

Background: GAC supplies for increased metabolic needs of tumors because of exclusive localization and kinetic properties.
Results: Higher than tetramer oligomers are the active form in in vitro and in cellular assays. Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide disrupts oligomers.
Conclusion: A novel molecular mechanism for GAC activation is proposed.
Significance: The data affect the development of therapies targeting GAC in tumors, with emphasis on allosteric inhibitors.
Keywords: Cancer, Enzyme Inhibitors, Enzyme Mechanisms, Glutaminase, Metabolism, Warburg Effect
Abstract
The phosphate-dependent transition between enzymatically inert dimers into catalytically capable tetramers has long been the accepted mechanism for the glutaminase activation. Here, we demonstrate that activated glutaminase C (GAC) self-assembles into a helical, fiber-like double-stranded oligomer and propose a molecular model consisting of seven tetramer copies per turn per strand interacting via the N-terminal domains. The loop 321LRFNKL326 is projected as the major regulating element for self-assembly and enzyme activation. Furthermore, the previously identified in vivo lysine acetylation (Lys311 in humans, Lys316 in mouse) is here proposed as an important down-regulator of superoligomer assembly and protein activation. Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide, a known glutaminase inhibitor, completely disrupted the higher order oligomer, explaining its allosteric mechanism of inhibition via tetramer stabilization. A direct correlation between the tendency to self-assemble and the activity levels of the three mammalian glutaminase isozymes was established, with GAC being the most active enzyme while forming the longest structures. Lastly, the ectopic expression of a fiber-prone superactive GAC mutant in MDA-MB 231 cancer cells provided considerable proliferative advantages to transformed cells. These findings yield unique implications for the development of GAC-oriented therapeutics targeting tumor metabolism.
Introduction
Cancer cells have a well established dependence on glutamine metabolism to support their highly proliferative status. Apart from acting as a source for nitrogen as well as reductive power, its anabolic carbon skeletons can be siphoned from the TCA cycle to be used as building blocks for the growing and dividing cells (1, 2). Both Myc and Rho GTPases have been shown to stimulate glutaminase C (GAC),5 an isozyme that possesses a distinct cellular localization as well as activation levels purported to provide the aforesaid proliferative advantage to cancer cells (3–5).
We have recently described the structural determinants of the phosphate-dependent activation mechanism of GAC, based on the tetramerization-induced lifting of a so-called gating loop (321LRFNKL326; NCBI sequence NP_001106854.1), which controls substrate accessibility to the active site. We showed that phosphate binds inside the catalytic pocket, resulting in allosteric stabilization of tetramers and facilitating substrate entry by outcompeting with the product, glutamate, to guarantee enzyme cycling (5). Additionally, recent publications have provided structural insights into glutaminase inhibition by making use of the small molecules BPTES and 968 (6–9). Nevertheless, the precise descriptions of their modes of inhibition are still lacking.
Renewed interest in cancer metabolism has prompted an innovative warfront against metabolic enzymes, aiming at the development of alternative and efficient therapeutic opportunities. Glutaminase C is a key target in this sense (2, 4, 10, 11), and the need for new and accurate biochemical and structural information to speed up and improve the development of successful therapies is therefore essential. In this regard, we now provide novel information demonstrating that the assembly of higher order, fiber-like GAC oligomers, henceforth termed the GAC superstructure, is necessary for proper enzyme activation both in vitro and in a cancer cell model. First, we demonstrate that the superstructure is mandatorily present when GAC is in the active form, as shown by negatively stained samples analyzed by transmission electron microscopy (TEM). The tendency toward the superstructure correlates well with the activation levels induced by phosphate among GAC and the other two mammalian glutaminase isozymes: the kidney-type glutaminase (KGA) and the liver-type glutaminase (LGA). Although LGA is a synonym for the GLS2 glutaminase, KGA and its splicing variant GAC are both usually indistinguishably referred to as GLS1. Moreover, we observe that the addition of the GLS1 inhibitor BPTES hampers protein polymerization by stabilizing inactive tetramers.
Further research identified a subset of key residues involved in the superstructure formation process. They are located in the gating loop, as well as at the N and C termini, which have been previously shown to be key structural features for enzyme activation (5, 12). One specific gating loop mutant, GAC.K325A, both assembles into the superstructure and shows a 600-fold enhancement in catalytic efficiency toward l-glutamine, even in the absence of phosphate. Conversely, GAC.R322A, also at the gating loop, abrogated protein activation and impeded superstructure formation. A previously identified in vivo post-translational modification of human GLS1 (13), the acetylation of Lys316 (equivalent to Lys311 in human), was also studied in this context. We show that the acetylation mimetic GAC.K316Q does not assemble into higher order oligomers, and this modification likely inhibits protein activity in cells. By combining data from point mutants, TEM, MS, and computational biology, we offer a low resolution model for the superstructure assembly. The superstructure is based on a double-stranded helix, with each strand containing tetramers interacting with each other via the N terminus domain. Lastly, we demonstrate that MDA-MB 231 cells silenced for the endogenous GAC expression and stably expressing an ectopic fiber-prone superactive mutant, proliferate more, consume higher amounts of glutamine, and grow bigger than the wild-type and mock transformed cells. Our results shed new light on the molecular mechanism of phosphate-dependent activation of the glutaminases and highlight the importance of the development of allosteric inhibitors when targeting GAC in tumors.
EXPERIMENTAL PROCEDURES
Protein Production, Enzymatic Assay, Size Exclusion Serial Dilution, and Site-directed Mutagenesis
Recombinant protein expression and purification, the streamlined glutaminase activity assay, and the size exclusion analysis of serial dilutions were performed as previously published (5). Point mutants were generated with the QuikChange II site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. The inhibition assays using BPTES (kindly provided by Dr. Chi Van Dang, Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA) were done with GAC or GAC.K325A at 5 nm and BPTES diluted in Me2SO, the last always at a 0.5% final concentration. Measurements were done in triplicate and analyzed using GraphPad Prism 5.00 (GraphPad Software) and Origin 8.1 (Originlab). The parameters of the dose-response curve were determined by fitting an error bar-weighted logistic function, in the form of y = A2 + (A1 − A2)/(1 + (x/x0) ^ p), where A1 is the initial apparent turnover rate (absence of inhibitor), A2 is the turnover rate at saturating concentrations of inhibitor, x0 is the half-maximum inhibitory concentration, or IC50, and p is the Hill slope.
Transmission Electron Microscopy
For visualization of negatively stained grids, protein samples, at a concentration ranging from 0.5 to 1 μm, were deposited onto glow-discharged holey carbon-coated grids for 60 s followed by two steps of blotting and staining with 2% uranyl acetate. Images were acquired between −1- and −3-μm defocus at 15,000–80,000× magnification using a Jeol JEM-2100 operating at 200 kV and recorded on a F-416 CMOS camera (Tietz Video and Image Processing Systems).
Crystallization and X-ray Crystallography
Crystallization experiments were performed at 291 K using the conventional sitting drop vapor diffusion technique. Drops were made by mixing three parts of protein (at 3.3 mg/ml) to one of well solution, containing 17% PEG 3350, 0.2 m NaCl, and 0.1 m Bis-Tris, pH 6.5. Clusters of plates were observed after 5 days and used as seeds for a standard streak seeding in a mother liquor solution containing 11% PEG 3350, 0.2 m NaCl, and 0.1 m Bis-Tris, pH 6.5. Before data collection at cryogenic temperature (100 K), harvested crystals were cryoprotected with 10% ethylene glycol added to the mother liquor. X-ray diffraction data were obtained at the D03B-MX1 Beamline at Laboratório Nacional de Luz Síncrotron in Brazil. The data were processed using Mosflm (14) and Scala (15). The first set of phases was obtained by the molecular replacement as implemented in Phaser (16), using the data set from the ligand-free form of GAC, available under Protein Data Bank code 3ss3 (5). Positional and B-factor refinement cycles were carried out with Phenix (17). Manual building of the extra portions and real space refinement, including Fourier electron density map inspection, were performed with Coot (18). The overall stereochemical quality of the final models and the agreements between them and experimental data were assessed by the program Molprobit (19) and the appropriate Coot routines.
Cross-linking Analysis of GAC Complexes by LC-MS/MS
GAC complexes were reduced (5 mm dithiothreitol, 25 min at 56 °C), alkylated (14 mm iodoacetamide, 30 min at room temperature in the dark), and digested with trypsin (Promega). The samples were lyophilized in a vacuum concentrator and reconstituted in 0.1% formic acid. Then 4.5 μl (1 μg) of the resulting peptide was analyzed on an LTQ Velos Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled with LC-MS/MS by an EASY-nlC system (Thermo Fisher Scientific) through a Proxeon nanoelectrospray ion source. Peptides were separated by a 2–90% acetonitrile gradient in 0.1% formic acid using a pre-column EASY-Column (2 cm × 100-μm inner diameter, 5-μm particle size) and an analytical column PicoFrit Column (20 cm - 75-μm inner diameter, 5-μm particle size; New Objective) at a flow rate of 300 nl/min over 65 min. The nanoelectrospray voltage was set to 2.2 kV, and the source temperature was 275 °C. The instrument methods in LTQ Velos Orbitrap were set up in the data-dependent acquisition mode of higher-energy collisional dissociation fragmentation. The full scan MS spectra (m/z 300–1,600) were acquired in the Orbitrap analyzer after accumulation to a target value of 1 × e6. The resolution in the Orbitrap system was set to r = 60,000, and the five most intense peptide ions with charge states of ≥2 were sequentially isolated to a target value of 50,000 and fragmented in HCD with normalized collision energy of 40% with the resolution in the Orbitrap system was set to r = 7,500 for MS/MS. The signal threshold for triggering an MS/MS event was set to 80,000 counts, and an activation time of 0.1 ms was used. Dynamic exclusion was enabled with exclusion size list of 400, exclusion duration of 60 s, and repeat count of 2. For cross-linking analysis, the raw data files generated by Xcalibur v.2.1 (Thermo Fisher Scientific) were converted to a peak list format (mgf) using Proteome Discoverer version 1.3 (Thermo Fisher Scientific). The mgf files were analyzed in MassMatrix software to automatically search chemical cross-linkage against database containing the GAC amino acid sequences. The parameters for cross-linking analysis were carbamidomethylation (+57.021460 Da) as fixed modification, oxidation of methionine (+15.99491 Da) as variable modifications, chemical cross-linked with disuccinimidyl suberate-DSS (138.06808 Da) noncleavable by enzymes, four trypsin missed cleavages, and a tolerance of 10 ppm for precursor and 0.02 Da for fragment ions (20). Potential cross-linked peptides were manually validated. This experiment was independently performed four times for the tetramer form and seven times for the superoligomer form. Unique lysine linkages (underlined in the following peptides) were only observed in the tetramer samples, between residues Lys181 and Lys207 (α-KCVQSNIVLLTQAFR and β-LKECMDMLR), Lys202 and Lys578 (α-LTLQTTSDGVMLDKDLFK and β-DTVWKK), and Lys403 and Lys512 (α-SGVAGGILLVVPNVMGMMCWSPPLDKMGNSVK and β-EKK).
Computational Biology
To obtain a model of the glutaminase fiber, we applied a previously developed rigid body docking algorithm restrained by experimental DSS cross-linking data (20). Initially, we reconstructed both the N- and C-terminal regions of the monomers using a loop reconstruction routine and placed a copy of the tetramer, namely the ligand domain, on the N-terminal of the receptor domain. Using only the exclusive intermonomer linkage that was observed in our reconstructed model (Lys181–Lys207), we selected the decoy that was in the lowest energy range and within 11 Å of lysine pair distance.
Cell Assays
MDA-MB 231 cells were purchased from ATCC (initial passage of 29) and cultivated in RPMI 1640 medium (Cultilab, Campinas, Brazil) supplemented with 10% fetal bovine serum (Cultilab). The pcDNA3.1/V5-His-hGAC (GAC-V5) clone was kindly provided by Dr. Richard Cerione (Cornell University, Ithaca, NY), and the mutation K320A (equivalent to K325A in the mouse gene) performed with the QuikChange II site-directed mutagenesis kit (GAC.K325A-V5) following the manufacturer's instructions. Lipofectamine-transfected cells were G418 selected for stable expression, which was confirmed by Western blotting. The mock control was obtained by transfecting cells with empty vector. To accomplish the endogenous GAC knockdown, the same cell clones were transduced with lentiviral particles containing pLKO-shGAC plasmid (forward, 5′-CCGGCCTCTGTTCTGTCAGAGTTCTCGAGAACTCTGACAGAACAGAGGTTTTTG-3′, and reverse, 5′-AATTCAAAAACCTCTGTTCTGTCAGAGTTCTCGAGAACTCTGACAGAACAGAGG-3′), and a pool of cells was selected for puromicin resistance. The cells were lysed with 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1 mm DTT, 1 mm NaVO4, 1 mm β-glycerol phosphate, 11 μg·ml−1 aprotinin, 11 μg·ml−1 pepstatin, and 1 mm PMSF. The lysates were resolved by SDS-PAGE, and the proteins were transferred to polyvinylidene fluoride membranes. The membranes were incubated overnight with the primary antibodies diluted in 20 mm Tris, 135 mm NaCl, and 0.1% Tween 20. The primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies followed by exposure to Pierce SuperSignal West Pico Substrate. We used anti-V5 1:10,000 (Invitrogen), anti-GAC custom made by Genescript as previously described in Ref. 5 at 1 μg·ml−1, anti-Vinculin 1:1,000 (Cell Signaling) and anti-VDAC 1:1,000 (Cell Signaling). As for the immunofluorescence and glutamine consumption studies, MDA-MB 231 cells were seeded in a 6-well plate (Becton Dickson) at a density of 2.5 × 10−5 cells·cm−2, cultivated for 48 h at 37 °C with 5% of CO2 and then fixated and permeabilized with 3.7% formaldehyde in PBS, 0.2% Triton X-100. The cells were blocked with 3% BSA in PBS, 0.8% Triton X-100, and incubated at room temperature for 1 h, with anti-V5 1:20,000 diluted in 3% BSA in PBS/0.8% Triton X-100. Goat secondary anti-mouse-Alexa 633 (Invitrogen), diluted in blocking solution (1:200) was used to reveal ectopically expressed V5-tagged GAC. The nuclei and actin were stained with 2.5 μg·ml−1 DAPI (Sigma) and phalloidin-Alexa 488 (1:40), respectively. Data were collected with the plate reader Fluorescence Microscope Operetta (PerkinElmer Life Sciences), and the cell number and area quantified were using the software Harmony 3.0. Glutamine consumption from the culture medium was assessed using the Bio-Profile Basic 4 (Nova Biomedical). BPTES-treated cells were allowed to attach for 24 h and then had the medium replaced by fresh medium added with 10 μm of the inhibitor for 48 h. For the whole cell lysate glutaminase activity assay, the cells were lysed by ressuspension in 25 mm Hepes, pH 8.0, 150 mm NaCl, 1 mm EDTA, 0.01% Triton X-100, and 1× Protease Inhibition Solution (Qiagen) followed by 20 strokes through an insulin needle. Glutaminase activity assay of the whole cell extracts was performed following the published streamlined assay (5), using 5 μg of lysate/well and l-glutamine concentration of 7.5 mm. Diameter of detached viable cells was measured by the Countess (Invitrogen) after trypsinization and trypan blue incubation.
In Cell Protein Cross-linking
Approximately 3 × 106 cells were seeded in 10-cm plates and allowed to adhere overnight. The next day, the medium was removed, and the cells were washed twice with PBS and incubated for 24 h with 2 mm l-photo-leucine and 4 mm l-photo-methionine (Pierce) in leucine- and methionine-free DMEM supplemented with 10% PBS-dialyzed FBS. After this incubation, the medium was removed, and cells were washed twice with PBS and then irradiated at 365 nm for 15 min in the Epi Chemi II Darkroom equipment (UVP Laboratory Products). The cells were harvested for lysis, and cross-linked proteins were resolved by 3–15% step gradient SDS-PAGE (3, 9, and 15% SDS-PAGE solutions prepared with 50:1 acrylamide:bisacrylamide mix, poured one after another in volume proportions of 1.5:2:1). The immunoblotting was performed with the anti-V5 1:10,000 (Invitrogen) as described in the preceding section.
Estimation of Intracellular Glutamine Concentration in Breast Tumor Cells
From metabolomics experiments on tumor samples, Yuneva et al. (21) reported a relationship of 40 to hundreds of nanomoles of glutamine for each gram of protein. We have found that unattached GAC.K325A-V5, GAC-V5, and mock transformed MD-MBA 231 cells have average diameters of 13.3, 10.9, and 10.6 μm, respectively. Assuming a spherical shape for these cells, the following cell volume can be calculated: 1.2 nl (GAC.K325A-V5 cells) and 0.6 nl (GAC-V5 and mock cells). Using the Pierce BCA protein assay kit (ThermoScientific), we quantified an average of 0.23 grams of protein for 2 × 106 cells, sampling from the three cell clones. Based on the lowest detected glutamine amount from Yuneva et al. (21) of 40 nmol Gln/g of protein, we estimate that these cells contained between 3.7 and 7.4 mm glutamine.
Quantitative PCR
RNA samples were extracted using the kit RNAeasy Mini (Qiagen) following the manufacturer's instructions. cDNA synthesis and PCR amplification were performed with the kits SuperScript III first strand synthesis system for RT-PCR (Invitrogen) and Power SYBR Green PCR Master Mix (Applied Biosystems), respectively, as instructed by the manufacturers. Samples were run on the Applied Biosystems 7500 real time PCR system and analyzed following the 2−ΔΔCT method. Primers used: (i) both endogenous and ectopic GAC: forward, 5′-GATCAAAGGCATTCCTTTGG-3′, and reverse, 5′-TACTACAGTTGTAGAGATGTCC-3′; (ii) ectopic GAC only: forward, 5′-AGAATGGAAAGTCTGGGAGAGAAA-3′, and reverse, 5′-CCGAGGAGAGGGTTAGGGATA-3′; (iii) SN2: forward, 5′-GGCTTGGTAGTGTTTGCCAT-3′, and reverse, 5′-GGGCAAAGAGTAAACCCACA-3; (iv) ASCT2: forward, 5′-ATGAAACACTTGGGCTAC-3′, and reverse, 5′-ATGACCGAAACAAGGAAA-3′; (v) rRNA 18 S: forward, 5′-ATTCCGATAACGAACGAGAC-3′, and reverse, 5′-TCACAGACCTGTTATTGCTC-3′.
RESULTS
Active GAC Forms a Rod-like Supratetrameric Structure
We have previously documented that wild-type GAC, when in the presence of its activator Pi, showed a tendency to form molecular species larger than tetramers and that this behavior was counteracted when the concentration of NaCl, a known glutaminase inhibitor, was increased in the protein solution (5, 12). Although the structural nature of the higher order species remained elusive at that time, it was curious to note that there was a significantly major shift toward higher than tetramer oligomers in the case of the activity-enhancer mutation F327S (GAC.F327S; Fig. 1A, top right panel).
FIGURE 1.

GAC polymerization is essential for enzymatic activation. A, size exclusion chromatography analysis of serial dilutions for wild-type and mutated GAC in the presence or absence of 20 mm phosphate. The green region delimits the expected Stokes radii between GAC dimers (D) and tetramers (T), calculated based on the crystal structure (3ss3) dimensions. The light purple region, delimited by V in the GAC.K325A graph, indicates the void volume of the gel-filtration column used. The asterisk indicates a previously published result for wild-type GAC (5). B, the software DigitalMicrograph (Gatan) was used to estimate the two orthogonal size distributions (short and long dimensions) of the wild-type and point mutant glutaminase particles, formed under the diverse conditions described in the main text (absence or presence of 20 mm phosphate and cross-linked versus non-cross-linked particles). C, supratetrameric organization of GAC upon the addition of 20 mm phosphate, a well known activator of GLS1 glutaminases. D, box plot and scatter representation of the two orthogonal dimensions for GAC wild-type and point mutants, as taken from the TEM micrographs. E, chemically cross-linked GAC superoligomers. Some of the observed filaments resemble lateral association between two simple filaments. F, GAC.K325A presents a much higher catalytic efficiency, already in the absence of phosphate, when compared with wild-type GAC and the inactive GAC.R322A. G, the high enzymatic efficiency of GAC.K325A correlates well with its tendency to self-assemble into rod-like polymers, regardless of the presence of the activator inorganic phosphate (left and middle panels) and DSS. Conversely, the catalytically inactive GAC.R322A protein remains in its tetrameric form, even in the presence of 20 mm phosphate (right panel). The scale bars represent 100 nm.
In the course of further studies of the molecular mechanism of activation of mammalian glutaminase, we used TEM to better understand the self-assembly process. First, recombinant GAC (in the absence of Pi) was used to set up negatively stained grids. Image analysis, after sampling ∼100 randomly chosen particles, allowed the determination of two perpendicular size distributions (exemplified in Fig. 1B): a short dimension, with a mean size of 7 ± 1 nm, and a longer one of 8 ± 1 nm (Fig. 1C, upper panel), both close to the expected dimensions for the dimer particle (5). In contrast, particles formed in the presence of 20 mm phosphate had a larger and more heterogeneous size distribution on the grid. Whereas the shorter dimension doubled on average (18 ± 3 nm), there was clearly favored growth across the long dimension, averaging 88 ± 31 nm (Fig. 1C, lower panel). The box chart with scatter plot for the size survey is shown in Fig. 1D.
The equilibrium between the GAC oligomeric species depends on protein concentration, phosphate, and NaCl (5). To circumvent the dissociative effects of protein dilution and work at appropriate protein concentrations for TEM characterization, superstructure assembly was stabilized by adding the cross-linking agent DSS in the presence of 20 mm phosphate and then purified by gel filtration. Under the conditions used, the superstructures eluted in the void volume of the column. The appropriate DSS concentration and time of incubation was previously established by a dose-response assay (data not shown). The negative-stained micrographs showed evidence for the formation of elongated, chain-like, nonbranched filaments, heterogeneous in length, that favored side-on adsorption to the carbon film (Fig. 1E). Some of the observed particles, such as those in the rightmost panels of Fig. 1E, seem to resemble side by side aggregation of simpler filaments (left and bottom panels).
We have previously identified the gating loop as essential for the phosphate-dependent activation process of the GLS1 isoforms (5). By replacing residues in the gating loop by alanine, we identified a point-mutation (GAC.K325A) that conferred a 670-fold increase in catalytic efficiency over that found for the wild-type enzyme, even in the absence of inorganic phosphate (GAC.K325A kcat-app/Km-app of 281.8 mm−1·s−1 compared with 0.42 mm−1·s−1 for GAC) (Fig. 1F). Both negative staining TEM (Fig. 1G, left panel) and size exclusion chromatography (Fig. 1A, bottom left panel) confirmed a much greater particle size distribution for this mutant, even in non-cross-linked samples. For example, TEM analysis showed dimensions of 17 ± 2 and 107 ± 79 nm, for the short and long dimensions, respectively, in the absence of phosphate (Fig. 1D). More importantly, this mutant assembles into oligomers visually similar to the ones formed with the wild-type protein (Fig. 1E, rightmost panels), guaranteeing that the mutation is only enhancing the oligomerization capability. The K325A mutation uncouples the protein dependence on phosphate for activation and further confirms the positive correlation between the superstructure assembly and enzyme activation. On the other hand, a second point mutation within the gating loop, affecting Arg322 (termed GAC.R322A), was enough to generate a catalytically inactive mutant irresponsive to phosphate (kcat-app/Km-app of 0.6 mm−1·s−1 at 20 mm Pi against 3.6 mm−1·s−1 for the wild type), which accordingly did not assemble into superoligomers (Fig. 1, F and G, right panel). The dimensions observed for particles of GAC.R322A in the presence of 20 mm Pi were very similar to those for the wild-type protein in the absence of Pi (7 ± 1 and 8 ± 2 nm, for the short and long dimensions, respectively) (Fig. 1D).
Superstructure among Glutaminase Isozymes
GAC is the most active of the three known mammalian glutaminase isozymes in response to inorganic phosphate (5). To further investigate the correlation between increased activity and larger oligomeric species, we observed the ability of KGA and LGA (also known as GLS2) to assemble into supratetrameric structures by TEM. Superstructure assembly was induced at 20 mm phosphate, stabilized by cross-linking with DSS, and subsequently gel filtration-purified. A tendency toward longer oligomers correlates well with the activity levels of the three isozymes. More specifically, GAC—the most active glutaminase at 20 mm Pi—forms the longest rod-shape oligomers (Fig. 2, top left panel), followed by KGA (Fig. 2, top right panel) and then LGA (Fig. 2, bottom left panel), which has been previously shown to be enzymatically insensitive to Pi (5, 22) and to not form the superstructure. The mean values for the shortest and longest dimensions of the particles are 16 ± 2 and 181 ± 108 nm for GAC, 14 ± 3 and 74 ± 26 nm for KGA, and 9 ± 2 and 12 ± 3 nm for LGA (Fig. 2, bottom right panel).
FIGURE 2.
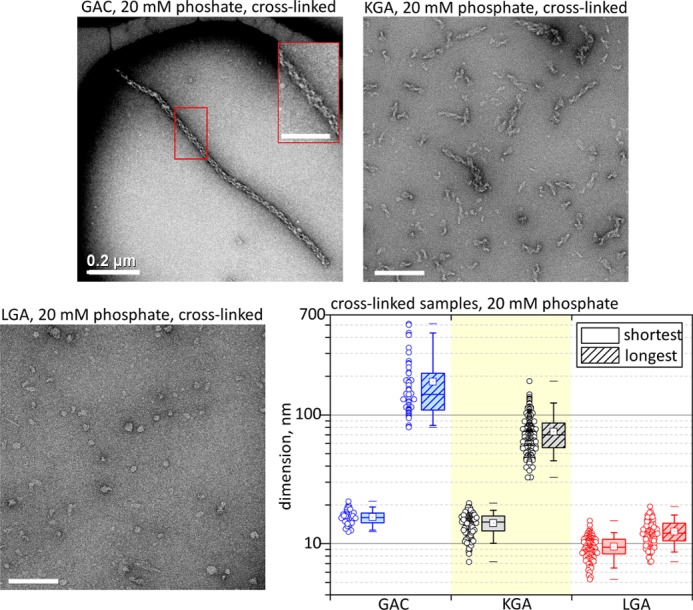
Glutaminase isozymes versus self-assembly tendency. TEM analysis of the three glutaminase isozymes, cross-linked after incubation with 20 mm phosphate. The average length of the superstructure correlates positively with the previously published glutaminase activity levels (5). GAC is the most active isozyme and forms the longest polymers (top left panel), followed by KGA (top right panel), with lower activity and shorter structures, and lastly LGA, which is insensitive to phosphate and accordingly does not form filaments (bottom left panel). Unless otherwise specified, the scale bars represent 100 nm.
BPTES Disrupts GAC Superstructure
Counterintuitive to the current dimer to tetramer model of activation for GLS1, the inhibitor BPTES has been shown to lock GLS1 into an inactive yet tetrameric form (6). Interested in deciphering the mode of inhibition by which BPTES exerts its effect and its relationship with the formation of the superstructure, we determined a detailed BPTES dose-response curve for GAC in the presence of 20 mm Pi. This phosphate concentration was chosen because it is closer to the physiological one under hypoxic conditions (23) and substantially stimulates GAC catalytic activity (5). We observed a strong inhibitory effect on GAC, where nanomolar levels of BPTES affected the maximum catalysis rate, kcat-app, without changing the Km-app for glutamine (Fig. 3A), a clear indication of an allosteric noncompetitive inhibitor as already published by others (6, 7). Curiously, beyond 200 nm BPTES, a secondary effect is observed and the inhibitor started affecting also the Km-app (a 3-fold increase compared with the absence of BPTES), redefining BPTES as a mixed mode inhibitor (Fig. 3A). By fitting an error bar-weighted logistic sigmoid function to explain the decrease in the turnover rates of the enzyme caused by the increased presence of inhibitor, we obtained an IC50 of 80.4 ± 7.2 nm for BPTES, which is consistent with the value provided by DeLaBarre et al. (7). Upper and lower limits for the catalytic rates are 16.4 and 7.9 s−1, respectively, with a Hill slope of 1.7 ± 0.2 (R2 = 0.99), indicating cooperative binding (Fig. 3A). Concerning the effect on Km-app, the upper and lower limits are 4.6 and 13.8 mm, with an IC50 of 629 ± 61 nm for BPTES (Hill slope of 2.0 ± 0.3, R2 = 0.99).
FIGURE 3.

BTPES inhibits GAC superstructure formation. A, dose-response profile of BPTES inhibition on GAC, assayed in the presence of 20 mm phosphate, showing two complementary effects on the apparent turnover rates and the Michaelis constant of the enzyme. B, BPTES traps the gating loop (black ribbons) in a rigid open conformation relative to the catalytic pocket of GAC (delimited by a green surface). C, stereographic view of a Fourier 2Fobs − Fcalc map (at 1 σ) confirming the proposed conformation in the refined crystallographic model. D, TEM analysis of the effects of BPTES on the formation of GAC superstructure. The filament formation is hindered regardless of whether BPTES is added to the protein solution prior to (middle panel) or after (bottom panel) incubation with 20 mm phosphate. E and F, conversely, GAC.K325A is catalytically insensitive to BPTES treatment (E), and this inhibitor is also incapable of disrupting the non-cross-linked GAC.K325A filaments (F).
Concomitant with the studies above, we have also determined the crystal structure of mouse GAC in complex with BPTES (2.77 Å resolution, Rfactor of 25.3%, and Rfree of 29.5%; Table 1). Although similar to previously reported BPTES-bound glutaminase structures, with two inhibitor molecules tightly accommodated at the tetramer interface and average backbone root mean square deviations of 0.44 and 0.56 Å when superposed to Protein Data Bank structures 3UO9 and 3VOZ, respectively (7, 8), our crystal form presents a key unique feature. As a consequence of the binding of the small molecule to the tetramer interface, the main chain of the gating loop—consistently poorly ordered across all the GAC crystallographic models—is in a stable open conformation in all four monomers. This is due especially to the contacts made mainly between BPTES and Lys325, and consequently the loop assumes the conformation of a one-turn helix (Fig. 3, B and C). Fifteen hydrogen bonds are made between a dimer of GAC and one BPTES molecule (data not shown). Eleven residues from each monomer share the interface with BPTES, occluding a combined total area of 620 Å2. Of the total solvent-accessible surface of BPTES (788 Å2), 80% is buried upon interaction with the enzyme.
TABLE 1.
X-ray crystallography data collection parameters and structure refinement statistics of BPTES-bound mouse glutaminase C (Protein Data Bank code 4jkt)
The data for the outer shell are shown in parentheses. LNLS, Laboratório Nacional de Luz Síncrotron.
| Data collection | |
| Beamline | D03B-MX1 at LNLS, Brazil |
| Wavelength (Å) | 1.608 |
| Space group | P212121 |
| Cell parameters a, b, c (Å) | 100.7, 140.2, 180.9 |
| Resolution range (Å) | 38.4–2.77 (2.82–2.77) |
| Unique reflections | 63,967 (2,039) |
| Multiplicity | 3.3 (2.6) |
| Rp.i.m. (%) | 11.0 (24.0) |
| Completeness (%) | 95.5 (46.2) |
| <I/σ(I)> | 4.5 (2.8) |
| Average mosaicity (o) | 1.0 |
| B-factor Wilson Plot (Å2) | 40.5 |
| Monomers/AU | 4 |
| Solvent content (%) | 59.2 |
| Matthews coefficient (A3/Da) | 3.01 |
| Model refinement | |
| Resolution range (Å) | 20.0–2.77 |
| Reflections (cross-validation) | 63,892 (5,952) |
| Rfactor/Rfree (%) | 25.3/29.5 |
| Average B-factor (Å2) | |
| Main chain (no. of residues) | 32.9/3.4 (1,563) |
| Side chain (no. of residues) | 32.8/4.3 (1,362) |
| BPTES (no. of molecules) | 44.1/9.2 (2) |
| Solvent (no. of molecules) | 24.4/5.9 (265) |
| Root mean square deviation from standard geometry | |
| Bond length (Å) | 0.003 |
| Bond angles (°) | 0.783 |
| Ramachandran plot | |
| Most favored (%) | 94.5 |
| Allowed (%) | 4.8 |
| Outlier (%) | 0.7 |
Next, we assessed the effects of BPTES on the formation of GAC superoligomers. TEM and size exclusion chromatography of wild-type GAC samples prepared in the presence of phosphate and the inhibitor and subsequently cross-linked showed that regardless of whether BPTES is added prior or after protein incubation in 20 mm phosphate; complete disruption of the superstructure is observed at a concentration of 30 μm of BPTES (Fig. 3D). The distribution of the disrupted particles on the grids is similar to the wild-type protein in the absence of phosphate and BPTES, as shown in Fig. 1A (top panel).
Finally, we observed that the mutant GAC.K325A, besides being hyperactive, is also catalytically insensitive to BPTES as determined by a dose-response curve (Fig. 3E). Accordingly, BPTES is incapable of disrupting non-cross-linked GAC.K325A oligomers, assembled in the presence or absence of phosphate (Fig. 3F).
Superstructure Characterization
Because of its heterogeneous length distribution, the GAC superstructure is intrinsically inappropriate for crystallization. However, by combining TEM, chemical cross-linking followed by MS analysis, and computational biology, we provide a low resolution model for the superstructure. The extended particles tend to adsorb to the microscopy grids in a very limited number of orientations. However, the best micrographs show a combination of translational and rotational symmetry elements lying parallel to the polymer, thus yielding a helical symmetry along its longitudinal axis. Detailed analysis of low pass filtered images provided its geometrical attributes. The observed spiral polymer, under particular staining conditions, resembles a right-handed double-stranded helix, with a rise per turn of ∼53 ± 2 nm, a strand inclination of 25°, and an average width for a single strand of 6.6 ± 0.7 nm (Fig. 4A).
FIGURE 4.

A model of GAC superoligomer assembly. A, the removal of high frequencies (strongly associated with noise) from the TEM micrographs highlights the right-handed double-strand nature of the superstructure, allowing the determination of its geometric features. B, cross-linked MS/MS spectra were manually validated for b and y ion series of the α and β chains of cross-linked peptides (between residues Lys181 and Lys207, Lys202 and Lys578, and Lys403 and Lys512. The ions are indicated by arrows with corresponding m/z value. C, relative position of DSS-linked lysine pairs in the tetramer, as identified by MS. The boundaries of the active sites are delimited by solid surfaces. †, Lys578 is not modeled in the crystal structure. The proposed single strand growth direction is across the longest axis of the tetramer, via an end to end interaction between pairs of N-terminal domains. Alternating orange and yellow colors facilitate the identification of the tetramers along the single strand. D, one lysine residue from each cross-linked pair was individually substituted by a glutamate and assayed for enzymatic activity (left panel) and superstructure formation (right panel). The N-terminal region mutants (GAC.K202E and GAC.K207E) enhanced both effects. On the other hand, GAC.K512E, within the glutaminase domain, generated a nonfunctional protein, similarly to the acetylation mimetic GAC.K316Q. The green region delimits the expected Stokes radii between GAC dimers (D) and tetramers (T), calculated based on the crystal structure dimensions. The asterisk indicates a previously published result for wild-type GAC (5). E, the double strand was manually modeled (panel i), using two entwined copies of the single strand from C and following the geometric restrictions of A. In panel ii, the calculated Fourier Fcalc map (with amplitudes and phases from the double strand model)—limited to 35 Å maximum resolution—is two-dimensionally projected, and the end result is shown in panel iii. All features are comparable to those observed in panel iv, concerning the presence of alternating high density narrow regions (indicated by filled triangles) and low density broad, disc-like regions (indicated by open triangles).
Simultaneously, size exclusion-purified cross-linked polymers—as well as GAC cross-linked in the tetrameric form (used as a control)—were digested with trypsin, and the resulting peptides were subjected to liquid chromatography coupled to tandem mass spectrometry. The cross-linked peptides were modeled based on the crystal structure of the tetramer, using a spacer arm length for DSS of ∼11.4 Å as a restraint for maximum distance between the side chain amine groups of linked lysines. This was necessary to distinguish between the different possible inter- and intramolecular cross-links. Unique linkages were only observed in the tetramer samples (spectra shown in Fig. 4B), between residues Lys181 and Lys207 (both located in the outer N-terminal portion of the same monomer), Lys202 and Lys578 (the latter located at the C terminus), and Lys403 and Lys512 (both within the glutaminase domains, flanking the active site of two different monomers) as depicted in Fig. 4C. The observation that these DSS-mediated links are unable to form upon polymerization suggests occlusion of their respective surfaces from the solvent when in the superstructure form. One lysine residue from each of the cross-linked pairs was then substituted by an oppositely charged glutamate residue, and the individually mutated proteins (termed GAC.K202E, GAC.K207E, and GAC.K512E) were tested for superoligomer-dependent glutaminase activity. Both N-terminal mutants, GAC.K202E and GAC.K207E, resulted in enzymes with enhanced activity (Fig. 4D, left panel), which coherently formed bigger particles, when compared with wild-type GAC (Fig. 4D, right panel). Conversely, the glutaminase domain mutant GAC.K512E fully lost the phosphate-dependent enzymatic capability and, accordingly, the ability to self-assemble into the superoligomers. The three point mutations reinforce the need for superoligomer formation for protein enzymatic activity and, most importantly, further show the importance of regions unrelated to and distant from the catalytic pocket for superoligomer formation and enzyme activation.
In the original biochemical characterizations of recombinant GLS1, Kenny et al. (12) demonstrated the importance of the GLS1 N-terminal portion for enzyme activity by generating a truncated, catalytically inactive, recombinant construct. Nevertheless, subsequent crystal structures of GAC clearly showed no direct participation of residues belonging to the N-terminal portion in catalysis or even their requirement for the proper folding or stabilization of the active site. Based on the dimensions observed in the GAC crystal structures, the average width of 66 Å for a single strand indicates that polymerization occurs along the longest axis of the tetramer, thus suggesting an end to end interaction via N-terminal regions. The growth is thus consistently favored in one direction and may occur indefinitely, because N-terminal sticky ends will always be available at both termini (computational docking of several tetramers simulating strand growth is shown in Fig. 4C, left panel). Therefore, because activity is dependent on filament assembly and the N-terminal portion is key for such, we provide a plausible explanation for the findings of Kenny et al. (12).
Next, to generate a model for the double-stranded filamentous structure, the coordinate files of two single strands were manually coiled around each other, using the helical parameters as restraints (Fig. 4E). The Gaussian low pass filter applied to the reference micrographs remove spatial frequencies higher than 0.028 Å−1, thus resulting in an estimated 35 Å resolution in real space. According to its overall dimensions, both length-wise and angle-wise, the estimated minimum repeating unit (one full helical turn) is composed of seven tetramers in each of the entwined strands. The intrinsic 2-fold dihedral symmetry of the tetramer (5) renders it unnecessary to determine whether the double-stranded helix is of a parallel or anti-parallel nature because each strand is apolar. To validate the model against the experimental data, an Fcalc Fourier electron density map was generated by the program FFT (24) (to 35 Å maximum resolution), using structure factors calculated from the three-dimensional double-stranded coordinate file and subsequently theoretically projected in two-dimensions using the electron microscopy software Imagic (25). The end result is shown in the right panel of Fig. 4E and presents features that are fully comparable to those observed in the representative micrographs of Fig. 1E (bottom panel), especially concerning the presence of an alternating pattern of narrow, compact regions of high density interspersed with broad, disc-like regions of low density.
Lastly, Lys311 in human GLS1 glutaminases was found to be a target for in vivo acetylation by high resolution mass spectrometry analysis (13). This lysine residue belongs to the glutaminase domain and is located on the opposite surface in relation to the catalytic pocket and the gating loop, with no direct participation in the active site. To evaluate the effects of this reversible, post-translational modification over superstructure formation and therefore protein activation, we have generated a point mutant in the equivalent residue of the mouse protein (GAC.K316Q), replacing it with a glutamine residue, so as to mimic the noncharged nature of acetylated lysine. Interestingly, the newly mutated protein was characterized as less sensitive to the activator phosphate with the concomitant lowered tendency to assemble into the superoligomers, keeping the protein in sizes compatible with only tetramers and dimers (Fig. 4D, left and right panels).
GAC Superstructure in Cell Model
To further demonstrate the importance of the superstructure assembly for GAC enzymatic activation, we generated stable clones of the MDA-MB 231 breast cancer cell line expressing either the human V5-tagged wild-type or the GAC.K325A mutant proteins, as well as cells transformed with a mock plasmid. Although in humans the appropriate numbering should be GAC.K320A (NCBI reference sequence AAD47056.1), we have decided to keep GAC.K325A (mouse GAC) for the sake of coherence throughout the paper. Cell clones were selected to present comparable levels of ectopic V5-tagged mRNA and protein while not presenting abnormally higher levels of ectopic expression (Fig. 5, A and B). Proper mitochondrial localization of the ectopic protein was confirmed by cell fractionation and immunoblotting (data not shown). Two distinct phenotypes were readily observed for the GAC.K325A-V5 cells, when compared with the GAC-V5 and mock counterparts. First, GAC.K325A-V5 cells were, on average, much larger and heterogeneous in size, as based on the measurement of cell area (Fig. 5C). More specifically, whereas the GAC-V5 and the mock transformed cells presented similar, normally distributed cell areas, peaking at 593 and 600 μm2, respectively, the GAC.K325A-V5 population was much more heterogeneous in size (relative standard deviation of 89%) and still ∼7% larger, averaging 637 μm2. Second, the mutant-transformed cells proliferated ∼10% more than the wild-type and mock transfected cells, as determined by the quantification of cell numbers (Fig. 5D). The average diameter of unattached (trypsinized) cells was also assessed to account for the effects of cell spreading on the area measurements. GAC.K325A-V5 cells presented the largest diameters, averaging at 13.3 μm (n = 178), when compared with the wild-type GAC-V5 (10.9 μm, n = 151) and mock cells (10.6 μm, n = 174) (data not shown).
FIGURE 5.
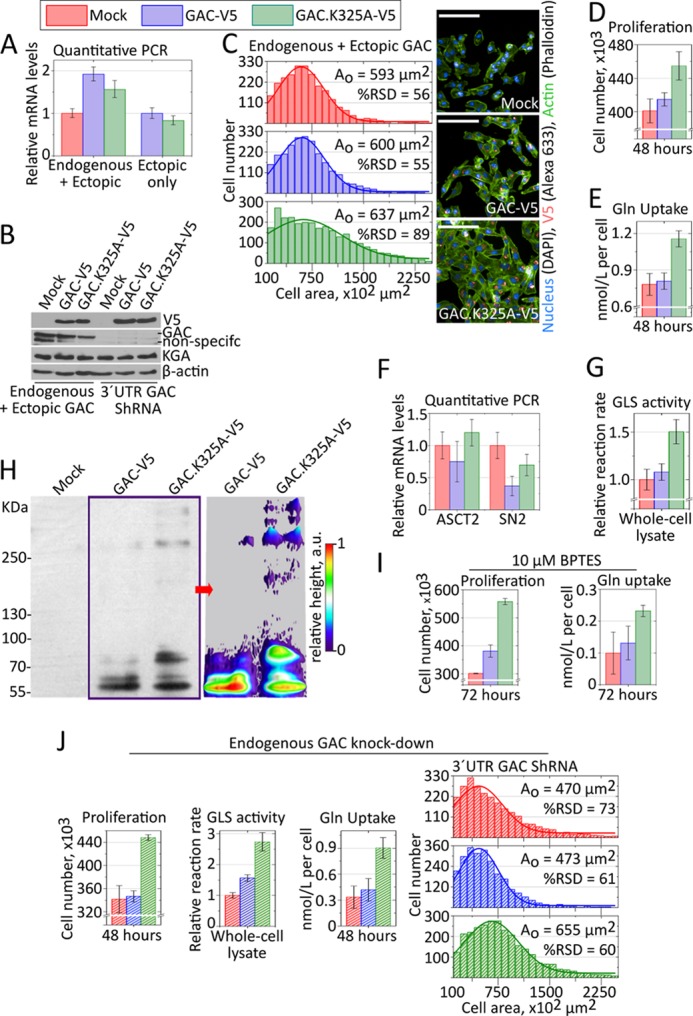
GAC superstructure in cell models. A and B, endogenous and V5-tagged ectopic proteins expressed to similar levels. C–E, a stable MDA-MB 231 clone selected after GAC.K325A-V5 transfection presented larger and more heterogeneous cell area (C), proliferated more (D), and consumed more glutamine from the culture media (E), all compared with cells bearing the V5-tagged wild-type protein or a mock plasmid. F, relative mRNA levels of ASCT2 and SN2 glutamine transporters, as defined by quantitative PCR using rRNA 18 S as a housekeeping gene, showing that the GAC.K325A-V5 cells do not overexpress these transporters. G, glutaminase activity from whole cell lysate, in the presence of 20 mm phosphate, showing consistently higher turnover rates for the GAC.K325A-V5 samples, against the physiological glutamine levels in tumors. H, left panel, step gradient SDS-PAGE (3–15%) followed by immunoblotting (anti-V5) of UV-induced cross-linked intracellular protein with incorporated photo-reactive amino acids, showing the tendency of the GAC.K325A-V5 to form higher molecular weight superstructures within the cells. The UV-induced cross-linking was performed in living, intact cells in culture. Right panel, densitometry was performed in conditions of nonsaturated signal, using ImageJ, to evidence the differential cross-linking of bigger species for GAC.K325A-V5. I, similar to what was observed for the recombinant protein, cells expressing the fiber-prone hyperactive GAC.K325 mutant (GAC.K325A-V5) were less sensitive to BPTES treatment, still proliferating more (left panel) and consuming more glutamine (right panel) than BPTES-treated counterparts. J, the knockdown of endogenous GAC favored the enhancing of the phenotypic differences observed above, better highlighting the outcome from GAC.K325A-V5 expression.
Next, glutamine uptake from the culture medium was evaluated after 48 h of plating. We found that although the glutamine consumption levels for the GAC and the mock transformed cells was deemed indistinguishable (0.78 ± 0.09 and 0.81 ± 0.07 nmol/liter per cell, respectively), GAC.K325A-V5 cells consumed 40% more glutamine (1.16 ± 0.07 nmol/liter per cell), as shown in Fig. 5E. In parallel, we showed that GAC.K325-V5 cells do not express more of the glutamine transporters ASCT2 and SN2 (Fig. 5F). To account for the observed increase in glutamine consumption, we assessed the glutaminase activity in a physiological level of l-glutamine (7.5 mm) and observed that the reaction rates for the GAC.K325A-V5 cells are 40–50% higher when compared with the other two clones (Fig. 5G). Faster turnover rates were also consistently observed for the recombinant mutant protein, as shown in the first section.
To inspect for higher than tetramer oligomer formation within the living cells, cells were grown in the presence of photo-reactive amino acids and then exposed to UV light (at 365 nm), followed by immunoblotting against the whole cell extract. The data clearly showed that GAC.K325A-V5 was cross-linked in cell into higher molecular weight species than GAC-V5 (Fig. 5H). Although the cross-linked species did not extend as far as those observed for the recombinant protein in vitro—likely because of the hetero-oligomerization between the V5-tagged mutant protein and endogenous wild-type GAC inside the mitochondria—the differential cross-linking pattern is evident. We have shown that the enzymatic activity of the recombinant mutant GAC.K325A is unaffected by BPTES treatment (Fig. 3E), which, accordingly, did not disrupt the superstructure (Fig. 3F). As a further confirmation that the superoligomer formation is important for the observed phenotypes, we treated cells with 10 μm of BPTES. As expected, GAC.K325A-V5 cells still grew more and consumed higher amounts of glutamine than BPTES-treated control cells (Fig. 5I). BPTES treatment has drastically decreased the glutamine consumption on all the cell clones because it likely disrupted hetero-oligomers (formed by endogenous wild-type GAC and ectopic GAC.K325A-V5) and because it is also inhibits KGA (6). Despite this, the ectopic expression of GAC.K325A was able to improve the proliferation and growth phenotype of the cells.
Lastly, to exclude the possibility that the difference in cell proliferation and growth, as well as Gln uptake for the GAC.K325A-V5 cells were due to higher amounts of endogenous enzyme (in comparison to control cells), as well as to highlight the ectopic protein activity, we knocked down endogenous GAC by using a shRNA target to its mRNA 3′-UTR (Fig. 5B). Not surprisingly, the previously observed phenotypic differences became enhanced. The mutant-transformed cells grew 30% more and were 40% bigger, while consuming ∼2.2 times more glutamine per cell, than the wild-type-expressing cells (Fig. 5J). Accordingly, the activity levels assessed from the whole cell extract in the presence of 7.5 mm l-glutamine were 75% higher than the control assays (Fig. 5J). Overall, this collection of results directly demonstrated that the phenotypic differences above resulted from the presence of an ectopically expressed, hyperactive, and higher molecular weight prone protein GAC.K325-V5, which presented the same biochemical features and the intrinsic tendency to assemble into a superstructure as the recombinant mutant protein.
DISCUSSION
Previous biochemical studies have established that GLS1 glutaminases are mainly found as inactive dimers and that the presence of phosphate correlates with changes leading to tetramerization and enzyme activation (26, 27). We provide here novel information in that regard and show that the catalytic activation of the GLS1 glutaminases is directly linked to a fiber-like supratetrameric assembly, which correlates well with activation levels of the three glutaminase isozymes. The first observations of this phenomenon were provided ∼40 years ago, using purified glutaminase from pig renal extract (28, 29). The formation of extended polymers was later used to purify the native enzyme by size exclusion, allowing its first biochemical and kinetic characterization (30), thus implying that our results are not an artifact of recombinantly expressed, truncated protein constructions, or even the chemically induced intermolecular cross-linking. Robinson et al. (6) have also documented larger oligomers for recombinant GLS1 in the presence of phosphate. The right-handed double-stranded molecular model that we propose here, when two-dimensionally projected using electron microscopy software, is not only in fully agreement with our experimental micrographs but also with the phosphate-borate induced form and the shadow casting reported by Olsen et al. (29) in 1973. Some of our micrographs suggest lateral association between two polymers, as seen, for instance in Figs. 1E (top right and middle panels) and 2 (top left box), as well as those of the GAC.K325A mutant (Figs. 1E and 3F). Regardless of these side by side associations, only the longitudinal growth seems to be connected to an increase in protein activity. Such a feature was also observed by Olsen et al. (29). Furthermore, it is also worth mentioning that with regard to the glutaminolytic pathway, polymerization is not exclusive to GLS1 glutaminases. Glutamic dehydrogenase from bovine liver has been shown to self-assemble into long multichain tubular structures under appropriate conditions (31). It is plausible then, that nonpathogenic polymer assembly may be a widespread process for the functioning of other metabolic enzymes.
Robinson et al. (6) first demonstrated biochemically that BPTES inhibits the GLS1 isoform (but not the liver-type isozyme, LGA/GLS2) by interfering with its phosphate-dependent allosteric activation and promoting the stabilization of the glutaminase into an inactive tetrameric form, later confirmed structurally (7, 8). Our collection of results indicates that BPTES inhibits GAC by trapping the gating loop at a rigid open conformation, which in turn prevents superoligomer formation. Although the tetramer- and phosphate-induced opening of this loop is necessary for enzyme activity, its intrinsic flexibility seems to play a major role on the enzymatic process. Indeed, we verified by molecular dynamics simulation on the tetramer that although phosphate binding to the catalytic site increases the gating loop flexibility, both l-glutamate (enzyme inhibition by l-glutamate has been reported for the kidney isoform of GLS1 (32)), and the presence of BPTES freezes the gating loop into a less mobile state (data not shown).
We observed from the kinetic studies with the GAC.K325A mutant that filament formation results in a drastic decrease in the Michaelis constant of the protein, over 100-fold, compared with the wild-type enzyme, in the absence of the activator Pi. A much greater accessibility of the active site to the substrate is suggested upon polymerization, as well as a key role of the gating loop in this process, a phenomenon that is fully reversed when Arg322 is replaced by alanine. Therefore, the present work unveils a new role for the gating loop, which besides regulating substrate accessibility to active site, also controls the reversible protein polymerization. Because of experimental limitations, which led to a low-resolution 35 Å model, we are unable to describe the specific interactions that hold both the single and the double strands together and result in a fully activated enzyme. However, based on the most diverse effects generated mainly by the individual point mutations, it is plausible to suggest that shape complementarity is the driving force for self-assembly and can only be fully achieved after specific surface charges are enhanced or neutralized, an outcome easily attainable in the presence of a polyanion such as the phosphate ion. This is especially true in the case of the gating loop, where the replacement of two closely positioned, long and positively charged residues (Arg322 and Lys325) by alanine had antagonic effects with regards to protein self-assembly and activation. Concomitantly, major torsions in the relative positions of the monomers inside the tetramer—that are still experimentally undetected, because all GAC crystal structures available to date are virtually identical—may be required for gating loop-mediated superoligomer formation. The binding of BPTES, which shares an extensive area and a large number of hydrogen bonds at the tetramer interface and which also holds the loop into a rigid conformation (besides reducing the flexibility of the tetramer), would also prevent the gating loop from making these contacts. Such torsions in the tetramer cannot be predicted from our manually built model but could possibly be described in the future from higher resolution experimental data, such as by cryoelectron microscopy.
Recently, Katt et al. (9) described a potential binding site for the GAC inhibitor 968. In the proposed model, the small molecule docks inside a concave surface region formed at the dimerization interface of two GAC glutaminase domains. The 968 binding region lies very close to the N-terminal portion of GAC, which is predicted here to be involved in the polymerization of the enzyme. The authors further showed that 968 is unable to bind and inhibit a previously phosphate-activated enzyme, which is in full consistency with what we present. Once GAC oligomers are assembled, because of the occlusion of the putative 968-binding site and the possible conformational changes in the N-terminal portion of the enzyme required for self-assembly, they cannot be reversed by the addition of 968, explaining its limited inhibitory capacity on an already activated enzyme. However, if 968 is bound to free tetramers, then such conformational changes cannot be subsequently achieved, stopping GAC activation via self-assembly. Given the complementary modes of inhibition for BPTES and 968, acting at different stages of enzyme assembly and polymerization, one might envision that a synergy in inhibition would result when both are administered to glutaminase sensitive transformed cell. To our knowledge, this still needs testing.
Our findings also suggest a role for the previously detected in vivo acetylation of Lys311 in human GLS1 glutaminases (13). We propose that this post-translational modification down-regulates the enzymatic levels by antagonizing the formation of the active superoligomers. Given that acetylation is a reversible post-translational modification, it is plausible to suppose that glutaminase superactivation may, at least in part, be regulated by acetyltransferases and deacetylases yet to be identified.
Finally, we need to state that we would not expect that micrometer-long GAC polymers assemble inside the mitochondria. Rather, a more tangible scenario might be the presence of shorter, heterogeneous filaments, similar to those observed in Fig. 1A (lower panel). Nonetheless, direct visualization of such in the mitochondria has proven a difficult task. We showed that the superactive mutant GAC.K325A, intrinsically bound to self-assemble, was capable of providing growth and proliferation advantages to cells. However, a more important conclusion that can be drawn from these experiments is that an increased level of GAC in the mitochondria by itself is not sufficient to increase cell proliferation. The protein must be in the active form for such a phenotype to become evident. In cells, phosphate accumulation induced by hypoxia, for instance (23, 33), or even post-translational modifications, such as phosphorylation, could trigger polymerization, suggesting the possibility of distinct therapeutic opportunities for 968-like and BPTES-like inhibitors. Overall, our results reinforce the importance in focusing on the development of allosteric over active site-targeted inhibitors, glutamine analog inhibitors, when targeting GAC in tumors. This would result in preferred isoform-specific inhibitors, because LGA (GLS2), necessary for glutamine metabolism in the liver and brain, does not assemble into superoligomers, as well as avoiding the undesirable cross-inhibition of amidotransferases (21).
Acknowledgments
We thank the Laboratório Nacional de Biociências for access to its facilities (Laboratório de Purificação de Proteínas, Laboratório de Espectroscopia e Calorimetria, Laboratório de Espectrometria de Massas, Laboratório de Bioensaios, Laboratório de Vetores Virais, and Robolab); Laboratório Nacional de Nanotecnologia for access to Laboratório de Microscopia Eletrônica; and Laboratório Nacional de Luz Síncrotron for access to D03B-MX1 Beamline. We thank Dr. Alessandra Girasole for secretarial and technical support of Annelize Aragão and Romênia Domingues for helping with the MS analysis. We thank Dr. Richard C. Garratt (Instituto de Física de São Carlos, Universidade de São Paulo) for critical reading of the manuscript.
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) under Grants 2009/10875-9 (to S. M. G. D.) and 2010/05003-0 and 2012/14298-9 (to A. L. B. A.) and Fellowships 2010/05987-0 (to A. P. S. F.), 2010/05987-0 (to A. C.), 2011/06654-7 (to K. A. G.), and 2009/54067-3 (to A. F. P. L.). This work was also supported in part by the Laboratório Nacional de Biociências.
The atomic coordinates and structure factors (code 4jkt) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GAC
- glutaminase C
- BPTES
- bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
- TEM
- transmission electron microscopy
- KGA
- kidney-type glutaminase
- LGA
- liver-type glutaminase
- DSS
- disuccinimidyl suberate.
REFERENCES
- 1. Gaglio D., Metallo C. M., Gameiro P. A., Hiller K., Danna L. S., Balestrieri C., Alberghina L., Stephanopoulos G., Chiaradonna F. (2011) Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 7, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Le A., Lane A. N., Hamaker M., Bose S., Gouw A., Barbi J., Tsukamoto T., Rojas C. J., Slusher B. S., Zhang H., Zimmerman L. J., Liebler D. C., Slebos R. J., Lorkiewicz P. K., Higashi R. M., Fan T. W., Dang C. V. (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao P., Tchernyshyov I., Chang T. C., Lee Y. S., Kita K., Ochi T., Zeller K. I., De Marzo A. M., Van Eyk J. E., Mendell J. T., Dang C. V. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang J. B., Erickson J. W., Fuji R., Ramachandran S., Gao P., Dinavahi R., Wilson K. F., Ambrosio A. L., Dias S. M., Dang C. V., Cerione R. A. (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cassago A., Ferreira A. P., Ferreira I. M., Fornezari C., Gomes E. R., Greene K. S., Pereira H. M., Garratt R. C., Dias S. M., Ambrosio A. L. (2012) Mitochondrial localization and structure-based phosphate activation mechanism of glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 1092–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robinson M. M., McBryant S. J., Tsukamoto T., Rojas C., Ferraris D. V., Hamilton S. K., Hansen J. C., Curthoys N. P. (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 406, 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeLaBarre B., Gross S., Fang C., Gao Y., Jha A., Jiang F., Song J. J., Wei W., Hurov J. B. (2011) Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50, 10764–10770 [DOI] [PubMed] [Google Scholar]
- 8. Thangavelu K., Pan C. Q., Karlberg T., Balaji G., Uttamchandani M., Suresh V., Schüler H., Low B. C., Sivaraman J. (2012) Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 7705–7710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katt W. P., Ramachandran S., Erickson J. W., Cerione R. A. (2012) Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol. Cancer Ther. 11, 1269–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vander Heiden M. G. (2011) Targeting cancer metabolism. A therapeutic window opens. Nat. Rev. Drug. Discov. 10, 671–684 [DOI] [PubMed] [Google Scholar]
- 11. Jones N. P., Schulze A. (2012) Targeting cancer metabolism. Aiming at a tumour's sweet-spot. Drug Discov. Today 17, 232–241 [DOI] [PubMed] [Google Scholar]
- 12. Kenny J., Bao Y., Hamm B., Taylor L., Toth A., Wagers B., Curthoys N. P. (2003) Bacterial expression, purification, and characterization of rat kidney-type mitochondrial glutaminase. Protein Expr. Purif. 31, 140–148 [DOI] [PubMed] [Google Scholar]
- 13. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets proteins complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 14. Leslie A. G. (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography 26, 27–33 [Google Scholar]
- 15. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 16. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser Crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aragão A. Z., Nogueira M. L., Granato D. C., Simabuco F. M., Honorato R. V., Hoffman Z., Yokoo S., Laurindo F. R., Squina F. M., Zeri A. C., Oliveira P. S., Sherman N. E., Paes Leme A. F. (2012) Identification of novel interaction between ADAM17 (a disintegrin and metalloprotease 17) and thioredoxin-1. J. Biol. Chem. 287, 43071–43082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuneva M. O., Fan T. W., Allen T. D., Higashi R. M., Ferraris D. V., Tsukamoto T., Matés J. M., Alonso F. J., Wang C., Seo Y., Chen X., Bishop J. M. (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Campos-Sandoval J. A., López de la Oliva A. R., Lobo C., Segura J. A., Matés J. M., Alonso F. J., Márquez J. (2007) Expression of functional human glutaminase in baculovirus system. Affinity purification, kinetic and molecular characterization. Int. J. Biochem. Cell Biol. 39, 765–773 [DOI] [PubMed] [Google Scholar]
- 23. Gorman M. W., He M. X., Hall C. S., Sparks H. V. (1997) Inorganic phosphate as regulator of adenosine formation in isolated guinea pig hearts. Am. J. Physiol. 272, H913–H920 [DOI] [PubMed] [Google Scholar]
- 24. Read R. J., Schierbeek A. J. (1988) A phased translation function. J. Appl. Crystallogr. 21, 490–495, DOI 10.1107/S002188988800562X [DOI] [Google Scholar]
- 25. Van Heel M., Portugal R., Rohou A., Linnemayr C., Bebeacua C., Schmidt R., Grant T. R., Schatz M. (2012) Four-dimensional Cryo Electron Microscopy at Quasi Atomic Resolution. IMAGIC 4D in International Tables for Crystallography Volume F. Crystallography of biological macromolecules (Arnold E., Himmel D. M., Rossmann M. G., eds) 2nd edition, pp. 624–628, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 26. Godfrey S., Kuhlenschmidt T., Curthoys P. (1977) Correlation between activation and dimer formation of rat renal phosphate dependent glutaminase. J. Biol. Chem. 252, 1927–1931 [PubMed] [Google Scholar]
- 27. Morehouse R. F., Curthoys N. P. (1981) Properties of rat renal phosphate-dependent glutaminase coupled to Sepharose. Evidence that dimerization is essential for activation. Biochem. J. 193, 709–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olsen B. R., Svenneby G., Kvamme E., Tveit B., Eskeland T. (1970) Formation and ultrastructure of enzymically active polymers of pig renal glutaminase. J. Mol. Biol. 52, 239–245 [DOI] [PubMed] [Google Scholar]
- 29. Olsen B. R., Torgner I. A., Christensen T. B., Kvamme E. (1973) Ultrastructure of pig renal glutaminase. Evidence for conformational changes during polymer formation. J. Mol. Biol. 74, 239–251 [DOI] [PubMed] [Google Scholar]
- 30. Curthoys N. P., Kuhlenschmidt T., Godfrey S. S. (1976) Regulation of renal ammoniagenesis, purification and characterization of phosphate-dependent glutaminase from rat kidney. Arch. Biochem. Biophys. 174, 82–89, DOI 10.1016/0003-9861(76)90326-X [DOI] [PubMed] [Google Scholar]
- 31. Josephs R., Borisy G. (1972) Self-assembly of glutamic dehydrogenase into ordered superstructures. Multichain tubes formed by association of single molecules. J. Mol. Biol. 65, 127–155 [DOI] [PubMed] [Google Scholar]
- 32. Curthoys N. P., Watford M. (1995) Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr. 15, 133–159 [DOI] [PubMed] [Google Scholar]
- 33. Fan T. W., Higashi R. M., Macdonald J. M. (1991) Emergence and recovery response of phosphate metabolites and intracellular pH in intact Mytilus edulis as examined in situ by in vivo 31P-NMR. Biochim. Biophys. Acta 1092, 39–47 [DOI] [PubMed] [Google Scholar]