

Background: The regulatory mechanisms of glycosyltransferase activity are still poorly understood.
Results: ENPP3 was identified as an inhibitory factor for N-acetylglucosaminyltransferase GnT-IX (GnT-Vb) in Neuro2a cells. The underlying basis for this inhibition is the ENPP3-catalyzed hydrolysis of the nucleotide sugar donor substrate.
Conclusion: ENPP3 is a regulator of glycosyltransferase activity.
Significance: A novel regulatory system for the cellular glycosylation process is proposed.
Keywords: Glycobiology, Glycosylation, Glycosyltransferases, Nucleoside Nucleotide Metabolism, Phosphodiesterases
Abstract
Our previous studies on a β1,6-N-acetylglucosaminyltransferase, GnT-IX (GnT-Vb), a homolog of GnT-V, indicated that the enzyme has a broad GlcNAc transfer activity toward N-linked and O-mannosyl glycan core structures and that its brain-specific gene expression is regulated by epigenetic histone modifications. In this study, we demonstrate the existence of an endogenous inhibitory factor for GnT-IX that functions as a key regulator for GnT-IX enzymatic activity in Neuro2a (N2a) cells. We purified this factor from N2a cells and found that it is identical to ectonucleotide pyrophosphatase/phosphodiesterase 3 (ENPP3), as evidenced by mass spectrometry and by the knockdown and overexpression of ENPP3 in cultured cells. Kinetic analyses revealed that the mechanism responsible for the inhibition of GnT-IX caused by ENPP3 is the ENPP3-mediated hydrolysis of the nucleotide sugar donor substrate, UDP-GlcNAc, with the resulting generation of UMP, a potent and competitive inhibitor of GnT-IX. Indeed, ENPP3 knockdown cells had significantly increased levels of intracellular nucleotide sugars and displayed changes in the total cellular glycosylation profile. In addition to chaperones or other known regulators of glycosyltransferases, the ENPP3-mediated hydrolysis of nucleotide sugars would have widespread and significant impacts on glycosyltransferase activities and would be responsible for altering the total cellular glycosylation profile and modulating cellular functions.
Introduction
Protein glycosylation is the most frequent post-translational modification in mammals (1). Glycans are structurally highly diverse and widely distributed on cell surfaces and in the extracellular and intracellular spaces, where they play important roles in various cellular processes, such as cell adhesion, molecular trafficking and clearance, receptor function, and signal transduction (2–5). A series of glycosyltransferases (6, 7) that reside in the endoplasmic reticulum, Golgi apparatus, nucleus, and cytoplasm are principally involved in the construction of glycans and play a role in generating the structurally diverse glycans that are found in nature. Such structural diversity is regulated by various factors encompassing the cell type-specific expression of glycosyltransferases, the availability of acceptor and nucleotide sugar donor substrates, and competition between glycosyltransferases for common acceptor substrates (2, 3, 8, 9). Additionally, several unique mechanisms for dictating the structural diversity of glycans are gradually being revealed. Some glycosyltransferases are regulated by the formation of heterocomplexes with other glycosyltransferases or by proteolytic cleavage for full or accelerated activities (10–14). Several other glycosyltransferases are known to be under the control of unique regulatory proteins that modulate protein folding, enzymatic activity, and functional localization in the intra-Golgi subcompartment (15–18). Furthermore, it has been speculated that several nucleoside diphosphatases or phosphodiesterases are implicated in the cellular glycosylation process because of their hydrolytic activities toward nucleotide sugars and their metabolites (19–23). Although the mechanisms for regulating the cellular glycosylation process are quite complicated, in order to understand the biosynthesis and functions of glycans, it will be necessary to develop a better understanding of specifically how each glycosyltransferase activity is regulated at the cellular level.
Previously, we and Pierce's group (24, 25) independently succeeded in cloning a brain-specific N-acetylglucosaminyltransferase GnT-IX4 (also designated as GnT-Vb), a closely related homolog of GnT-V. The acceptor substrate specificity of this enzyme is broader than that for GnT-V and catalyzes the transfer of a GlcNAc residue in a β1,6-linkage from UDP-GlcNAc to α-mannose residues in both N-linked and O-mannosyl glycan core structures (26). Recent reports on the functional characteristics of GnT-IX suggest that the enzyme acts almost exclusively on the O-mannosyl glycan as an acceptor substrate in vivo that results in the formation of a brain-specific β1,6-branched O-mannosyl glycan (27, 28). The β1,6-branched O-mannosyl glycan was proposed to be involved in neural cell adhesion and migration through β-catenin signaling (29). In addition, our quite recent work has demonstrated that the β1,6-branched O-mannosyl glycan in the corpus callosum plays a role in inhibiting remyelination in the cuprizone-induced demyelination mouse model (28). These accumulated observations suggest that GnT-IX has a specific and significant role in the nervous system. We also demonstrated that the brain-specific gene expression of GnT-IX is under the control of epigenetic histone modification (30); however, the regulatory mechanisms of the enzymatic activity of GnT-IX remain poorly understood.
In the present study, we report on a novel regulatory mechanism for glycosyltransferase activity, based on purifying, characterizing, and identifying a cellular proteinaceous inhibitor for GnT-IX as ENPP3 (ectonucleotide pyrophosphatase/phosphodiesterase 3), which catalyzes the hydrolysis of the nucleotide sugar donor substrate UDP-GlcNAc, which results in a reduction in its cellular level and simultaneously generates UMP, which exerts a strong inhibitory activity.
EXPERIMENTAL PROCEDURES
Materials
Materials were obtained as follows: para-nitrophenyl (pNP)-5′-thymidine monophosphate (TMP), anti-FLAG (M2) antibody, and FLAG peptides from Sigma-Aldrich; HiTrap Q HP, Sephadex G-150, HiTrap Chelating HP, Superdex 200 10/300 GL, Mono Q 5/50 GL, nickel-Sepharose 6 Fast Flow, and NHS-activated Sepharose 4 Fast Flow from GE Healthcare; hydroxyapatite from Seikagaku Co. (Tokyo, Japan); Trypsin Gold from Promega (Madison, WI); lysylendopeptidase (Achromobacter proteinase I) from Wako Pure Chemicals (Osaka, Japan); Anti-c-Myc (9E10) antibody from Abcam (Cambridge, UK); and horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody from Jackson ImmunoResearch (West Grove, PA). Anti-human GnT-V (24D11) antibody was generously supplied by Fujirebio (Tokyo, Japan), and 4-fluoro-7-nitro-2,1,3-benzoxadiazole (NBD-F) was from Tokyo Chemical Industry Co. (Tokyo, Japan).
Construction of Plasmids
All PCR steps were carried out using PrimeStar HS DNA polymerase (Takara, Shiga, Japan). The C-terminally FLAG-tagged human GnT-IX was constructed as follows. The previously constructed GnT-IX expression plasmid (24) was used as a PCR template to obtain the brain form of GnT-IX (25) with the following primer set: forward primer, 5′-CTGGAAGGGGAAGGAGAAGTTCCTGGGCATCCTGAA-3′; reverse primer, 5′-TCCTTCCCCTTCCAGATGCTCGCCTCCTTGCCGTA-3′. The resulting PCR product was cloned in Escherichia coli. The resulting clone, GnT-IX(−LQ), was further amplified by PCR to add FLAG sequence on the C terminus with the following primer set: forward primer, 5′-CACCATGATCACCGTCAACCCCGA-3′; reverse primer, 5′-AGGCGAATTCACTTGTCGTCATCGTCTTTGTAGTCCAGACAGCCCTGGCACAAGGCCACCT-3′. The PCR product was cloned into pENTR-D-TOPO (Invitrogen). The expression plasmid for GnT-IX was finally constructed by conversion of the insert from pENTR to pcDNA6.2-DEST vector (designated as pcDNA6.2-hGnT-IX-FLAG).
The C-terminally Myc-His-tagged human GnT-V (31) was prepared as follows. The GnT-V coding sequence was amplified by PCR on the plasmid pCXN2-hGnT-V (32) as a template with the following primer set: forward primer, 5′-AGGGTACCATGGCTCTCTTCACTCC-3′ (KpnI site underlined); reverse primer, 5′-AGGCAGTCTTTGCAGAGAGC-3′. The PCR product was digested with KpnI and ligated between the KpnI and EcoRV sites of pcDNA3.1/Myc-His version C (Invitrogen) (designated as pcDNA3.1-hGnT-V-Myc-His).
Mouse ENPP3 cDNA was cloned by RT-PCR on total RNA from mouse brain tissue with the following primer set: forward primer, 5′-ACGGGAACAATGGATTCCAG-3′; reverse primer, 5′-CCCCATTTTGTCAAATGGCT-3′. The resulting PCR product was subcloned into the EcoRV site of the pBluescript II SK vector (Stratagene, La Jolla, CA) (designated as pBS-mENPP3).
The C-terminally Myc-His-tagged mouse ENPP3 was constructed as follows. The ENPP3 coding sequence was amplified by PCR on the pBS-mENPP3 as the template with the following primer set: forward primer (M13-RV), 5′-GGAAACAGCTATGACCATG-3′; reverse primer (mENPP3-R1), 5′-ATAATGGTTTCAAACGTGGGCA-3′. The PCR product was digested with BamHI and ligated between the BamHI and EcoRV sites of pcDNA3.1/Myc-His version C (designated as pcDNA3.1-mENPP3-Myc-His).
The C-terminally Myc-His-tagged and catalytically inactive mouse ENPP3 was constructed as follows. The upstream half and downstream half of the mouse ENPP3 coding sequences were separately amplified by PCR on the pBS-mENPP3 as the template with the following primer sets, which were specifically designed to mutate the threonine 205 catalytic center into alanine: forward primer M13-RV and reverse primer mENPP3-R2 (5′-GATTTGGGAAGGCTTTGGTGGGAT-3′) (site of the mutation is underlined) and forward primer mENPP3-F1 (5′-ATCCCACCAAAGCCTTCCCAAATC-3′) (site of the mutation underlined) and reverse primer mENPP3-R1, respectively. The two separately amplified PCR products were combined and then used as the template for the final PCR with the following primer set: forward primer, M13-RV; reverse primer, mENPP3-R1. The amplified PCR product was digested with BamHI and then ligated between the BamHI and EcoRV sites of pcDNA3.1/Myc-His version C (designated as pcDNA3.1-mENPP3(T205A)-Myc-His). The coding sequences of all the constructs were verified by DNA sequencing with a Dye Terminator Cycle Sequencing Kit and ABI Prism 3100 Genetic Analyzer (PerkinElmer Life Sciences).
Preparation of Fluorescent Acceptor Substrates
Fluorescent acceptor substrates for GnT activity assays, including pyridylaminated (PA)-agalactobiantennary sugar chain (GnGnbi-PA, GlcNAcβ1–2Manα1-3(GlcNAcβ1-2Manα1–6)Manβ1-4GlcNAcβ1–4GlcNAc-PA), PA-agalactotriantennary sugar chain (Gn3-tri′-PA, GlcNAcβ1–2Manα1–3(GlcNAcβ1–2(GlcNAcβ1–6)Manα1–6)Manβ1–4GlcNAcβ1–4GlcNAc-PA), and 7-nitro-2,1,3-benzoxadiazole-4-yl (NBD)-Ser-linked O-mannosyldisaccharide (GnM-S-NBD, GlcNAcβ1–2Manα1-O-Ser-NBD), were prepared as follows.
GnGnbi-PA was prepared from the hen's egg yolk sialylglycopeptide, which contains an N-linked disialobiantennary sugar chain as glycan moiety (33). The N-glycan was liberated from the sialylglycopeptide by hydrazinolysis and pyridylaminated, as described previously (34). The resulting PA-N-glycan was digested using a mixture of Arthrobacter ureafaciens sialidase (Nacalai Tesque, Kyoto, Japan) and jack bean β-galactosidase (Seikagaku Biobusiness, Tokyo, Japan) to remove sialic acid and galactose residues. The digested product, GnGnbi-PA, was then purified by reversed phase HPLC with a TSKgel ODS-80TM column (0.78 × 30 cm, Tosoh, Tokyo, Japan). The elution was performed isocratically at 55 °C at a flow rate of 3 ml/min using 20 mm ammonium acetate (pH 4.0) containing 0.15% 1-butanol as the eluate, and fluorescence was monitored at excitation and emission wavelengths of 320 and 400 nm, respectively.
Gn3-tri′-PA was enzymatically synthesized from GnGnbi-PA by reaction with UDP-GlcNAc catalyzed by GnT-V, which had been overexpressed in COS-1 cells. The resulting Gn3-tri′-PA was purified by reversed phase HPLC under the same conditions as described above.
GnM-S-NBD was prepared as follows. The Ser-linked O-mannosyltrisaccharide (GGnM-S, Galβ1–4GlcNAcβ1–2Manα1-O-Ser) was chemically synthesized as described previously (35). The α-amino group of Ser in the synthetic GGnM-S was fluorescently labeled with NBD-F as described previously (36, 37) with minor modifications. Briefly, 40 μmol of NBD-F was dissolved in 200 μl of acetonitrile and then added to 200 nmol of GGnM-S that had been dissolved in 200 μl of 50 mm borate buffer (pH 8.0). The reaction was allowed to proceed for 15 min at 50 °C, and the reaction mixture was then extracted with 400 μl of ethyl acetate to remove the large excess of free labeling reagent, followed by centrifugation at 20,000 × g for 1 min. The resulting lower layer (aqueous phase) containing the NBD-labeled GGnM-S (GGnM-S-NBD) was collected and subjected to two more extractions and centrifugations. The collected GGnM-S-NBD was then purified by reversed phase HPLC with a TSKgel ODS-80TM column (0.78 × 30 cm). The elution was performed isocratically at 55 °C using 20 mm ammonium acetate buffer (pH 4.0) containing 7% acetonitrile at a flow rate of 3 ml/min, and the fluorescence was monitored at excitation and emission wavelengths of 470 and 530 nm, respectively. The purified GGnM-S-NBD was digested with jack bean β-galactosidase to remove a galactose residue, and the resulting GnM-S-NBD was purified by reversed phase HPLC under the same conditions as described above. The content of the fluorescent acceptor substrate was determined by spectrofluorometry using free NBD-F as a standard.
Purification of GnT-IX-inhibitory Factor
All purification steps were carried out at 4 °C unless otherwise stated. The buffers used were as follows, with the pH adjusted at room temperature: buffer A, 10 mm Tris-HCl buffer (pH 7.4), 0.25 m sucrose, and proteinase inhibitors (Complete EDTA-free, Roche Applied Science); buffer B, 10 mm Tris-HCl buffer (pH 7.4), 20% glycerol, 1% Triton X-100, and proteinase inhibitors (Complete EDTA-free); buffer C, 10 mm Tris-HCl buffer (pH 8.0) and 0.1% Triton X-100; buffer D, 20 mm sodium phosphate buffer (pH 6.8), 0.1 m NaCl, and 0.05% Triton X-100; buffer E, 50 mm sodium phosphate buffer (pH 6.8) and 0.05% Triton X-100; buffer F, 20 mm Tris-HCl buffer (pH 8.0), 0.5 m NaCl, and 0.05% Triton X-100; buffer G, 20 mm Tris-HCl buffer (pH 8.0), 0.15 m NaCl, and 0.05% Triton X-100; and buffer H, 20 mm Tris-HCl buffer (pH 8.0) and 0.05% Triton X-100.
The cultured N2a cells (180 × 15-cm diameter dishes) were harvested after the cells reached confluence. The harvested cells were homogenized in 85 ml of buffer A using a Dounce homogenizer, followed by centrifugation at 600 × g for 10 min. The resulting supernatant was collected, and the residual pellet was subjected to one additional extraction and centrifugation, after which all of the supernatants were combined (165 ml). After centrifugation at 8,000 × g for 10 min, the precipitate was collected (designated as P8), the supernatant was further centrifuged at 105 × g for 1 h, and the precipitate was collected (designated as P100). The P8 and P100 fractions were then separately powdered with cold acetone (−20 °C). An inhibitory factor for GnT-IX could be purified from both the P8 and P100 fractions by basically the same protocol as described below. Thus, the purification procedures for the P8 fraction are described below.
Step 1
The P8 acetone powder was suspended in 20 ml of buffer B, gently rotated for 1 h, and then centrifuged at 105 × g for 1 h. The resulting supernatant was collected, and the residual pellet was subjected to one additional extraction and ultracentrifugation, after which all of the supernatants were combined (36 ml).
Step 2
Two volumes of buffer C were added to the extract from Step 1 and then applied to two tandemly connected HiTrap Q HP columns (5 ml × 2) that had been equilibrated with buffer C. Fractions of 5 ml were collected during this column chromatography. The column was washed with 5 column volumes of buffer C, and the bound proteins were eluted with a linear gradient (20 column volumes) of 0–0.8 m NaCl in buffer C. Fractions containing GnT-IX-inhibitory activity were combined and then concentrated to 5 ml with an Amicon Ultra 15 centrifugal filter device (30K, Millipore, Billerica, MA).
Step 3 (Performed at Room Temperature)
The concentrate from Step 2 was applied to a Sephadex G-150 column (2.5 × 97.5 cm) that had been equilibrated with buffer D. Fractions of 5.5 ml were collected during this column chromatography. The elution was performed with the same buffer. Fractions containing the inhibitory activity were combined.
Step 4 (Performed at Room Temperature)
The combined fraction from Step 3 was directly applied to a hydroxyapatite column (2.5 × 15.5 cm) that had been equilibrated with buffer E. Fractions of 15 ml were collected during this column chromatography. The elution was performed with the same buffer, and the flow-through fractions containing the inhibitory activity were combined. The buffer in the combined fraction was replaced by buffer F with an Amicon Ultra 15 centrifugal filter device.
Step 5
The concentrate (7.6 ml) from Step 4 was applied to a HiTrap Zn2+-chelating HP column (5 ml) that had been equilibrated with buffer F. Fractions of 1.8 ml were collected during this column chromatography. The elution was performed with the same buffer, and the flow-through fractions containing the inhibitory activity were combined. The combined fraction was concentrated to 5.6 ml with an Amicon Ultra 4 centrifugal filter device (30K, Millipore).
Step 6
The concentrate from Step 5 was applied to a HiTrap Cu2+-chelating HP column (1 ml) that had been equilibrated with buffer F. Fractions of 1 ml were collected during this column chromatography. The column was washed with 5 column volumes of buffer F, and the bound proteins were eluted with a linear gradient (20 column volumes) of 0–0.2 m glycine in buffer F. Fractions containing the inhibitory activity were combined and then concentrated to 0.3 ml with an Amicon Ultra 4 centrifugal filter device.
Step 7
The concentrate from Step 6 was applied to a Superdex 200 10/300 GL column that had been equilibrated with buffer G. Fractions of 0.4 ml were collected during this column chromatography. The elution was performed with the same buffer. Fractions containing the inhibitory activity were combined, and the buffer in the fraction was replaced by buffer H with an Amicon Ultra 4 centrifugal filter device.
Step 8
The concentrate (1.8 ml) from Step 8 was applied to a Mono Q 5/50 GL column that had been equilibrated with buffer H. Fractions of 0.5 ml were collected during this column chromatography. The column was washed with 5 column volumes of buffer H, and the elution was carried out with a linear gradient (20 column volumes) of 0–0.5 m NaCl in buffer H. Fractions containing the inhibitory activity were combined, and the buffer in the fraction was replaced by 20 mm Tris-HCl buffer (pH 8.0), concentrated with an Amicon Ultra 4 centrifugal filter device, and then collected as the final purified protein fraction (0.4 ml). This final fraction was predicted to contain less than 0.4% Triton X-100 because the detergent contained in buffer H had been concentrated at the final ultrafiltration step.
Identification of Inhibitory Factor
The purified proteins were digested with PNGase F (Roche Applied Science) at 37 °C overnight to remove N-glycan moieties, and the digested products were subjected to SDS-PAGE under reducing conditions on an 8.5% gel. The protein bands were visualized using a SilverQuest silver staining kit (Invitrogen). The stained bands were excised, reduced, and S-carbamoylmethylated and then in-gel digested using a mixture of 1.3 μg of trypsin and 0.6 μg of lysylendopeptidase (Achromobacter proteinase I) in 100 μl of 50 mm ammonium hydrogen carbonate solution containing 0.1% n-octyl-β-d-glucoside at 37 °C overnight. The resulting peptides were extracted from the gel with 50% acetonitrile, 0.1% TFA, followed by secondary extraction with 70% acetonitrile, 0.1% TFA, after which all of the extracts were combined, evaporated, and then lyophilized. After redissolving the lyophilizate with 20 μl of 2% acetonitrile, 0.1% TFA, tandem mass spectra were obtained on a Finnigan LCQ Deca XP ion trap mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray ionization device (AMR, Tokyo, Japan), which was connected to a Paradigm MS4 μHPLC system (Michrom BioResources, Auburn, CA). Reversed phase HPLC was performed on a Magic C18 column (5 μm, 0.2 × 50 mm; Michrom BioResources) equipped with a FortisTip capillary needle (AMR). In order to identify the proteins, the tandem mass spectra obtained were compared with the data in the NCBI-nr database using MASCOT Daemon software (Matrix Science, London, UK).
Protein Expression System in Cultured Cells
All of the cell lines used were grown and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin G, and 0.1 mg/ml streptomycin at 37 °C under a humidified atmosphere of 95% air and 5% CO2. The HEK 293T human embryonic kidney cells or Neuro2a (N2a) mouse neuroblastoma cells were transiently transfected with pcDNA6.2-hGnT-IX-FLAG, pCXN2-hGnT-V, pcDNA3.1-mENPP3-Myc-His, pcDNA3.1-mENPP3(T205A)-Myc-His, or pcDNA3.1-hGnT-V-Myc-His using a Lipofectamine 2000 reagent (Invitrogen). Two days after transfection, the cells were harvested and used in various experiments.
ENPP3 Knockdown in Cultured Cells
Stealth RNAi Enpp3 siRNAs (MSS277508 and MSS209891, Invitrogen) or stealth RNAi Negative Control Duplex (Invitrogen) were transiently transfected to N2a cells at 20 nm concentration using a Lipofectamine RNAiMAX reagent (Invitrogen). Two days after transfection, the cells were harvested and used in various experiments.
Preparation of Cell Lysate
The harvested cells were suspended in the lysis buffer consisting of 10 mm HEPES buffer (pH 7.5), 150 mm NaCl, 1% Triton X-100, and proteinase inhibitors (Complete EDTA-free) and sonicated for 5 min in an ice-chilled water bath using BioRuptor sonicator (Cosmobio, Tokyo, Japan). After centrifugation at 20,000 × g for 10 min, the resulting supernatant was collected and used in various experiments. The protein concentration was determined with a BCA protein assay kit (Pierce) using bovine serum albumin (BSA) as a standard.
Preparation of Recombinant Proteins
HEK 293T cells (three 10-cm diameter dishes) were transiently transfected with pcDNA3.1-mENPP3-Myc-His, pcDNA3.1-mENPP3(T205A)-Myc-His, pcDNA3.1-hGnT-V-Myc-His, or pcDNA6.2-hGnT-IX-FLAG using Lipofectamine 2000 reagent, and, after 2 days of culture, the cells were harvested. The harvested cells were homogenized with a Dounce homogenizer in 1.5 ml of 10 mm HEPES buffer (pH 7.5) containing 150 mm NaCl and proteinase inhibitors (Complete EDTA-free), followed by centrifugation at 600 × g for 10 min. The resulting supernatant was collected, and the residual pellet was subjected to one additional extraction and centrifugation, after which all of the supernatants were combined. After centrifugation at 105 x g for 1 h, the resulting precipitate was collected as the membrane fraction.
The intact and mutated ENPP3-Myc-His recombinant proteins were purified as follows. The membrane fraction from the transfectant for each construct was solubilized in 1 ml of 20 mm Tris-HCl buffer (pH 8.0) containing 0.5 m NaCl, 1% Triton X-100, and 25 mm imidazole for 1 h with gentle rotation, followed by centrifugation at 105 × g for 1 h. The resulting supernatant was collected and incubated with Ni2+-chelating Sepharose 6 Fast Flow resin (50 μl) that had been equilibrated with 20 mm Tris-HCl buffer (pH 8.0), 0.5 m NaCl, 0.1% Triton X-100, and 25 mm imidazole. After washing the resin with the same buffer, the bound proteins were eluted with 20 mm Tris-HCl buffer (pH 8.0), 0.5 m NaCl, 0.1% CHAPS, and 0.25 m imidazole.
The GnT-IX-FLAG and GnT-V-Myc-His recombinant proteins were purified using immunoaffinity resins as follows. Anti-FLAG (M2) antibody or anti-c-Myc (9E10) antibody was introduced into NHS-activated Sepharose 4 Fast Flow resin according to the manufacturer's instructions. The membrane fraction from the transfectant for each construct was solubilized in 1 ml of 10 mm HEPES buffer (pH 7.5) containing 0.15 m NaCl, 1% Triton X-100, and proteinase inhibitors (Complete, EDTA-free) for 1 h with gentle rotation. After centrifugation at 105 × g for 1 h, the resulting supernatant was collected. The GnT-IX-FLAG protein was pulled down from the supernatant using the anti-FLAG-Sepharose 4 Fast Flow resin (50 μl) that had been equilibrated with 10 mm HEPES buffer (pH 7.5) containing 0.15 m NaCl and 0.1% Triton X-100, and after washing the resin with the same buffer, the bound proteins were eluted with 0.1 mm FLAG peptide dissolved in 10 mm HEPES buffer (pH 7.5) containing 0.15 m NaCl and 0.1% CHAPS. The GnT-V-Myc-His protein was pulled down from the supernatant using the anti-c-Myc-Sepharose 4 Fast Flow resin (50 μl) that had been equilibrated with 10 mm HEPES buffer (pH 7.5) containing 0.15 m NaCl and 0.1% Triton X-100, and after washing the resin with the same buffer, the bound proteins were eluted with 0.1 m glycine-HCl buffer (pH 2.8) containing 0.1% CHAPS, and the pH of the eluate was rapidly neutralized with a one-tenth volume of 1 m Tris-HCl buffer (pH 9.0). Finally, the buffer of each eluate was replaced with 10 mm HEPES buffer (pH 7.5) containing 0.15 m NaCl and 0.1% CHAPS using an Amicon Ultra 0.5 centrifugal filter device (30K, Millipore) and then used in various experiments.
GnT Activity Assays
GnT-IX activity was assayed using Gn3-tri′-PA as a fluorescent acceptor substrate unless otherwise stated. The enzyme was incubated at 37 °C in a final volume of 10 μl in a reaction mixture consisting of 50 mm HEPES buffer (pH 7.5), 10 mm MnCl2, 0.5% Triton X-100, 1 mg/ml BSA, 20 mm UDP-GlcNAc, 5 μm Gn3-tri′-PA, and 2.5 μl of enzyme source. After 4 h of incubation, 40 μl of water was added, and the enzyme reaction was terminated by boiling for 2 min, followed by centrifugation at 20,000 × g for 5 min. The resulting supernatant was collected and injected into a TSKgel ODS-80TM column (0.46 × 25 cm) to separate and quantitatively determine the product. The elution was performed isocratically at 55 °C at a flow rate of 1 ml/min using 20 mm ammonium acetate buffer (pH 4.0) containing 0.1% 1-butanol. The fluorescence was monitored using excitation and emission wavelengths of 320 and 400 nm, respectively.
After establishing a complete assay protocol for GnT-IX activity using the Gn3-tri′-PA acceptor substrate, as described above, we found that the O-mannose-type substrate, GnM-S-NBD, was a better acceptor substrate for GnT-IX, and the enzyme activity was about 30-fold higher than that for Gn3-tri′-PA. Thus, in the second half of this study, GnT-IX was assayed using the GnM-S-NBD acceptor substrate so as to achieve a more sensitive measurement of the enzyme activity and for minimizing the consumption of the enzyme source. The separation and quantification of the product derived from the GnM-S-NBD was performed under a second set of reversed phase HPLC conditions using a TSKgel ODS-80TM column (0.46 × 15 cm). The elution was performed isocratically at 55 °C at a flow rate of 1 ml/min using 20 mm ammonium acetate buffer (pH 4.0) containing 7% acetonitrile, and the fluorescence was monitored using excitation and emission wavelengths of 470 and 530 nm, respectively.
GnT-V activity was assayed in the same reaction mixture as for GnT-IX except that the fluorescent acceptor substrate used was 5 or 25 μm GnGnbi-PA. The conditions used for separating and quantitating the product by reversed phase HPLC have been reported previously (38).
Inhibitory Activity Assay
GnT activity was assayed in the reaction mixture described above in the presence or absence of 2.5 μl of inhibitory factor fraction, and the enzyme activities between the above two measured conditions were compared. The inhibitory activity was calculated according to our tentative unit definition as follows. One unit of inhibitory factor is defined as the amount required for causing a 50% inhibition of GnT activity.
ENPP3 Activity Assay
The phosphodiesterase activity of ENPP3 was measured using pNP-5′-TMP as the substrate, as described previously (39, 40) with minor modifications. Briefly, the ENPP3 reaction was carried out in a final volume of 200 μl in a reaction mixture consisting of 50 mm Tris-HCl buffer (pH 8.5), 5 mm KCl, 140 mm NaCl, 1 mm CaCl2, 0.5 mm pNP-5′-TMP, and 20 μl of enzyme source. After incubation at 37 °C for 15 min, 200 μl of 0.2 m NaOH was added to terminate the reaction, and the amount of generated p-nitrophenol was measured colorimetrically at 400 nm using a molar extinction coefficient of 18,320 m−1·cm−1 (41).
Real-time PCR
Total RNA was isolated from cultured cells using RNAiso PLUS reagent (Takara). One microgram of total RNA was reverse-transcribed using the Transcriptor First Strand cDNA Synthesis kit (Roche Applied Science) with an oligo(dT)18 primer. Real-time PCR was performed on the reverse-transcribed cDNA using THUNDERBIRD SYBR quantitative PCR mix (TOYOBO, Tsuruga, Japan) with the following primer sets: ENPP3, forward primer 5′-CCATTGTCACGGGTTTGTATC-3′ and reverse primer 5′-GGGATTGCTTTTCTCCACTG-3′; GAPDH, forward primer 5′-CACAATTTCCATCCCAGACC-3′ and reverse primer 5′-GGGTGCAGCGAACTTTATTG-3′. Normalization of the data were carried out using GAPDH mRNA levels.
Western Blot Analyses
The protein fraction was resolved by SDS-PAGE according to the method of Laemmli (42) and transferred to nitrocellulose membrane (Bio-Rad). After blocking with 5% skim milk in Tris-buffered saline containing 0.05% Tween 20 (TBST), the membrane was incubated with the primary antibody (anti-c-Myc (9E10, 1:5,000), anti-FLAG (M2, 1:5,000), or anti-GnT-V (24D11, 1:10,000)) for 1 h at room temperature. After washing with TBST, the membrane was incubated with the secondary antibody, HRP-conjugated anti-mouse IgG (1:5,000) for 30 min at room temperature, followed by washing with TBST. Immunoreactive proteins were finally visualized by chemiluminescence using an ECL system reagent (GE Healthcare).
UDP-GlcNAc Hydrolytic Activity of ENPP3
The purified recombinant ENPP3 protein was incubated in a final volume of 10 μl in a reaction mixture consisting of 50 mm HEPES buffer (pH 7.5), 10 mm MnCl2, 0.5% Triton X-100, 20 mm UDP-GlcNAc, and 100 ng of the recombinant enzyme. After incubation at 37 °C for 4 h, 40 μl of ice-chilled water and 150 μl of cold ethanol (−20 °C) were added to terminate the enzyme reaction. After centrifugation at 20,000 × g for 10 min at 4 °C, the supernatant was collected and taken to dryness using a centrifugal evaporator. The dried sample was redissolved in water and analyzed by our previously developed ion pair reversed phase HPLC (43) with minor modifications. Briefly, the water-soluble materials were injected into an Inertsil ODS-4 HPLC column (3 μm, 0.46 × 15 cm, GL Science, Tokyo, Japan) to separate and determine the product at 40 °C at a flow rate of 1 ml/min. The eluents used were 100 mm potassium phosphate buffer (pH 6.4) containing 8 mm tetrabutylammonium hydrogen sulfate (eluent A) and a mixture of eluent A and acetonitrile (70:30) (eluent B). The column was equilibrated with 100% eluent A, and after sample injection, eluent A was held at 100% for 25 min, and then solvent B was increased to 100% in 1 min and then held at 100% for 4 min. The progress of the elution was monitored at 254 nm.
Kinetics
The purified GnT recombinant protein was incubated in a reaction mixture of 10 μl consisting of 50 mm HEPES buffer (pH 7.5), 10 mm MnCl2, 0.5% Triton X-100, 1 mg/ml BSA, 25 μm GnM-S-NBD for GnT-IX or 25 μm GnGnbi-PA for GnT-V and various concentrations of UDP-GlcNAc in the presence or absence of UMP at 37 °C for 4 h. In all assays, the consumption of the acceptor substrate was kept below 20% to ensure accurate initial velocity measurement.
Simultaneous Determination of Intracellular Nucleotide Sugars in Cultured Cells
To determine the level of nucleotide sugars in the cultured cells, our previously developed ion pair reversed phase HPLC method was used. The experimental procedures, including the extraction of nucleotide sugars from cultured cells, purification of the extracted nucleotide sugars, and separation and quantification by ion pair reversed phase HPLC, have already been reported in detail previously (43).
Preparation of PA-Glycans from Cultured Cells
In order to determine the total cellular glycosylation profile, cultured cells were homogenized in ice-chilled phosphate-buffered saline (PBS), and the protein fraction (1 mg) was precipitated with 3 volumes of cold acetone (−20 °C). The precipitated proteins were redissolved in water, evaporated, and then lyophilized. The N- and O-glycans were simultaneously liberated from the lyophilizate by hydrazinolysis at 60 °C for 16 h, and the reducing ends of the liberated glycans were pyridylaminated as described previously (34). The resulting PA-glycans were purified with a graphite carbon column (GL-Pak Carbograph 300 mg, GL Science) according to the method of Natsuka et al. (44) and then used for HPLC analysis.
HPLC for PA-Glycans
Normal phase HPLC was performed as described previously (45). Reversed phase HPLC was performed at 30 °C on a TSKgel ODS-80TS column (0.2 × 15 cm, Tosoh) at a flow rate of 0.2 ml/min. The eluents used were 50 mm triethylamine acetate (pH 6.0) (eluent A) and eluent A containing 20% acetonitrile (eluent B). The column was equilibrated with eluent A, and after sample injection, eluent B was linearly increased to 18% over 54 min and then increased to 100% in 16 min. Fluorescence was monitored using excitation and emission wavelengths of 315 and 400 nm, respectively.
MS Analysis of PA-Glycans
Mass spectra of PA-oligosaccharides were obtained as described previously (45).
RESULTS
N2a Cells Possess an Intrinsic Inhibitory Factor for GnT-IX
In the course of our establishing a high expression system for GnT-IX in several mammalian cell lines, including COS-1, HEK 293, and N2a cells, we found evidence for an endogenous inhibitory activity for GnT-IX specifically in N2a cells and not among the other cell lines. The apparent GnT-IX enzymatic activity was significantly reduced only in lysates from N2a cells in which GnT-IX was being overexpressed, although comparative levels of GnT-IX transcript expression were detected among those cell lines. This caused us to hypothesize that N2a cells intrinsically produce an inhibitor of GnT-IX.
To examine this aspect further, we prepared the cell lysate from GnT-IX-overexpressing HEK 293T cells as the GnT-IX enzyme source and measured the enzymatic activity in the presence of an N2a cell lysate. We found that GnT-IX was inhibited by the addition of an N2a cell lysate in a dose-dependent manner (Fig. 1A). Moreover, we found that this inhibition was heat-labile (Fig. 1A). Contrary to this, GnT-V, a homolog of GnT-IX, was not inhibited in a range of quantities of N2a cell lysate (Fig. 1B). Collectively, these findings indicate that a GnT-IX-inhibitory factor was endogenously present in N2a cells. Additionally, we found that the GnT-IX-inhibitory activity was exclusively detected in the membrane fraction of N2a cells and could be efficiently solubilized with detergents such as Triton X-100 and sodium deoxycholate, indicating that the GnT-IX-inhibitory factor is membrane-bound. When we treated an N2a cell lysate with PNGase F, the GnT-IX-inhibitory activity in the cell lysate was completely abolished (Fig. 1C), indicating that the GnT-IX-inhibitory factor in N2a cells is probably a glycoprotein.
FIGURE 1.
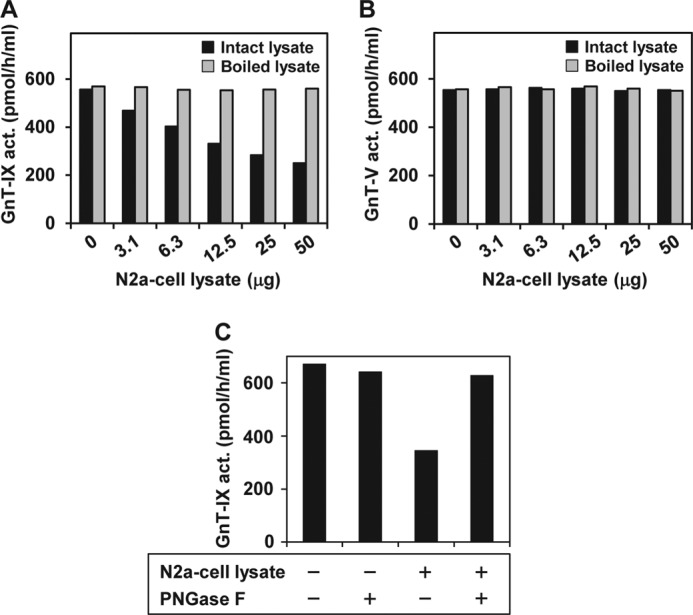
Identification of GnT-IX-inhibitory activity in N2a cells. Effects of N2a cell lysate on GnT-IX (A) and GnT-V (B) activities, respectively. The lysate of the HEK 293T cells transfected with each construct was used as the enzyme source. Enzymatic activity was assayed in the presence of various quantities of N2a cell lysate with or without boiling for 3 min prior to assays. C, effects of de-N-glycosylation of the N2a cell lysate on its endogenous GnT-IX-inhibitory activity. N2a cell lysate (∼50 μg) was treated with PNGase F (0.5 unit) for 37 °C overnight prior to the assay and then assessed for its endogenous GnT-IX-inhibitory activity. All data are presented as the mean of duplicate measurements. Act., activity.
ENPP3 Is a GnT-IX-inhibitory Factor in N2a Cells
Protein purification is one of the most effective ways to identify such an unidentified proteinaceous factor. GnT-IX-inhibitory activity was measured according to our tentative unit definition (see “Experimental Procedures”). When the GnT-IX-inhibitory activity contained by an N2a cell lysate was assayed, the inhibitory activity was found to be proportional to the quantity of cell lysate examined, at least up to 5 μg. As a result, our assay method for GnT-IX inhibitory activity was judged to be sufficient for monitoring the GnT-IX-inhibitory factor in its purification.
We prepared the membrane fraction from N2a cells that were cultured on a large scale as the starting material, and after an acetone treatment and solubilization with Triton X-100, the resulting extract was subjected to seven successive column chromatography steps (Fig. 2A). GnT-IX-inhibitory activity was measured and the active fractions from each column chromatography step were pooled and then loaded onto the next column. Finally, the purification of the GnT-IX-inhibitory factor was completed by Mono Q anion exchange column chromatography. SDS-PAGE analysis of the final purified protein fraction showed the presence of two bands with molecular weights of >250,000 and 145,000 under both reducing and non-reducing conditions. The molecular weights of the two bands were shifted from >250,000 and 145,000 to 250,000 and 116,000, respectively, as the result of PNGase F digestion, conclusively demonstrating that they are N-glycosylated proteins.
FIGURE 2.
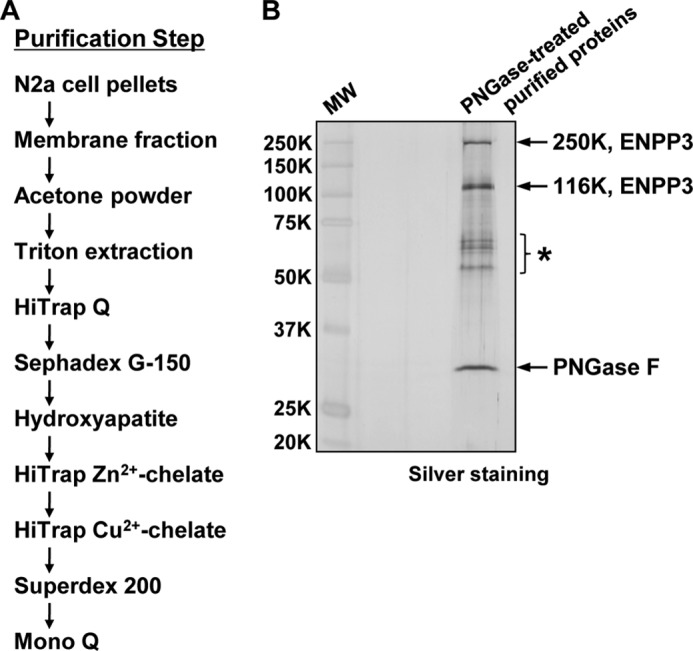
Purification and identification of ENPP3 as an inhibitory factor for GnT-IX in N2a cells. A, schematic representation of the protocol for the purification of the GnT-IX-inhibitory factor from N2a cells. B, SDS-PAGE of the de-N-glycosylated purified proteins, which were then subjected to mass spectrometry analysis for protein identification. A 1:20 aliquot of the final purified protein fraction was pretreated with PNGase F (0.5 unit) for 37 °C overnight and then subjected to SDS-PAGE under reducing conditions followed by silver staining. Protein identification was carried out by LC-electrospray ionization-ion trap-MS/MS analysis on the in-gel digested proteins. MW, molecular weight markers. *, bands representing contaminating keratins.
To identify the purified inhibitor proteins, after PNGase F digestion, they were subjected to SDS-PAGE and visualized by silver staining (Fig. 2B). The resulting 250,000 and 116,000 bands were excised from the gel and analyzed by mass spectrometry. The findings indicate that they were derived from a single protein and were identified as ENPP3, a member of the ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) enzyme family, which is composed of seven members, ENPP1–7. The 250,000 band may have been due to the non-reducible dimerization of the enzyme. ENPP3 is a typical type II transmembrane glycoprotein with a small N-terminal intracellular domain, a single-spanning transmembrane domain, and a large catalytic C-terminal extracellular domain and is localized to the cell surface, where the enzyme is thought to hydrolyze pyrophosphate or phosphodiester bonds in extracellular nucleotides and their derivatives by the following catalytic reaction (46, 47, 48): NTP → NMP + PPi.
To verify that ENPP3 is indeed a GnT-IX-inhibitory factor in N2a cells, we first engineered ENPP3 knockdown (KD) cells by introducing siRNAs into N2a cells. As shown in Fig. 3, A and B, the introduction of ENPP3 siRNAs into N2a cells resulted in a significant reduction in the levels of ENPP3 mRNA and phosphodiesterase activity to about 50% compared with the control cells. This indicates that the alteration in the ENPP3 enzymatic activity was entirely consistent with its mRNA level in this cell line. We confirmed that the GnT-IX-inhibitory activity was also significantly reduced in the ENPP3-KD cells to about 50%, in good agreement with the levels of ENPP3 mRNA and enzymatic activity (Fig. 3C), indicating that ENPP3 is a GnT-IX-inhibitory factor in N2a cells.
FIGURE 3.

Confirmation of ENPP3-mediated GnT-IX inhibition. A–C, endogenous ENPP3 in N2a cells was knocked down (KD) by introducing siRNAs. siControl, siRNA negative control duplex; siENPP3_1 and 2, specific Enpp3 siRNAs, MSS277508 and MSS209893, respectively (Invitrogen). A, ENPP3 mRNA level in the KD cells. Real-time PCR was performed on the reverse-transcribed total RNA from each KD cell. The ENPP3 mRNA level was normalized to the corresponding GAPDH mRNA level, and the percentage level relative to the control is shown. B, phosphodiesterase activity in the KD cells. Enzyme activity was assayed using pNP-5′-TMP as the substrate on the cell lysate from each KD cell. C, GnT-IX-inhibitory activity in the KD cells. The GnT-IX-inhibitory activity was assayed on the cell lysate from each KD cell. The GnT-IX-transfected HEK 293T-cell lysate was used as the enzyme source. D–F, the C-terminally Myc-His-tagged intact and T205A-mutated ENPP3s were transiently transfected to HEK 293T cells. D, ENPP3 protein level in the transfected cells. Western blot analysis was performed on the cell lysate (5 μg) from each transfectant (n = 3). E, phosphodiesterase activity in the transfected cells. The activity was assayed using pNP-5′-TMP as the substrate on the cell lysate from each transfectant. F, GnT-IX-inhibitory activity in the transfected cells. The inhibitory activity of the cell lysate from each transfectant was assayed. The GnT-IX-transfected HEK 293T-cell lysate was used as the enzyme source. N.D., not detected. All data except in D are shown as the mean ± S.D. (error bars) (n = 3).
To further strengthen this conclusion, we next engineered ENPP3-overexpressing cells and enzymatically characterized their inhibitory activity for GnT-IX. It is well known that the ENPP family members have a structurally related catalytic domain and that catalysis proceeds via the formation of a covalent catalytic intermediate with a conserved threonine residue (serine in the case of ENPP6) in the catalytic site (46, 49). We constructed intact ENPP3 and mutated ENPP3 in which the threonine 205 active site was point-mutated to alanine (T205A), which would be expected to render the molecule catalytically inactive. We then transiently transfected them into HEK 293T cells and confirmed that the intact and T205A-mutated ENPP3s were expressed in the transfected cells at equivalent levels, as evidenced by Western blotting (Fig. 3D). Phosphodiesterase activity assays using the transfected cell lysate revealed that the intact ENPP3 exhibited a substantial enzymatic activity, whereas the T205A-mutated ENPP3 was indeed catalytically inactive (Fig. 3E). We found that the GnT-IX-inhibitory activity was significantly increased in the intact ENPP3-transfected cells, whereas this was not the case in the mock- or T205A-mutated ENPP3-transfected cells (Fig. 3F), again indicating that ENPP3 is indeed a GnT-IX-inhibitory factor in N2a cells and, further, that the enzymatic activity of ENPP3 is essential for exerting its GnT-IX-inhibitory activity. Additionally, several other in vitro studies on the characteristics of ENPP3 using the transfected cell lysate as enzyme source revealed that 1) the phosphodiesterase and GnT-IX-inhibitory activities of ENPP3 were completely abolished by boiling or by PNGase F digestion and 2) ENPP3 potently inhibited GnT-IX but hardly inhibited GnT-V in vitro. All of these characteristics of ENPP3 are completely consistent with those observed at the level of an N2a cell lysate (Fig. 1) and provide further support for our conclusion that ENPP3 is a GnT-IX-inhibitory factor in N2a cells.
ENPP3 Hydrolyzes UDP-GlcNAc to UMP and GlcNAc 1-Phosphate
Because GnT-IX requires UDP-GlcNAc as a nucleotide sugar donor substrate, we examined the issue of whether ENPP3 is capable of catalyzing the hydrolysis of UDP-GlcNAc. We partially purified the intact and T205A-mutated ENPP3 recombinant proteins from the transfectant for each construct (see “Experimental Procedures”) and confirmed their enzymatic functionality by assessing the phosphodiesterase and GnT-IX- and GnT-V-inhibitory activities in vitro. As shown in Fig. 4A, when the intact ENPP3 reacted with UDP-GlcNAc and the resulting reaction mixture was subjected to ion pair reversed phase HPLC analysis (see “Experimental Procedures”), UMP was newly detected, and the level of UDP-GlcNAc was correspondingly decreased. In contrast, no changes were observed in reaction mixtures of T205A-mutated ENPP3 (Fig. 4A). These findings conclusively indicate that ENPP3 catalyzes the hydrolysis of UDP-GlcNAc to UMP and GlcNAc 1-phosphate (Fig. 4B), although the latter product was not directly detected in the HPLC analysis because of its lack of effective absorbance.
FIGURE 4.

ENPP3-mediated UDP-GlcNAc hydrolysis. The intact and T205A-mutated ENPP3 recombinant proteins were generated and partially purified (see “Experimental Procedures”). A, ion pair reversed phase HPLC profile on the hydrolysate of UDP-GlcNAc by ENPP3. The reaction was carried out by mixing 100 ng of the partially purified intact or T205A-mutated recombinant ENPP3 with 20 mm UDP-GlcNAc, and the resulting product was analyzed by ion pair reversed phase HPLC, as described under “Experimental Procedures.” The arrows indicate the elution positions of authentic compounds, UMP, UDP-GlcNAc, and UDP. B, deduced reaction of ENPP3-catalyzed hydrolysis of UDP-GlcNAc. GlcNAc-1-P, GlcNAc 1-phosphate.
GnT-IX Is Highly Sensitive to UMP and Is More Strongly Inhibited by UMP than GnT-V
The obtained data indicate that the reduction in UDP-GlcNAc level caused by the ENPP3 enzymatic activity represents one of the underlying mechanisms for the inhibition of GnT-IX. This could not, however, explain the specificity of inhibition in which ENPP3 potently inhibits GnT-IX but not GnT-V. We next focused on the inhibition of GnTs caused by the two hydrolysis products of UDP-GlcNAc by ENPP3 (i.e. UMP and GlcNAc 1-phosphate). We partially purified the GnT-IX and -V recombinant proteins from the transfectant for each construct (see “Experimental Procedures”), and using these recombinant enzymes, we evaluated the inhibition of GnT-IX and -V caused by UMP or GlcNAc 1-phosphate. As shown in Fig. 5, A and B, UMP, but not GlcNAc 1-phosphate, inhibited both GnTs and more strongly affected the GnT-IX reaction even at low concentrations, compared with that for GnT-V. A kinetic analysis of the inhibition of GnT-IX and -V caused by UMP revealed the existence of competitive inhibition, and the Ki value for UMP of GnT-IX (0.07 mm) was determined to be about 64 times lower than that of GnT-V (4.49 mm) (Fig. 5, C and D), revealing the enzymatic basis for why GnT-IX is more sensitive to inhibition caused by UMP than GnT-V. We conclude that a marked difference in the Ki value for UMP between GnT-IX and -V satisfactorily explains why GnT-IX is more potently inhibited by ENPP3 than GnT-V at least in vitro.
FIGURE 5.

Mechanism of ENPP3-mediated GnT-IX inhibition. The partially purified recombinant GnT-IX and -V (see “Experimental Procedures”) were used as the enzyme sources. Shown is inhibition of GnT-IX (A) and GnT-V (B) activities caused by UMP and GlcNAc 1-phosphate (GlcNAc-1-P). Enzymatic activity was assayed at a fixed concentration of 2 mm UDP-GlcNAc and 25 μm acceptor substrate, GnM-S-NBD and GnGnbi-PA for GnT-IX and -V, respectively. Shown is competitive inhibition of GnT-IX (C) and GnT-V (D), respectively, caused by UMP. Data are shown in double-reciprocal plots. Enzymatic activity was assayed at a fixed concentration of 25 μm acceptor substrate as described above and various concentrations of UDP-GlcNAc. Ki values for UMP of GnT-IX and -V are indicated in the panels. Apparent Km values for UDP-GlcNAc of GnT-IX and -V estimated from the plots were 1.5 and 7.6 mm, respectively. All data are shown as the mean of duplicate measurements.
ENPP3 Regulates the Level of Intracellular Nucleotide Sugars
We next addressed the intracellular function of ENPP3 on the regulation of GnT-IX activity. Because no probe for detecting GnT-IX-glycan products, such as lectins or antibodies, is available, we instead monitored intracellular nucleotide sugar levels, which reflect the first step of the ENPP3-mediated GnT-IX inhibition. We again engineered ENPP3-KD N2a cells by introducing siRNA and confirmed the reduction (∼−60%) in the phosphodiesterase activity in the ENPP3-KD cells compared with that in the control cells. As shown in Fig. 6, the level of intracellular UDP-GlcNAc was significantly increased in the ENPP3-KD cells compared with the control cells, an observation that is consistent with our in vitro observation on the ENPP3-catalyzed hydrolysis of UDP-GlcNAc (Fig. 4). These findings indicate that ENPP3 controls the intracellular UDP-GlcNAc level and further suggest that the ENPP3 latently regulates the intracellular GnT-IX activity.
FIGURE 6.
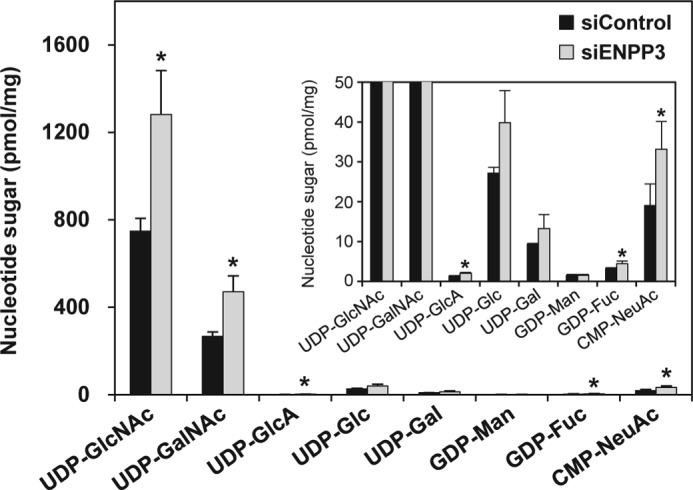
The level of intracellular nucleotide sugars in ENPP3-KD cells. Endogenous ENPP3 in N2a cells was knocked down by introducing siRNAs, negative control duplex, and specific Enpp3 siRNA (MSS277508). Intracellular nucleotide sugars were extracted, purified, and separated and quantitatively determined by ion pair reversed phase HPLC as described under “Experimental Procedures.” The inset exhibits the magnified figure to clearly demonstrate the changes in the minor nucleotide sugars. All data are shown as the mean ± S.D. (error bars) (n = 3). Significant difference versus control cells as determined by Student's t test is shown. *, p < 0.05.
In addition, we found that the level of several other intracellular nucleotide sugars, including UDP-GalNAc, CMP-NeuAc, GDP-Fuc, and UDP-GlcA, was significantly increased in the ENPP3-KD cells (Fig. 6), suggesting that ENPP3 may catalyze the hydrolysis of a broader spectrum of nucleotide sugars and may have impacts on a variety of glycosyltransferase activities through dictating the availability of intracellular nucleotide sugars.
ENPP3 Modulates the Cellular Glycosylation Profile
Finally, we examined whether ENPP3 is capable of modulating the glycan biosynthesis in vivo through its hydrolytic activity toward the intracellular nucleotide sugars. We again engineered ENPP3-KD N2a cells as described above. The N- and O-glycans were simultaneously liberated from the ENPP3-KD and control cells by hydrazinolysis at 60 °C for 16 h, and the reducing ends of the liberated glycans were fluorescently labeled by pyridylamination (see “Experimental Procedures”). As shown in Fig. 7A, the prepared PA-glycans were first separated into six fractions, F1–F6, by normal phase HPLC, the PA-oligosaccharides being separated in accordance with their molecular size. Each collected fraction was then further analyzed by reversed phase HPLC, the PA-oligosaccharides being separated according to their oligosaccharide structure. As shown in Fig. 7, B–G, comparison of the PA-glycan profiles from the ENPP3-KD cells with those from the control cells revealed small but distinct changes in the HPLC elution profiles of the PA-glycans in the ENPP3-KD cells. We picked up 21 characteristic peaks, G1–G21, whose expression changes were caused by ENPP3 knockdown in N2a cells and confirmed that they are all PA-glycans by mass spectrometry (Table 1). Fifteen glycans, G1–G12, G16, G20, and G21, among the selected glycans displayed an increase in the expression levels in the ENPP3-KD cells, and the other glycans, G13–G15 and G17–G19, conversely displayed a reduction in the expression levels by ENPP3 knockdown in N2a cells. From these findings, we conclude that ENPP3 is able to function as a modifier of the glycan biosynthesis in vivo.
FIGURE 7.

Comparison of HPLC elution profiles of the PA-glycans from ENPP3-KD and control cells. ENPP3-KD N2a cells were engineered as described in the legend to Fig. 6. A, normal phase HPLC profile of the PA-glycans from ENPP3-KD and control cells. Fractions F1–F6 were collected as indicated by the partitioned bars, and each collected fraction was further analyzed by reversed phase HPLC (B–G). Although many peaks were found to show changes in their expression level in the ENPP3-KD cells, we picked up 21 peaks, G1–G21, which displayed characteristic changes in the expression level and were judged to be quantitatively sufficient for subsequent MS analysis. The numbered arrowheads in A indicate the elution position of PA-glucose oligomers with the corresponding degree of polymerization. The inset in C shows an enlargement to clearly demonstrate the changes in the elution profile. Peaks eluted at less than 40 min and 15 min in A and B–G, respectively, were due to contaminating materials or by-products of the pyridylamination reaction.
TABLE 1.
Mass analysis of the representative PA-glycans that displayed characteristic changes in expression level in ENPP3-KD N2a cells
| Peak | Mass |
Estimated compositiona | |
|---|---|---|---|
| Observed | Calculated | ||
| G1 | 948.3 | 948.4 [M + H]+ | Hex4HexNAc1-PA |
| G2 | 1151.5 | 1151.4 [M + H]+ | Hex4HexNAc2-PA |
| G3 | 1151.5 | 1151.4 [M + H]+ | Hex4HexNAc2-PA |
| G4 | 1110.2 | 1110.4 [M + H]+ | Hex5HexNAc1-PA |
| G5 | 1110.5 | 1110.4 [M + H]+ | Hex5HexNAc1-PA |
| G6 | 1231.4 | 1231.4 [M + H]+ | Hex7-PA |
| G7 | 1442.4 | 1442.5 [M + H]+ | NeuAc1Hex4HexNAc2-PA |
| G8 | 1272.4 | 1272.5 [M + H]+ | Hex6HexNAc1-PA |
| G9 | 1272.4 | 1272.5 [M + H]+ | Hex6HexNAc1-PA |
| G10 | 1272.4 | 1272.5 [M + H]+ | Hex6HexNAc1-PA |
| G11 | 1906.4 | 1906.7 [M + H]+ | Hex4HexNAc5dHex1-PA |
| G12 | 2198.1 | 2197.8 [M + H]+ | NeuAc1Hex4HexNAc5dHex1-PA |
| G13 | 2010.4 | 2010.8 [M + H]+ | NeuAc1Hex5HexNAc4-PA |
| G14 | 1906.8 | 1906.7 [M + H]+ | Hex4HexNAc5dHex1-PA |
| G15 | 1678.1 | 1678.6 [M + H]+ | Hex6HexNAc3-PA |
| G16 | 2359.4 | 2359.9 [M + H]+ | NeuAc1Hex5HexNAc5dHex1-PA |
| G17 | 2649.7 | 2651.0 [M + H]+ | NeuAc2Hex5HexNAc5dHex1-PA |
| G18 | 2650.2 | 2651.0 [M + H]+ | NeuAc2Hex5HexNAc5dHex1-PA |
| G19 | 1970.0 | 1969.7 [M + H]+ | NeuAc1Hex6HexNAc3-PA |
| G20 | 3088.6 | 3090.2 [M + H]+ | NeuAc1Hex7HexNAc7dHex1-PA |
| G21 | 3102.9 | 3104.1 [M + H]+ | NeuAc3Hex6HexNAc5dHex1-PA |
a A typical saccharide spaced ladder signal was fully or partially confirmed by MS/MS analysis with respect to these PA-glycans.
DISCUSSION
In this study, we identified ENPP3 as a GnT-IX-inhibitory factor in N2a cells (Figs. 2 and 3) and revealed the mechanism of the ENPP3-mediated GnT-IX inhibition. The underlying basis for this inhibition was found to be the ENPP3-catalyzed hydrolysis of the nucleotide sugar donor substrate, UDP-GlcNAc, and the resulting generation of UMP, a potent and competitive inhibitor of GnT-IX (Figs. 4 and 5). We also reported herein an important link of ENPP3 to the regulatory system for dictating the level of intracellular nucleotide sugars (Fig. 6) that would be possible to modulate the broad spectrum glycosyltransferase activities and to alter the total cellular glycosylation profile (Figs. 7 and 8 and Table 1).
FIGURE 8.
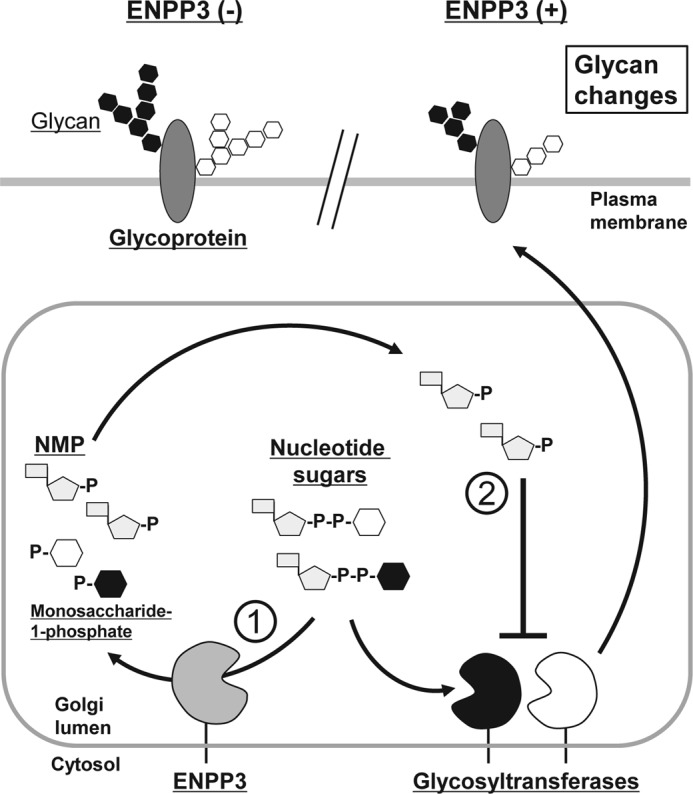
A hypothetical model of the ENPP3-mediated modulation of glycan biosynthesis. A schematic representation of the ENPP3-mediated nucleotide sugar degradation system in the regulation of glycan biosynthesis is shown. ENPP3 is predicted to regulate the cellular glycosylation process potentially through the following two mechanisms: 1) ENPP3-catalyzed hydrolysis of the intracellular nucleotide sugars that would reduce the availability of the donor substrates for glycosyltransferases and act on the enzymes in an inhibitory manner and 2) the inhibition of glycosyltransferases caused by a hydrolysate of nucleotide sugars by ENPP3, such as NMP, which can be a potent inhibitor for certain kinds of glycosyltransferases with high sensitivity (low Ki value) toward it. P, phosphate.
ENPP3 was initially determined to be a 130-kDa glycoprotein (gp130RB13-6) recognized by the monoclonal antibody RB13-6 on a specific subset of rat brain glial precursor cells (50), which were later reported to differentiate in vitro into radial-glial, astrocytic, and ependymal cells (51), a finding suggesting that ENPP3 plays a role in the glial cell differentiation process. This expression of ENPP3 in the brain is consistent with that of GnT-IX and is not inconsistent with our proposing the ENPP3-mediated GnT-IX inhibition in vivo; however, attention should be paid to the reported findings on the spatiotemporal expression of ENPP3 in rat brain development (52, 53). Namely, ENPP3-positive cells are distributed throughout the neuroepithelium in early rat brain development, whereas the spatiotemporal expression is characteristic in the germinal layers of the ventricular zone during later development (51). On the other hand, GnT-IX was recently reported to be distributed throughout the neuroepithelium in early mouse brain development, a finding that is consistent with rat ENPP3, whereas the enzyme was relatively absent from the ventricular zone but was highly expressed in the subventricular zone during later development (27). Thus, it will be necessary to clarify the physiological significance of the ENPP3-mediated GnT-IX inhibition from a spatiotemporal viewpoint in the future.
ENPP3 has a type II transmembrane topology, in common with the majority of glycosyltransferases cloned to date, and is known to be a typical ectoenzyme that is localized to the cell surface, where the enzyme plays a role in metabolizing extracellular nucleotides and their derivatives (46). Thus, it has been proposed that ENPP3 has an extracellular role in modulating nucleotide-mediated signal transduction, a process that is known as purinergic signaling (47, 52, 53); however, an intracellular role of ENPP3 has not been elucidated. During its transport to the cell surface, newly synthesized ENPP3 is folded in the endoplasmic reticulum and then transported to the Golgi apparatus, where the catalytic domain of ENPP3 faces the lumen and would be predicted to catalyze the hydrolysis of nucleotide sugars. Indeed, the present study provides the first demonstration that ENPP3 catalyzes the hydrolysis of the nucleotide sugar UDP-GlcNAc in vitro (Fig. 4) and also shows that the levels of several intracellular nucleotide sugars, including UDP-GlcNAc, UDP-GalNAc, CMP-NeuAc, GDP-Fuc, and UDP-GlcA, are significantly increased by knocking down endogenous ENPP3 in N2a cells (Fig. 6). These findings strongly suggest that ENPP3 has an intracellular function in which it plays a role in modulating the level of intracellular nucleotide sugars, possibly through its catalytic activity. We may say that the intracellular nucleotide sugars, at least as indicated above, are candidates for physiological substrates of ENPP3, although an additional in vitro examination will be needed in the future regarding nucleotide sugars other than UDP-GlcNAc.
There are many lines of evidence to indicate that the availability of intracellular nucleotide sugars regulates glycosyltransferase activities and controls the total cellular glycosylation profile (54–59). Thus, the ENPP3-mediated alteration of intracellular nucleotide sugar levels would modulate the activity of a broad spectrum of glycosyltransferases and consequently alter the total cellular glycosylation profile. Indeed, in this study, we demonstrated that the ENPP3-KD N2a cells have the altered cellular glycosylation profiles compared with those for the control cells (Fig. 7 and Table 1), conclusively indicating that ENPP3 is one of the regulatory factors for the glycan biosynthesis in vivo. We found that ENPP3 elicits a different action in each glycan (i.e. the levels of some glycans are increased and others are decreased by ENPP3 knockdown in N2a cells). With respect to the increase in the level of the glycans caused by ENPP3 knockdown, we think that this could be explained by the increase in the level of the intracellular nucleotide sugars and/or the reduction in the level of the inhibitory nucleotides, such as UMP. On the other hand, two possibilities may be considered with regard to the decrease in the level of the glycans by ENPP3 knockdown. The first possibility is that the intracellular environment of N2a cells (i.e. the predicted low level of the intracellular nucleotide sugars caused by ENPP3) could be originally disadvantageous to the glycosyltransferases with a high Km value for nucleotide sugars. In that milieu, the activity of the glycosyltransferases with a low Km value for nucleotide sugars would be expected to be high. When ENPP3 is knocked down in N2a cells, the resulting increase or amelioration in the level of the intracellular nucleotide sugars could become advantageous to the high Km glycosyltransferases, which could result in the relative reduction of the availability of the common nucleotide sugars for the low Km glycosyltransferases and also in the reduction in the low Km enzyme product glycans. The second possibility is that ENPP3 may have the ability to modulate glycosyltransferase gene expression, which results in alterations of the repertoire of the glycosyltransferases and glycan structures expressed in the cells, although these issues remain to be examined in the future. Furthermore, additional experiments will be needed to uncover the full repertoire of the affected nucleotide sugars, glycosyltransferases, and glycan structures in the future.
The findings reported herein indicate that ENPP3 strongly inhibits GnT-IX but only negligibly inhibits GnT-V (Fig. 1), and this result can be attributed to the marked difference in the Ki value for UMP, a hydrolysate of UDP-GlcNAc by ENPP3, between both enzymes (i.e. the Ki value for GnT-V is 64 times higher than that for GnT-IX (Fig. 5)). These findings provide a way to rationalize the sensitivity of GnT-IX and -V to the ENPP3-mediated inhibition in vitro. However, the in vivo ENPP3-mediated GnT-V inhibition is uncertain. Because the glycosyltransferase reaction in vitro was performed under extremely artificial conditions containing a large excess (20 mm) of the donor substrate UDP-GlcNAc, the level of the donor substrate was probably not decreased by ENPP3 to a degree that it would strongly affect GnT-V activity, although GnT-IX was inhibited by the generated UMP even at low concentrations. On the other hand, for example, the intra-Golgi concentration of UDP-GlcNAc was calculated to be at least less than 1 mm (60, 61). Thus, the possibility that the ENPP3-mediated reduction of the nucleotide sugar donor substrates more potently affects various glycosyltransferases in the Golgi apparatus cannot be excluded.
In order to fully establish the validity of this novel nucleotide sugar degradation system in the regulation of cellular glycosylation process, the detailed mechanism responsible for the ENPP3-mediated inhibition of glycosyltransferases and its molecular relevance to cellular functions need to be clarified in the future.
Acknowledgments
We thank Fumi Ota and Tomoko Hasegawa for technical assistance. We also thank Drs. Yasuhiko Kizuka (RIKEN), Masahiko Yabu (Osaka Medical Center for Cancer and Cardiovascular Diseases), Hidehiko Shogomori (Seikagaku Corp.), Tomohiko Taguchi (University of Tokyo), and Norihiro Kotani (Saitama Medical University) for valuable suggestions and discussions. We also thank Dr. Milton S. Feather for aid in editing the content of the manuscript.
This work was supported by Grant-in-aid for Scientific Research (A) 20249018 (to N. T.) and the Global Centers of Excellence Program of Osaka University (to N. T.) funded by the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by the Program for Promotion of Fundamental Studies in Health Science (to N. T.) of the National Institute of Biomedical Innovation, Japan. This work was also supported by incentive research projects (to H. K.) of RIKEN.
- GnT
- N-acetylglucosaminyltransferase
- Fuc
- fucose
- Man
- mannose
- Hex
- hexose
- HexNAc
- N-acetylhexosamine
- PNGase F
- peptide N-glycanase F
- PA
- pyridylaminated
- pNP
- para-nitrophenyl
- GnGnbi
- agalactobiantennary sugar chain
- Gn3-tri′
- agalactotriantennary sugar chain
- NBD
- 7-nitro-2,1,3-benzoxadiazole-4-yl
- NBD-F
- 4-fluoro-7-nitro-2,1,3-benzoxadiazole
- GGnM-S
- Ser-linked O-mannosyltrisaccharide
- ENPP
- ectonucleotide pyrophosphatase/phosphodiesterase
- KD
- knockdown.
REFERENCES
- 1. Apweiler R., Hermjakob H., Sharon N. (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 [DOI] [PubMed] [Google Scholar]
- 2. Ohtsubo K., Marth J. D. (2006) Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 [DOI] [PubMed] [Google Scholar]
- 3. Moremen K. W., Tiemeyer M., Nairn A. V. (2012) Vertebrate protein glycosylation. Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taniguchi N., Miyoshi E., Gu J., Jianguo G., Honke K., Matsumoto A. (2006) Decoding sugar functions by identifying target glycoproteins. Curr. Opin. Struct. Biol. 16, 561–566 [DOI] [PubMed] [Google Scholar]
- 5. Hart G. W., Slawson C., Ramirez-Correa G., Lagerlof O. (2011) Cross talk between O-GlcNAcylation and phosphorylation. Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Glycosyltransferases. Structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 7. Taniguchi N., Honke K., Fukuda M. (2002) Handbook of Glycosyltransferases and Related Genes, Springer, Tokyo [Google Scholar]
- 8. Dennis J. W., Granovsky M., Warren C. E. (1999) Protein glycosylation in development and disease. BioEssays 21, 412–421 [DOI] [PubMed] [Google Scholar]
- 9. Taniguchi N. (2009) From the γ-glutamyl cycle to the glycan cycle. A road with many turns and pleasant surprises. J. Biol. Chem. 284, 34469–34478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seko A., Yamashita K. (2008) Activation of β1,3-N-acetylglucosaminyltransferase-2 (β3Gn-T2) by β3Gn-T8. Possible involvement of β3Gn-T8 in increasing poly-N-acetyllactosamine chains in differentiated HL-60 cells. J. Biol. Chem. 283, 33094–33100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akasaka-Manya K., Manya H., Nakajima A., Kawakita M., Endo T. (2006) Physical and functional association of human protein O-mannosyltransferases 1 and 2. J. Biol. Chem. 281, 19339–19345 [DOI] [PubMed] [Google Scholar]
- 12. Kouno T., Kizuka Y., Nakagawa N., Yoshihara T., Asano M., Oka S. (2011) Specific enzyme complex of β-1,4-galactosyltransferase-II and glucuronyltransferase-P facilitates biosynthesis of N-linked human natural killer-1 (HNK-1) carbohydrate. J. Biol. Chem. 286, 31337–31346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kizuka Y., Matsui T., Takematsu H., Kozutsumi Y., Kawasaki T., Oka S. (2006) Physical and functional association of glucuronyltransferases and sulfotransferase involved in HNK-1 biosynthesis. J. Biol. Chem. 281, 13644–13651 [DOI] [PubMed] [Google Scholar]
- 14. Sugimoto I., Futakawa S., Oka R., Ogawa K., Marth J. D., Miyoshi E., Taniguchi N., Hashimoto Y., Kitazume S. (2007) β-Galactoside α2,6-sialyltransferase I cleavage by BACE1 enhances the sialylation of soluble glycoproteins. A novel regulatory mechanism for α2,6-sialylation. J. Biol. Chem. 282, 34896–34903 [DOI] [PubMed] [Google Scholar]
- 15. Ju T., Cummings R. D. (2002) A unique molecular chaperone Cosmc required for activity of the mammalian core 1 β3-galactosyltransferase. Proc. Natl. Acad. Sci. U.S.A. 99, 16613–16618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ju T., Aryal R. P., Stowell C. J., Cummings R. D. (2008) Regulation of protein O-glycosylation by the endoplasmic reticulum-localized molecular chaperone Cosmc. J. Cell Biol. 182, 531–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang H. H., Stanley P. (2010) A testis-specific regulator of complex and hybrid N-glycan synthesis. J. Cell Biol. 190, 893–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sasai K., Ikeda Y., Ihara H., Honke K., Taniguchi N. (2003) Caveolin-1 regulates the functional localization of N-acetylglucosaminyltransferase III within the Golgi apparatus. J. Biol. Chem. 278, 25295–25301 [DOI] [PubMed] [Google Scholar]
- 19. Ohkubo I., Ishibashi T., Taniguchi N., Makita A. (1980) Purification and characterization of nucleoside diphosphatase from rat-liver microsomes. Evidence for metalloenzyme and glycoprotein. Eur. J. Biochem. 112, 111–118 [DOI] [PubMed] [Google Scholar]
- 20. Ohkubo I., Taniguchi N., Mitsuyama T., Tsukada Y., Makita A. (1982) Comparative studies on nucleoside diphosphatase of rat ascites hepatoma and rat liver. Activity level, purification and properties. Int. J. Biochem. 14, 1075–1081 [DOI] [PubMed] [Google Scholar]
- 21. Yanagisawa K., Resnick D., Abeijon C., Robbins P. W., Hirschberg C. B. (1990) A guanosine diphosphatase enriched in Golgi vesicles of Saccharomyces cerevisiae. Purification and characterization. J. Biol. Chem. 265, 19351–19355 [PubMed] [Google Scholar]
- 22. Rebbe N. F., Tong B. D., Finley E. M., Hickman S. (1991) Identification of nucleotide pyrophosphatase/alkaline phosphodiesterase I activity associated with the mouse plasma cell differentiation antigen PC-1. Proc. Natl. Acad. Sci. U.S.A. 88, 5192–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang M., Shen Z., Huang S., Zhao L., Chen S., Mak T. W., Wang X. (2010) The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 143, 711–724 [DOI] [PubMed] [Google Scholar]
- 24. Inamori K., Endo T., Ide Y., Fujii S., Gu J., Honke K., Taniguchi N. (2003) Molecular cloning and characterization of human GnT-IX, a novel β1,6-N-acetylglucosaminyltransferase that is specifically expressed in the brain. J. Biol. Chem. 278, 43102–43109 [DOI] [PubMed] [Google Scholar]
- 25. Kaneko M., Alvarez-Manilla G., Kamar M., Lee I., Lee J. K., Troupe K., Zhang W., Osawa M., Pierce M. (2003) A novel β(1,6)-N-acetylglucosaminyltransferase V (GnT-VB). FEBS Lett. 554, 515–519 [DOI] [PubMed] [Google Scholar]
- 26. Inamori K., Endo T., Gu J., Matsuo I., Ito Y., Fujii S., Iwasaki H., Narimatsu H., Miyoshi E., Honke K., Taniguchi N. (2004) N-Acetylglucosaminyltransferase IX acts on the GlcNAc β1,2-Manα1-Ser/Thr moiety, forming a 2,6-branched structure in brain O-mannosyl glycan. J. Biol. Chem. 279, 2337–2340 [DOI] [PubMed] [Google Scholar]
- 27. Lee J. K., Matthews R. T., Lim J. M., Swanier K., Wells L., Pierce J. M. (2012) Developmental expression of the neuron-specific N-acetylglucosaminyltransferase Vb (GnT-Vb/IX) and identification of its in vivo glycan products in comparison with those of its paralog, GnT-V. J. Biol. Chem. 287, 28526–28536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanekiyo K., Inamori K., Kitazume S., Sato K., Maeda J., Higuchi M., Kizuka Y., Korekane H., Matsuo I., Honke K., Taniguchi N. (2013) Loss of branched O-mannosyl glycans in astrocytes accelerates remyelination. J. Neurosci. 33, 10037–10047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abbott K. L., Matthews R. T., Pierce M. (2008) Receptor tyrosine phosphatase β (RPTPβ) activity and signaling are attenuated by glycosylation and subsequent cell surface galectin-1 binding. J. Biol. Chem. 283, 33026–33035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kizuka Y., Kitazume S., Yoshida M., Taniguchi N. (2011) Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J. Biol. Chem. 286, 31875–31884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gu J., Nishikawa A., Tsuruoka N., Ohno M., Yamaguchi N., Kangawa K., Taniguchi N. (1993) Purification and characterization of UDP-N-acetylglucosamine. α-6-d-mannoside β1–6N-acetylglucosaminyltransferase (N-acetylglucosaminyltransferase V) from a human lung cancer cell line. J. Biochem. 113, 614–619 [DOI] [PubMed] [Google Scholar]
- 32. Nakahara S., Saito T., Kondo N., Moriwaki K., Noda K., Ihara S., Takahashi M., Ide Y., Gu J., Inohara H., Katayama T., Tohyama M., Kubo T., Taniguchi N., Miyoshi E. (2006) A secreted type of β1,6N-acetylglucosaminyltransferase V (GnT-V), a novel angiogenesis inducer, is regulated by γ-secretase. FASEB J. 20, 2451–2459 [DOI] [PubMed] [Google Scholar]
- 33. Seko A., Koketsu M., Nishizono M., Enoki Y., Ibrahim H. R., Juneja L. R., Kim M., Yamamoto T. (1997) Occurence of a sialylglycopeptide and free sialylglycans in hen's egg yolk. Biochim. Biophys. Acta 1335, 23–32 [DOI] [PubMed] [Google Scholar]
- 34. Natsuka S., Hase S. (1998) Analysis of N- and O-glycans by pyridylamination. Methods Mol. Biol. 76, 101–113 [DOI] [PubMed] [Google Scholar]
- 35. Seifert J., Ogawa T., Kurono S., Ito Y. (2000) Syntheses of α-dystroglycan derived glycosyl amino acids carrying a novel mannosyl serine/threonine linkage. Glycoconj. J. 17, 407–423 [DOI] [PubMed] [Google Scholar]
- 36. Kurihara T., Min J. Z., Toyo'oka T., Fukushima T., Inagaki S. (2007) Determination of fluorescence-labeled asparaginyl-oligosaccharide in glycoprotein by reversed-phase ultraperformance liquid chromatography with electrospray ionization time-of-flight mass spectrometry. Anal. Chem. 79, 8694–8698 [DOI] [PubMed] [Google Scholar]
- 37. Min J. Z., Toyo'oka T., Kawanishi H., Fukushima T., Kato M. (2005) Novel fluorescent asparaginyl-N-acetyl-d-glucosamines (Asn-GlcNAc) for the resolution of oligosaccharides in glycopeptides, based on enzyme transglycosylation reaction. Anal. Chim. Acta 550, 173–181 [Google Scholar]
- 38. Sasai K., Ikeda Y., Eguchi H., Tsuda T., Honke K., Taniguchi N. (2002) The action of N-acetylglucosaminyltransferase-V is prevented by the bisecting GlcNAc residue at the catalytic step. FEBS Lett. 522, 151–155 [DOI] [PubMed] [Google Scholar]
- 39. Rücker B., Almeida M. E., Libermann T. A., Zerbini L. F., Wink M. R., Sarkis J. J. (2007) Biochemical characterization of ecto-nucleotide pyrophosphatase/phosphodiesterase (E-NPP, E.C. 3.1.4.1) from rat heart left ventricle. Mol. Cell. Biochem. 306, 247–254 [DOI] [PubMed] [Google Scholar]
- 40. Lévesque S. A., Lavoie E. G., Lecka J., Bigonnesse F., Sévigny J. (2007) Specificity of the ecto-ATPase inhibitor ARL67156 on human and mouse ectonucleotidases. Br. J. Pharmacol. 152, 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kelly S. J., Butler L. G. (1975) Enzymic hydrolysis of phosphonate esters. Biochem. Biophys. Res. Commun. 66, 316–321 [DOI] [PubMed] [Google Scholar]
- 42. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 43. Nakajima K., Kitazume S., Angata T., Fujinawa R., Ohtsubo K., Miyoshi E., Taniguchi N. (2010) Simultaneous determination of nucleotide sugars with ion-pair reversed-phase HPLC. Glycobiology 20, 865–871 [DOI] [PubMed] [Google Scholar]
- 44. Natsuka S., Hirohata Y., Nakakita S., Sumiyoshi W., Hase S. (2011) Structural analysis of N-glycans of the planarian Dugesia japonica. FEBS J. 278, 452–460 [DOI] [PubMed] [Google Scholar]
- 45. Misonou Y., Shida K., Korekane H., Seki Y., Noura S., Ohue M., Miyamoto Y. (2009) Comprehensive clinico-glycomic study of 16 colorectal cancer specimens. Elucidation of aberrant glycosylation and its mechanistic causes in colorectal cancer cells. J. Proteome Res. 8, 2990–3005 [DOI] [PubMed] [Google Scholar]
- 46. Stefan C., Jansen S., Bollen M. (2005) NPP-type ectophosphodiesterases. Unity in diversity. Trends Biochem. Sci. 30, 542–550 [DOI] [PubMed] [Google Scholar]
- 47. Stefan C., Jansen S., Bollen M. (2006) Modulation of purinergic signaling by NPP-type ectophosphodiesterases. Purinergic Signal. 2, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakanaga K., Hama K., Aoki J. (2010) Autotaxin. An LPA producing enzyme with diverse functions. J. Biochem. 148, 13–24 [DOI] [PubMed] [Google Scholar]
- 49. Gijsbers R., Ceulemans H., Stalmans W., Bollen M. (2001) Structural and catalytic similarities between nucleotide pyrophosphatases/phosphodiesterases and alkaline phosphatases. J. Biol. Chem. 276, 1361–1368 [DOI] [PubMed] [Google Scholar]
- 50. Deissler H., Lottspeich F., Rajewsky M. F. (1995) Affinity purification and cDNA cloning of rat neural differentiation and tumor cell surface antigen gp130RB13-6 reveals relationship to human and murine PC-1. J. Biol. Chem. 270, 9849–9855 [DOI] [PubMed] [Google Scholar]
- 51. Blass-Kampmann S., Kindler-Röhrborn A., Deissler H., D'Urso D., Rajewsky M. F. (1997) In vitro differentiation of neural progenitor cells from prenatal rat brain. Common cell surface glycoprotein on three glial cell subsets. J. Neurosci. Res. 48, 95–111 [DOI] [PubMed] [Google Scholar]
- 52. Goding J. W., Grobben B., Slegers H. (2003) Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim. Biophys. Acta 1638, 1–19 [DOI] [PubMed] [Google Scholar]
- 53. Zimmermann H. (2006) Nucleotide signaling in nervous system development. Pflugers Arch. 452, 573–588 [DOI] [PubMed] [Google Scholar]
- 54. Lübke T., Marquardt T., Etzioni A., Hartmann E., von Figura K., Körner C. (2001) Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat. Genet. 28, 73–76 [DOI] [PubMed] [Google Scholar]
- 55. Smith P. L., Myers J. T., Rogers C. E., Zhou L., Petryniak B., Becker D. J., Homeister J. W., Lowe J. B. (2002) Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J. Cell Biol. 158, 801–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sasai K., Ikeda Y., Fujii T., Tsuda T., Taniguchi N. (2002) UDP-GlcNAc concentration is an important factor in the biosynthesis of β1,6-branched oligosaccharides. Regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology 12, 119–127 [DOI] [PubMed] [Google Scholar]
- 57. Noda K., Miyoshi E., Gu J., Gao C. X., Nakahara S., Kitada T., Honke K., Suzuki K., Yoshihara H., Yoshikawa K., Kawano K., Tonetti M., Kasahara A., Hori M., Hayashi N., Taniguchi N. (2003) Relationship between elevated FX expression and increased production of GDP-l-fucose, a common donor substrate for fucosylation in human hepatocellular carcinoma and hepatoma cell lines. Cancer Res. 63, 6282–6289 [PubMed] [Google Scholar]
- 58. Lau K. S., Partridge E. A., Grigorian A., Silvescu C. I., Reinhold V. N., Demetriou M., Dennis J. W. (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129, 123–134 [DOI] [PubMed] [Google Scholar]
- 59. Taniguchi N. (2007) A sugar-coated switch for cellular growth and arrest. Nat. Chem. Biol. 3, 307–309 [DOI] [PubMed] [Google Scholar]
- 60. Waldman B. C., Rudnick G. (1990) UDP-GlcNAc transport across the Golgi membrane. Electroneutral exchange for dianionic UMP. Biochemistry 29, 44–52 [DOI] [PubMed] [Google Scholar]
- 61. Traynor A. J., Hall E. T., Walker G., Miller W. H., Melançon P., Kuchta R. D. (1996) Inhibition of UDP-N-acetylglucosamine import into Golgi membranes by nucleoside monophosphates. J. Med. Chem. 39, 2894–2899 [DOI] [PubMed] [Google Scholar]