

Background: Itk is considered an important target for anti-inflammatory drug discovery, particularly asthma.
Results: We have designed an irreversible covalent inhibitor of the kinase by targeting Cys-442 in the ATP pocket.
Conclusion: We have produced a potent, selective inhibitor with a long duration of action.
Significance: We describe a novel drug discovery strategy with specific design features for inhaled delivery.
Keywords: Asthma, Drug Discovery, Medicinal Chemistry, Nonreceptor Tyrosine Kinase, T Cell, IL-2-inducible Tyrosine Kinase, Kinase Inhibitor
Abstract
IL-2-inducible tyrosine kinase (Itk) plays a key role in antigen receptor signaling in T cells and is considered an important target for anti-inflammatory drug discovery. In order to generate inhibitors with the necessary potency and selectivity, a compound that targeted cysteine 442 in the ATP binding pocket and with an envisaged irreversible mode of action was designed. We incorporated a high degree of molecular recognition and specific design features making the compound suitable for inhaled delivery. This study confirms the irreversible covalent binding of the inhibitor to the kinase by x-ray crystallography and enzymology while demonstrating potency, selectivity, and prolonged duration of action in in vitro biological assays. The biosynthetic turnover of the kinase was also examined as a critical factor when designing irreversible inhibitors for extended duration of action. The exemplified Itk inhibitor demonstrated inhibition of both TH1 and TH2 cytokines, was additive with fluticasone propionate, and inhibited cytokine release from human lung fragments. Finally, we describe an in vivo pharmacodynamic assay that allows rapid preclinical development without animal efficacy models.
Introduction
IL-2-inducible tyrosine kinase (Itk)4 is a member of the Tec family of non-receptor tyrosine kinases that includes Tec, Itk, Btk, Txk/Rlk, and Bmx/Etk. These kinases are mainly expressed in hematopoietic cells and act downstream of Src and Syk kinases in antigen receptor complexes in T cells, B cells, and mast cells. They play an important role in signal transduction from antigen receptors by activating PLCγ-1, which cleaves phosphatidylinositol 4,5-bisphosphate into the second messengers inositol 1,4,5-trisphosphate and diacylglycerol, which are critical for cellular activation in response to antigen. Itk is expressed in T cells and mast cells, and studies in knock-out mice (Itk−/−) have shown that it plays an important role in the development and function of T cells (1). Itk has been reported to play a selective role in the function of TH2 cells (2–7), suggesting that Itk inhibitors may be useful therapeutics for the treatment of allergic inflammatory diseases such as asthma. Indeed, Itk−/− mice showed greatly attenuated symptoms in models of allergic asthma (6–9), and these findings have prompted many research laboratories to initiate drug discovery campaigns relating to the modification of Itk action, although no drugs have, as yet, reached the clinic. A number of chemotypes have been described from screening and structure-based design approaches, but there have been significant challenges in achieving the desired potency and selectivity against other kinases, in addition to a failure to achieve acceptable activity in whole cells (10–15). Although a selective reduction in TH2 responses has been widely reported in Itk−/− studies (2–7), attenuation of TH1 responses is also seen under certain experimental conditions (3, 16), and the functions of CD8 (17) and NKT cells (18–19) are also impaired. Therefore, we have chosen to develop an inhaled Itk inhibitor to limit the immunosuppression risk associated with systemic T cell inhibition. Recently, there have been attempts to enhance the potency and selectivity of kinase inhibitors by developing electrophilic inhibitors that irreversibly target a nucleophilic cysteine residue located in the ATP pocket. Cysteines in this specific position within the ATP pocket are rare and are found in only 11 kinases, including Itk. Irreversible inhibitors targeting Cys-773 in the EGFR have been developed (20) and are in clinical trials (21), and a similar approach has been used to target Cys-481 in Btk (22). In an attempt to develop Itk inhibitors with acceptable properties for the treatment of allergic asthma by inhaled delivery, we describe the discovery and biological characterization of a novel irreversible inhibitor targeting Cys-442.
EXPERIMENTAL PROCEDURES
Chemicals and Biological Reagents
All compounds were synthesized in house as described in the supplemental material. All other reagents are described in the supplemental material.
Irreversible Inhibition
See Scheme 1 for a description of time-dependent irreversible inhibition.
SCHEME 1.
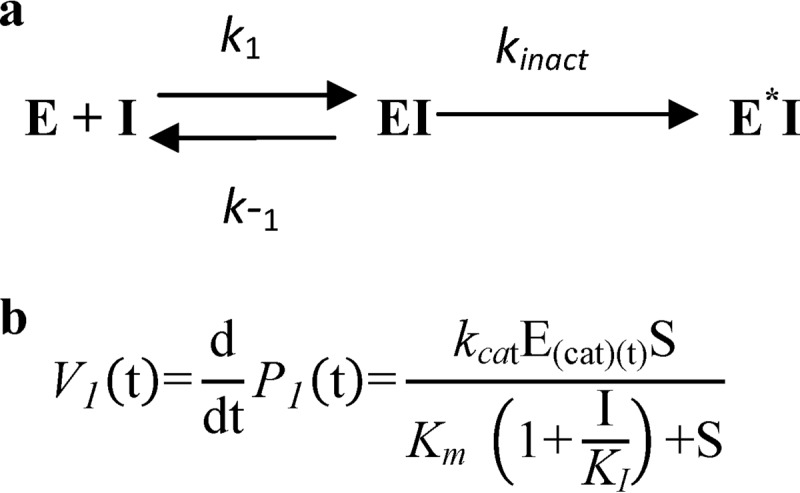
Time-dependent irreversible inhibition. a, in addition to competitive inhibition, irreversible inhibition also involves a reaction that irreversibly transforms the inhibitor-enzyme complex into an “inactive“ form (denoted by E*I, where kinact describes the rate at which inhibitor-enzyme complex is irreversibly transformed into E*I). The parameter KI is the inhibitor concentration that results in half the maximal rate of inactivation (23). b, the velocity of product formation approximated for irreversible inhibition is denoted, where S represents substrate concentration, and I is inhibitor concentration. Due to the irreversible formation of E*I, the functional catalytic enzyme concentration E(cat)(t) decreases in time, resulting in a time-dependent maximal velocity Vmax(t) = kcat·E(cat)(t).
Assessing Initial Rates of Binding of Compounds to Itk Using the Invitrogen LanthaScreen Kinase Binding Assay
This assay is based on the binding and displacement of Alexa-fluor 647-labeled ATP-competitive kinase inhibitor tracer 236. Binding of the tracer is detected using a europium-labeled anti-tag antibody that binds to the purified recombinant tagged Itk protein. Binding of the tracer 236 is reversible and rapid, and simultaneous binding of the tracer and antibody results in fluorescence resonance energy transfer (FRET). Displacement of the tracer upon inhibitor binding results in a decrease of FRET. The assay can be read continuously, enabling rates of binding (and displacement) to be monitored. The reagents were all purchased from Invitrogen, and full details are given in the supplemental material.
Assessing the Reversibility of Compound Binding Itk through Rapid Dilution into an Excess of Competing Ligand (“Jump-dilution”)
This method is necessary to complement the data generated in the kinetic binding assay, because the two-step time-dependent binding profiles could also be consistent with slow binding reversible inhibition. The protocol is identical to the kinetic method, with the modification described in the supplemental material. Reactivity and reversibility of binding of compounds to glutathione (GSH) was determined using a modification of the GSH-GloTM assay (Promega) as described in the supplemental material.
Washout Assay in Peripheral Blood Mononuclear Cells (PBMCs)
Blood was obtained by venipuncture from human volunteers into heparin (10 units/ml). All donors provided written informed consent for use of their samples, and the collection and use of the samples received Institutional Ethics Committee approval. PBMCs were purified from human volunteers and incubated for 1 h with varying concentrations of drug or DMSO as control. The cells were washed and then further incubated for periods between 2 and 19 h prior to stimulation with anti-CD3/CD28-coated beads for 2 h. Media from the cell supernatants were collected, and IL-2 production was quantified using a Meso Scale Discovery (MSD) assay. Detailed methods are described in the supplemental material.
Rat Pharmacodynamic (PD) Model
All animal procedures were reviewed and approved by the GlaxoSmithKline Animal Care and Use Committee and were performed in accredited facilities in accordance with institutional guidelines and the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council). Male Wistar Han rats, 235–255 g (Charles River), were used in all experiments. Crystalline compound 12 (difumarate salt) was particle size-reduced by micronization to give a particle size of ∼1.6 μm and was administered as an inhaled dose to rats via an inhalation tower connected to an aerosol generator, over an exposure time of 20 min. This protocol delivered a calculated dose of 2.32 mg/kg/rat based on the amount of compound deposited on an in-line filter (24). Control animals received only air in the inhalation tower. At sacrifice, lungs were perfused free of blood and removed, and a cell suspension was prepared. T cells in this lung cell preparation were activated with anti-CD3 over 18 h, and their activation state was quantified by measuring CD25 staining on the surface of CD45+ CD4+ cells by flow cytometry. Detailed methods are given in the supplemental material.
Human Lung Parenchyma Assay
The effect of compound 12 and fluticasone propionate (FP) on cytokine release by phytohemagglutinin A (PHA)-activated fragments of human lung parenchyma was investigated as described previously (25). Human lung was obtained from the National Research Disease Interchange, with ethical approval. Briefly, fragments of lung parenchyma (1 mm3) were incubated with compound for 1 h prior to stimulation with PHA (10 μg/ml). Following a 72-h incubation, supernatants were removed and assayed for IL-2, IFNγ, and IL-17 by Meso Scale Discovery (MSD) assay.
Itk Pulse-Chase Kinetics
Jurkat cells were pulsed with [35S]methionine for 30 min, followed by chase periods of up to 24 h with unlabeled methionine. The cells were stimulated with anti-CD3/CD28-coated beads for 1 h prior to biosynthetic labeling. Stimulated cells were additionally treated with or without compound 12. Cell extracts were prepared at the end of various chase periods and immunoprecipitated with an anti-Itk monoclonal antibody prior to analysis by SDS-PAGE and fluorography. Parallel non-radiolabeled cultures were prepared, and Itk levels were determined in cell extracts by SDS-PAGE and Western blotting using the same anti-Itk monoclonal antibody. Actin staining was included as a loading control. Detailed methods are provided in the supplemental material.
Protein Expression and Purification
Itk (residues 357–620, Y512E) kinase domain protein expression and purification were carried out according to published methods (26).
Protein Crystallization and Structure Determination
Co-crystallization was attempted for the purified protein with compound 7 according to a published method (26), but this was unsuccessful. However, it was possible to grow Itk co-crystals with a tool compound from a structurally related chemical series (26) under comparable conditions. Replacement soaking of compound 7 into one of these co-crystals was carried out in a soaking buffer comprising 1 m ammonium sulfate, 20% glycerol, 0.5 mm compound 7, and 0.5% DMSO, for 3 days. X-ray diffraction data were collected from the soaked crystal at 100 K at the Diamond synchrotron (station I03) using an ADSC Q315 CCD detector. Data were processed using MOSFLM (27) and SCALA (28) (within the CCP4 programming suite (29)). The complex was solved by Fourier synthesis using Itk protein coordinates determined in house (data not shown). Refinement was carried out using REFMAC (30) and model building using COOT (31). The final Rfactor and Rfree achieved for the structure were 16.7 and 20.1%, respectively. Data and model statistics are given in supplemental Table 1, and the structure has been deposited in the Protein Data Bank with accession number 4KIO.
RESULTS
Design of Irreversible Inhibitors
Using conventional kinase inhibitor drug discovery strategies, we were able to identify a number of highly potent Itk inhibitor chemotypes; unfortunately, all suffered from a lack of kinase selectivity to varying extents. Additionally, we were unable to drive cellular potency down into the low nanomolar range, a highly desirable feature for an inhaled drug. In part, this is due to the Km,app(ATP) of Itk being low (∼5 μm); maintaining inhibitor potency in a cellular environment where ATP concentration is ∼1–2 mm is challenging for reversible ATP-competitive compounds (23). Consequently, we changed strategy to build cellular potency and selectivity by making irreversible inhibitors, using compound 1 as a starting point. This compound, a reversible Itk inhibitor, was chosen due its acceptable physicochemical properties (clogP (the logarithm of the partition coefficient of a solute between n-octanol and water, log(coctanol/cwater)) = 1.8, chromatographic logD (the logarithm of the chromatographically determined distribution of a solute between octanol and a pH 7.4 buffered aqueous solution) = 2.0, aqueous solubility >400 μm), good biochemical potency (pIC50 of 6.9 at 1 mm ATP against purified Itk), and reasonable cellular activity (pIC50 of 7 for the inhibition of IFNγ secretion in a human PBMC assay). Additionally, this compound displayed a reasonable overall kinase selectivity profile, including evidence of selectivity among the other 10 kinases bearing a non-catalytic cysteine residue at this position (>200-fold selectivity in biochemical assays versus Btk and EGFR; data not shown). We believe the ability to discriminate between these kinases through reversible molecular recognition to be a critical foundation on which to append the covalent binding moiety. Examination of x-ray structural data from related inhibitors revealed that the cyclohexanol moiety was in reasonable proximity (<5 Å) to Cys-442 at the end of the C-lobe α-helix (26). Because there is precedent that acrylamide groups can form a covalent bond with a cysteine residue when held in an appropriate position via non-covalent molecular recognition (32), we replaced the cyclohexanol by a series of acrylamide groups (compounds 2–7) that molecular modeling suggested would place the reactive electrophilic terminal carbon atom of the acrylamide in a proximal position to the cysteine sulfur.
Compounds 2–7 all bind to Itk in the enzyme assay conducted at high ATP concentration (1 mm), and all display good levels of cellular activity in the PBMC activation assay, parallel activity in both enzyme and cell-based assays being a key feature we sought to maintain throughout the lead optimization phase (Table 1).
TABLE 1.
Itk enzyme inhibition, cell potency (inhibition of CytoStim-induced IFNγ release from PBMC), and kinetic binding data for a series of analogues probing covalent interaction with Cys-442 in Itk
Stereochemistry is relative for compounds 1, 4, 5, and 6. n.d., not determined.
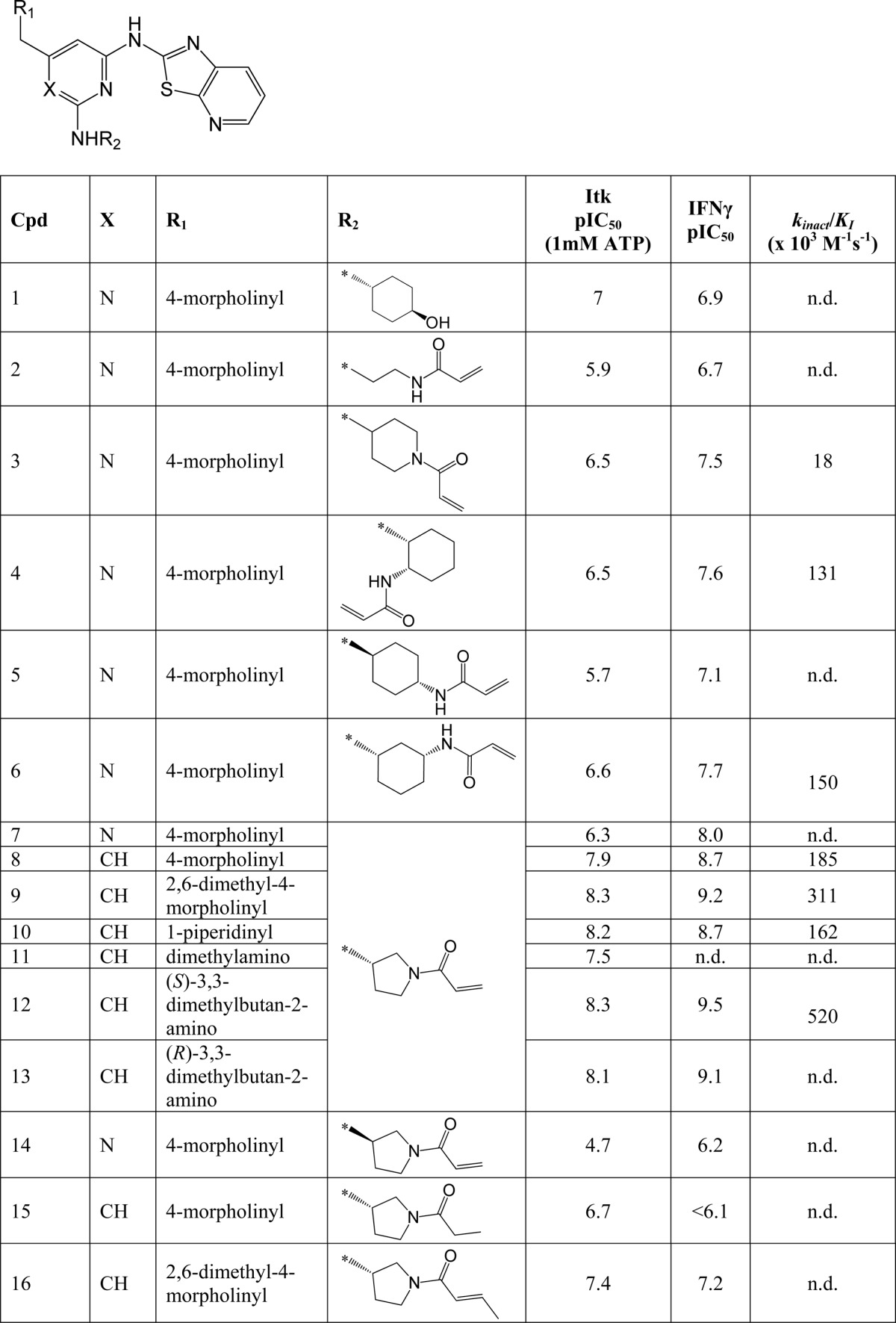
Irreversible binding of a drug to a target protein (Scheme 1a) shows a characteristic time dependence due to the irreversible formation of E*I leading to a decrease in the functional catalytic enzyme concentration (E(cat)(t)) over time. This results in a time-dependent reduction in maximal velocity (Vmax(t) = kcat·E(cat)(t); Scheme 1b). Therefore, to confirm that these compounds were indeed binding irreversibly to Itk, we developed a LanthascreenTM kinetic binding assay. We chose a direct binding assay rather than an activity assay in order to avoid complications from the lag period routinely observed in Itk activity assays (data not shown). The binding profiles for compounds 3, 4, 6, 8, 9, 10, and 12 all show an initial, rapid (<1-min) binding event, followed by a slower onset of increased levels of inhibition (representative data for compound 12 is shown in Fig. 1a). This two-step, time-dependent inhibition is consistent with either slow onset reversible inhibition (the slower phase representing the rate of enzyme isomerization) or irreversible inhibition (the slower phase representing inactivation of the enzyme). However, the non-zero y intercept of the Kitz-Wilson analysis plot in Fig. 1b is consistent with two-step irreversible binding (kinact = 0.0026 s−1, Ki = 5 nm) (33). In order to confirm further the mechanism of binding, jump-dilution studies were employed. Because rapid dilution of the E·I complex into excess competitor tracer 236 did not result in increased binding of the latter over a period of >6 h (data not shown), this is consistent with irreversible complex formation between the inhibitor and the enzyme. Furthermore, the measured pIC50 values were significantly higher than predicted under the conditions of the assay according to the Cheng-Prusoff equation (supplemental Equation 3), again consistent with time-dependent, irreversible inhibition. Although compound 5 possesses a similar acrylamide moiety, it does not show time-dependent binding, because the profile (Fig. 1c) indicates a single binding event that maintains a stable level of inhibition over time; moreover, jump-dilution studies resulted in rapid recovery of the tracer 236 binding, and the measured pIC50 value was consistent with the predicted value (data not shown). This correlated well with the initial molecular modeling, which suggested that compound 5 placed the terminal carbon of the acrylamide further away from the cysteine sulfur than the other acrylamide moieties. To confirm the binding mode and the presence of a covalent linkage for our irreversible inhibitors, an x-ray crystal structure of compound 7 complexed with the Itk kinase domain (Y512E) at 2.18 Å resolution was obtained by replacement soaking a co-crystal of the kinase domain complexed with a non-covalently bound tool compound (24). Fig. 2a shows the bound conformation of compound 7 with the covalent bond formed to Cys-442. Further details can be seen from the omit map electron density (Fig. 2b), which reveals that compound 7 has displaced the tool compound (most conclusively in Complex-C of the asymmetric unit) and is consistent with Cys-442 adopting two rotamer conformations, one of which is covalently bound to compound 7. Compound 14 is the enantiomer of compound 7 and shows significantly lower activity, indicating a clear preference in the stereochemistry on the pyrrolidine ring. With an effective irreversible inhibitor in hand, further modification focused on improving the non-covalent recognition and solubility of the inhibitors. Switching the pyrimidine core to a pyridine core afforded a significant increase in potency (compound 8), and this was used in all further analogues.
FIGURE 1.
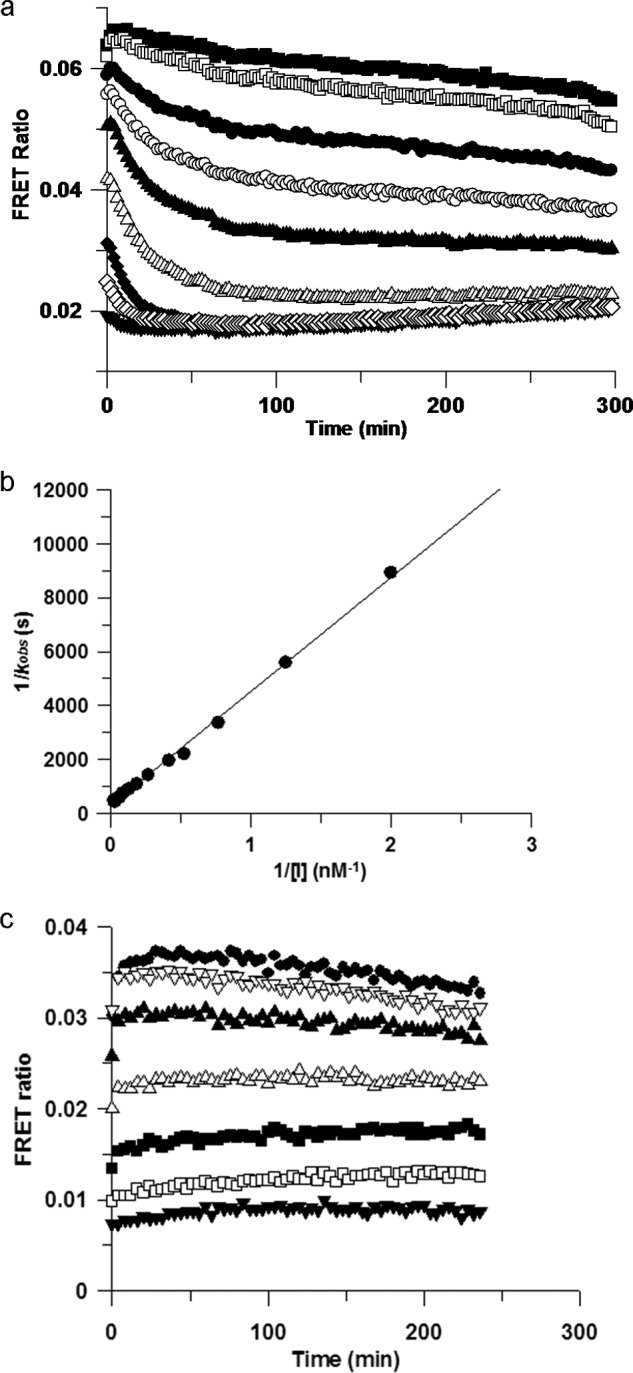
a, inhibition of Itk by compound 12 is time-dependent. The rate of binding of compound 12 to Itk was assessed in the LanthaScreen binding assay. Concentrations of compound 12 were as follows: 0 nm (■), 0.3 nm (□), 0.8 nm (●) 1.3 nm (○), 1.9 nm (▴), 3.7 nm (▵), 8 nm (♦), and 17 nm (♢), and 50 nm (▾). A rapid initial binding event is followed by a slower onset of increased inhibition. b, Kitz-Wilson plot of compound 12. Shown is Kitz-Wilson analysis for compound 12 binding to Itk (kobs data derived from Fig. 1a). The kinact and KI parameters can be derived from the reciprocals of the y and x intercepts, respectively. The non-zero y intercept is consistent with a two-step inactivation event. c, inhibition of Itk by compound 5 is not time-dependent. The rate of binding of compound 5 to Itk was assessed in the LanthaScreen binding assay. Concentrations of compound 5 were as follows: 0 nm (●), 15 nm (▿), 46 nm (▴), 137 nm (▵), 412 nm (■), 1,235 nm (□), and 3,704 nm (▾). A single, rapid binding event is consistent with simple reversible binding.
FIGURE 2.
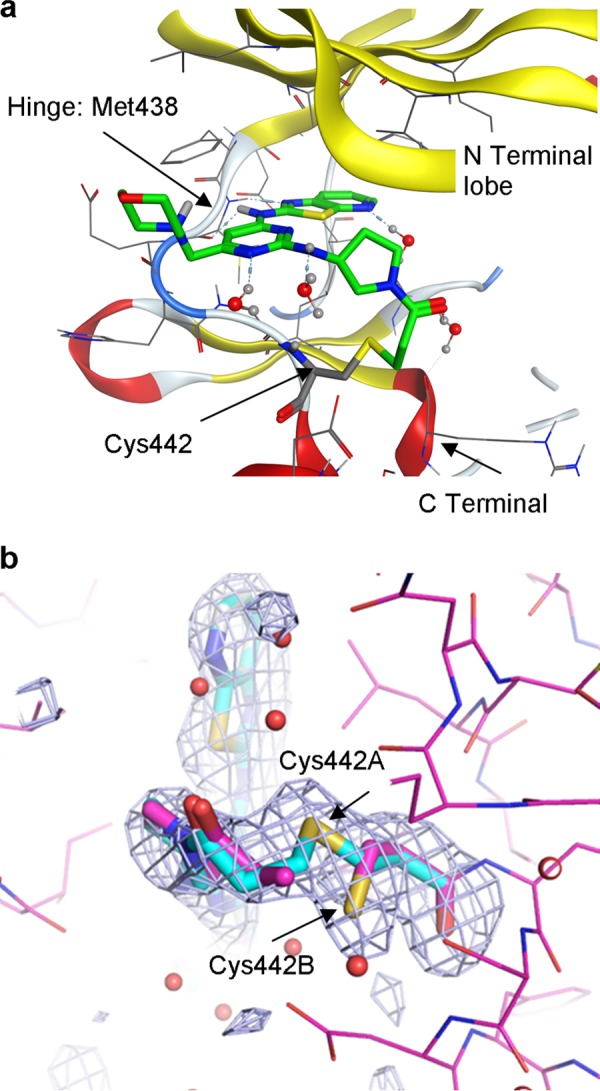
a, x-ray crystallography of drug-kinase complex. Shown is the x-ray crystal structure of compound 7 covalently complexed with the Itk kinase domain (Y512E) at 2.18 Å resolution obtained by replacement soaking a co-crystal of the kinase domain complexed with a non-covalently bound tool compound. b, an Fo − Fc difference “omit” map contoured at 3σ (gray mesh) highlighting the active site of complex C (of the four complexes (A–D) in the asymmetric unit) and calculated after omitting compound 7 and Cys-442 from the model. It reveals that compound 7 has displaced the tool compound (conclusively in this site) and is consistent with Cys-442 adopting two rotamer conformations, one of which is covalently bound to compound 7.
Compounds 9 and 12 both represent highly potent irreversible inhibitors with subnanomolar levels of cellular potency. Compound 16 has a methyl group added to the terminus of the acrylamide when compared with compound 8. The addition of this methyl group resulted in only a modest drop in potency in the biochemical assay of 0.5 log units. Kinetic studies revealed, however, that this compound was binding reversibly, the methyl group presumably inhibiting the formation of the covalent bond due to both steric and electronic factors. This switch to a reversible binding mode, although only manifesting itself as a modest drop in binding affinity in the biochemical assay, resulted in a significant 1.5 log unit drop in potency in the cellular assay. As expected, saturating the double bond (compound 15) of the acrylamide resulted in a reversible compound that had significantly reduced enzyme and cellular potency (Table 1).
We went on to demonstrate selectivity of compound 12 over a panel of other kinases where only reversible binding is possible and kinases that contain a similar cysteine in the ATP pocket as Itk (Table 2). The pIC50 of compound 12 against Itk at 1 mm ATP is 8.3, whereas in T cell activation assays, its pIC50 is 9.5. Biochemical assays against other kinases were conducted at an ATP concentration equal to the Km for ATP. This underestimates the degree of IC50 separation between Itk and the other kinases; nevertheless, there was clear selectivity over Btk and EGFR, which both contain a cysteine residue in the analogous position to Itk, and even greater selectivity over other kinases. Given that inhibition by irreversible inhibitors can be time-dependent, there can be concerns about the validity of such measurements even under standardized conditions unless more detailed studies, such as those described in this paper, are performed. Therefore, to confirm the selectivity over Btk, compound 12 was tested in a primary B cell assay, measuring up-regulation of CD69 following activation with F(ab′)2 anti-IgM. Although compound 12 did produce a concentration-dependent inhibition of CD69 expression with a pIC50 of 7.25 ± 0.04 (S.E.), this is ∼100-fold less potent than its effect on IFNγ production in PBMCs activated by CytoStim and 20-fold less potent than inhibition of anti-CD3/CD28-induced IL-2 release (Fig. 3). Therefore, compound 12 demonstrates a satisfactory degree of selectivity for T cells over B cells.
TABLE 2.
Kinase selectivity panel
Compound 12 was tested for inhibition of a range of kinases in biochemical assays conducted at an ATP concentration equal to the Km for each individual kinase. Results are shown as the pIC50 value.
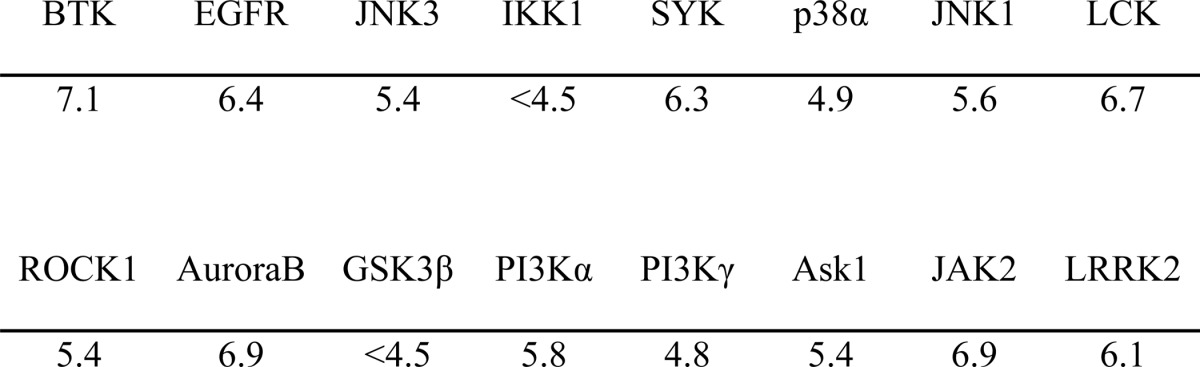
FIGURE 3.
T cell versus B cell selectivity. Compound 12 shows inhibition T cell response (CytoStim (●) or anti-CD3/CD28 (■)) with ∼20–100-fold greater potency than B cell response (anti-IgM (▴)). Inhibition of CD3/CD28- or CytoStim-induced IFNγ production was used as a marker of T cell inhibition. Inhibition of anti-IgM-induced CD69 was used as a marker of B cell inhibition. Results are shown as a mean ± S.E. (error bars) of seven donors for CytoStim, four donors for anti-CD3/CD28, and three donors for anti-IgM. Note that different donors were used for each of the three assay types.
An additional concern for irreversible compounds of this type is their general reactivity toward other proteins, which may result in “off target” effects and potentially toxicity. We addressed this in two ways; first, we determined their reactivity with GSH and, second, their inhibitory activity toward a panel of unrelated target proteins of different classes. The glutathione assay is a modification of the GSH-GloTM assay, which is based on the conversion of a luciferin derivative into luciferin catalyzed by glutathione S-transferase (GST) in the presence of GSH. The drug was either preincubated for 20 min with GSH and GST prior to the addition of the luciferin substrate or co-incubated with GST, GSH, and the substrate. We compared a matched pair of compounds, one with the acrylamide, which is irreversible (compound 8), and one where the double bond has been replaced by an ethyl group, which renders it reversible (compound 15). Both compounds showed the same reversible reactivity with GSH, demonstrating that the acrylamide does not confer a significant degree of general thiol reactivity (supplemental Table 2). We also tested compound 12 against >40 targets from a range of target classes, including G protein-coupled receptors, ion channels, and nuclear receptors in either agonist or antagonist mode (supplemental Table 3). No significant off target activity was detected to raise concerns about the reactivity of compound 12.
Drugs delivered topically to the lungs have requirements for certain physicochemical properties, one key feature being solubility in the fluid lining the lungs. Consequently, the basic compound 12 was selected for further study over compound 9 due its significantly better solubility as a difumarate salt in simulated lung fluid (412 versus 105 μg/ml). Measurement of the permeability of this compound through MDCK cells afforded a relatively low exact permeability coefficient (Pexact) of 37 nm/s, which encouraged us to believe that, following inhaled dosing, the compound would only pass slowly through the lungs, a highly desirable design feature to retain duration of action of an inhaled drug. A rat intravenous/oral pharmacokinetic study revealed negligible oral bioavailability (F < 5%) and moderate clearance (47 ml/min/kg). Both of these parameters were consistent with a compound designed for inhaled delivery.
In Vitro Biological Characterization
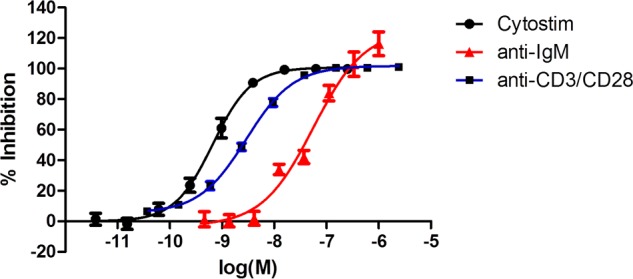
With our potent and selective Itk inhibitor, we went on to investigate aspects of Itk biology important for validating it as a therapeutic target for allergic inflammatory disease because Itk has been reported to play a selective role in the function of TH2 cells. We activated human PBMCs with CytoStim (which cross-links the T cell receptor (TCR) to a major histocompatibility complex (MHC) molecule on an antigen-presenting cell) or a mixture of anti-CD2, -CD3, and -CD28 conjugated to beads, this alternative triple stimulus being less sensitive to glucocorticoid inhibition and therefore considered to model more severe inflammatory situations. Measuring the production of four cytokines revealed that under both stimulation regimes, compound 12 was equally effective and gave 100% inhibition. However, it was most potent at inhibiting IL-2, followed by IFNγ, and least potent at inhibiting IL-13 and IL-17 (Fig. 4). We went on to test compound 12 in a PBMC activation assay in combination with a standard glucocorticoid, FP (Fig. 5). FP was a potent inhibitor of production of all four cytokines; however, in contrast to compound 12, it did not produce complete inhibition of IL-2 or IL-17, and this was particularly evident using the CD2/3/28 activation. However, co-administration of compound 12 showed marked additive inhibition.
FIGURE 4.
Compound 12 does not selectively inhibit TH2 cytokine release. Compound 12 produced a concentration-dependent inhibition of IL-2 (■), IFNγ (●), IL-17 (▴), and IL-13 (▾) release following activation of human PBMCs with either CytStim (a) or CD2/CD3/CD28 (b) for 24 h. Results are shown as mean ± S.E. (error bars) of seven donors.
FIGURE 5.

Compound 12 shows additive inhibition with a glucocorticoid. Human PBMCs were stimulated with either CytoStim- or anti-CD2/CD3/CD28-coated beads in the presence of increasing concentrations of glucocorticoid (FP) alone (●) or FP combined with increasing concentrations of compound 12 (0.2 nm (■), 1 nm (▴), or 5 nm (▾)). Co-administration of FP with compound 12 produced an upward shift of the FP concentration response curve, indicative of an additive effect. Inhibition by vehicle or Compound 12 alone is shown in open symbols. Results are shown as percentage inhibition of cytokine release following 24-h incubation, expressed as mean ± S.E. (error bars) of four donors.
Finally, we compared compound 12 with FP at suppressing cytokine production from human lung fragments following stimulation with PHA (Fig. 6). Compound 12 produced a concentration-dependent inhibition of IFNγ, IL-2, and IL-17 release from human lung, reaching complete inhibition but with weaker potency than observed with this compound in PBMC assays. Compound 12 was less potent than FP in the lung fragments but demonstrated a greater maximal level of inhibition.
FIGURE 6.
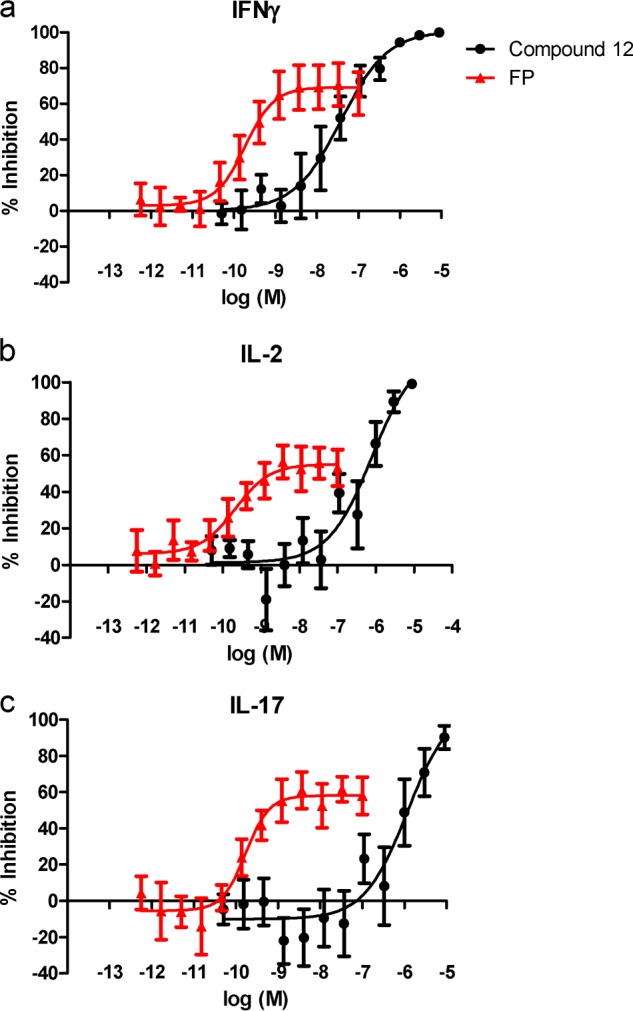
Compound 12 inhibits IL-2, IFNγ, and IL-17 release from fragments of human lung parenchyma. Compound 12 (●) and glucocorticoid (FP) (▴) produced a concentration-dependent inhibition of IL-2 (a), IFNγ (b), and IL-17 (c) release from fragments of human lung parenchyma following a 72-h incubation with PHA. Results are expressed as mean ± S.E. of 5–6 donors.
Compound 12 demonstrated potent inhibition (pIC50 = 9.5) of IFNγ release from CytoStim-activated PBMC and was progressed to wash out studies to investigate the duration of action. For washout studies, anti-CD3/CD28-coated beads were used to activate PBMC because this method gives a robust activation that can be detected by IL-2 release within 2 h. This short incubation period provides an assay that can discriminate between compounds with differing cellular retention properties, which was not possible with CytoStim. Itk compounds show a reduced potency in the anti-CD3/CD28 assay compared with the CytoStim assay; however, because this is seen for both reversible and irreversible compounds and because Itk is not essential for CD28 signaling (34), this decrease in potency is likely to be due to the method of activating cells rather than the 2-h incubation. Purified PBMCs were activated in vitro with anti-CD3/CD28, and although there was donor to donor variation, all donors produced detectable levels of IL-2 in the range of 100–500 pg/ml. In the absence of anti-CD3/CD28 activation, IL-2 levels were below the limit of detection (2.4 pg/ml). Treatment of the PBMCs with compound 12 had no adverse effect on viability, and unactivated cells were 97.6 ± 0.6% (S.E.) viable following a 48-h incubation at 2.4 μm of the drug compared with 98.4 ± 0.3% (S.E.) for cells treated with a DMSO control. Of note, the 48-h incubation time and the concentration of drug used for viability measurement are much greater than those used in subsequent assays to provide a wide margin of confidence.
To determine cellular activity and duration of action of reversible and irreversible inhibitors, we incubated PBMCs with increasing concentrations of compounds for 1 h followed by extensive washing in fresh medium to attempt to wash out the drug. Washed cells were further incubated for 2 or 19 h prior to stimulation with anti-CD3/28 for 2 h and determination of IL-2 secretion. The reversible compound 5, when tested without a washout, showed a full inhibition curve with a pIC50 of 6.39 ± 0.08 (S.E.); however, all inhibitory activity was lost upon washing, even at 2 h. In contrast, the irreversible compound 12 showed potent cellular activity with a pIC50 of 8.57 ± 0.05 (S.E.) for the unwashed control and this reducing only slightly to 7.96 ± 0.13 (S.E.) at 2 h postwashout and to 7.61 ± 0.13 (S.E.) at 19 h postwashout (Fig. 7). These data are consistent with compound 5 being freely reversible, because there is no retention of activity upon washout. Conversely, compound 12 shows strong retention of activity following washout, which is consistent with this compound being an irreversible inhibitor. However, there is a small reduction in activity with washout, which may reflect a reversible component to the inhibition or protein turnover and de novo synthesis of Itk. As shown previously, the crystal structure for compound 7 provides evidence for both reversible and irreversible binding.
FIGURE 7.
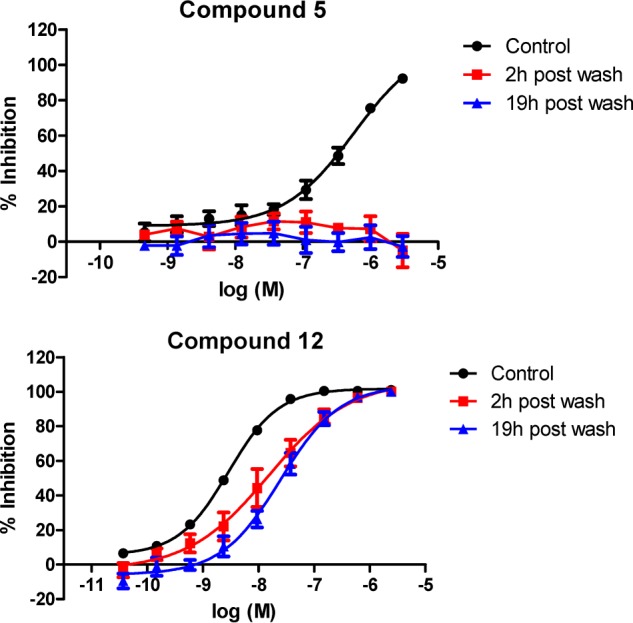
Compound 12 retains long duration of action activity following washout. The washout profile was determined for both reversible (compound 5) and irreversible (compound 12) Itk inhibitors. Control curves show the concentration-dependent inhibition of IL-2 release from PBMC in response to activation by anti-CD3/CD28 in the presence of compound. Cells were washed to remove free compound and incubated for a further 2 or 19 h before activation with anti-CD3/CD28. Results are shown as mean ± S.E. (error bars) of four donors.
Itk Turnover
To investigate the half-life of Itk and the influence of binding an irreversible inhibitor, we performed [35S]methionine pulse-chase experiments in Jurkat cells. Cells were stimulated with anti-CD3/28 for 1 h in the presence or absence of 20 nm compound 12. Cells were then pulsed for 30 min with [35S]methionine, followed by a cold methionine chase period of up to 24 h. Biosynthetically radiolabeled Itk was immunoprecipitated from cell extracts and detected by SDS-PAGE and fluorography (Fig. 8). Itk has a long half-life in the absence of drug; however, treatment of the cells with compound 12 appeared to increase the rate of turnover of biosynthetically labeled Itk. Identical experiments were performed without [35S]methionine labeling, and cell extracts were prepared and analyzed by Western blotting to determine the mass of kinase in the cell at each time point. The data revealed that the total mass of Itk in each cell extract remained constant over the chase period under all conditions. We established that under these pulse-chase conditions, viability of the cells was not significantly affected. In addition, we measured IL-2 secretion from stimulated and drug-treated cultures to confirm stimulation by anti-CD3/CD28 and inhibition by drug (supplemental Table 4).
FIGURE 8.
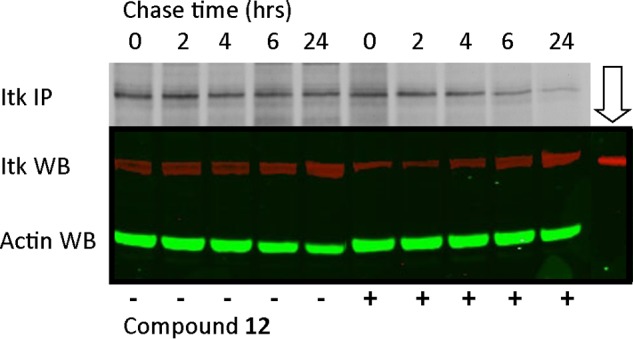
Itk pulse-chase. Jurkat cells, in the presence of absence of compound 12, were biosynthetically pulse-labeled with [35S]methionine for 30 min, followed by a cold methionine chase for 0–24 h. Cell extracts were prepared, and Itk was immunoprecipitated and analyzed by SDS-PAGE and fluorography (Itk IP). Identical cell extracts were prepared in parallel from non-radiolabeled Jurkat cells and analyzed by Western blotting with antibodies to Itk (Itk WB) and actin (Actin WB) as a loading control. In lane 11 (downward arrow) was loaded 20 ng of purified Itk protein as a control.
In Vivo Biological Characterization
Having demonstrated potency and long duration of action of compound 12 in a cellular assay, we wished to test the compound in an animal PD model. Compound 12, as a crystalline difumarate salt, was micronized to give a particle size of ∼1.6 μm, and a calculated dose of 2.32 mg/kg, based on the amount of compound deposited on an in-line filter (24), was administered by inhalation to rats via an inhalation tower over an exposure time of 20 min. Control animals received only air. Animals were sacrificed, and their lungs were perfused in situ and then removed prior to tissue disruption and preparation of a cell suspension as described under “Experimental Procedures.” The concentration of drug in the lung homogenate postperfusion was 2.9 ± 0.8 μg/g (S.D.) (6 μm); however, the majority of this was not cell-associated. Therefore, once the single cell suspension was prepared, the concentration of compound was estimated at 4 ng/ml (8 nm). The cell suspension was stimulated with anti-CD3 for 18 h, whereas controls received no stimulation. Following this 18-h incubation, T cell activation was determined by flow cytometry. Following exclusion of dead cells, CD25 staining on CD4+/CD45+ T cells was quantified and is shown in Fig. 9. In control, air-treated animals, the median fluorescent intensity of CD25 staining without CD3 activation was 13.78 ± 11.6 (S.E.), and following activation, this rose to 2038.7 ± 689.5 (S.E.). In drug-treated animals, CD25 staining without CD3 activation was 5.8 ± 2.7 (S.E.), and following CD3 activation, this rose to 27.8 ± 6.6 (S.E.). Consequently, compound 12, when dosed by a single 20-min inhalation, produced an almost complete inhibition of the ability of T cells to be activated by an 18-h stimulation with anti-CD3.
FIGURE 9.

Rat PD model following inhaled administration of compound 12. Lung cell suspensions were prepared from rats who had inhaled air alone (a) or compound 12 (b). Cells were unstimulated (control) or activated ex vivo with anti-CD3, followed by measurement of CD25 expression by flow cytometry on CD4+ cells. Specificity of the anti-CD25 staining is demonstrated with anti-CD3 activated cells by the isotype control (isotype). The CD4+/CD25+ cells are boxed, and the percentage of CD4+ cells with increased CD25 staining is shown.
DISCUSSION
The Tec kinases are a family of non-receptor tyrosine kinases that are mainly expressed in hematopoietic cells and act downstream of T and B cell receptors and Fc receptors for IgE and IgG. Itk is expressed in T cells, NK cells, and mast cells, and in T cells, Itk plays an important role in the signaling cascade following activation of the T cell receptor. Evidence from Itk knock-out mice shows reduced cytokine release in response to TCR activation (2–9, 16, 35). In NK cells, Itk plays a role in the response to FcγRIIIa activation (36), whereas in mast cells, Itk is activated following cross-linking of FcϵRI but plays both positive and negative roles in the signaling events (37). Based on studies in knock-out mice, the main interest in Itk seems to be that it is critical for the development of TH2 immunity. Itk−/− mice are protected in models of allergic asthma (6–9) and are more susceptible to parasitic worm infections (2, 3), both of which are attributed to a failure to produce TH2 cytokines (1, 2, 5–8). Consequently, there is a widely held belief that Itk inhibition is an attractive and well validated target for allergic diseases such as asthma. Indeed, an Itk inhibitor (38) and a peptide (39) that prevents the binding of Itk to SLP-76 (and therefore recruitment of Itk to the signaling complex and the subsequent activation of Itk) have been shown to be effective in animal models of asthma. Recent studies have suggested that an explanation for the suppressed TH2 response in Itk−/− mice is that Itk negatively regulates TH1 genes, such as IFNγ. Itk knockout resulted in increased IFNγ expression, which suppressed TH2 differentiation (40). However, a question of major importance is how well do developmental defects in transgenic mice predict the behavior of a small molecule Itk inhibitor, and consequently, what is the real therapeutic potential of inhibiting Itk? There are two issues to consider, the first being how to compare T cell development in a knock-out mouse with inhibition of peripheral T cell activation; the second is how to compare a knockout with a small molecule inhibitor because Itk plays a key role in the spatiotemporal assembly and organization of the receptor TCR signaling complex, and knockout of Itk causes disruption to multiple components of the TCR complex (41). A small molecule binding to the catalytic site of the kinase is unlikely to result in the same degree of disruption of the whole TCR complex.
Although there are a number of potent in vitro Itk inhibitors exemplified in the literature, none have been used to address the question of TH subtype specificity, although a few studies have gone on to describe in vivo activity (11, 38). However, there have been no reported studies investigating the inhaled route of delivery. Our findings importantly show that inhibition of Itk blocks production of TH1, TH2, and TH17 cytokines from human PBMCs and human lung fragments activated with different stimuli. This finding has been replicated using a structurally diverse range of small molecule inhibitors of Itk (data not shown). We conclude from this that an Itk inhibitor is unlikely to specifically suppress activation of TH2 cells in allergic disease but will have broad T cell-inhibitory activity. We also explored the ability of Itk inhibitors to suppress T cell activation when used together with glucocorticoids (FP), because they are the most widely used anti-inflammatory drug, and potentially any new medicine might be used therapeutically in combination with them. It is well known that glucocorticoids exert their effects through transcriptional regulation; however, it has come to light that they also influence T cell activation by regulating the assembly and function of the TCR complex (42, 43). Consequently, the combined actions of glucocorticoids and Itk inhibitors is of interest both from basic and applied perspectives. We demonstrate that compound 12 is effective in the presence of a steroid and is additive with FP for inhibiting the release of IFNγ, IL-2, and IL-17 and, to a lesser extent, IL-13 from activated PBMCs. Finally, we have demonstrated that compound 12 is effective at inhibiting IFNγ, IL-2, and IL-17 from lung parenchyma fragments following activation with PHA, and although the potency was lower than predicted from PBMC studies, it did produce maximal inhibition of cytokine release. In this assay, glucocorticoid treatment, while being ∼100–1000-fold more potent than compound 12, does not fully inhibit cytokine release, as has been shown previously (44, 45).
Producing potent and selective kinase inhibitors is not novel; however, inhaled drug delivery is particularly challenging, and unlike with oral drugs, there is no accepted set of rules defining ideal druglike properties. The conventional strategy in drug discovery is to design reversible inhibitors. Although irreversible inhibitors are more prevalent than widely appreciated, there is a risk of immunogenicity, which may only be seen in late stage development. These risks are greatest for compounds that are weakly selective and highly reactive or that generate highly reactive electrophilic drug metabolites. Recently, there have been efforts to generate targeted covalent inhibitors, where a specific non-covalent interaction within the enzyme active site orientates a weakly reactive covalent warhead to a specific non-catalytic residue (46). The recent progression of irreversible kinase inhibitors in oncology goes some way to attenuating these safety concerns, and we chose a similar approach for an inhaled Itk inhibitor. An irreversible binding mechanism represents an attractive and novel option for inhaled delivery because it allows the dissociation between efficacy and persistent elevated drug levels in the lung. Inhaled drug design has often developed compounds of low solubility to enhance duration of action through lung retention, although this can lead in certain circumstances to an undesirable inflammatory macrophage response. The irreversible inhibitor approach described herein allowed the identification of highly potent inhibitors with persistent inhibition yet with high aqueous solubility.
Rational structure based design allowed the rapid optimization of the irreversible series. High levels of non-covalent molecular recognition represent an important aspect of the approach and enable selectivity to be achieved over other kinases, such as Btk, which contain a reactive Cys in an analogous position. Our most potent and selective irreversible inhibitors maximize their non-covalent recognition and couple this with optimal placement of the electrophilic acrylamide with respect to Cys-442 (compounds 9 and 12), enabling their potency to be driven beyond that conventionally predicted by their ligand efficiency. Indeed, the extreme tight binding of the compounds provided challenges in accurately characterizing their true enzymatic potency, and we were forced to assay our most potent compounds at 1 mm rather than at a Km concentration of ATP in order to decrease the sensitivity of the assay and make quantitative determinations. This significant increase in potency, evident at both enzyme and cellular levels, also proved highly beneficial from a selectivity perspective. Furthermore, the slower (secondary) covalent mechanism allowed the compound's duration of action to be mediated by the rate of protein turnover while retaining selectivity due to the requirement for target proximity for reaction. Clearly, making an irreversible inhibitor with such a mechanism raises concerns about reactivity with non-target proteins. We tested this possibility by measuring the reactivity with GSH of two closely matched compounds that were reversible and irreversible. Our observation that both compounds showed identical reversibility with GSH gave us confidence that general thiol reactivity was not an issue with these compounds. Moreover, profiling compound 12 against a panel of >40 unrelated targets confirmed that it was not a nonspecific reactive molecule.
We set out to make an irreversible inhibitor of Itk to generate a long duration of action; however, such a strategy would have had limited or no benefit if the kinase itself had a rapid turnover. It would also be difficult to reconcile the long duration of action we observed with our inhibitors if the kinase had a short half-life. Studies on HA-tagged Itk overexpressed in HEK293 cells revealed a half-life of 107 min, and this was reduced by 25–69% in certain Itk mutants (47). A study in human T cells suggested that Itk had a very short half-life, in the region of 1 h, and that in the presence of a covalent Itk inhibitor, this was increased to 22 h (48). In our study in Jurkat cells, we found Itk to have a very long half-life (>24 h), which was shortened by compound 12. However, although the drug appeared to enhance the rate of kinase turnover, the total mass of Itk per cell remained unchanged, suggesting a compensatory resynthesis. Even in the presence of drug, the slow turnover of Itk is consistent with the prolonged duration of action of compound 12 that we observed in vitro and in vivo being mediated by an irreversible component to its mechanism of action.
Having generated both reversible (compound 5) and irreversible (compound 12) Itk inhibitors, we went on to examine their properties both in vitro and in vivo. In human PBMC assays in vitro, we were able to show that compound 12 shows excellent retention of cellular activity 19 h following an extensive washout, whereas the inhibitory activity of compound 5 was rapidly washed out of cells. This was a key feature we wished to build into the molecule to deliver improved lung retention that would translate into greater in vivo efficacy. To examine the in vivo properties of our inhibitors, we developed a PD model that revealed the irreversible binding of the Itk inhibitor. Rats were dosed with compounds by inhalation, and cell suspensions were prepared from the lungs, followed by extensive washing to remove any free compound prior to activation in vitro with anti-CD3. Using this protocol, it is possible to demonstrate both the degree and duration of Itk inhibition by our compounds. This PD model has advantages over “disease” models of allergic inflammation in that it measures the direct response to activation of CD3 on CD4+ cells rather than the complexity of cellular interactions that are evoked following sensitization and challenge with antigen. Furthermore, it does not require a lengthy sensitization regime, thereby reducing and refining animal experimentation. This is a pure PD model that does not seek to explore the role of T cells or Itk activation in asthma but is a streamlined approach to drug discovery that enables determination of the pharmacokinetic/pharmacodynamic (PK/PD) relationship. The PK/PD model offers an alternative to animal “disease” models, which may have limited ability to predict efficacy in humans (49). The PD model utilizes naive Wistar Han rats, the preferred species for toxicology studies, thereby enabling a direct determination of the therapeutic index. Furthermore, compound 12 was administered as a dry powder via an inhalation tower connected to an aerosol generator, which is the same method of drug delivery used for toxicology with any future human studies also using a dry powder formulation. The PD model used in this study is translational because the ex vivo activation of bronchial alveolar lavage cells or bronchial biopsy may be a possible method to determine engagement of the Itk mechanism and establish a PK/PD correlation for human studies.
In summary, we have described the generation of a potent and selective irreversible inhibitor of Itk, building in many design principles that we believe to be favorable for inhaled drug delivery. Itk has a long half-life and is therefore a good target for irreversible inhibitors. We have shown in both in vitro and in vivo models that our compounds are potent and long lasting, and we describe an accelerated translational route forward, avoiding animal efficacy studies. Finally, we have shown that small molecule Itk inhibitors block the activation of TH1, TH2, and TH17 cells and show additivity when combined with a glucocorticoid.
Acknowledgments
We thank Lindsay Butterfield and Doug Ball for conducting the rat in vivo dosing studies, Bob Gibbon for formulation support and solubility analysis, Ian Smith for expertise in medicinal chemistry, Carol Harris and Chris Mooney for expression and purification of the Itk enzyme and Itk (residues 357–620, Y512E) kinase domain protein, Vikki Barrett for assay development of the human lung parenchyma assay, Nikolai Belyaev for help with preparation of figures, Fiona Lucas for testing of compounds in the CytoStim PBMC assay, and the Department of Screening and Compound Profiling for carrying out the Itk enzyme assay.

This article contains supplemental material, including supplemental Equations 1–3 and Tables 1–4.
The atomic coordinates and structure factors (code 4KIO) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Itk
- IL-2-inducible tyrosine kinase
- pIC50
- minus logarithm to base 10 of the half-maximal inhibitory concentration
- EGFR
- EGF receptor
- PBMC
- peripheral blood mononuclear cell
- PD
- pharmacodynamic
- PK
- pharmacokinetic
- FP
- fluticasone propionate
- PHA
- phytohemagglutinin A
- TCR
- T cell receptor.
REFERENCES
- 1. August A., Ragin M. J. (2012) Regulation of T-cell responses and diseases by Tec kinase Itk. Int. Rev. Immunol. 31, 155–165 [DOI] [PubMed] [Google Scholar]
- 2. Fowell D. J., Shinkai K., Liao X. C., Beebe A. M., Coffman R. L., Littman D. R., Locksley R. M. (1999) Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity 11, 399–409 [DOI] [PubMed] [Google Scholar]
- 3. Schaeffer E. M., Yap G. S., Lewis C. M., Czar M. J., McVicar D. W., Cheever A. W., Sher A., Schwartzberg P. L. (2001) Mutation of Tec family kinases alters T helper cell differentiation. Nat. Immunol. 2, 1183–1188 [DOI] [PubMed] [Google Scholar]
- 4. Miller A. T., Wilcox H. M., Lai Z., Berg L. J. (2004) Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity 21, 67–80 [DOI] [PubMed] [Google Scholar]
- 5. Au-Yeung B. B., Katzman S. D., Fowell D. J. (2006) Cutting edge. Itk-dependent signals required for the CD4+ T cells to exert, but not gain, Th2 effector function. J. Immunol. 176, 3895–3899 [DOI] [PubMed] [Google Scholar]
- 6. Ferrara T. J., Mueller C., Sahu N., Ben-Jebria A., August A. (2006) Reduced airway hyperresponsiveness and tracheal responses during allergic asthma in mice lacking tyrosine kinase inducible T-cell kinase. J. Allergy Clin. Immunol. 117, 780–786 [DOI] [PubMed] [Google Scholar]
- 7. Sahu N., Mueller C., Fischer A., August A. (2008) Differential sensitivity to Itk kinase signals for T helper 2 cytokine production and chemokine-mediated migration. J. Immunol. 180, 3833–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mueller C., August A. (2003) Attenuation of immunological symptoms of allergic asthma in mice lacking the tyrosine kinase ITK. J. Immunol. 170, 5056–5063 [DOI] [PubMed] [Google Scholar]
- 9. Forssell J., Sideras P., Eriksson C., Malm-Erjefält M., Rydell-Törmänen K., Ericsson P. O., Erjefält J. S. (2005) Interleukin-2-inducible T cell kinase regulates mast cell degranulation and acute allergic responses. Am. J. Respir. Cell Mol. Biol. 32, 511–520 [DOI] [PubMed] [Google Scholar]
- 10. Das J., Furch J. A., Liu C., Moquin R. V., Lin J., Spergel S. H., McIntyre K. W., Shuster D. J., O'Day K. D., Penhallow B., Hung C. Y., Doweyko A. M., Kamath A., Zhang H., Marathe P., Kanner S. B., Lin T. A., Dodd J. H., Barrish J. C., Wityak J. (2006) Discovery and SAR of 2-amino-5-(thioaryl)thiazoles as potent and selective Itk inhibitors. Bioorg. Med. Chem. Lett. 16, 3706–3712 [DOI] [PubMed] [Google Scholar]
- 11. Riether D., Zindell R., Kowalski J. A., Cook B. N., Bentzien J., Lombaert S. D., Thomson D., Kugler S. Z., Jr., Skow D., Martin L. S., Raymond E. L., Khine H. H., O'Shea K., Woska J. R., Jr., Jeanfavre D., Sellati R., Ralph K. L., Ahlberg J., Labissiere G., Kashem M. A., Pullen S. S., Takahashi H. (2009) 5-Aminomethylbenzimidazoles as potent ITK antagonists. Bioorg. Med. Chem. Lett. 19, 1588–1591 [DOI] [PubMed] [Google Scholar]
- 12. Jurcak J. G., Barrague M., Gillespy T. A., Edwards M. L., Musick K. Y., Weintraub P. M., Du Y., Dharanipragada R. M., Parkar A. A. (March 24, 2005) Preparation of thienopyrazoles as inhibitors of interleukin-2 inducible tyrosine kinase for treating diseases involving overproduction of Th2 cytokine like asthma. International Patent WO2005026175
- 13. Herdemann M., Weber A., Jonveaux J., Schwoebel F., Stoeck M., Heit I. (2011) Optimisation of ITK inhibitors through successive iterative design cycles. Bioorg. Med. Chem. Lett. 21, 1852–1856 [DOI] [PubMed] [Google Scholar]
- 14. Charrier J.-D., Miller A., Kay D. P., Brenchley G., Twin H. C., Collier P. N., Ramaya S., Keily S. B., Durrant S. J., Knegtel R. M., Tanner A. J., Brown K., Curnock A. P., Jimenez J. M. (2011) Discovery and structure-activity relationship of 3-aminopyrid-2-ones as potent and selective interleukin-2 inducible T-cell kinase (Itk) inhibitors. J. Med. Chem. 54, 2341–2350 [DOI] [PubMed] [Google Scholar]
- 15. McLean L. R., Zhang Y., Zaidi N., Bi X., Wang R., Dharanipragada R., Jurcak J. G., Gillespy T. A., Zhao Z., Musick K. Y., Choi Y. M., Barrague M., Peppard J., Smicker M., Duguid M., Parkar A., Fordham J., Kominos D. (2012) X-ray crystallographic structure-based design of selective thienopyrazole inhibitors for interleukin-2-inducible tyrosine kinase. Bioorg. Med. Chem. Lett. 22, 3296–3300 [DOI] [PubMed] [Google Scholar]
- 16. Schaeffer E. M., Debnath J., Yap G., McVicar D., Liao X. C., Littman D. R., Sher A., Varmus H. E., Lenardo M. J., Schwartzberg P. L. (1999) Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science 284, 638–641 [DOI] [PubMed] [Google Scholar]
- 17. Atherly L. O., Brehm M. A., Welsh R. M., Berg L. J. (2006) Tec kinases Itk and Rlk are required for CD8+ T cell responses to virus infection independent of their role in CD4+ T cell help. J. Immunol. 176, 1571–1581 [DOI] [PubMed] [Google Scholar]
- 18. Au-Yeung B. B., Fowell D. J. (2007) A key role for Itk in both IFNγ and IL-4 production by NKT cells. J. Immunol. 179, 111–119 [DOI] [PubMed] [Google Scholar]
- 19. Felices M., Berg L. J. (2008) The Tec kinases Itk and Rlk regulate NKT cell maturation, cytokine production, and survival. J. Immunol. 180, 3007–3018 [DOI] [PubMed] [Google Scholar]
- 20. Li D., Ambrogio L., Shimamura T., Kubo S., Takahashi M., Chirieac L. R., Padera R. F., Shapiro G. I., Baum A., Himmelsbach F., Rettig W. J., Meyerson M., Solca F., Greulich H., Wong K. K. (2008) BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27, 4702–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carpenter R. L., Lo H.-W. (2012) Dacomitinib, an emerging HER-targeted therapy for non-small cell lung cancer. J. Thorac. Dis. 4, 639–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pan Z., Scheerens H., Li S. J., Schultz B. E., Sprengeler P. A., Burrill L. C., Mendonca R. V., Sweeney M. D., Scott K. C., Grothaus P. G., Jeffery D. A., Spoerke J. M., Honigberg L. A., Young P. R., Dalrymple S. A., Palmer J. T. (2007) Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem 2, 58–61 [DOI] [PubMed] [Google Scholar]
- 23. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–4108 [DOI] [PubMed] [Google Scholar]
- 24. Alexander D. J., Collins C. J., Coombs D. W., Gilkison I. S., Hardy C. J., Healey G., Karantabias G., Johnson N., Karlsson A., Kilgour J. D., McDonald P. (2008) Association of inhalation toxicologists (AIT) working party recommendation for standard delivered dose calculation and expression in non-clinical aerosol inhalation toxicology studies with pharmaceuticals. Inhal. Toxicol. 20, 1179–1189 [DOI] [PubMed] [Google Scholar]
- 25. Rice L. V., Bax H. J., Russell L. J., Barrett V. J., Walton S. E., Deakin A. M., Thomson S. A., Lucas F., Solari R., House D., Begg M. (2013) Characterization of selective calcium-release activated calcium channel blockers in mast cells and T-cells from human, rat, mouse and guinea-pig preparations. Eur. J. Pharmacol. 704, 49–57 [DOI] [PubMed] [Google Scholar]
- 26. Alder C. M., Ambler M., Campbell A. J., Champigny A. C., Deakin A. M., Harling J. D., Harris C. A., Longstaff T., Lynn S., Maxwell A. C., Mooney C. J., Scullion C., Singh O. M. P, Smith I. E. D., Somers D., Tame C. J., Wayne G., Wilson C., Woolven J. M. (2013) The identification of a novel and selective series of Itk inhibitors via a template-hopping strategy. ACS Med Chem. Lett. 10.1021/ml400206q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leslie A. G. W., Powell H. R. (2007) Processing diffraction data with mosflm. in Evolving Methods for Macromolecular Crystallography (Read R. J., Sussman J. L., eds) pp. 41–51, Springer, New York [Google Scholar]
- 28. Evans P. (1993) Data reduction. Data collection and processing. in Data Collection and Processing, Proceedings of the CCP4 Study Weekend, 29–30 January 1993 (Sawyer L., Isaac N., Bailey S.) pp. 114–122, CLRC Daresbury Laboratory, Daresbury, UK [Google Scholar]
- 29. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 31. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 32. Fry D. W., Bridges A. J., Denny W. A., Doherty A., Greis K. D., Hicks J. L., Hook K. E., Keller P. R., Leopold W. R., Loo J. A., McNamara D. J., Nelson J. M., Sherwood V., Smaill J. B., Trumpp-Kallmeyer S., Dobrusin E. M. (1998) Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 95, 12022–12027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitz R., Wilson I. B. (1962) Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 237, 3245–3249 [PubMed] [Google Scholar]
- 34. Li C.-R., Berg L. J. (2005) Cutting edge. Itk is not essential for CD28 signaling in naive T cells. J. Immunol. 174, 4475–4479 [DOI] [PubMed] [Google Scholar]
- 35. Liao X. C., Littman D. R. (1995) Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity 3, 757–769 [DOI] [PubMed] [Google Scholar]
- 36. Khurana D., Arneson L. N., Schoon R. A., Dick C. J., Leibson P. J. (2007) Differential regulation of human NK cell-mediated cytotoxicity by the tyrosine kinase Itk. J. Immunol. 178, 3575–3582 [DOI] [PubMed] [Google Scholar]
- 37. Iyer A. S., August A. (2008) The Tec family kinase, IL-2-inducible T cell kinase, differentially controls mast cell responses. J. Immunol. 180, 7869–7877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin T.-A., McIntyre K. W., Das J., Liu C., O'Day K. D., Penhallow B., Hung C. Y., Whitney G. S., Shuster D. J., Yang X., Townsend R., Postelnek J., Spergel S. H., Lin J., Moquin R. V., Furch J. A., Kamath A. V., Zhang H., Marathe P. H., Perez-Villar J. J., Doweyko A., Killar L., Dodd J. H., Barrish J. C., Wityak J., Kanner S. B. (2004) Selective Itk inhibitors block T-cell activation and murine lung inflammation. Biochemistry 43, 11056–11062 [DOI] [PubMed] [Google Scholar]
- 39. Guimond D. M., Cam N. R., Hirve N., Duan W., Lambris J. D., Croft M., Tsoukas C. D. (2013) Regulation of immune Responsiveness in vivo by disrupting an early T-cell signaling event using a cell-permeable peptide. PLoS One 8, e63645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kannan A. K., Sahu N., Mohanan S., Mohinta S., August A. (2013) IL-2-inducible T-cell kinase modulates TH2-mediated allergic airway inflammation by suppressing IFN-γ in naïve CD4+ T cells. J. Allergy Clin. Immunol. 10.1016/j.jaci.2013.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singleton K. L., Gosh M., Dandekar R. D., Au-Yeung B. B., Ksionda O., Tybulewicz V. L., Altman A., Fowell D. J., Wülfing C. (2011) Itk controls the spatiotemporal organization of T cell activation. Sci. Signal. 4, ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Laethem F., Baus E., Smyth L. A., Andris F., Bex F., Urbain J., Kioussis D., Leo O. (2001) Glucocorticoids attenuate T cell receptor signaling. J. Exp. Med. 193, 803–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Löwenberg M., Verhaar A. P., van den Brink G. R., Hommes D. W. (2007) Glucocorticoid signaling. A nongenomic mechanism for T-cell immunosuppression. Trends Mol. Med. 13, 158–163 [DOI] [PubMed] [Google Scholar]
- 44. Bochner B. S., Rutledge B. K., Schleimer R. P. (1987) Interleukin 1 production by human lung tissue. II. Inhibition by anti-inflammatory steroids. J. Immunol. 139, 2303–2307 [PubMed] [Google Scholar]
- 45. Kato M., Schleimer R. P. (1994) Antiinflammatory steroids inhibit granulocyte/macrophage colony-stimulating factor production by human lung tissue. Lung 172, 113–124 [DOI] [PubMed] [Google Scholar]
- 46. Tsou H. R., Mamuya N., Johnson B. D., Reich M.F., Gruber B. C., Ye F., Nilakantan R., Shen R., Discafani C., DeBlanc R., Davis R., Koehn F. E., Greenberger L. M., Wang Y. F., Wissner A. (2001) 6-Substituted-4-(3-bromophenylamino)quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity. J. Med. Chem. 44, 2719–2734 [DOI] [PubMed] [Google Scholar]
- 47. Linka R. M., Risse S. L., Bienemann K., Werner M., Linka Y., Krux F., Synaeve C., Deenen R., Ginzel S., Dvorsky R., Gombert M., Halenius A., Hartig R., Helminen M., Fischer A., Stepensky P., Vettenranta K., Köhrer K., Ahmadian M. R., Laws H. J., Fleckenstein B., Jumaa H., Latour S., Schraven B., Borkhardt A. (2012) Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia 26, 963–971 [DOI] [PubMed] [Google Scholar]
- 48. Zapf C. W., Gerstenberger B. S., Xing L., Limburg D. C., Anderson D. R., Caspers N., Han S., Aulabaugh A., Kurumbail R., Shakya S., Li X., Spaulding V., Czerwinski R. M., Seth N., Medley Q. G. (2012) Covalent inhibitors of interleukin-2 inducible T cell kinase (itk) with nanomolar potency in a whole-blood assay. J. Med. Chem. 55, 10047–10063 [DOI] [PubMed] [Google Scholar]
- 49. Holmes A. M., Solari R., Holgate S. T. (2011) Animal models of asthma. Value, limitations and opportunities for alternative approaches. Drug Discov. Today 16, 659–670 [DOI] [PubMed] [Google Scholar]