

Background: IR/IGF-1R kinase inhibitors are promising therapeutic agents in cancer.
Results: Irfin1, a compound closely related to the ERK inhibitor FR180204, inhibits IR/IGF-1R family kinases.
Conclusion: Irfin1 is a remarkably selective inhibitor for the inactive states of IR/IGF-1R kinases.
Significance: Broad spectrum kinase inhibitor profiling can be exploited to uncover novel targets of small-molecule compounds.
Keywords: Enzyme Inhibitors, ERK, High Throughput Screening (HTS), Receptor Tyrosine Kinase, X-ray Crystallography, Insulin Receptor, Insulin-like Growth Factor 1 Receptor, Kinase Inhibitor, Small-molecule Inhibition
Abstract
Dual inhibitors of the closely related receptor tyrosine kinases insulin-like growth factor 1 receptor (IGF-1R) and insulin receptor (IR) are promising therapeutic agents in cancer. Here, we report an unusually selective class of dual inhibitors of IGF-1R and IR identified in a parallel screen of known kinase inhibitors against a panel of 300 human protein kinases. Biochemical and structural studies indicate that this class achieves its high selectivity by binding to the ATP-binding pocket of inactive, unphosphorylated IGF-1R/IR and stabilizing the activation loop in a native-like inactive conformation. One member of this compound family was originally reported as an inhibitor of the serine/threonine kinase ERK, a kinase that is distinct in the structure of its unphosphorylated/inactive form from IR/IGF-1R. Remarkably, this compound binds to the ATP-binding pocket of ERK in an entirely different conformation to that of IGF-1R/IR, explaining the potency against these two structurally distinct kinase families. These findings suggest a novel approach to polypharmacology in which two or more unrelated kinases are inhibited by a single compound that targets different conformations of each target kinase.
Introduction
The insulin-like growth factor 1 receptor (IGF-1R)4 is a receptor tyrosine kinase that is activated by binding the ligands insulin-like growth factor 1 and 2 (IGF-1 and IGF-2). Signaling pathways downstream of IGF-1R activation are involved in glucose metabolism and cell survival. IGF-1R has been implicated in cancers in several different tissues and has thus been explored as a target for cancer therapeutics (1–3).
IGF-1R and the insulin receptor (IR) are closely related by amino acid sequence, sharing 57% overall sequence identity and 82% sequence identity between their kinase domains. In their basal, inactive forms, IGF-1R and IR adopt similar inhibited conformations in which the unphosphorylated activation loop partially occludes the ATP binding pocket, disrupting the architecture of residues critical for catalysis and substrate binding (4, 5). In the inactive state of IR and IGF-1R, the conserved Asp-Phe-Gly (DFG; Asp-1150, Phe-1151, and Gly-1152 in IR) motif in the activation loop is described as being in the “DFG-out” conformation due to the fact that the Asp residue points away from the ATP-binding site. Activation involves sequential auto-phosphorylation of three tyrosine residues (Tyr-1158, Tyr-1162, and Tyr-1163 in IR) within the activation loop leading to a conformational rearrangement that displaces the activation loop into the canonical position observed for most active kinases (6). In this active state, the Asp of the DFG motif points toward the ATP-binding site, and this conformation of the activation loop is termed “DFG-in.”
IGF-1R and IR can also form signaling-competent heterodimers, and indeed, it has been argued that co-targeting of both receptors is advantageous for potential therapeutics (7, 8). Small-molecule kinase inhibitors discriminate poorly between the two (9), making these compounds ideal for pharmacological use in co-targeting IGF-1R and IR. A disadvantage to using small-molecule ATP competitive inhibitors is that these compounds can also inhibit other kinases in addition to intended targets. Recently, several large-scale kinase inhibitor screens have shown that many small-molecule kinase inhibitors do not exclusively inhibit their intended targets (10–12). This promiscuity arises from the fact that the ATP binding pockets of activated kinases are highly conserved, making selective inhibition of a single target or a few selected targets very challenging.
One promising approach that has been proposed to yield more selective small-molecule kinase inhibitors is to target the inactive state of kinases (13). The features of the ATP binding pockets of the inactive states of kinases are less conserved than those of activated kinases; in activated kinases, catalysis dictates the arrangement of residues, whereas in inactive kinases, there are no such restrictions. Targeting the inactive states of kinases is also advantageous because it results in the trapping of the inactive state, preventing kinase activation. Here, we report a compound family that displays remarkable selectivity for the inactive states of the IGF-1R and IR kinases.
EXPERIMENTAL PROCEDURES
Kinase Selectivity Profiling
Kinase selectivity profiling was carried out exactly as described previously using the “HotSpot” assay platform (10). The final ATP concentration was 10 μm. Irfin1 and FR180204 were screened against the panel of kinases at a concentration of 0.5 μm. OSI-906 and BMS-754807 were screened at 18.6 nm and 1.82 nm, respectively. Kinase activity data were expressed as the percent remaining kinase activity in test samples compared with vehicle (DMSO) reactions. All measurements were performed in duplicate.
Outlier Detection
For outlier detection, all negative values were truncated to zero, and values in excess of 100 were retained. In addition, all kinase-inhibitor pairs with identical values across duplicates were removed from this analysis. The coefficient of variation (CV) and the difference (D) in duplicate observations for each kinase-inhibitor pair were used in detecting and removing outlying pairs after accounting for inherent noise in the assay measurements. Kernel density plots and quantile-quantile plots were used to empirically determine the distributions of these quantities, and maximum likelihood methods were used to estimate the parameters. Observations within one S.D. of the empirical distribution for D (determined to be double exponential) were considered to be within acceptable limits and excluded from further analysis. The middle 98% of the remainder of the data were fit to the quantile-quantile plot positions of the empirical distribution of CV (determined to be log-normal, R2 = 0.89). A test was then performed to determine whether extreme observations are outliers by computing the threshold beyond which exactly one observation is expected. Any observation that lies above this threshold (estimated to be 0.489) has a very low probability (3.3 × 10−4) of being generated by a log-normal model. Observations that lay above this threshold and beyond one S.D. of the empirical distribution of D were identified to be outliers. More details on this approach can be found in Anastassiadis et al. (10). Computations were performed in the R statistical language and environment using libraries VGAM and extreme values (10, 14–16).
IR/IGF-1R Kinase Domain Expression and Purification
The baculovirus encoding the human IR kinase domain (residues 978–1283) with one amino acid substitution (C981S) was a generous gift from Dr. Stevan Hubbard. Amplified viruses were used to infect a 1-liter culture of Sf9 cells. At 65 h post-infection, cells were harvested at 200 × g and lysed by sonication into a buffer containing 20 mm Tris, pH 7.5, and 100 mm NaCl. Cell debris was pelleted at 30,000 × g. The IR kinase domain was purified by anion exchange chromatography on a Source Q column (GE Healthcare) and then by size exclusion chromatography on a Superdex75 column (GE Healthcare). Purity was assessed by SDS-PAGE to be >95%. The kinase domain of IGF-1R was purchased from BPS Bioscience, Inc.
Phosphorylation and Purification of IR and IGF-1R
Recombinant, unphosphorylated IR was incubated with ATP and allowed to undergo partial activation and autophosphorylation. Unphosphorylated, monophosphorylated, diphosphorylated, and triphosphorylated forms of the IR kinase were generated and purified as described previously for the IGF-1R kinase (17).
In Vitro Dose-response Measurements for Irfin1 and FR180204
Irfin1 or FR180204 stocks were prepared in DMSO. For dose-response measurements, the compound was diluted in DMSO so that the final concentration of DMSO in the reaction mixture was 1%. To this diluted compound, a buffer resulting in a final concentration of 20 mm HEPES, pH 7.4, 1 mm EGTA, 100 μm Na3VO4, 1 mm DTT, 2 mm MnCl2, 0.02 mg/ml BSA, and 10 mm MgCl2 was added. A mixture of ATP and [γ-32P]ATP was added to initiate each reaction. The final concentration of ATP was 1 mm. Reactions were incubated at 30 °C for 1 h and were quenched by freezing the reaction with dry ice. Reactions were spotted on P81 chromatography paper, washed in cold 0.5% phosphoric acid four times for 5 min each, and then washed in acetone for 5 min. Filter paper was dried and then exposed to a phosphorimaging screen overnight. The screen was scanned and quantified using a Fuji BAS-2000 image analyzer. All measurements were performed in triplicate. IC50 values were calculated using Graphpad Prism.
Crystallization, Data Collection, and Structure Determination
Unphosphorylated IR kinase domain was concentrated to 15 mg/ml in a buffer containing 20 mm Tris-HCl, pH 7.5, and 100 mm NaCl and mixed with Irfin1 at a 1:2 molar ratio. Crystals of the IR kinase domain in complex with Irfin1 were grown at 20 °C by hanging drop vapor diffusion in drops containing 1 μl of a solution of IR kinase domain and Irfin1 and 1 μl of the reservoir solution containing 0.1 m sodium cacodylate, pH 6.5, 23% (w/v) polyethylene glycol 4000, 10% (w/v) ethylene glycol, and 0.03 m glycyl-glycyl-glycine. The crystals belong to space group P212121 with two complexes per asymmetric unit (unit cell dimensions: a = 50.84, b = 89.21, and c = 142.19 Å; α = β = γ = 90°). Crystals were flash frozen in liquid nitrogen prior to data collection.
Diffraction data were collected at the beamline X4C at the National Synchrotron Light Source (Brookhaven National Laboratory). Data were processed with HKL2000 (18). The structure was solved by molecular replacement by MOLREP (19) with a structure of IR kinase and kinase regulatory loop-binding (KRLB) region complex (Protein Data Bank code 3BU5) (20) as the search model using data in the resolution range of 50–3 Å. The best solution had a correlation coefficient of 0.532 and an Rfactor of 0.554. The next most favored solution had a correlation coefficient of 0.378 and an Rfactor of 0.629. Irfin1 and the activation loop of IR kinase domain were built into 2FO − FC and FO − FC electron density maps using COOT (21). The stereochemical library description for Irfin1 was generated using Sketcher (22). The model was refined using REFMAC (23). Ramachandran plot analysis reveals 97.4% of the residues have backbone torsion angles in preferred regions, and 2.6% are in allowed regions. Atomic coordinates for this structure have been deposited in the Protein Data Bank with the accession code 4IBM.
Generation of IR Activation Loop Mutations
The pEF-IR plasmid (human IR cDNA cloned in a mammalian expression vector with an EF-1 promoter) was a generous gift from Dr. Stevan Hubbard. The pEF-IR plasmid was digested with BamHI and XbaI restriction enzymes (Invitrogen) and a 2.7-kb fragment was isolated by gel extraction (Qiagen). This fragment was cloned into pFastBacHTB (Invitrogen). M1153L, T1154S, T1154A, M1153A/T1154A, M1153L/T1154S, and M1153L/T1154A mutations were generated using the Quikchange site-directed mutagenesis kit (Stratagene). IR fragments containing the mutations were subcloned into digested pEF vector and fully sequenced.
Kinase Assay of WT IR and IR Activation Loop Mutations in CHO-K1 Lysates
CHO-K1 cells were cultured in Ham's F12 medium containing l-glutamine and 10% FBS. For transfection, CHO-K1 cells were seeded into 6-cm plates and grown overnight to 80–90% confluency. Cells were transfected with 8 μg of DNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After incubation with lipid·DNA complexes for 6 h, the medium of the cells was replaced with Ham's F12 medium containing l-glutamine and 10% FBS. 24 h after transfection, the medium of the cells was replaced with medium lacking FBS. Cells were grown for an additional 4 h. Lysates were prepared in a buffer containing 20 mm Hepes, pH 7.4, 1 mm EGTA, 20 μm Na3VO4, 1 mm DTT, 2 mm MnCl2, 0.02 mg/ml BSA, 10 mm MgCl2, 1 mm ATP, 1% Triton X-100, and Complete EDTA-free (Roche Applied Science). Cells were treated with either DMSO or 10 μm Irfin1 for 30 min on ice and then were stimulated with 100 nm insulin for 10 min. Kinase assays were quenched upon the addition of Laemmli buffer. Auto-phosphorylated IR and GAPDH were visualized by Western blot using anti-phospho-IR/IGF-1R and anti-GAPDH antibodies (Cell Signaling Technology). Quantification was carried out using NIH ImageJ software (24).
RESULTS
Irfin1 Selectively Inhibits Activation of IR/IGF-1R
In an ongoing effort to elucidate the kinase selectivity of known kinase inhibitors, we have tested >175 compounds for their ability to inhibit the catalytic activity of a panel ∼300 recombinant human protein kinases (10). To identify inhibitors with high target selectivity, we calculated uni-specificity scores for each compound to quantitatively rank them based on their selectivity for a single kinase over all other kinases in the panel (10). Strikingly, among the most uni-specific inhibitors was a pyrazolopyridazine compound (Fig. 1A), which inhibited the IR by 73% at the screening concentration of 500 nm while inhibiting no other kinase in the panel by >21% (Fig. 1A; complete data presented in supplemental Table 1). On the basis of the activity of this compound as an insulin receptor family inhibitor, we designated this compound Irfin1. Irfin1 (FR180209) was originally reported as an inactive control compound for the ERK inhibitor FR180204 (Fig. 1B) (25), which was also in our compound collection. Interestingly, FR180204 also inhibited IR in addition to ERK, although more weakly and much less selectively than Irfin1 (Fig. 1, A and B). To understand how Irfin1 achieves such selective kinase inhibition, we investigated its mechanism of action.
FIGURE 1.
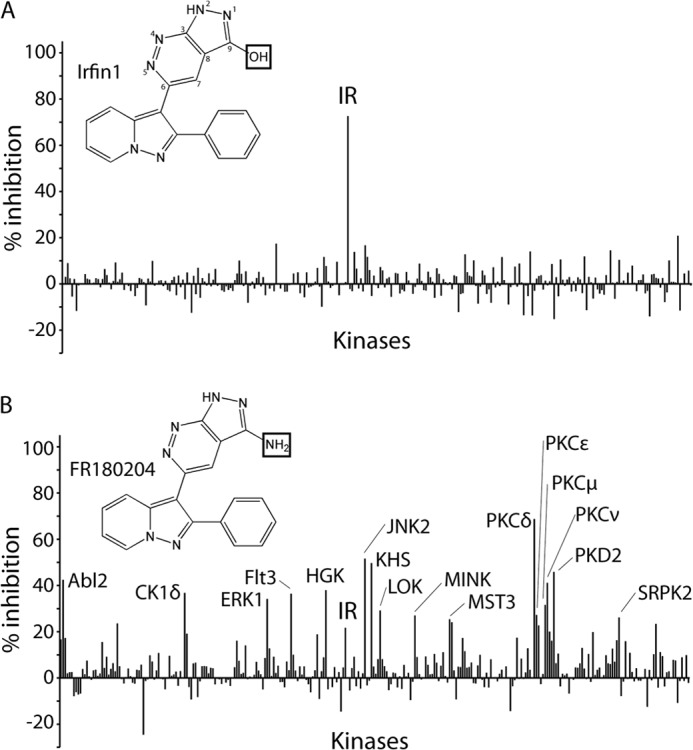
Structure and selectivity of pyrolopyridazine compounds. A, kinase selectivity of Irfin1. Percent inhibition relative to solvent control reactions is plotted for each of 290 kinases incubated with 0.5 μm Irfin1. Kinases inhibited by >25% are indicated. The chemical structure of Irfin1 is shown with the positions of the pyrazolopyridine ring numbered. B, kinase selectivity of FR180204. Reactions were conducted and are presented as described in A. The complete data set is provided in supplemental Table 1. The boxes in the chemical structures of Irfin1 and FR180204 indicate structural differences between the compounds.
Surprisingly, Irfin1 showed no inhibitory activity against the closely related IGF-1R kinase in the primary screen (Fig. 1A and supplemental Table 1), and we hypothesized that this could be due to differences in the activation state of IR and IGF-1R, which were unphosphorylated (inactive) and phosphorylated (activated), respectively, in the original screen. Indeed, when Irfin1 was tested in dose-response experiments against the inactive forms of IR and IGF-1R in vitro, we observed IC50 values of 1.8 and 10.2 μm, respectively (Fig. 2A). This 6-fold difference in potency is most likely due to slightly different N-terminal lobe conformations that arise from a few amino acid differences between the IR and IGF-1R sequences. The slight quantitative difference in the potency of Irfin1 inhibition of IR between the primary screening data (73% inhibition with 0.5 μm) and dose-response experiments (IC50, 1.8 μm) is likely due to differences in the protein constructs used in the two assays. To test whether this inhibitory activity was selective for the unphosphorylated form of IR/IGF-1R, the catalytic activity of unphosphorylated IR and each of the three phospho-forms (monophosphorylated, diphosphorylated, and triphosphorylated) was monitored in the presence of Irfin1. Although unphosphorylated IR was potently inhibited by Irfin1, consistent with our screening results, monophosphorylated, diphosphorylated, and triphosphorylated forms of IR became increasingly resistant to inhibition by Irfin1 (Fig. 2B, left). This result indicates that Irfin1 selectively binds and inhibits inactive, unphosphorylated, and singly phosphorylated IR. Consistent with these results for Irfin1, we found that the structurally related ERK inhibitor FR180204 also showed much more potent inhibition of un- and monophosphorylated forms of IR compared with the di- and triphosphorylated forms (Fig. 2B, right). FR180204 inhibits unphosphorylated IR and IGF-1R with observed IC50 values of 5.4 and 32.5 μm, respectively (Fig. 2A). These figures are only marginally different from the IC50 values with Irfin1 (1.8 and 10.2 μm) and likely reflect similar binding modes for the Irfin1-IR·IGF-1R and FR180204-IR·IGF-1R complexes.
FIGURE 2.

Dose- and activation state-dependent inhibition of IR/IGF-1R. A, in vitro dose-response measurements of inhibition of IR and IGF-1R catalytic activity by Irfin1 and FR180204. Remaining kinase activity is shown relative to solvent controls. Each data point represents the mean of three replicate measurements. Error bars indicate S.D. B, left: Irfin1 inhibition of activation intermediates of IR. Unphosphorylated (0p), mono (1p)-, di (2p)-, and tri (3p)-phosphorylated forms of IR were purified and subjected to in vitro kinase reactions in the presence of 2, 10, and 100 μm Irfin1. Right: FR180204 inhibition of activation intermediates of IR. Measurements were performed as on the left.
We also tested the ability of Irfin1 and FR180204 to inhibit insulin-dependent autophosphorylation of IR/IGF-1R in cultured cells. FR180204 shows weak inhibition of IR activation in insulin-stimulated CHO-IR cells (data not shown). Irfin1 did not display inhibition of the receptors in intact cells but did display robust inhibition of IR autophosphorylation in detergent lysates of CHO-IR cells (data not shown; see also Fig. 4), suggesting that the cell permeability of Irfin1 is poorer than that of FR180204. Collectively, these findings identify the pyrazolopyridazine scaffold of Irfin1 and FR180204 as a promising basis for the design of inhibitors against inactive, unphosphorylated IR/IGF-1R.
FIGURE 4.
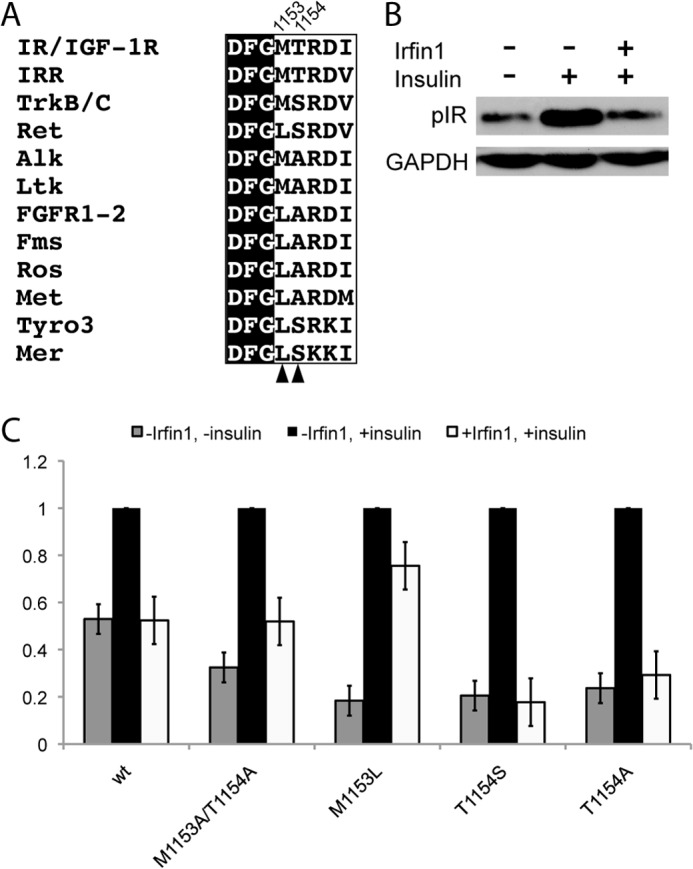
Activation loop residues contribute to the kinase specificity of Irfin1. A, activation loop sequences of insulin receptor-related (IRR) and IR/IGF-1R are aligned with analogous residues from the 10 most closely related human kinases. Arrowheads and residue numbers indicate the residues that are the subject of the mutagenesis experiments. B, inhibition of IR by Irfin1 in cell lysates. Lysates of CHO-K1 cells expressing wild type IR were pretreated (or not, as indicated) with 10 μm Irfin1 and stimulated with insulin. IR autophosphorylation was monitored by Western blotting for phospho-IR (pIR). C, the experiment in B was repeated for wild type and mutant IR constructs and quantified. Each set of bars was normalized to the vehicle-treated, insulin-stimulated phospho-IR Western blot signal. Error bars represent the S.E. from three replicates.
Structural Basis for Irfin1 Selectivity and Comparison with FR180204
The inhibition of the inactive form of IR/IGF-1R by FR180204 was particularly striking since this compound binds and inhibits ERK (25), a kinase that, unlike IR/IGF-1R, does not adopt the DFG-out activation loop conformation (26). To understand the structural basis for the selectivity of this compound class and its ability to inhibit two highly divergent kinases with two distinct activation loop conformations, we solved the crystal structure of IR kinase domain (IRK) bound to Irfin1 at 1.8 Å resolution and compared it to the previously reported structure of ERK2 bound to FR180204 (25). Two Irfin1·IRK complexes are present in the asymmetric unit. Data collection and refinement statistics are shown in Table 1.
TABLE 1.
X-ray data collection and refinement statistics
| Parameter | Result for complex of IR kinase domain and Irfin1 |
|---|---|
| Space group | P212121 |
| Unit cell | a = 50.84, b = 89.21, and c = 142.19 Å; α = 90, β = 90, and γ = 90° |
| Resolution range (Å)a | 50–1.8 |
| No. of observed reflections | 405,939 |
| No. of unique reflections | 60,964 |
| Redundancy | 6.7 (4.6) |
| Completeness (%) | 99.8 (97.8) |
| Rsymb | 0.075 (0.502) |
| Mean I/σI | 25.6 (2.1) |
| Refinement statistics | |
| Resolution range (Å) | 50–1.8 |
| Reflections | 57,490 |
| Rcryst/Rfree (%) | 18.2/21.7 |
| No. of atoms refined | 5310 |
| Protein | 4706 |
| Water | 554 |
| Irfin1 | 50 |
| r.m.s.d. from ideal geometryc | |
| Bond lengths (Å) | 0.010 |
| Bond angles | 1.38° |
| Mean B-factors (Å2) | 17.3 |
| Protein (Å2) | 16.4 |
| Water (Å2) | 25.6 |
| Irfin1 (Å2) | 14.1 |
| Wilson B-factor (Å2) | 17.1 |
a Data in parentheses correspond to the outermost resolution shell.
b Rsym = Σ(hkl)Σ(j)|Ij − 〈I〉 is the mean intensity of j observations from a reflection hkl and its symmetry equivalents.
c r.m.s.d. indicates root mean square deviation.
Consistent with the biochemical results, Irfin1 binds to an inactive conformation of IRK (Fig. 3A, upper panel, teal schematic) that is similar, but not identical, to its unliganded and unphosphorylated form (5). The N-terminal lobe is rotated as a rigid body about the kinase hinge region by ∼15° (as measured by the angle formed between the helix normal of the C helix of the Irfin1·IRK complex and that of the apo form of IRK, Protein Data Bank code 1IRK (5)), “clamping” down on Irfin1. A similar rotation of the N-terminal lobe has been observed in other kinase-inhibitor co-crystals. A total surface area (inhibitor and kinase) of 723.4 Å2 is buried in the complex. The binding pocket is bounded by elements of the ATP-binding site as well as the kinase activation loop (shown in pink in Fig. 3A, upper panel). The activation loop partially obscures the ATP-binding pocket as in many inactive kinases. Although FR180204 occupies an analogous position in ERK (Fig. 3A, upper panel, green schematic), the orientation of the compounds in the two structures differs by an almost 90° rotation (Fig. 3A, lower panel). In addition, the activation loop of ERK (shown in red) is shifted well away from the ATP-binding site and only Asp-167 of the activation loop contacts FR180204. Hereafter, we will refer to the binding mode of Irfin1 to IRK as mode 1 and that of FR180204 bound to ERK2 as mode 2.
FIGURE 3.

The crystal structure of Irfin1-IR and comparison with the structure of FR180204-ERK2. A, upper panel: a structural alignment of the crystal structures of Irfin1-IRK (teal) and FR180204-ERK2 (green) (Ref. 25) in ribbon representation. Activation loops (residues 1150–1171 in IR, 167–188 in ERK2) are colored pink (IR) and red (ERK2). Catalytic loops (residues 1130–1137 in IR and residues 147–154 in ERK2) are colored tan (IR) and orange (ERK2). Irfin1 (carbon atoms are yellow, nitrogen atoms are blue, and oxygen atoms are red) and FR180204 (carbon atoms are orange and nitrogen atoms are blue) are shown in stick representation. Lower panel: a close-up view of the relative orientation of the each compound in the alignment as well as the omit density map for Irfin1 (blue mesh). B, stereoview of the interactions between Irfin1 and IR. Atoms and residues are colored according to the scheme described in A. The side chains of residues 1077 and 1078 were omitted for simplicity. An ordered water molecule is shown as a red sphere, and select hydrogen bonds are represented by dashed lines. C, schematic representation of the Irfin1-IRK interactions (left) and the FR180204-ERK2 interactions (right) from Ref. 25. Residues making van der Waals contacts (≤3.9 Å) with Irfin1 are colored according to the scheme described in A.
Strikingly, similar to the adenine base of ATP, both Irfin1 and FR180204 make hydrogen bonds to analogous main chain amide groups in the kinase hinge region (Glu-1077 and Met-1079 in IR, and Asp-106 and Met-108 in ERK2) (Fig. 3, B and C). These interactions are mediated by different nitrogen atoms in each binding mode. In mode 1, the nitrogen atoms at the 4- and 5- positions of Irfin1 form hydrogen bonds with Met-1079 in IR, whereas in mode 2, the nitrogen atoms in the 1- and 2-positions of FR180204 form hydrogen bonds with the analogous Met-108 in ERK2. Additionally, in mode 1, the nitrogen in position 2 of Irfin1 forms a hydrogen bond with Glu-1077, whereas in mode 2, the amino group in position 9 of FR180204 forms a hydrogen bond with the analogous Asp-106. Irfin1 forms an additional water-mediated hydrogen bond to the carboxylate of Asp-1083 and a bidentate hydrogen bond via the hydroxyl group at position 9 and nitrogen at position 1 to the carboxylate of Asp-1150 in the canonical DFG motif. Several additional hydrophobic contacts with Irfin1 are mediated by residues of the ATP-binding pocket (blue in Fig. 3C) as well as with Met-1153 and Thr-1154 of the activation loop (pink in Fig. 3C).
The Irfin1-IRK structure suggests that several features are responsible for the exquisite selectivity of Irfin1 for inactive IR. Perhaps most important to selectivity are the six hydrogen bonds described above involving Asp-1083, Asp-1150, Glu-1077, and Met-1079. The mode 1 interaction is also likely favored by the high shape complementarity of a preexisting pocket in IR as well as the contribution of two non-conserved activation loop residues (Met-1153 and Thr-1154) to inhibitor binding. Unliganded, unphosphorylated IR and Irfin1-bound IR exhibit only 1.98 Å root mean square deviation over 291 Cα atoms. Much of this difference is attributable to a rotation of the N-terminal lobe (described above) as well as to alterations in the activation loop. The backbone of Asp-1150 and Phe-1151 of the DFG motif at the N-terminal end of the activation loop is rotated by ∼180°, switching the phenyl side chain from the DFG-out position typical of inactive IRK into the pocket it normally occupies in the DFG-in/active conformation. Indeed, the Phe-1151 side chain would sterically clash with the hydroxyl of Irfin1 in the DFG-out conformation. Thus, mode 1 binding likely drives DFG rotation and is further stabilized by the formation of a hydrogen bond between Asp-1150 and Irfin1. Despite this “active-like” positioning of Phe-1151, the remainder of the activation loop occupies a position similar to that in inactive, unliganded IRK and the side chains of activation loop residues Met-1153 and Thr-1154 cradle the phenyl ring of Irfin1, precluding ATP binding.
To further examine the contribution of Met-1153 and Thr-1154 to Irfin1 binding, we used site-directed mutagenesis to replace these residues with Ala. In CHO-K1 cells transfected with WT IR, a 2-fold increase in tyrosine phosphorylation of IR is observed upon stimulation with insulin (Fig. 4B). This activation of IR is abrogated when lysates were incubated with 10 μm Irfin1 prior to insulin stimulation (Fig. 4B). The M1153A/T1154A IR double mutant also displayed increased tyrosine phosphorylation upon stimulation with insulin (Fig. 4C), but Irfin1 only partially suppressed IR autophosphorylation, suggesting that removal of the side chains of these residues confers some resistance to Irfin1.
We then examined the effects of mutating Met-1153 and Thr-1154 to other amino acids that are found in these positions in the 10 kinases most closely related to IR by amino acid sequence (Fig. 4A). Met-1153 is most often substituted with a Leu in these kinases. The M1153L mutation in IR is largely resistant to Irfin1, suggesting that this residue plays a significant role in the selectivity of Irfin1 toward IR (Fig. 4C). This result is consistent with our crystal structure of the IRK·Irfin1 complex. Leu has a bulky side chain and is also more rigid than Met. Thus, the M1153L mutation likely sterically hinders Irfin1 binding. Surprisingly, mutation of Thr-1154 to either Ser or Ala was not incompatible with Irfin1 binding despite the fact that this residue is conserved in IR, IGF-1R, and the insulin receptor-related kinase but not in the other related kinases. Ser and Ala both contain smaller side chains than Thr and are thus unlikely to hinder Irfin1 binding. However, it is likely that substitution of Thr-1154 with a bulkier residue would interfere in Irfin1 binding. These results suggest that the multiple points of contact between Irfin1 and the activation loop in mode 1 contribute to the selectivity of the mode 1 interaction. Furthermore, all of the IR residues that contact Irfin1 are absolutely conserved in IGF-1R with the exception of Ala-1080 (Thr-1053 in IGF-1R), explaining the comparable potency of Irfin1 against these highly homologous kinase domains.
Our structure also suggests why Irfin1, the inactive control compound for the ERK inhibitor FR180204 (25), shows no significant mode 2 inhibition of ERK1 or ERK2 (Fig. 1A). The primary amine of FR180204 makes a bidentate hydrogen bond to the main chain amide of Asp-106 and the side chain of Asn-105 in ERK2 (Fig. 3C). The inability of the analogous functional group in Irfin1, a hydroxyl, to act as a donor in two hydrogen bonds likely contributes to its poor ability to inhibit ERK in mode 2. We found that FR180204 shows significant inhibitory activity against unphosphorylated and monophosphorylated IR but, unlike Irfin1, FR180204 also shows additional weak inhibitory activity against di- and triphosphosphorylated IR (Fig. 2B), implying that FR180204 inhibits unphosphorylated IR in mode 1 but inhibits triphosphorylated IR in mode 2. Consistent with this hypothesis, FR180204 can be modeled into our crystal structure of mode 1 without steric clashes, replicating the interactions of Irfin1. The weak but significant inhibition of triphosphorylated IR by FR180204 demonstrates that this compound, unlike Irfin1, can also bind to active IR, likely in mode 2. Thus, although Irfin1 is restricted to mode 1 binding, FR180204 binds ERK2 in mode 2 and can bind IR in both modes 1 and 2. The variety of binding modes for FR180204 is consistent with the much poorer selectivity of this compound compared with Irfin1 (Fig. 1B).
Comparison with Other IR/IGF-1R Inhibitors
To compare the binding mode of Irfin1 to that of other IR/IGF-1R inhibitors, we identified structures of IR or IGF-1R bound to small-molecule inhibitors previously reported in the Protein Data Bank. Sixteen crystal structures have been reported for ∼14 small-molecule inhibitor classes bound to IR or IGF-1R (supplemental Table 2) (28–41). Only one of these inhibitors, compound 3, does not bind to the ATP-binding site of the kinase domain. In nine of these structures, the activation loop of IR/IGF-1R adopts a conformation similar to that observed in the Irfin1-IR structure. Small changes in the position of the activation loop or the degree of closure of the ATP binding pocket in these structures demonstrate an inherent plasticity to this pocket in IR/IGF-1R that is exploited by these structurally diverse compounds. Although none of the inhibitors are structurally related to Irfin1, the activation loop conformations of IGF-1R bound to compound 2 (BMS-754807) and compound 6 most closely resemble the conformation of IR bound by Irfin1.
To assess the selectivity of Irfin1 as compared with that of BMS-754807 (40, 42) and OSI-906/linsitinib (43), two IR/IGF-1R inhibitors in clinical development, we screened each inhibitor against a panel of ∼300 protein kinases using the Reaction Biology Corporation Kinase HotSpot platform (Fig. 5A). Under the conditions of the initial screen, 500 nm Irfin1 inhibits IR activity by 73% (Fig. 5B). We therefore screened BMS-754807 and OSI-906 at their respective inhibitory concentrations for 73% inhibition (IC73) of IR to control for potency as inhibitor selectivity is generally related to potency (44). Dose-response measurements were performed for each compound under the same conditions as those used for the screen, and the IC73 was determined, yielding 18.6 and 1.82 nm, respectively, for BMS-754807 and OSI-906.
FIGURE 5.

Inhibitor selectivity profiling. Inhibitors were screened against a panel containing 290 human protein kinases at their IC73 for IR. A, selectivity data are represented in a radial plot where the lengths of radii correspond to the percent inhibition of kinases in the panel. Kinases with negative inhibition values were truncated to 0. Kinases are arranged in a clockwise fashion and are ranked in order of kinase druggability as defined previously (Ref. 10). The black square indicates kinases that were not found to be sensitive to small-molecule inhibition, and the direction of increasing druggability is indicated by the arrow. Concentric circles denote the level of inhibition. The center of all of the circles denotes 0% inhibition. Kinases that are inhibited by >25% are indicated on each plot. Complete screening data is provided in supplemental Table 1. B, Irfin1. (Note that this radial plot and the linear plot in Fig. 1A are generated from the same data.) C, BMS-754807. D, OSI-906.
Next, we screened each inhibitor at its respective IC73 for IR. As expected, both BMS-754807 and OSI-906 inhibited the unphosphorylated IR in the screening panel (Fig. 5, C and D). Remarkably, both compounds also showed inhibition of IGF-1R and insulin receptor-related kinase (Fig. 5, C and D), whereas Irfin1 did not (Fig. 5B), presumably because both IGF-1R and insulin receptor-related are both fully phosphorylated in the screen. It is not surprising that OSI-906 inhibits both unphosphorylated and fully-phosphorylated IR/IGF-1R kinases as a close relative of OSI-906, cis-3-[3-(4-methyl-piperazin-l-yl)-cyclobutyl]-1-(2-phenyl-quinolin-7-yl)-imidazo[1,5-a]pyrazin-8-ylamine (PQIP), binds to an intermediate conformation of the kinase domain where the activation loop is extended, reminiscent of the active state, and where the C helix is in a conformation reminiscent of the inactive state (41, 43). However, it was somewhat surprising that BMS-754807 inhibits fully phosphorylated IGF-1R because BMS-754807 was crystallized in complex with unphosphorylated IGF-1R (40). The inhibition of fully phosphorylated IGF-1R can be rationalized by the fact that the conformation of BMS-754807 bound to unphosphorylated IGF-1R (40) can be docked into the structure of the active form of IGF-1R without any clashes (data not shown). In contrast, Irfin1 only inhibited inactive IR in our screen. Our structure suggests that the conformation of the activation loop determines Irfin1 binding and is restricted to an inactive activation loop conformation.
In our screen, Irfin1 exhibited no off-target activity, defined as inhibition of non-IR/IGF-1R family kinases by >25% (Fig. 4B). In contrast, both BMS-754807 and OSI-906 exhibited a number of off-target effects (Fig. 5, C and D). Nearly all off-targets for BMS-754807 that were detected in our screen were consistent with those found using another kinase selectivity profiling platform (42). This study also found additional targets, likely due to differences in screening methodologies. Prior screening data for OSI-906 is limited, but our finding of GSK-3β as an off-target does contradict previous findings (43). Interestingly, BMS-754807 and OSI-906 shared both GSK-3β and CAMKK2 as targets. These kinases are Ser/Thr protein kinases and share only weak sequence homology with IR/IGF-1R family kinases. Both CAMKK2 and GSK-3β are more sensitive to small-molecule inhibition than most kinases in the panel (10). Irfin1 inhibited neither GSK-3β nor CAMKK2. Collectively, these data demonstrate that Irfin1 is an unusually selective inhibitor of inactive IR.
DISCUSSION
Kinases are often mutated in cancer and are well validated drug targets. Recently, we and others (10–12) have shown that many small molecule kinase inhibitors display a number of unintended off-target effects, perhaps due to the conserved nature of the ATP binding site in the active state of kinases. It has been argued that compounds that bind to inactive states of kinases may be more selective because the conformations of inactive kinases vary more widely than the conformations of activated kinases (13).
We conducted a large scale screen of 175 compounds against a panel of 300 recombinant human protein kinases. This screen was performed at a compound concentration of 500 nm. This concentration was chosen to capture off-target effects as it was much higher than the average IC50 of each compound against its intended target, 66 nm (10). In this screen, Irfin1 was found to selectively target the inactive state of the highly homologous IR and IGF-1R kinases. Upon binding Irfin1, the N-terminal lobe of the kinase domain of IR is rotated ∼15° from the basal, inactive state. Interestingly, the DFG motif of the activation loop also shifts so that it is no longer in the DFG-out conformation reminiscent of inactive protein kinases. Instead, the activation loop has the Asp side chain of the DFG motif pointing “in” and the Phe side chain of the DFG motif pointing “out.” This activation loop conformation, however, is distinct from the DFG-in conformation observed in structures of the active states of kinases. This conformational change may explain the uniquely selective nature of Irfin1 as the structurally related FR180204 is unable to induce this conformation and is far less selective than Irfin1. Similar conformational changes in the activation loop are observed for other IR/IGF-1R family inhibitors against the inactive state (28, 38–40). The diversity of conformations that are observed for these IR/IGF-1R·inhibitor complexes as well as the structural diversity of the inhibitors themselves reveals that the activation loop of the IR/IGF-1R family of kinases displays a great deal of structural plasticity. We have demonstrated how this plasticity can accommodate a variety of inhibitor scaffolds.
We have also demonstrated the potential of broad-spectrum kinase inhibitor profiling for the discovery of novel kinase inhibitor scaffolds. The traditional method of drug discovery is to seek inhibitors against a specific target, screen a number of compounds, select a lead compound, and then optimize that lead compound for selectivity and potency (45). In contrast, broad-spectrum kinase inhibitor profiling is not centered on one particular target; many inhibitors are screened against many kinases to identify novel scaffolds that target particular kinases (45). Irfin1 was discovered as a selective inhibitor of IR in a screen of ∼180 compounds against ∼300 human protein kinases (10). In the same screen, we also showed that the structurally related FR180204 can inhibit this kinase family (10), suggesting that the pyrazolopyridazine scaffold is a promising lead to pursue in drug discovery efforts for the IR/IGF-1R family kinases.
We also showed that the screening of kinases in multiple activation states can reveal opportunities for more selective kinase inhibition. Because our screening panel included IR/IGF-1R family members in both inactive and active forms, we were able to identify Irfin1 as a highly selective inhibitor of the inactive form of IR/IGF-1R. These results suggest that comprehensive protein kinase panels for use in kinase inhibitor discovery should include both active and inactive forms of kinases, which often differ in the conformations of their activation loops. Furthermore, we observed that FR180204 can inhibit IR and ERK by binding these kinases in different conformations.
It has been proposed that broad spectrum kinase inhibitor profiling could be an efficient route to identify single compounds that target a limited number of kinases of interest but do so selectively (45). Such a polypharmacological approach is very promising in cancer where multiple kinase-regulated signaling pathways are often hyper-activated and where selective inhibition of single targets may be insufficient to down-regulate an entire pathway. Identification of compound scaffolds with the desired multi-target spectrum is, however, a significant challenge. As we illustrate here, untargeted compound profiling can allow the serendipitous discovery of compounds, similar to Irfin1, that show a favorable target spectrum against even unrelated kinases. Subsequent efforts can then be made to further optimize these promising lead compounds against desired targets. As broad-spectrum profiling becomes more cost-effective and readily available, we expect it will play an increasingly important role in early-stage kinase inhibitor discovery efforts.
Acknowledgments
We thank Drs. Samarjit Patnaik and Juan Marugan (National Institutes of Health) for providing FR180204 and Cynthia Myers (Fox Chase Cancer Center) for assistance in organic synthesis. We also thank Dr. Stevan Hubbard (New York University) for providing the IR kinase domain construct and for use of the x-ray facility at New York University. We are also grateful to the staff at beamline X4C of the National Synchrotron Light Source at Brookhaven National Laboratory.
This work was supported by the National Institutes of Health Grants GM083025 (to J. R. P.), T32 CA009035-36 (to K. C. D.), and P30 CA006927 (to the Fox Chase Cancer Center) and by a W. W. Smith Charitable Trust Award. This work was also supported by the FCCC Keystone Program in Head and Neck Cancer.
The atomic coordinates and structure factors (code 4IBM) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Tables 1 and 2.
- IGF-1R
- insulin-like growth factor 1 receptor
- IR
- insulin receptor
- IRK
- insulin receptor kinase domain
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Larsson O., Girnita A., Girnita L. (2005) Role of insulin-like growth factor 1 receptor signalling in cancer. Br J. Cancer 92, 2097–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller B. S., Yee D. (2005) Type I insulin-like growth factor receptor as a therapeutic target in cancer. Cancer Res. 65, 10123–10127 [DOI] [PubMed] [Google Scholar]
- 3. Hartog H., Wesseling J., Boezen H. M., van der Graaf W. T. (2007) The insulin-like growth factor 1 receptor in cancer: old focus, new future. Eur. J. Cancer 43, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 4. Munshi S., Hall D. L., Kornienko M., Darke P. L., Kuo L. C. (2003) Structure of apo, unactivated insulin-like growth factor-1 receptor kinase at 1.5 A resolution. Acta Crystallogr. D Biol. Crystallogr 59, 1725–1730 [DOI] [PubMed] [Google Scholar]
- 5. Hubbard S. R., Wei L., Ellis L., Hendrickson W. A. (1994) Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372, 746–754 [DOI] [PubMed] [Google Scholar]
- 6. Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin M., Wang J., Buck E., Mulvihill M. J. (2012) Small-molecule ATP-competitive dual IGF-1R and insulin receptor inhibitors: structural insights, chemical diversity and molecular evolution. Future Med. Chem. 4, 315–328 [DOI] [PubMed] [Google Scholar]
- 8. Vincent E. E., Elder D. J., Curwen J., Kilgour E., Hers I., Tavaré J. M. (2013) Targeting non-small cell lung cancer cells by dual inhibition of the insulin receptor and the insulin-like growth factor-1 receptor. PLoS One 8, e66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fidler M. J., Shersher D. D., Borgia J. A., Bonomi P. (2012) Targeting the insulin-like growth factor receptor pathway in lung cancer: problems and pitfalls. Ther. Adv. Med. Oncol 4, 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anastassiadis T., Deacon S. W., Devarajan K., Ma H., Peterson J. R. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 [DOI] [PubMed] [Google Scholar]
- 12. Fabian M. A., Biggs W. H., 3rd, Treiber D. K., Atteridge C. E., Azimioara M. D., Benedetti M. G., Carter T. A., Ciceri P., Edeen P. T., Floyd M., Ford J. M., Galvin M., Gerlach J. L., Grotzfeld R. M., Herrgard S., Insko D. E., Insko M. A., Lai A. G., Lélias J. M., Mehta S. A., Milanov Z. V., Velasco A. M., Wodicka L. M., Patel H. K., Zarrinkar P. P., Lockhart D. J. (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336 [DOI] [PubMed] [Google Scholar]
- 13. Liu Y., Gray N. S. (2006) Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364 [DOI] [PubMed] [Google Scholar]
- 14. R Development Core Team (2010) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 15. Yee T. W. (2010) The VGAM package for categorical data analysis. J. Stat. Softw. 32, 1–34 [Google Scholar]
- 16. van der Loo M. P. (2010) extremevalues: a package for outlier detection in univariate data. R package, version 2.2, Statistics Netherlands, The Hague, Netherlands [Google Scholar]
- 17. Favelyukis S., Till J. H., Hubbard S. R., Miller W. T. (2001) Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 8, 1058–1063 [DOI] [PubMed] [Google Scholar]
- 18. Otwinowski Z., Minor W., Charles W. Carter., Jr. (1997) Processing of X-ray diffraction data collected in oscillation mode in Methods in Enzymology, pp. 307–326, Academic Press, New York, NY: [DOI] [PubMed] [Google Scholar]
- 19. Vagin A., Teplyakov A. (1997) MOLREP: an Automated Program for Molecular Replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 20. Wu J., Tseng Y. D., Xu C. F., Neubert T. A., White M. F., Hubbard S. R. (2008) Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat. Struct. Mol. Biol. 15, 251–258 [DOI] [PubMed] [Google Scholar]
- 21. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 22. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 24. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohori M., Kinoshita T., Okubo M., Sato K., Yamazaki A., Arakawa H., Nishimura S., Inamura N., Nakajima H., Neya M., Miyake H., Fujii T. (2005) Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 336, 357–363 [DOI] [PubMed] [Google Scholar]
- 26. Hari S. B., Merritt E. A., Maly D. J. (2013) Sequence determinants of a specific inactive protein kinase conformation. Chem. Biol. 20, 806–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deleted in proof [Google Scholar]
- 28. Buchanan J. L., Newcomb J. R., Carney D. P., Chaffee S. C., Chai L., Cupples R., Epstein L. F., Gallant P., Gu Y., Harmange J. C., Hodge K., Houk B. E., Huang X., Jona J., Joseph S., Jun H. T., Kumar R., Li C., Lu J., Menges T., Morrison M. J., Novak P. M., van der Plas S., Radinsky R., Rose P. E., Sawant S., Sun J. R., Surapaneni S., Turci S. M., Xu K., Yanez E., Zhao H., Zhu X. (2011) Discovery of 2,4-bis-arylamino-1,3-pyrimidines as insulin-like growth factor-1 receptor (IGF-1R) inhibitors. Bioorg. Med. Chem. Lett. 21, 2394–2399 [DOI] [PubMed] [Google Scholar]
- 29. Chamberlain S. D., Redman A. M., Wilson J. W., Deanda F., Shotwell J. B., Gerding R., Lei H., Yang B., Stevens K. L., Hassell A. M., Shewchuk L. M., Leesnitzer M. A., Smith J. L., Sabbatini P., Atkins C., Groy A., Rowand J. L., Kumar R., Mook R. A., Jr., Moorthy G., Patnaik S. (2009) Optimization of 4,6-bis-anilino-1H-pyrrolo[2,3-d]pyrimidine IGF-1R tyrosine kinase inhibitors towards JNK selectivity. Bioorg. Med. Chem. Lett. 19, 360–364 [DOI] [PubMed] [Google Scholar]
- 30. Heinrich T., Gradler U., Bottcher H., Blaukat A., Shutes A. (2010) Allosteric IGF-1R Inhibitors. ACS Medicinal Chemistry Letters 1, 199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katayama N., Orita M., Yamaguchi T., Hisamichi H., Kuromitsu S., Kurihara H., Sakashita H., Matsumoto Y., Fujita S., Niimi T. (2008) Identification of a key element for hydrogen-bonding patterns between protein kinases and their inhibitors. Proteins 73, 795–801 [DOI] [PubMed] [Google Scholar]
- 32. Lesuisse D., Mauger J., Nemecek C., Maignan S., Boiziau J., Harlow G., Hittinger A., Ruf S., Strobel H., Nair A., Ritter K., Malleron J. L., Dagallier A., El-Ahmad Y., Guilloteau J. P., Guizani H., Bouchard H., Venot C. (2011) Discovery of the first non-ATP competitive IGF-1R kinase inhibitors: advantages in comparison with competitive inhibitors. Bioorg. Med. Chem. Lett. 21, 2224–2228 [DOI] [PubMed] [Google Scholar]
- 33. Mayer S. C., Banker A. L., Boschelli F., Di L., Johnson M., Kenny C. H., Krishnamurthy G., Kutterer K., Moy F., Petusky S., Ravi M., Tkach D., Tsou H. R., Xu W. (2008) Lead identification to generate isoquinolinedione inhibitors of insulin-like growth factor receptor (IGF-1R) for potential use in cancer treatment. Bioorg. Med. Chem. Lett. 18, 3641–3645 [DOI] [PubMed] [Google Scholar]
- 34. Miller L. M., Mayer S. C., Berger D. M., Boschelli D. H., Boschelli F., Di L., Du X., Dutia M., Floyd M. B., Johnson M., Kenny C. H., Krishnamurthy G., Moy F., Petusky S., Tkach D., Torres N., Wu B., Xu W. (2009) Lead identification to generate 3-cyanoquinoline inhibitors of insulin-like growth factor receptor (IGF-1R) for potential use in cancer treatment. Bioorg. Med. Chem. Lett. 19, 62–66 [DOI] [PubMed] [Google Scholar]
- 35. Nemecek C., Metz W. A., Wentzler S., Ding F. X., Venot C., Souaille C., Dagallier A., Maignan S., Guilloteau J. P., Bernard F., Henry A., Grapinet S., Lesuisse D. (2010) Design of potent IGF1-R inhibitors related to bis-azaindoles. Chem. Biol. Drug. Des. 76, 100–106 [DOI] [PubMed] [Google Scholar]
- 36. Parang K., Till J. H., Ablooglu A. J., Kohanski R. A., Hubbard S. R., Cole P. A. (2001) Mechanism-based design of a protein kinase inhibitor. Nat. Struct. Biol. 8, 37–41 [DOI] [PubMed] [Google Scholar]
- 37. Patnaik S., Stevens K. L., Gerding R., Deanda F., Shotwell J. B., Tang J., Hamajima T., Nakamura H., Leesnitzer M. A., Hassell A. M., Shewchuck L. M., Kumar R., Lei H., Chamberlain S. D. (2009) Discovery of 3,5-disubstituted-1H-pyrrolo[2,3-b]pyridines as potent inhibitors of the insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase. Bioorg. Med. Chem. Lett. 19, 3136–3140 [DOI] [PubMed] [Google Scholar]
- 38. Sampognaro A. J., Wittman M. D., Carboni J. M., Chang C., Greer A. F., Hurlburt W. W., Sack J. S., Vyas D. M. (2010) Proline isosteres in a series of 2,4-disubstituted pyrrolo[1,2-f][1,2,4]triazine inhibitors of IGF-1R kinase and IR kinase. Bioorg Med. Chem. Lett. 20, 5027–5030 [DOI] [PubMed] [Google Scholar]
- 39. Velaparthi U., Wittman M., Liu P., Stoffan K., Zimmermann K., Sang X., Carboni J., Li A., Attar R., Gottardis M., Greer A., Chang C. Y., Jacobsen B. L., Sack J. S., Sun Y., Langley D. R., Balasubramanian B., Vyas D. (2007) Discovery and initial SAR of 3-(1H-benzo[d]imidazol-2-yl)pyridin-2(1H)-ones as inhibitors of insulin-like growth factor 1-receptor (IGF-1R). Bioorg. Med. Chem. Lett. 17, 2317–2321 [DOI] [PubMed] [Google Scholar]
- 40. Wittman M. D., Carboni J. M., Yang Z., Lee F. Y., Antman M., Attar R., Balimane P., Chang C., Chen C., Discenza L., Frennesson D., Gottardis M. M., Greer A., Hurlburt W., Johnson W., Langley D. R., Li A., Li J., Liu P., Mastalerz H., Mathur A., Menard K., Patel K., Sack J., Sang X., Saulnier M., Smith D., Stefanski K., Trainor G., Velaparthi U., Zhang G., Zimmermann K., Vyas D. M. (2009) Discovery of a 2,4-disubstituted pyrrolo[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. J. Med. Chem. 52, 7360–7363 [DOI] [PubMed] [Google Scholar]
- 41. Wu J., Li W., Craddock B. P., Foreman K. W., Mulvihill M. J., Ji Q. S., Miller W. T., Hubbard S. R. (2008) Small-molecule inhibition and activation-loop trans-phosphorylation of the IGF1 receptor. EMBO J. 27, 1985–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carboni J. M., Wittman M., Yang Z., Lee F., Greer A., Hurlburt W., Hillerman S., Cao C., Cantor G. H., Dell-John J., Chen C., Discenza L., Menard K., Li A., Trainor G., Vyas D., Kramer R., Attar R. M., Gottardis M. M. (2009) BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 8, 3341–3349 [DOI] [PubMed] [Google Scholar]
- 43. Mulvihill M. J., Cooke A., Rosenfeld-Franklin M., Buck E., Foreman K., Landfair D., O'Connor M., Pirritt C., Sun Y., Yao Y., Arnold L. D., Gibson N. W., Ji Q. S. (2009) Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 1, 1153–1171 [DOI] [PubMed] [Google Scholar]
- 44. Knight Z. A., Shokat K. M. (2005) Features of selective kinase inhibitors. Chem. Biol. 12, 621–637 [DOI] [PubMed] [Google Scholar]
- 45. Goldstein D. M., Gray N. S., Zarrinkar P. P. (2008) High-throughput kinase profiling as a platform for drug discovery. Nat. Rev. Drug. Discov. 7, 391–397 [DOI] [PubMed] [Google Scholar]