

Background: Fatty acyl-AMP ligase FadD33 is required for mycobactin biosynthesis.
Results: FadD33 catalyzes a two-step kinetic mechanism, and FadD33 activity is regulated by post-translational acetylation.
Conclusion: Mycobactin FadD33 activity is reversibly regulated by Pat (acetylation) and DAc1 (deacetylation).
Significant: Post-translational regulation via acetylation of enzymes can modulate siderophore biosynthesis.
Keywords: Cyclic AMP (cAMP), Enzyme Mechanisms, Kinetics, Mycobacteria, Siderophores, Acetylation, FadD33, Mycobactin, Ping-Pong
Abstract
Mycobacterial siderophores are critical components for bacterial virulence in the host. Pathogenic mycobacteria synthesize iron chelating siderophores named mycobactin and carboxymycobactin to extract intracellular macrophage iron. The two siderophores differ in structure only by a lipophilic aliphatic chain attached on the ϵ-amino group of the lysine mycobactin core, which is transferred by MbtK. Prior to acyl chain transfer, the lipophilic chain requires activation by a specific fatty acyl-AMP ligase FadD33 (also known as MbtM) and is then loaded onto phosphopantetheinylated acyl carrier protein (holo-MbtL) to form covalently acylated MbtL. We demonstrate that FadD33 prefers long chain saturated lipids and initial velocity studies showed that FadD33 proceeds via a Bi Uni Uni Bi ping-pong mechanism. Inhibition experiments suggest that, during the first half-reaction (adenylation), fatty acid binds first to the free enzyme, followed by ATP and the release of pyrophosphate to form the adenylate intermediate. During the second half-reaction (ligation), holo-MbtL binds to the enzyme followed by the release of products AMP and acylated MbtL. In addition, we characterized a post-translational regulation mechanism of FadD33 by the mycobacterial protein lysine acetyltransferase in a cAMP-dependent manner. FadD33 acetylation leads to enzyme inhibition, which can be reversed by the NAD+-dependent deacetylase, MSMEG_5175 (DAc1). To the best of our knowledge, this is the first time that bacterial siderophore synthesis has been shown to be regulated via post-translational protein acetylation.
Introduction
Mycobacterium tuberculosis, the etiologic agent of tuberculosis, has developed during its evolution successful tools for living in the harsh macrophage environment. After bacterial inhalation and infection, alveolar macrophages act as key players for the host immune defense by engulfing and degrading the pathogen within phagolysosomes (1). Pathogenic mycobacteria have developed complex pathways to subvert host-cell signaling to prevent a fatal fusion of the initial phagosome with the lysosome (2) and to elicit immune responses leading to the formation of granuloma (3). A common strategy is the production of cAMP by mycobacterial adenylate cyclases to stimulate production of host cytokines in M. tuberculosis (4, 5) or to stop phagosome maturation as seen for Mycobacterium microti (6). The M. tuberculosis H37Rv genome contains an unusually high number of class III adenylate cyclases (16) and cAMP-binding proteins (10), highlighting the central role of cAMP in manipulating host immunity and coordinating its own intracellular regulation (7, 8). Functions of cAMP-binding proteins and their downstream effectors are mostly unknown with the exception of two cAMP receptor protein family transcription factors (9, 10) and a protein acetyltransferase (Pat)2 (11, 12). Pat was recently structurally characterized and shows a unique structural feature with a C-terminal GCN5-related N-acetyl-transferase domain fused to an N-terminal cAMP-binding domain (13). This bi-domain fusion allows Pat to inactivate acetyl-CoA synthase (ACS) via acetylation of a conserved lysine ACS residue (Lys-617) in a cAMP-dependent manner (12). ACS is an important enzyme for C2 carbon assimilation and metabolism. Moreover, ACS is also part of large family of acyl-activating enzyme, which includes FadDs enzymes along with non-ribosomal peptide synthetases (14).
During mycobacterial invasion, iron is an essential nutrient for bacterial growth and for virulence. The host is constantly depleting iron using serum transferrin (15), and the macrophage is keeping iron extremely low through the natural resistance-associated macrophage protein Nramp1 (16), decreasing the expression of transferrin receptor and intracellular ferritin (15). Despite iron restriction defense strategies developed by the host, mycobacteria successfully obtain intracellular iron using lipophilic siderophores capable of membrane diffusion (17). To do so, M. tuberculosis synthesize two siderophores, the lipophilic mycobactin and soluble carboxymycobactin, which share the same core iron-chelating mycobactin scaffold. The core mycobactin is synthesized by the mbt-1 gene cluster (MbtA to J) (18, 19), whereas the acyl transferase (MbtK) encoded within the mbt-2 gene cluster (MbtK to N) will transfer a lipophilic aliphatic chain to the ϵ-amino group of the lysine mycobatin core (Fig. 1) (20). Prior to lipophilic acyl chain transfer, the fatty acid chain needs to be activated by the fatty acyl-AMP ligase FadD33 (also known as MbtM) and then loaded onto the acyl carrier protein MbtL for final transfer by MbtK (Fig. 2) (20).
FIGURE 1.

Summary of mycobactin and carboxymycobatin biosynthesis. 10 proteins are involved in the mycobactin core formation (mbt-1 gene cluster) and four other proteins catalyzed the mycobactin formation (mbt-2 gene cluster). R1 = (CH2)nCH3 with n = 16–19 or R1 = (CH2)xCH = CH(CH2)yCH3 with x + y = 14–17 for lipophilic mycobactin and R1 = (CH2)nCOOCH3/COOH with n = 1–7 or R1 = (CH2)xCH = CH(CH2)yCOOCH3/COOH with x + y = 1–5 hydrophilic carboxymycobactin.
FIGURE 2.
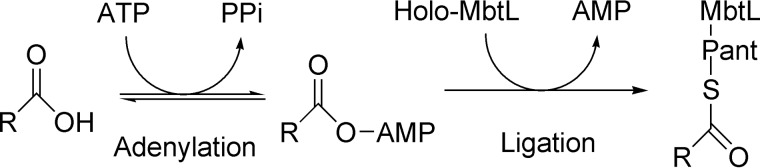
Adenylation- ligation catalyzed by FadD33 (MbtM).
In this study, we have carried out a detailed enzymatic characterization of the fatty acid AMP ligase, FadD33, and demonstrated that FadD33 can be reversibly inactivated by Pat acetylation in a cyclic AMP-dependent manner and reactivated by a specific deacetylase. In eubacteria, post-translational regulation via acetylation of enzymes involved in siderophore biosynthesis has no precedent.
MATERIALS AND METHODS
Materials
Molecular biology reagents were from Invitrogen, New England Biolabs, Novagen, Qiagen, or Stratagene. All chemicals were purchased from Sigma-Aldrich, unless otherwise noted.
Cloning, Expression, and Purification of FadD33
The fatty acyl AMP ligase, FadD33 (MSMEG_2131) was amplified from Mycobacterium smegmatis mc2155 genomic DNA using the primers pairs MSMEG_2131_F and MSMEG_2131_R containing NdeI and HindIII sites, respectively (Table 1). The PCR amplicon was ligated into the pYUB1062N vector and then transformed into Escherichia coli DH5α competent cells to create FadD33-pYUB1062 plasmid. A sequence-verified construct was transformed in Mycobacterium smegmatis mc24517 for protein expression. A 1-ml preculture was used to inoculate 1 liter of 7H9 medium (BD), supplemented with 0.2% glycerol, 0.025% Tween 20, 5% albumin-dextrose-catalase supplement (Difco), 25 μg/ml kanamycin, and 100 μg/ml hygromycin. The culture was grown to midlog phase (A600 ∼ 0.6) at 37 °C and then induced by the addition of 2.25 g/liter of acetamide. After 20 h of additional incubation (37 °C), cells were harvested by centrifugation (6,000 × g, 20 min) and stored at −80 °C. The cell pellet was thawed and resuspended in lysis buffer (50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl, 10 mm imidazole) supplemented with DNase I (0.1 μg/ml) and protease inhibitors (Roche Diagnostics). Resuspended cells were disrupted using an Avestin homogenizer (10,000 psi for 2.30 min in a closed circuit), and cellular debris was removed from the lysate by centrifugation at 38,000 × g for 1 h. To eliminate any residual debris, the supernatant was filtered using a 0.45-μm filter (Corning). The tagged proteins were purified from the clarified supernatant by Ni2+ column chromatography using Ni-NTA Superflow resin according to the manufacturer's instructions (Qiagen). After a 10-column volume wash with lysis buffer, the bound protein was eluted with a linear imidazole gradient (30–150 mm) at a flow rate of 1 ml/min. Pure fractions, as determined by SDS-PAGE were pooled together for buffer-exchange into 50 mm sodium phosphate, 150 mm NaCl, pH 8.0. The protein was concentrated using a 10-kDa cut-off centrifugal filter device (Millipore) to a final concentration of 300 μm.
TABLE 1.
Oligonucleotide sequences
Restriction sites are in boldface type, whereas mutated sites are underlined.
| MSMEG_2131_F | 5′-GGAATTCCATATGAACGTGCTCTCCGCGGCGCT-3′ |
| MSMEG_2131_R | 5′-CCCAAGCTTTCCTTTCGATTCCTCCAACTGTC-3′ |
| MSMEG_2131_K260A_F | 5′-CGGCCTGATCGGCGCGTACTCGCGCCGG-3′ |
| MSMEG_2131_K260A_R | 5′-CCGGCGCGAGTACGCGCCGATCAGGCCG-3′ |
| MSMEG_2131_K511A_F | 5′-CACCTCTTCGGGCGCGCTGCGCCGCCTG-3′ |
| MSMEG_2131_K511A_R | 5′-CAGGCGGCGCAGCGCGCCCGAAGAGGTG-3′ |
| MSMEG_2132_F | 5′-GGAATTCCATATGCAGTCAACATCCCCTGA-3′ |
| MSMEG_2132_R | 5′-CCCAAGCTTTCACTGGTTGCTCTGCTCC-3′ |
| Myokinase_F | 5′-CCTCGGCCATATGCGTATCATTCTGCTTGG-3′ |
| Myokinase_R | 5′-GCCAAGCAGAATGATAAGCTTTTAGCCGAG-3′ |
Cloning, Expression, and Purification of MbtL
The acyl carrier protein, MbtL (MSMEG_2132) was amplified from M. smegmatis mc2155 genomic DNA using the primer pairs MSMEG_2132_F and MSMEG_2132_R, which contained NdeI and HindIII restriction sites, respectively (Table 1). The PCR amplicon was ligated into a pET-28a(+) vector and then transformed into E. coli DH5α competent cells to create the pET-28a(+)::MbtL N-terminally His6 tag plasmid. A sequenced verified construct was transformed into E. coli T7 express lysy/Iq for protein expression. A 4-ml preculture was used to inoculate 1 liter of Luria-Bertani medium supplemented with 50 μg/ml kanamycin. The culture was grown to mid log phase (A600 ∼ 0.8) at 37 °C and then induced by the addition of 1 mm isopropyl-1-thio-β-d-galactopyranoside. After 18 h of additional incubation (25 °C), cells were harvested by centrifugation (6,000 × g, 20 min) and stored at −80 °C. MbtL was purified using Ni-NTA affinity as described above for FadD33. Pure fractions, as determined by SDS-PAGE were pooled together, and the MbtL protein was concentrated using a 3-kDa cut-off centrifugal filter device (Millipore). A second step of purification via exclusion chromatography was performed on a S75 column (GE Healthcare) equilibrated with 100 mm HEPES, 250 mm NaCl, pH 7.8, and controlled by an Akta FPLC pump. Fractions containing MbtL were determined by SDS-PAGE and concentrated as described above to a final concentration of 1000 μm.
Phosphopantetheinylation of MbtL
To convert the apo form of MbtL to the phosphopantetheinylated form, holo-MbtL, the phosphopantetheinyl transferase Sfp was used. The plasmid containing the Bacillus subtilis sfp gene (kind gift from C. T. Walsh) was transformed in E. coli, expressed, and purified as described by Yin and co-workers (21, 22). 250 μm apo-MbtL was incubated with 2.5 μm Sfp in 50 mm sodium phosphate, pH 7, with 10 mm MgCl2, 5 mm DTT, 500 μm coenzyme A (CoASH) in a 15-ml reaction volume. The reaction was allowed to proceed at 25 °C for 4 h, before buffer-exchange into 100 mm HEPES, 250 mm NaCl, pH 7.8. Phosphopantetheinylated holo-MbtL was confirmed by Fourier transform mass spectrometry.
Cloning, Expression, and Purification of Myokinase
The adenylate kinase, myokinase was amplified from E. coli genomic DNA using the primer pairs Myokinase_F and Myokinase_R, which contained NdeI and HindIII resctriction sites, respectively (Table 1). The PCR amplicon was ligated into the pET-28a(+) vector and then transformed into E. coli DH5α competent cells to create pET-28a(+)::MK N-terminally His6 tag plasmid. A sequenced-verified construct was transformed in E. coli T7 express lysy/Iq for protein expression. A 4-ml preculture was used to inoculate 1 liter of Luria-Bertani medium supplemented with 50 μg/ml kanamycin. The culture was grown to mid log phase (A600 ∼ 0.8) at 37 °C and then induced by addition of 1 mm isopropyl-1-thio-β-d-galactopyranoside. After 18 h of additional incubation (30 °C), cells were harvested by centrifugation (6,000 × g, 20 min) and stored at −80 °C. Myokinase was purified using Ni-NTA affinity as described above for FadD33. Pure fractions, as determined by SDS-PAGE were pooled together for buffer-exchange into 50 mm sodium phosphate, 150 mm NaCl, pH 8. The protein was concentrated using 10-kDa cut-off centrifugal filter devices (Millipore) to a final concentration of 400 μm and then diluted to 100 μm in 3.2 m (NH4)2SO4 and 1 μm EDTA, pH 6.
Protein Concentrations
The concentrations of MbtL, myokinase, and FadD33 were determined using the Bio-Rad protein assay using bovine serum albumin as a standard.
Measurement of Enzymatic Activity
The enzymatic activity of FadD33 was determined spectrophotometrically by coupling the formation of AMP to the reactions of myokinase, pyruvate kinase, and lactate dehydrogenase as described previously (23, 24). Reactions were performed in 100 mm HEPES, pH 7.8, 10 mm MgCl2, 250 mm NaCl, 1 mm PEP, 0.15 mm NADH, 200 μm holo-MbtL, 18 units of myokinase, 18 units of pyruvate kinase, and 18 units of lactate dehydrogenase in a final volume of 100 μl. Magnesium is a cofactor required by pyruvate kinase and by FadD ATP-dependent enzymes. Typically, 1 μm FadD33 was used and incubated 5 min at 25 °C with the reaction mix prior to reaction initiation by substrate addition with fatty acid. The reaction was monitored for ∼5 min at 340 nm (Δϵ340 = 6220 m−1 cm−1) using a Shimadzu spectrophotometer (UV-2450).
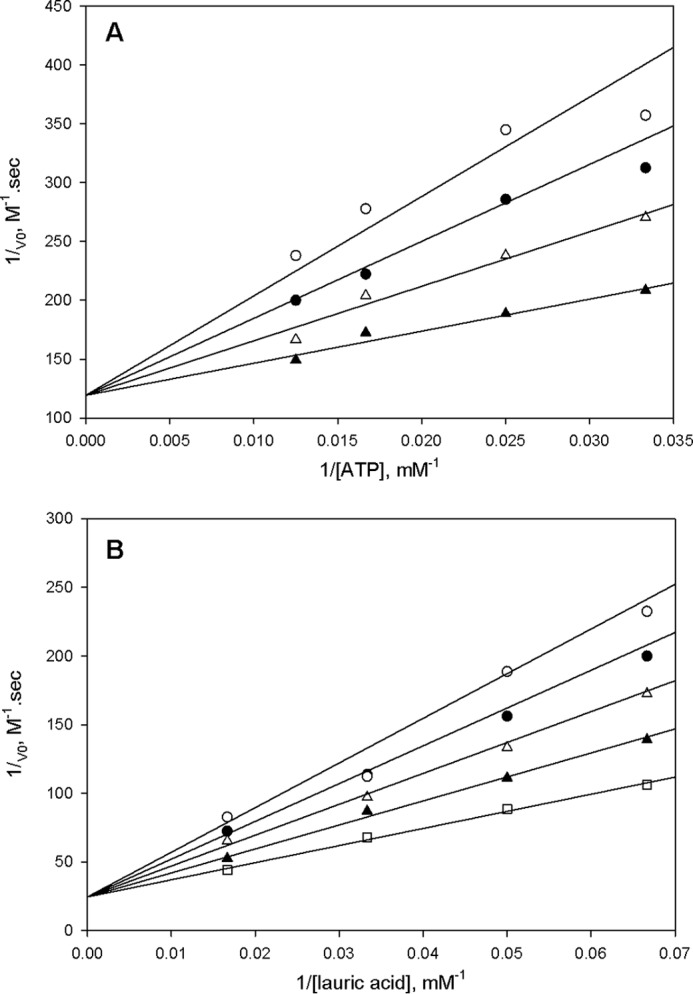
Initial Velocity Experiments
Initial kinetic parameters (kcat and Km) were obtained by saturating with two substrates and varying the concentration of a third substrate. For ATP kinetic constant determination, both holo-MbtL (500 μm) and lauric acid (C12; 100 μm) were kept at fixed saturating concentrations, and ATP (10–80 μm) was at variable concentrations. For holo-MbtL kinetic constant determination, both ATP (100 μm) and C12 (100 μm) were kept at fixed saturating concentrations, and holo-MbtL (25–400 μm) was kept at variable concentrations. For lauric acid kinetic constant determination, both holo-MbtL (500 μm) and ATP (100 μm) were kept at fixed saturating concentrations, and lauric acid (5–80 μm) was kept at variable concentrations. Individual substrate saturation kinetic data were fitted to Equation 1,
where V is the maximal velocity, A is the substrate concentration, and K is the Michaelis-Menten constant (Km). To probe the kinetic mechanism of FadD33, initial velocity patterns were determined at various concentrations of one substrate in the presence of different fixed concentrations of a second substrate with a saturating and constant concentration of the third substrate. Initial velocity data were fitted to Equation 2 for a parallel pattern and to Equation 3 for an intersecting pattern using Sigma Plot software (version 11.0),
where KA and KB are the Michaelis constants for the varied substrates A and B, respectively, and KiA is the dissociation constant for substrate A.
Inhibition Experiments
Product inhibition studies with pyrophosphate were performed at varying concentrations of lauric acid, holo-MbtL (500 μm), and 400 μm or 30 μm ATP. Product inhibition kinetic data were fitted to Equation 4 for an uncompetitive pattern and Equation 5 for a noncompetitive pattern using Sigma Plot software (version 11.0),
where I represents the concentration of inhibitor and Kis and Kii are the inhibition constants for the slope and intercept terms, respectively.
Bisubstrate Analog Inhibition Experiments
The bi-substrate, hydrolytically stable adenylate analog, 5′-O-(N-(palmitoic) sulfamoyl)adenosine (C16-AMS), kind gift of Prof. Courtney Aldrich, was tested as an inhibitor of FadD33 (Fig. 3). The C16-AMS analog was tested as an inhibitor versus both lauric acid (80 μm ATP and 300 μm MbtL) and ATP (40 μm lauric acid and 300 μm MbtL). Inhibition kinetic data were fitted to Equation 6 for a competitive pattern.
FIGURE 3.
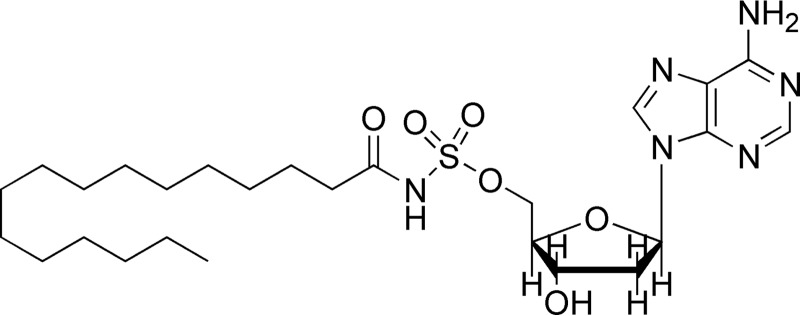
Structure of bisubstrate analog C16-AMS.
Expression and Purification of MSMEG_5458, MSMEG_5175, and MSMEG_4620
The acetyltranferase, MsPat (MSMEG_5458) and the two deacetylases, DAc1 and DAc2 (MSMEG_5175 and MSMEG_4620) were purified as described previously (12).
Site-directed Mutagenesis and Purification of FadD33-K260A and FadD33-K511A and Double Mutant FadD33-K260A/K511A
The two independent mutations K260A and K511A and the double mutant K260A/K511A were introduced into pYUB1062::FadD33 plasmid using the QuikChange mutagenesis kit (Stratagene) with the primers listed in Table 1. The mutations were confirmed by DNA sequencing. The expression and purification of FadD33 mutants were the same as described for the wild type.
In Vitro Acetylation Assay
10 μm FadD33 or mutant proteins were incubated with 1 mm cAMP, 100 μm acetyl-CoA, and 1 μm MsPat at 37 °C for 4 h. Samples were then analyzed by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane. Western blots were performed using an anti-acetyllysine antibody (Cell Signaling Technology, Inc. and ImmuneChem Pharmaceuticals, Inc., dilution of 1:2000) and goat anti-rabbit IgG AP conjugate (Bio-Rad Laboratories, dilution of 1:2000). Development was carried out according to the manufacturer's instructions.
Mass Spectrometric Analysis of Acetylated FadD33
The mass spectrometric analysis was performed at the Laboratory for Macromolecular Analysis and Proteomics of the Albert Einstein College of Medicine. Acetylated FadD33 was analyzed by SDS-PAGE. The single protein band corresponding to FadD33 was excised from the gel. After trypsin digestion, the digests were analyzed by LC-MS/MS using a LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific) equipped with a TriVersa NanoMate nanoelectrospray source (Advion BioSciences, Ithaca, NY). HPLC was conducted with the UltimatePlus nano-HPLC system using a PepMap C18 column (2 μm, 100 Å, 75 μm × 15 cm, Dionex). After 15 min of desalting with the solvent A (2% acetonitrile, 0.1% formic acid in water), a gradient elution was performed at 300 nl/min and increasing solvent B (80% acetonitrile, 0.1% formic acid in water) from 0–35% in 40 min, 35–50% in 5 min, 50–90% in 5 min, and then held at 90% B for 5 min. The 10 most intense ions that have a charge state between +2 to +4, determined from an initial survey scan from 300 to 1800 m/z, were selected for fragmentation (MS/MS). MS/MS was performed using an isolation width of 2 m/z and a normalized collision energy of 35%.
The MS/MS data were searched against the M. smegmatis protein database (NCBI) with carbamidomethylation (Cys), deamidation (Asn and Gln), pyro-Glu (Glu and Gln), oxidation (Met), and acetylation (Lys) as variable modifications using the Mascot search engine (Matrix Science). Up to two missed trypsin cleavage sites were allowed. The program-identified peptides were verified manually.
Time-dependent Inactivation of FadD33 and FadD33-K260A
20 μm FadD33 or 20 μm FadD33-K260A were incubated with 1 mm cAMP and 100 μm acetyl-CoA with 5 μm MsPat in 50 mm HEPES, 100 mm NaCl, pH 7.5, at 37 °C. Aliquots of the reaction mixture were withdrawn every hour. The residual activity of FadD33 and FadD33-K260A were measured as described above. Control experiments were performed when MsPat or acetyl-CoA were not included.
Time-dependent Reactivation of Acetylated FadD33 and FadD33-K260A
FadD33 enzymes were acetylated by MsPat as described above. The acetylated FadD33 enzymes were then purified by Ni-NTA chromatography and used in the deacetylation assay. A typical reaction mixture contained 10 μm acetylated FadD33, 2 mm NAD+, and 2 μm MSMEG_5175 (DAc1) or MSMEG_4620 (DAc2). The FadD33 and mutant activities were measured every hour. Control experiments were performed without adding NAD+ or the deacetylase.
RESULTS
Cloning, Expression, and Purification of FadD33
In a previous report, FadD33 expressed in E. coli was claimed to be present predominantly in inclusion bodies (20). To avoid this problem, FadD33 was expressed in M. smegmatis. PCR amplification of the FadD33 gene yielded a single fragment of the expected length (1575 bp). After DNA verification by sequencing, homologous expression in M. smegmatis yielded satisfactory quantities of soluble FadD33; 10 mg per liter of culture. As judged by SDS-PAGE (data not shown), FadD33 was purified to homogeneity after Ni2+-column affinity chromatography with an apparent molecular mass of ∼57,000 Da. The observed FadD33 molecular mass is consistent with the predicted molecular weight of 57,400 calculated from the amino acid sequence, including the His6 tag.
Cloning, Expression, and Purification of MbtL
PCR amplification of the MbtL gene yielded a single fragment of the expected length (270 bp). After sequence verification, heterologous expression in E. coli yielded satisfactory quantities of soluble apo-MbtL; 12.5 mg per liter of culture. As judged by SDS-PAGE (data not shown), apo-MbtL was 95% homogeneous, after Ni2+-column affinity chromatography and Superdex-200 gel filtration with an apparent molecular mass of ∼11,000 Da, consistent with the molecular weight of 11670 calculated from the amino acid sequence including the His6 tag. For the subsequent kinetic analysis with FadD33, apo-MbtL is required to be phosphopantetheinylated (holo-MbtL form). Sfp, a phosphopantetheinyl transferase from B. subtillis was used to covalently transfer the 4′-phosphopantetheinyl group from coenzyme A onto apo-MbtL as described under “Materials and Methods.” The conversion of apo-MbtL to holo-MbtL was confirmed by Fourier transform mass spectral analysis (data not shown). Apo-MbtL with a calculated molecular mass of 11,521 Da, without the first methionine, was converted to holo-MbtL with the corresponding 11,861 Da matching the addition of the phosphopantetheine moiety (340 Da). Kinetic data revealed that the apo form of MbtL is not a substrate for FadD33, thus revealing the necessity of phosphopantetheinyl modification of MbtL for FadD33 substrate activity. In addition, holo-acyl carrier protein (holo-ACP) from the M. tuberculosis PpsA module, which is involved in phenolic glycolipid and phthiocerol dimycocerosate biosynthesis, was tested as a substrate for the FadD33 reaction. Holo-ACP was not recognized as a substrate by FadD33 (data not shown), thus revealing a specific interaction of FadD33 with its natural substrate MbtL.
Cloning, Expression, and Purification of Myokinase
Due to an interruption of commercial myokinase, E. coli myokinase was cloned. PCR amplification of the myokinase gene yielded a single fragment of the expected length (705 bp). After DNA sequence verification, heterologous expression in E. coli gave large quantities of soluble myokinase, 30 mg per liter of culture. As judged by SDS- PAGE (data not shown), myokinase was 95% homogeneous, after Ni2+-NTA column affinity chromatography with an apparent molecular mass of ∼25,000 Da, consistent with the molecular weight of 25,767 calculated from the amino acid sequence, including the His6 tag. The enzyme activity was equivalent to the commercial myokinase.
Substrate Specificity of FadD33
The enzymatic activity of FadD33 was followed by monitoring the formation of AMP as described under “Materials and Methods.” FadD33 displays typical Michaelis-Menten profiles. To investigate the substrate specificity of FadD33, the protein was incubated with different length fatty acids ranging from C6 to C16. The enzyme exhibits measurable activity with all the substrates tested (Table 2) with a strong preference for palmitic acid (C16). Apparent Km values of FadD33 for fatty acids decrease as the chain length increases from C6 to C16. According to the kcat/Km values, the enzyme prefers the fatty acids with longer length acyl chain as observed previously for the lysine acetyl Nϵ-acyltransferase (MbtK), which is the enzyme transferring the ACP bound fatty acyl chain onto the Nϵ-lysine of the mycobactin core (25). The preference for the C16 substrate indicates that FadD33 is primarily involved in the biosynthesis of the lipophilic mycobactin but raises the following question. Does FadD33 utilize smaller fatty acids or dicarboxylic acids for carboxymycobactin formation? We therefore tested several compounds: butyric acid (C4), succinic acid (C4), caproic acid (C6), and the mono-methyl caproic acid (C6), but none of the small acids were a substrate for FadD33. For all subsequent experiments, lauric acid (C12) was used instead of the preferred C16 substrate to avoid potential micelle formation at high concentrations of C16.
TABLE 2.
FadD33 wild type and FadD33-K260A mutant acyl chain length selectivity and kinetic parameters
Initial velocities were measured spectrophotometrically in 100 mm HEPES, pH 7.8, 10 mm MgCl2, and 250 mm NaCl as described under “Materials and Methods.”
| Enzyme/Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | μm−1 min−1 | |
| FadD33 wild type | |||
| Hexanoic acid (C6) | 129 ± 25 | 0.11 ± 0.01 | 0.0008 ± 0.0001 |
| Decanoic acid (C10) | 121 ± 22 | 1.5 ± 0.1 | 0.012 ± 0.004 |
| Lauric acid (C12) | 21 ± 4 | 2.4 ± 0.1 | 0.110 ± 0.003 |
| Palmitic acid (C16) | 1.9 ± 0.3 | 4.2 ± 0.1 | 2.3 ± 0.1 |
| ATP | 15 ± 1 | 1.3 ± 0.2 | 0.085 ± 0.008 |
| MbtL-holo | 106 ± 19 | 2.0 ± 0.1 | 0.018 ± 0.006 |
| FadD33-K260A | |||
| Lauric acid (C12) | 3.4 ± 0.5 | 0.51 ± 0.01 | 0.15 ± 0.01 |
Kinetic Mechanism
The kinetic mechanism of FadD33 was determined by initial velocity experiments using the coupled assay described under “Materials and Methods.” The parallel lines observed in double reciprocal plots of the initial velocity using either ATP or lauric acid (C12) as varied substrates and holo-MbtL as the fixed substrate suggest a ping-pong kinetic mechanism for FadD33 (Fig. 4, A and B). The intersecting lines observed in double-reciprocal plots of the initial velocity with varying concentrations of ATP and lauric acid at saturating concentration of holo-MbtL are indicative of the formation of a ternary E-C12-Mg2+-ATP complex, where E is free enzyme (Fig. 4C). These data were globally fitted to Equation 2 or 3, yielding Km values for ATP, lauric acid, and holo-MbtL (Table 2). Pyrophosphate (PPi) was used as a product inhibitor versus lauric acid and ATP to further probe the nature of the ping-pong kinetic mechanism. When pyrophosphate was used versus lauric acid at non-saturating concentration of ATP, an noncompetitve inhibition pattern was observed (Fig. 5A), yielding to inhibition constants of 50 ± 18 μm and 91 ± 46 μm for Kis and Kii, respectively. However, when pyrophosphate was used versus lauric acid, an uncompetitve inhibition pattern was observed at a saturating concentration of ATP (Fig. 5B), yielding a Kii value of 55 ± 6 μm. These product inhibition patterns suggest that lauric acid binds to FadD33 prior to ATP and that pyrophosphate is released after both lauric acid and ATP have bound to the enzyme, but before holo-MbtL binds. The data collected from initial velocity and product inhibition experiments, are characteristic of a Bi Uni Uni Bi ping-pong kinetic mechanism for FadD33. As proposed in Fig. 6, lauric acid first binds to the free enzyme followed by the binding of Mg2+-ATP to form an E-C12-Mg2+-ATP ternary complex, where E is free enzyme. The next step is the formation of the lauryl-adenylate with subsequent release of pyrophosphate from the enzyme. Holo-MbtL binds to the enzyme lauryl-adenylate and forms a thioester bond between C12 and the reactive terminal thiol group of the phosphopantetheine group of MbtL. Finally, the two newly formed products, AMP and lauryl-MbtL, are released from the enzyme. Unfortunately, due to the type of coupling enzyme assay used, it was not possible to establish with certainty the order of products released from the enzyme.
FIGURE 4.
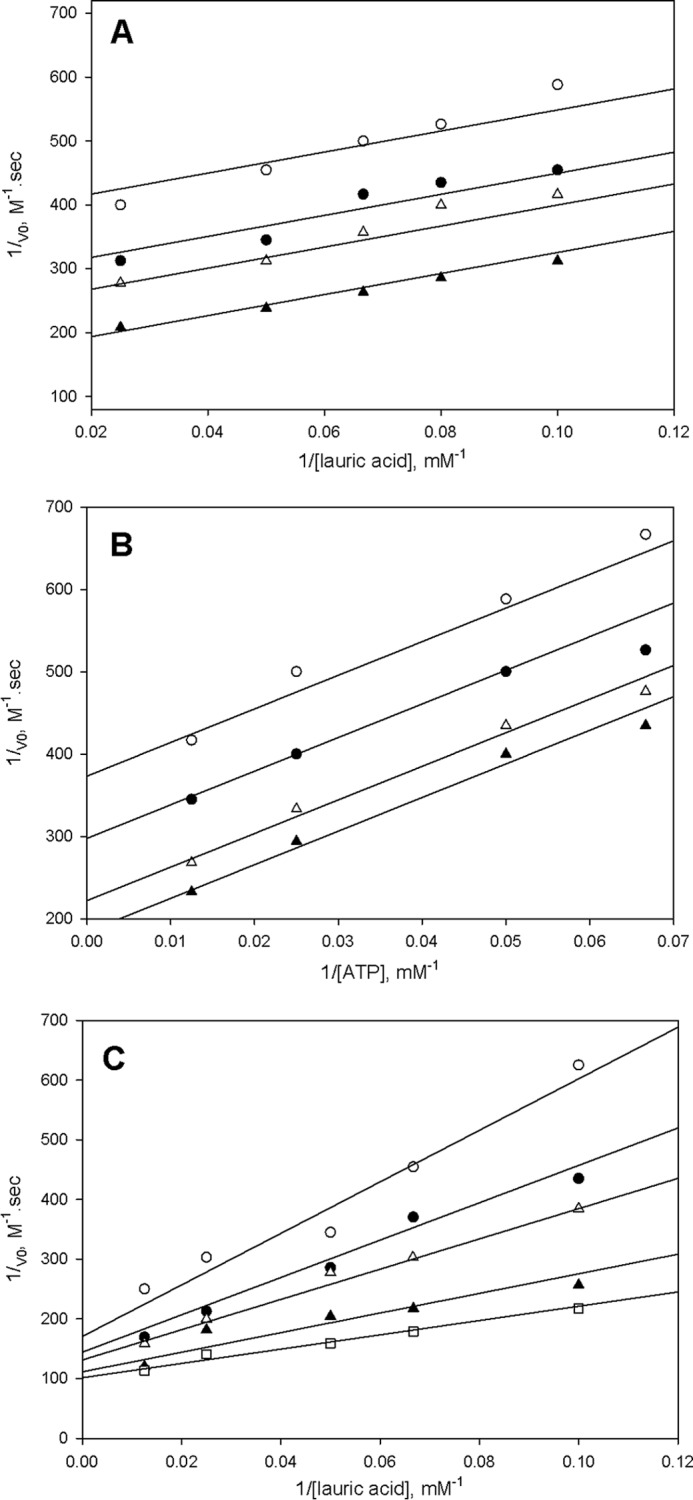
Kinetic mechanism of FadD33. A, assays were performed at varying concentrations of lauric acid (10–45 μm), a saturated concentration of ATP (400 μm), and fixed concentrations of MbtL (40 (○), 60 (●), 80 (▵), and 160 (▴) μm). B, assays were performed at varying concentrations of ATP (15–80 μm), a saturated concentration of lauric acid (100 μm), and fixed concentrations of MbtL (60 (○), 80 (●), 120 (▵), and 160 (▴) μm). C, assays were performed at varying concentrations of lauric acid (10–80 μm), a saturated concentration of MbtL (500 μm), and fixed concentrations of ATP (10 (○), 15 (●), 20 (▵), 40 (▴), and 80 (□) μm). Activity assays were measured spectrophotometrically in 100 mm HEPES, pH 7.8, 10 mm MgCl2, and 250 mm NaCl as described under “Materials and Methods.” The lines are fits of the data to Equations 2 for A and B, and Equation 3 for C.
FIGURE 5.
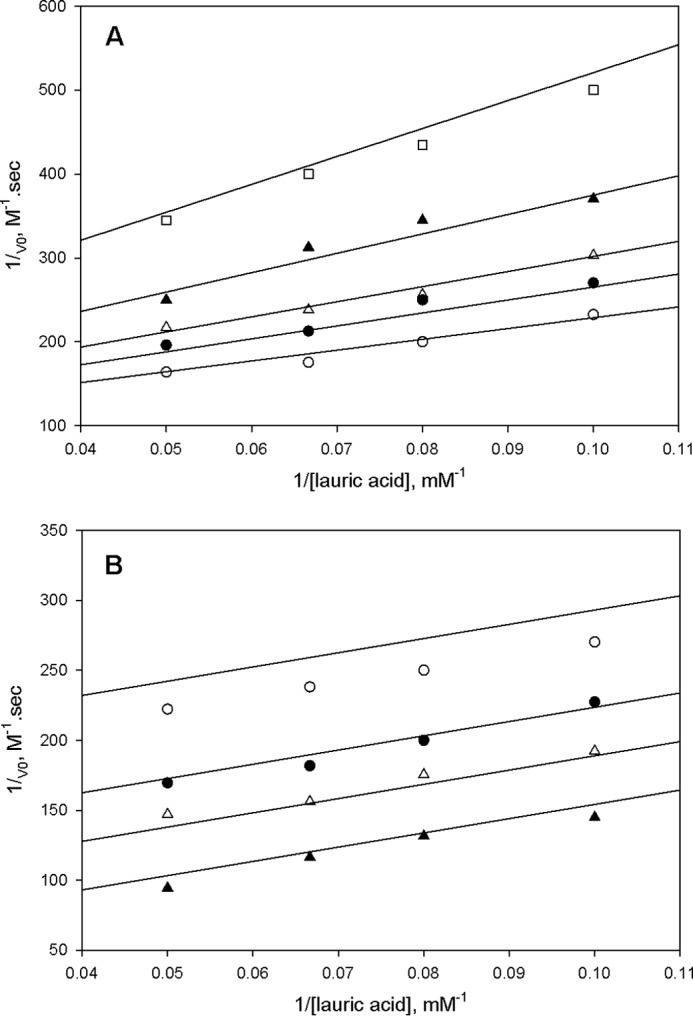
Product inhibition studies of FadD33 by pyrophosphate at 30 μm ATP (A) and (B) 400 μm ATP. In A, the following assays were performed: 0 (○), 10 (●), 20 (▵), 40 (▴), and 80 (□) μm pyrophosphate. In B, the following assays were performed: 0 (○), 20 (●), 40 (▵), and 80 (▴) μm pyrophosphate. Activity assays were measured spectrophotometrically in 100 mm HEPES, pH 7.8, 10 mm MgCl2, and 250 mm NaCl as described under “Materials and Methods.” The lines are fits of the data to Equations 5 and 4 for A and B, respectively.
FIGURE 6.
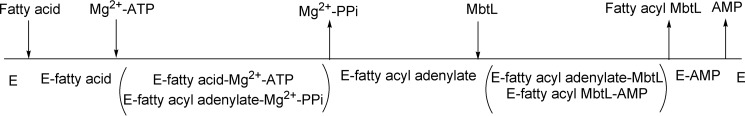
Proposed Bi Uni Uni Bi ping-pong kinetic mechanism for FadD33. E, free enzyme.
Bi-substrate Analog Inhibition Experiments
The bi-substrate, hydrolytically stable adenylate analog, C16-AMS (Fig. 3) was synthesized as described previously (26) and tested as an inhibitor of FadD33 because it is a structural analog of the C12-adenylate. The 5′-O-(N-(palmitoyl) sulfamoyl)adenosine acts as a competitive inhibitor versus both ATP and lauric acid with apparent Ki values of 2.8 ± 0.4 μm and 2.1 ± 0.3 μm, respectively (Fig. 7, A and B). These data are consistent with the Bi Uni Uni Bi ping-pong kinetic mechanism illustrated in Fig. 6.
FIGURE 7.
Inhibition studies of FadD33 with 5′-O-(N-(palmitoyl) sulfamoyl)adenosine as inhibitor. In A, assays were performed at varying concentrations: 0 (▴), 2 (▵), 4 (●), and 6 (○) μm of 5′-O-(N-(palmitoyl) sulfamoyl)adenosine; 40 μm lauric acid; and varying concentrations of ATP (30–80 μm). In B, assays were performed at varying concentrations: 0 (□), 2 (▴), 4 (▵), 6 (●), and 8 (○) μm of 5′-O-(N-(palmitoyl) sulfamoyl)adenosine; 80 μm ATP; and varying concentrations of lauric acid (15–80 μm). Activity assays were measured spectrophotometrically in 100 mm HEPES, pH 7.8, 10 mm MgCl2, 250 mm NaCl as described under “Materials and Methods.” The lines are fits of the data to Equation 6 for A and B.
Specific Acetylation of FadD33 by Protein Lysine Acetyltransferase
In addition to investigating the enzymatic mechanism of FadD33, we also looked at the possibility of fatty acid adenylate ligase regulation on the post-translational level. Recently, Pat has been identified as a cAMP sensor in mycobacteria and regulator of a metabolic enzyme ACS (12). FadD33 catalyzes a similar reaction to ACS, adenylation followed by a thioesteration (thiol ligation). Moreover, the acetylation site in ACS (Lys-617) is conserved in FadD33 (Lys-511, Fig. 8). By examining the amino acid sequence around the acetylation site in ACS, we noted the presence of a glycine residue preceding the acetylation site that likely minimizes the steric hindrance between the acetyltransferase and the protein substrate and also two nearby basic residues that likely will reduce the pKa value of the ϵ-NH2 group of Lys-511, facilitating acetylation (Fig. 8). Because this sequence pattern is highly conserved in FadD33, we believed that FadD33 may also be substrate for Pat. According to a Western blot-based acetylation assay using anti-acetyllysine antibody, FadD33 was acetylated by Pat in the presence of acetyl-CoA and cAMP, but not acetylated when either acetyl-CoA or cAMP were absent (Fig. 9, lanes 1–4). This observation strongly suggests that FadD33 is a substrate for Pat. To determine the site(s) of acetylation in FadD33, in vitro-acetylated FadD33 was separated by SDS-PAGE from Pat, and the extracted FadD33 band was analyzed by mass spectrometry. Although 80% sequence coverage was achieved for FadD33 (data not shown), the tryptic peptide (TSSGKLR) containing the predicted acetylation site lysine 511 was not identified in the mass spectrometric analysis, possibly due to glycosylation and/or phosphorylation on the serine/threonine residues within that particular peptide. Despite the lack of mass spectrometric evidence, we observed a dramatic decrease in acetylation in the Western blot-based assay when lysine 511 was mutated to alanine (Fig. 9, lane 6), suggesting Lys-511 is the major acetylation site in FadD33. The fact that the acetylation was not eliminated for the K511A mutant indicates that Lys-511 is not the only acetylation site. This is supported by the mass spectrometric analysis that showed that lysine 260 is also acetylated (Fig. 10). Interestingly, the sequence around Lys-260 shares the same pattern described above for ACS, with a glycine residue preceding the acetylation site and two basic residues in close proximity (Fig. 8). Consistently, a slight decrease in acetylation was observed for the FadD33-K260A mutant in comparison with wild type FadD33 (Fig. 9), and acetylation was eliminated for the FadD33 double mutant (K260A/K511A) (Fig. 9, lane 7). Therefore, Pat acetylates both Lys-260 and Lys-511 in FadD33 with Lys-511 being the major acetylation site.
FIGURE 8.

Partial alignments of FadD33 proteins from M. smegmatis (M. smeg; MSMEG_2131) and M. tuberculosis (M. tb; Rv1345) versus M. tuberculosis ACS (M. tb ACS; Rv3667). M. smegmatis FadD33-acetylated lysines, Lys-260 and Lys-511, are indicated in boldface, and conserved residues flanking acetylated sites are underlined.
FIGURE 9.
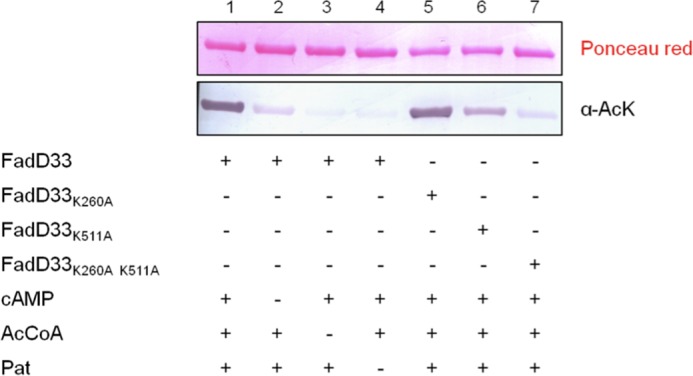
Acetylation pattern of FadD33 and FadD33 mutants by Pat. FadD33 were analyzed by Western blot (bottom panel) with acetyl-lysine antibody (α-AcK), and the total protein was determined by Ponceau red (top panel). AcCoA, acetyl-CoA.
FIGURE 10.
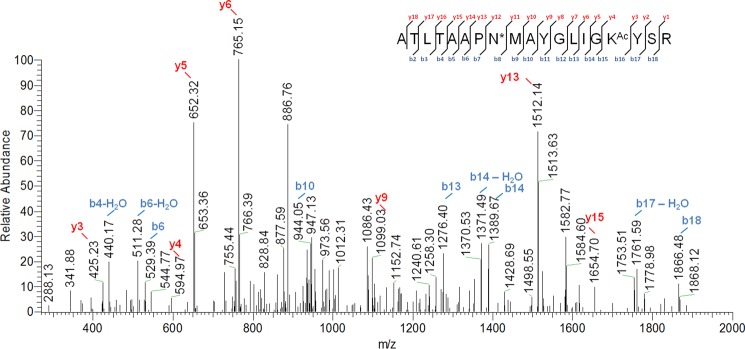
Identification of FadD33 Lys-260 acetylation site by MS/MS. Shown is an MS/MS spectrum of a triply charged tryptic peptide from FadD33 (YGLIGKAcYSRR) bearing an acetylated lysine. The acetylated lysine is indicated as KAc. Most of the major fragmentation ions in the mass spectrum match the predicted b or y ions.
Effect of Acetylation on FadD33 Activity
To identify the functional consequences of acetylation on Lys-260 and Lys-511, we measured the activity and kinetic parameters of the FadD33 mutants. Comparison of the FadD33-K260A mutant versus the wild type FadD33 revealed only a 4-fold reduction of the kcat (Table 2), suggesting that lysine 260 is not required for catalysis. However, the activity of the FadD33-K511A mutant is completely abolished. It was shown in the FadD13 structure that the equivalent residue to FadD33 Lys-511, FadD13 Lys-487 faces the catalytic pocket in the acyl-adenylate-forming conformation (27) and that the FadD13-K487A mutant exhibits a 30-fold reduction of the kcat (28). Acetylation on Lys-511 is therefore likely to have functional consequences on FadD33 activity. To investigate the effects of acetylation, the activity of FadD33 was monitored overtime in the presence of Pat. Wild type FadD33 and FadD33-K260A gradually lost activity during Pat-mediated acetylation (Fig. 11A), and the loss of FadD33 activity is dependent on both Pat and acetyl-CoA. Two known deacetylases, DAc1 and DAc2, are present in the M. smegmatis genome and were tested using acetylated FadD33. In Fig. 11B, gradual reactivation of FadD33 activity is observed only with DAc1 and NAD+. In contrast, DAc2 was unable to reverse Pat acetylation. FadD33-K260A activity could also be rescued by DAc1 deacetylase. These results suggest that the catalytic activity of FadD33 is regulated by Pat and DAc1 via the reversible acetylation and deacetylation of Lys-511. Acetylation of Lys-260 is minor and irrelevant for this regulation.
FIGURE 11.
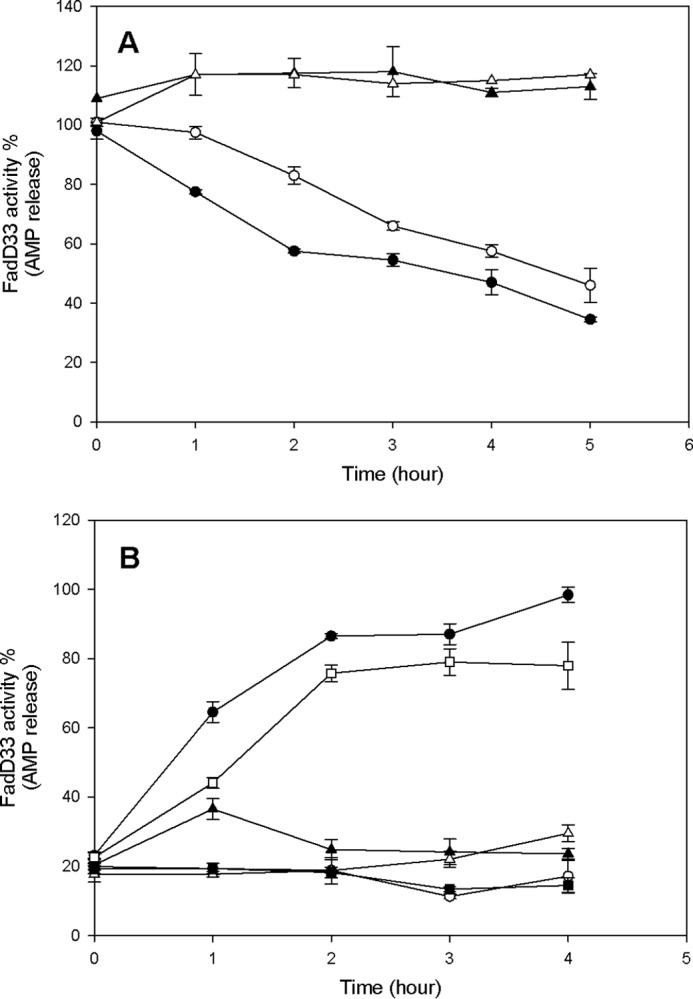
Effect of acetylation and deacetylation on FadD33 and FadD33-K260A activities. A, time-dependent inactivation of FadD33 and FadD33-K260A by acetylation. Enzymes activities were monitored at different time intervals with the following components: ● (FadD33), 1 mm cAMP, 5 μm Pat, and 100 μm acetyl-CoA; ○ (FadD33-K260A), 1 mm cAMP, 10 μm Pat, and 100 μm acetyl-CoA; ▴ (FadD33), 1 mm cAMP and 100 μm acetyl-CoA; ▵ (FadD33), 1 mm cAMP and 5 μm Pat. B, time-dependent reactivation of acetylated FadD33 and FadD33-K260A by deacetylation. After acetylation by Pat, FadD33, or mutant were then incubated with the following components: ● (Ac-FadD33), 2 mm NAD+, 2 μm DAc1; ○ (Ac-FadD33), 2 mm NAD+; ▴ (Ac-FadD33), 2 μm DAc1; ▵ (Ac-FadD33), 2 μm DAc2, 2 mm NAD+; □ (Ac-FadD33-K260A), 2 mm NAD+, 2 μm DAc1; and ■ (Ac-FadD33-K260A), 2 mm NAD+. Enzyme activities were monitored spectrophotometrically in 100 mm HEPES, pH 7.8, 10 mm MgCl2, and 250 mm NaCl as described under “Materials and Methods.”
DISCUSSION
Siderophores play an important role in the survival of mycobacteria by chelating iron from the environment or the host and are absolutely required for virulence in pathogenic mycobacterial strains (29). The enzymes involved in mycobactin biosynthesis have significant potential as drug targets for anti-mycobacterial treatment. In this study, we have performed a detailed kinetic and mechanistic characterization of M. smegmatis FadD33 and have shown that FadD33 activity can be regulated by reversible post-translational acetylation.
Steady-state Kinetic Mechanism
Substrate specificity experiments revealed that FadD33 is a fatty acid AMP ligase with a strong substrate preference for palmitic acid, likely arising from the type II fatty acid biosynthesis. This selectivity correlates well with the substrate specificity found for the lysine N-acetyltransferase (MbtK), which also prefers long acyl chain-CoA substrates (25). Lysine N-acetyltransferase was shown in vitro to transfer MbtL-tethered long chain fatty acid onto the mycobactin core lysine to form the lipophilic mycobactin (20). The fact that FadD33 is unable to activate short fatty acid or dicarboxylic substrates suggest that an unidentified short chain fatty acid AMP ligase may be required for the formation of the soluble carboxymycobactin. Alternatively, the lysine N-acetyltransferase could directly react with a short fatty acid-CoA. Krithika et al. (20) demonstrated that MbtK could transfer a lauroyl-CoA substrate even though MbtK has 50-fold higher activity with lauroyl-ACP substrate. Interestingly, structural studies of carboxymycobactin indicate that the short alkyl chain length not only varies by even increments, as observed for mycobactin variants, but also varies by odd increments (30, 31). An alternative hypothesis for dicarboxylic chain formation of the carboxymycobactin was postulated by Ratledge (32) as simply arising from the cleavage of the long mycobactin alkyl chain by an unknown enzyme, which could result in the formation of odd increments of the alkyl chain and oxidation to generate the carboxylic acid moiety.
The results of initial velocity, product, and bi-substrate analog inhibition experiments clearly demonstrated that FadD33 proceeds via a Bi Uni Uni Bi ping-pong kinetic mechanism (Fig. 6). Fatty acid AMP ligase catalyzes a two-step reaction: in the adenylation reaction, the fatty acid binds the free enzyme prior to Mg2+-ATP to form an E-fatty acid-Mg2+-ATP ternary complex, followed by pyrophosphate release and formation of fatty acid adenylate, and in the ligation reaction, ACP binds the enzyme to form a thioester bond with the fatty acid, followed by a sequential product release postulated to be fatty acid tethered-ACP prior to AMP. The order of product release could not be determined because the coupling system used to monitor the reaction was relying on AMP formation. Based on previous work on ATP-dependent acyl and aryl thio ligases (33), we propose fatty acid-tethered ACP release prior to AMP release.
Reversible Post-translational Modification of FadD33
Experiments probing FadD33 post-translational modification and regulation have led to the discovery of a rapid inactivation of FadD33 activity via lysine acetylation by Pat acetyltransferase. Two acetylation sites have been identified; lysine 260 and lysine 511, both flanked by a glycine residue and two basic residues, respectively, before and after the acetylation site. Lys-260 is a secondary acetylation site because the FadD33-K260A mutant was active but could be catalytically inactivated by Pat. On the other hand, Lys-511 is required for catalysis and is likely to be involved in the first adenylation half reaction because the corresponding residue in FadD13 was shown to interact with ATP in the FadD13 crystal structure (27). Consistent with this, the K511A mutant form of FadD33 was inactive. Therefore, the inactivation of FadD33 is caused by the acetylation of Lys-511, and FadD33 inactivation was shown to be a reversible event because rapid deacetylation and reactivation occurs exclusively with the DAc1 deacetylase in the presence of NAD+. During the preparation of this manuscript, Nambi et al. (34) demonstrated that other mycobacterial FadDs can be inactivated by Pat acetylation at position equivalent to lysine Lys-511 in ACS and reactivated by Rv1151c, the DAc1 homolog in M. tuberculosis. They also predicted that FadD33 should not be post-translationally acetylated. Inactivation of FadD33 by Pat will reduce the production of mycobactin when the level of cAMP is high in the bacterial cytoplasm. This inactivation may lead to the accumulation of the non-acylated mycobactin core, which could be either degraded by a siderophore esterase, even though none has yet been identified in M. tuberculosis, or be used as intracellular ion storage molecule. Recently, it was shown that the host increases the macrophage vacuole concentration of zinc and copper, as a novel immune response, to “poison” the bacteria with metal ions (35, 36). It is reasonable to speculate that M. tuberculosis, in addition to specific metal efflux pumps, is using the mycobactin or the mycobactin core to neutralize excess copper and zinc as seen in Pseudomonas aeruginosa (37) and pathogenic strains of E. coli (38). To consolidate this hypothesis, Wagner et al. (39) have measured zinc content in bacteria within the infected macrophage vacuole at 1 and 24 h post infection with either M. tuberculosis H37Rv or M. tuberculosis H37Rv siderophore knock-out strains. After infection with M. tuberculosis H37Rv, the zinc content inside the bacteria within the vacuole was only slightly higher than found in the non-infected macrophage. However, the zinc level inside of the siderophore knock-out M. tuberculosis H37Rv within the vacuole was significantly higher, showing that mycobactin plays a role in intracellular zinc regulation.
In summary, we have revealed the detailed enzymatic mechanism of FadD33, which is applicable to any mycobacterial FadDs and will serve as a platform for future FadD inhibitor design. Furthermore, we have demonstrated for the first time that mycobactin FadD33 activity can be reversibly regulated by post-translational acetylation by Pat in a cAMP-dependent manner and reactivated by DAc1 deacetylation. This finding highlights the critical role of the Pat cAMP-binding protein as a central regulatory switch involved in the regulation of central carbon metabolism and the production of the mycobactin virulence factor.
Acknowledgments
We thank Prof. Courtney Aldrich for providing the C16-AMS bi-substrate analog. We are grateful to Jennifer T. Aguilan (Albert Einstein College of Medicine) for assistance in mass spectrometry.
This work was supported by National Institutes of Health Grant AI60899 (to J. S. B.).
- Pat
- protein acetyltransferase
- ACS
- acetyl-CoA synthase
- Ni-NTA
- nickel-nitrilotriacetic acid
- MsPat
- M. smegmatis Pat
- C16-AMS
- 5′-O-(N-(palmitoic) sulfamoyl)adenosine
- ACP
- acyl carrier protein
- CoASH
- coenzyme A
- E
- free enzyme.
REFERENCES
- 1. Deretic V. (2005) Autophagy in innate and adaptive immunity. Trends Immunol. 26, 523–528 [DOI] [PubMed] [Google Scholar]
- 2. Koul A., Herget T., Klebl B., Ullrich A. (2004) Interplay between mycobacteria and host signalling pathways. Nat. Rev. Microbiol. 2, 189–202 [DOI] [PubMed] [Google Scholar]
- 3. Ramakrishnan L. (2012) Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 12, 352–366 [DOI] [PubMed] [Google Scholar]
- 4. Agarwal N., Lamichhane G., Gupta R., Nolan S., Bishai W. R. (2009) Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature 460, 98–102 [DOI] [PubMed] [Google Scholar]
- 5. Kalamidas S. A., Kuehnel M. P., Peyron P., Rybin V., Rauch S., Kotoulas O. B., Houslay M., Hemmings B. A., Gutierrez M. G., Anes E., Griffiths G. (2006) cAMP synthesis and degradation by phagosomes regulate actin assembly and fusion events: consequences for mycobacteria. J. Cell Sci. 119, 3686–3694 [DOI] [PubMed] [Google Scholar]
- 6. Lowrie D. B., Aber V. R., Jackett P. S. (1979) Phagosome-lysosome fusion and cyclic adenosine 3′:5′-monophosphate in macrophages infected with Mycobacterium microti, Mycobacterium bovis BCG or Mycobacterium lepraemurium. J. Gen. Microbiol. 110, 431–441 [DOI] [PubMed] [Google Scholar]
- 7. McDonough K. A., Rodriguez A. (2012) The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat. Rev. Microbiol. 10, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shenoy A. R., Visweswariah S. S. (2006) New messages from old messengers: cAMP and mycobacteria. Trends Microbiol. 14, 543–550 [DOI] [PubMed] [Google Scholar]
- 9. Bai G., McCue L. A., McDonough K. A. (2005) Characterization of Mycobacterium tuberculosis Rv3676 (CRPMt), a cyclic AMP receptor protein-like DNA binding protein. J. Bacteriol. 187, 7795–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gazdik M. A., Bai G., Wu Y., McDonough K. A. (2009) Rv1675c (cmr) regulates intramacrophage and cyclic AMP-induced gene expression in Mycobacterium tuberculosis-complex mycobacteria. Mol. Microbiol. 71, 434–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nambi S., Basu N., Visweswariah S. S. (2010) cAMP-regulated protein lysine acetylases in mycobacteria. J. Biol. Chem. 285, 24313–24323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu H., Hegde S. S., Blanchard J. S. (2011) Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry 50, 5883–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee H. J., Lang P. T., Fortune S. M., Sassetti C. M., Alber T. (2012) Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat. Struct. Mol. Biol. 19, 811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duckworth B. P., Nelson K. M., Aldrich C. C. (2012) Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top. Med. Chem. 12, 766–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Byrd T. F., Horwitz M. A. (1993) Regulation of transferrin receptor expression and ferritin content in human mononuclear phagocytes. Coordinate upregulation by iron transferrin and downregulation by interferon gamma. J. Clin. Invest. 91, 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cellier M. F., Courville P., Campion C. (2007) Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect. 9, 1662–1670 [DOI] [PubMed] [Google Scholar]
- 17. Luo M., Fadeev E. A., Groves J. T. (2005) Mycobactin-mediated iron acquisition within macrophages. Nat. Chem. Biol. 1, 149–153 [DOI] [PubMed] [Google Scholar]
- 18. Chavadi S. S., Stirrett K. L., Edupuganti U. R., Vergnolle O., Sadhanandan G., Marchiano E., Martin C., Qiu W. G., Soll C. E., Quadri L. E. (2011) Mutational and phylogenetic analyses of the mycobacterial mbt gene cluster. J. Bacteriol. 193, 5905–5913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quadri L. E., Sello J., Keating T. A., Weinreb P. H., Walsh C. T. (1998) Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 5, 631–645 [DOI] [PubMed] [Google Scholar]
- 20. Krithika R., Marathe U., Saxena P., Ansari M. Z., Mohanty D., Gokhale R. S. (2006) A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2069–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lambalot R. H., Gehring A. M., Flugel R. S., Zuber P., LaCelle M., Marahiel M. A., Reid R., Khosla C., Walsh C. T. (1996) A new enzyme superfamily — the phosphopantetheinyl transferases. Chem. Biol. 3, 923–936 [DOI] [PubMed] [Google Scholar]
- 22. Yin J., Lin A. J., Golan D. E., Walsh C. T. (2006) Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 1, 280–285 [DOI] [PubMed] [Google Scholar]
- 23. Pfleiderer G., Kreiling A., Wieland T. (1960) [On pantothenic acid synthetase from E. coli. II. Quantitative enzymatic microdetermination of pantoyl acid, β-alanine and pantothenic acid]. Biochem. Z 333, 308–310 [PubMed] [Google Scholar]
- 24. Zheng R., Blanchard J. S. (2001) Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry 40, 12904–12912 [DOI] [PubMed] [Google Scholar]
- 25. Frankel B. A., Blanchard J. S. (2008) Mechanistic analysis of Mycobacterium tuberculosis Rv1347c, a lysine Nϵ-acyltransferase involved in mycobactin biosynthesis. Arch. Biochem. Biophys. 477, 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Somu R. V., Boshoff H., Qiao C., Bennett E. M., Barry C. E., 3rd, Aldrich C. C. (2006) Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 49, 31–34 [DOI] [PubMed] [Google Scholar]
- 27. Andersson C. S., Lundgren C. A., Magnúsdóttir A., Ge C., Wieslander A., Martinez Molina D., Högbom M. (2012) The Mycobacterium tuberculosis very-long-chain fatty acyl-CoA synthetase: structural basis for housing lipid substrates longer than the enzyme. Structure 20, 1062–1070 [DOI] [PubMed] [Google Scholar]
- 28. Goyal A., Verma P., Anandhakrishnan M., Gokhale R. S., Sankaranarayanan R. (2012) Molecular basis of the functional divergence of fatty acyl-AMP ligase biosynthetic enzymes of Mycobacterium tuberculosis. J. Mol. Biol. 416, 221–238 [DOI] [PubMed] [Google Scholar]
- 29. De Voss J. J., Rutter K., Schroeder B. G., Su H., Zhu Y., Barry C. E. (2000) The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A. 97, 1252–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gobin J., Moore C. H., Reeve J. R., Jr., Wong D. K., Gibson B. W., Horwitz M. A. (1995) Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. U.S.A. 92, 5189–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lane S., Marshall P., Upton R., Ratledge C. (1998) Isolation and characterization of carboxymycobactins as the second extracellular siderophores in Mycobacterium smegmatis. Biometals 11, 13–20 [Google Scholar]
- 32. Ratledge C. (2004) Iron, mycobacteria and tuberculosis. Tuberculosis 84, 110–130 [DOI] [PubMed] [Google Scholar]
- 33. Kim Y. S., Kang S.W. (1994) Steady-state kinetics of malonyl-CoA synthetase from Bradyrhizobium japonicum and evidence for malonyl-AMP formation in the reaction. Biochem. J. 297, 327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nambi S., Gupta K., Bhattacharyya M., Ramakrishnan P., Ravikumar V., Siddiqui N., Thomas A. T., Visweswariah S. S. (2013) Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J. Biol. Chem. 288, 14114–14124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Botella H., Stadthagen G., Lugo-Villarino G., de Chastellier C., Neyrolles O. (2012) Metallobiology of host-pathogen interactions: an intoxicating new insight. Trends Microbiol. 20, 106–112 [DOI] [PubMed] [Google Scholar]
- 36. Rowland J. L., Niederweis M. (2012) Resistance mechanisms of Mycobacterium tuberculosis against phagosomal copper overload. Tuberculosis 92, 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brandel J., Humbert N., Elhabiri M., Schalk I. J., Mislin G. L., Albrecht-Gary A. M. (2012) Pyochelin, a siderophore of Pseudomonas aeruginosa: Physicochemical characterization of the iron(iii), copper(ii) and zinc(ii) complexes. Dalton Trans. 41, 2820–2834 [DOI] [PubMed] [Google Scholar]
- 38. Chaturvedi K. S., Hung C. S., Crowley J. R., Stapleton A. E., Henderson J. P. (2012) The siderophore yersiniabactin binds copper to protect pathogens during infection. Nat. Chem. Biol. 8, 731–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wagner D., Maser J., Lai B., Cai Z., Barry C. E., 3rd, Höner Zu Bentrup K., Russell D. G., Bermudez L. E. (2005) Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J. Immunol. 174, 1491–1500 [DOI] [PubMed] [Google Scholar]