R., McKee M., Brown D. (2007) Heat shock protein 70 interacts with aquaporin-2 and regulates its trafficking. J. Biol. Chem. 282, 28721–28732 [DOI] [PubMed] [Google Scholar]
- 52. Yui N., Okutsu R., Sohara E., Rai T., Ohta A., Noda Y., Sasaki S., Uchida S. (2009) FAPP2 is required for aquaporin-2 apical sorting at trans-Golgi network in polarized MDCK cells. Am. J. Physiol. Cell Physiol. 297, C1389–C1396 [DOI] [PubMed] [Google Scholar]
- 53. Calebiro D., Nikolaev V. O., Gagliani M. C., de Filippis T., Dees C., Tacchetti C., Persani L., Lohse M. J. (2009) Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 7, e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lencer W. I., Brown D., Ausiello D. A., Verkman A. S. (1990) Endocytosis of water channels in rat kidney: cell specificity and correlation with in vivo antidiuresis. Am. J. Physiol. 259, C920–C932 [DOI] [PubMed] [Google Scholar]
- 55. Li C., Wang W., Summer S. N., Westfall T. D., Brooks D. P., Falk S., Schrier R. W. (2008) Molecular mechanisms of antidiuretic effect of oxytocin. J. Am. Soc. Nephrol. 19, 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shi M., Bennett T. A., Cimino D. F., Maestas D. C., Foutz T. D., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2003) Functional capabilities of an N-formyl peptide receptor-Gαi2 fusion protein: assemblies with G proteins and arrestins. Biochemistry 42, 7283–7293 [DOI] [PubMed] [Google Scholar]