

Background: Constitutively active mutations (CAMs) affect the dynamics of G protein-coupled receptors (GPCRs), through undefined mechanisms.
Results: Site-directed fluorescence labeling (SDFL) studies find a CAM (M257Y) alters the dynamics of the GPCR ligand-free opsin.
Conclusion: The M257Y CAM alters the dynamics of the conversion between active and inactive opsin conformations.
Significance: SDFL can be used to gain insights into how CAMs affect the dynamics of G protein-coupled receptors.
Keywords: Fluorescence, G Protein-coupled Receptors (GPCR), Protein Dynamics, Receptor Structure-Function, Rhodopsin, Conformational Transitions, Energy Landscapes, Protein Energetics, Site-directed Fluorescence Labeling (SDFL), Tryptophan-induced Quenching (TrIQ)
Abstract
G protein-coupled receptors (GPCRs) undergo dynamic transitions between active and inactive conformations. Usually, these conversions are triggered when the receptor detects an external signal, but some so-called constitutively activating mutations, or CAMs, induce a GPCR to bind and activate G proteins in the absence of external stimulation, in ways still not fully understood. Here, we investigated how a CAM alters the structure of a GPCR and the dynamics involved as the receptor transitions between different conformations. Our approach used site-directed fluorescence labeling (SDFL) spectroscopy to compare opsin, the ligand-free form of the GPCR rhodopsin, with opsin containing the CAM M257Y, focusing specifically on key movements that occur in the sixth transmembrane helix (TM6) during GPCR activation. The site-directed fluorescence labeling data indicate opsin is constrained to an inactive conformation both in detergent micelles and lipid membranes, but when it contains the M257Y CAM, opsin is more dynamic and can interact with a G protein mimetic. Further study of these receptors using tryptophan-induced quenching (TrIQ) methods indicates that in detergent, the CAM significantly increases the population of receptors in the active state, but not in lipids. Subsequent Arrhenius analysis of the TrIQ data suggests that, both in detergent and lipids, the CAM lowers the energy barrier for TM6 movement, a key transition required for conversion between the inactive and active conformations. Together, these data suggest that the lowered energy barrier is a primary effect of the CAM on the receptor dynamics and energetics.
Introduction
G protein-coupled receptors (GPCRs)3 transmit extracellular signals to intracellular signaling cascades via activation of heterotrimeric G proteins. During this process they undergo dynamic transitions between inactive and active (signaling) states (1, 2). It is thought that the “signal” stabilizes the active conformation(s) of a GPCR, resulting in elevated G protein activity.
What is different about the active GPCR conformation that enables it to bind and activate G proteins? In the case of the light sensing GPCR rhodopsin, early insights came from cross-linking and spin and fluorescence labeling studies, which found that light activation induces an outward movement of sixth transmembrane helix (TM6) (3–7), which is preceded by several sequential intra-molecular events (8). This movement was proposed to expose a “hydrophobic patch” that couples with the C terminus of the G protein α-subunit (Gα) (9), an interaction confirmed in subsequent crystal structures (10, 11) (see Fig. 1). TM6 movement is observed in activation of various rhodopsins (12) and ligand-binding GPCRs (13, 14), suggesting it may play a key, universal role in GPCR activation.
FIGURE 1.
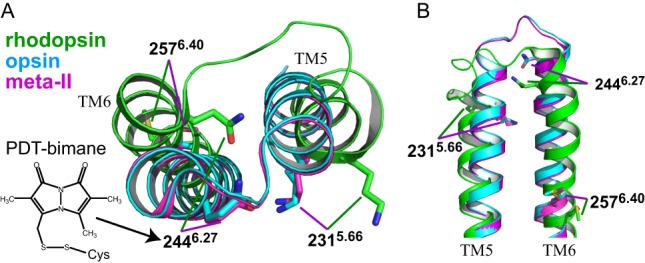
Comparison of TM5 and TM6 in the crystal structures of opsin, rhodopsin, and photoactivated (meta-II) form. Cytoplasmic view (A) and side view (B) showing the relative displacement of TM5 and especially TM6 in the active (meta-II) and inactive forms of rhodopsin. Note that the crystal structure of opsin appears more like the active form of rhodopsin. Also indicated are the sites of the bimane labeling (position 2446.27), the quencher tryptophan (position 2315.66) used for the TrIQ studies discussed in the text, and a constitutively activating mutation M257Y (position 2576.40). The superscripts of the respective positions refer to Ballesteros-Weinstein numbering (18). The models were constructed using the structures of bovine opsin (light blue, PDB code 3CAP) (22), rhodopsin in the dark (green, PDB code 1GZM) (31), and rhodopsin in meta-II state (magenta, PDB code 3PQR) (10).
What energetic and dynamic aspects control the transition of a GPCR into an active structure? The answer is not yet clear. Although it has been known for some time that empty GPCRs are in equilibrium between inactive and active conformations, it has been difficult to quantify this dynamic aspect of their behavior using conventional methods. Instead, traditionally the relative distribution of each form has been assessed by measuring the basal level of G protein activity observed for the apoprotein form (1). Currently more and more GPCR crystal structures are becoming available that reveal various static conformations (15). However, it is also becoming increasingly clear that studies using other biophysical, spectroscopic, and computational methods will be needed to develop a full understanding of the role of structural dynamics in GPCR (3, 7, 16–18).
Opsin, the retinal-free form of rhodopsin, illustrates the difficulty of assessing structural dynamics in GPCRs from biochemical and crystallographic data. Opsin is almost exclusively present in an inactive conformation, as indicated by its extremely low G protein activity (19, 20) and infrared spectroscopy studies that find its spectra are like dark state rhodopsin at physiological pH, and unlike the photoactivated state metarhodopsin II (meta-II) (21). In contrast, the crystal structure of opsin has the characteristics of an active state receptor, including an outward displacement of TM6 (see Fig. 1A) (22). Thus, the true conformation of TM6 for opsin in detergent solution and lipid bilayers is not yet resolved.
Another example of the fluid nature of GPCR structures is the existence of mutations that elevate G protein activity of the empty receptor. These so-called constitutively activating mutations, or CAMs, have been identified in numerous GPCRs, and some are linked to genetic diseases (23). Although CAMs are generally assumed to somehow affect the dynamics and energetics of ligand-free receptor conformation(s), the mechanism through which they do so is still unclear.
Thus, in this article we set out to gain insight into an empty GPCR transition between inactive and active states, and to resolve the issues discussed above. Our approach was to use site-directed fluorescence labeling (SDFL). First, we placed a fluorescent probe (bimane, see Fig. 1A) on TM6 of opsin and on opsin containing a CAM, M257Y (24), and then compared their spectral properties to look for mutation-linked changes in the structure. Our results show that both in detergent micelles and in lipid conditions (using so-called nanodiscs, or NABBs) (25–27), opsin is constrained to an inactive conformation, whereas the CAM converts into a more active shape.
We further resolved the effect of the CAM on TM6 movement through use of a novel SDFL method called TrIQ (tryptophan-induced-quenching), which involved assessing the ability of a tryptophan residue on TM5 to quench the bimane probe on TM6. Interestingly, the TrIQ studies find that CAM M257Y causes a significantly increased population of receptor with a rearranged TM6, but only in detergent.
Importantly, the TrIQ studies also provide a key insight into how the CAM induces activation of opsin. Arrhenius analysis of the TrIQ data measured at different temperatures suggests that the CAM reduces the energy barrier for TM6 movement, both in detergent and lipids. Based on these results, we discuss the mechanism how a CAM can affect the conformational dynamics of a ligand-free GPCR.
EXPERIMENTAL PROCEDURES
Materials
The Gα-peptide 23V (NH2-VLEDLKSVGLF-COOH) used in this study is a variant of the high affinity analogues of transducin C-terminal 11 residues 340–350 (9, 28). The peptide was synthesized by GenScript. PDT-bimane ((2-pyridyl)dithiobimane) (see Fig. 1A) was purchased from Toronto Research Chemicals. All of the remaining materials used in this study are similar to those described previously (9, 12, 26).
Construction and Expression of Opsin Mutants
The background opsin (rhodopsin) mutant θ (C140S/C316S/C322S/C323S) was prepared as described previously (6). The mutations N2C, K231W, Q244C, M257Y, or D282C were introduced into θ using the QuikChange mutagenesis kit (Stratagene). The mutants were transiently expressed in COS-1 cells, and the cells were harvested 48 h after transfection.
PDT-bimane Labeling
Purification of the samples labeled with PDT-bimane was carried out on the 1D4 antibody Sepharose as described previously (9, 12), except all procedures were carried out in the absence of retinal. Label incorporation in opsin was estimated by comparing its absorbance at ∼380 nm before addition of 11-cis-retinal for the bimane amount and using the absorbance at ∼500-nm after the addition of 11-cis-retinal to quantify the amount of opsin. An excitation coefficient of ϵ380 = 5,000 for bimane and ϵ500 = 40,000 for 11-cis-retinal bound opsin (rhodopsin) were used for all calculations. The labeling efficiencies for 244B-opsin, M257Y/244B-opsin, 231W/244B-opsin, and M257Y/231W/244B-opsin were estimated as ∼1.1, ∼1.2, ∼1.1, and ∼1.2 label/opsin, respectively. The background opsin mutant was labeled with bimane at less than 0.1 label/opsin (Fig. 2, A–C). Labeled samples were prepared in 0.05% DDM, 5 mm MES, 40 mm NaCl, pH 6.
FIGURE 2.

PDT-bimane labeling on opsin mutants. A-C, example of specific incorporation of PDT-bimane label into position 244 in opsin mutant. Opsin mutants 244C/2C/282C/θ and 2C/282C/θ were labeled with PDT-bimane as described under “Experimental Procedures.” A, absorption spectrum of 2C/282C/θ opsin in DDM after labeling with PDT-bimane. B, absorption spectrum of 244C/2C/282C/θ opsin in DDM, after labeling with PDT-bimane (244B-opsin). C, fluorescence emission spectra using the same amount (500 nm) of both labeled opsins. Spectra clearly show that fluorescence of labeled 244C/2C/282C/θ opsin (244B-opsin, black) is much greater than that of labeled 2C/282C/θ opsin (red). This result indicates that PDT-bimane is specifically incorporated into Cys-244 not Cys-2/Cys-282 in opsin. Note that nonspecific bimane labeling on 2C/282C/θ opsin is less than 0.1 bimane/opsin. D, schematic representation of reduction of disulfide linkage between PDT-bimane and protein by a reducing reagent TCEP. E and F, normalization of fluorescence emission spectra by comparison to bimane emission after release from protein due to reduction of disulfide linkage. Solid lines indicate fluorescence emission spectra of 244B-opsin (black) and M257Y/244B-opsin (red) in DDM (E) and in nanodiscs (F). The red and black broken lines indicate the spectra of these same samples after release of the bimane label from the protein by reduction of the disulfide bond by TCEP (see “Experimental Procedures”). All fluorescence spectra are normalized to the fluorescence intensity after TCEP treatment.
Preparation and Purification of Nanodiscs
Nanodisc samples were prepared as described previously, using a lipid mixture previously found in our hands to produce optimal rhodopsin functional and spectroscopic properties (26, 27). Briefly, labeled sample (solubilized in 0.05% DDM), MSP, and lipid (mixture of 1-palmitoyl-2-oleoyl phosphatidylcholine and 1-palmitoyl-2-oleoyl phosphatidylglycerol at a 3:2 ratio solubilized in 0.5 m sodium cholate) were mixed with ∼2/3 volume of Bio-Beads SM-2 (Bio-Rad) overnight at 4 °C. The molar ratio of Rh:MSP:lipid was set to 0.1:1:75. The Bio-Beads were removed by centrifugation. After reconstitution into nanodiscs, the samples were injected onto a Superdex 200 column (GE Healthcare) with a column volume of 23.55 ml. The fraction containing nanodiscs that corresponds to a diameter of ∼12 nm was collected (26). The diameter of nanodisc was determined by a Gel Filtration Calibration Kit HMW (GE Healthcare). The collected sample was concentrated by Amicon Ultra 0.5-ml Centrifugal Filters (10,000 MWCO, Millipore). Nanodisc samples were prepared in 5 mm MES, 40 mm NaCl, pH 6.
Spectrophotometry
Absorption spectra of samples were recorded at 10 °C with a Shimadzu UV-1601 spectrophotometer, and photoactivation of the rhodopsin samples were carried out as previously described (26).
Steady-state Fluorescence Measurements
Fluorescence emission spectra were recorded using a PTI steady-state fluorescence spectrophotometer. Emission spectra were measured by excitation at 380 nm (0.5-nm slit) while scanning the fluorescence intensity of 405 to 605 nm (12-nm slit).
Release of PDT-bimane from Labeled Samples to Enable Normalization and Comparison of Spectra
The disulfide bond between PDT-bimane and rhodopsin/opsin was reduced using TCEP as previously described (Fig. 2, D–F) (9). This procedure enabled comparison between samples by normalizing the spectra to that of the TCEP reduced sample.
Fluorescence Lifetime Measurements
Fluorescence lifetimes for labeled opsins were measured using a FluoTime 200 TCSPC system (PicoQuant), with excitation from a 405-nm diode laser. Emission was monitored at 480-nm using a 1-nm slit and a >470-nm long pass filter. The instrument response function was determined using a solution of Ludox, with a FWHM (full-width at half-maximum) of ∼60 ps.
The fluorescence decays were analyzed by three different ways using the FluoFit program (PicoQuant), with all three approaches yielding χ2 values ranging from 0.9 to 1.3. For use in calculating the amount of static and dynamic quenching, the data were subjected to a three-exponential fit, and the average fluorescence lifetimes 〈τ〉, calculated as: 〈τ〉 = α1τ1 + α2τ2 + α3τ3, where α1, α2, and α3 are the fractional amplitudes of each lifetime τ1, τ2, and τ3, respectively. For the Arrhenius analysis, the data were fit to a single lifetime using a stretched exponential function (I(t) = αexp(−t/τ)β) (29). For comparison, the decay data were also fit using bimodal Lorentzian distribution, and the longer lifetime component (∼10 ns) used for subsequent Arrhenius analysis (data not shown).
Calculations of Parameters for TrIQ Study and Arrhenius Analysis
Detailed theory and method to calculate the parameters (Figs. 5–7) are described in Ref. 35. Briefly, the relative fraction of non-static fluorescence quenching components is defined as (Fw/Fo)(〈τo〉/〈τw〉), and the static quenching fraction is simply calculated as (1 − (Fw/Fo)(〈τo〉/〈τw〉)). The fraction of the dynamic quenching component is calculated as (Fw/Fo)(〈τo〉/〈τw〉) − Fw/Fo = Fw/Fo(〈τo〉/〈τw〉 − 1). In these calculations, the 〈τo〉 and 〈τw〉 values represent the averaged lifetimes obtained from the three exponential fits (see above). Note that Fw and 〈τw〉 represent the fluorescence intensity and average lifetime in the presence of the quenching tryptophan, and Fo and 〈τo〉 represent the corresponding values in the absence of the tryptophan.
FIGURE 5.
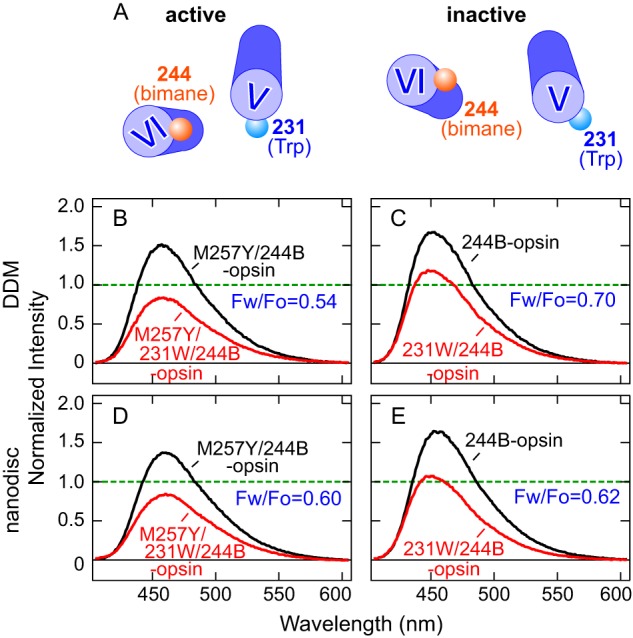
TrIQ studies detect that opsin with a constitutively activating mutation induces movement of TM6 toward TM5. A, a bimane label at position 2446.27 (on TM6) and a tryptophan at position 2315.66 (on TM5) are expected to show more TrIQ in the active form (and thus decreased fluorescence intensity), based on the respective crystal structures (Protein Data Bank codes 1GZM, 3CAP, and 3PQR). B-E, the emission spectra show ligand-free-labeled opsins in the absence (black) and presence of a Trp at position 2315.66 (red). B, M257Y/244B-opsin in DDM; C, 244B-opsin in DDM; D, M257Y/244B-opsin in nanodiscs; E, 244B-opsin in nanodiscs. The ratio values of fluorescence intensity in the absence and presence of 231W (Fw/Fo) are also indicated. The experiments were carried out at 10 °C.
FIGURE 6.
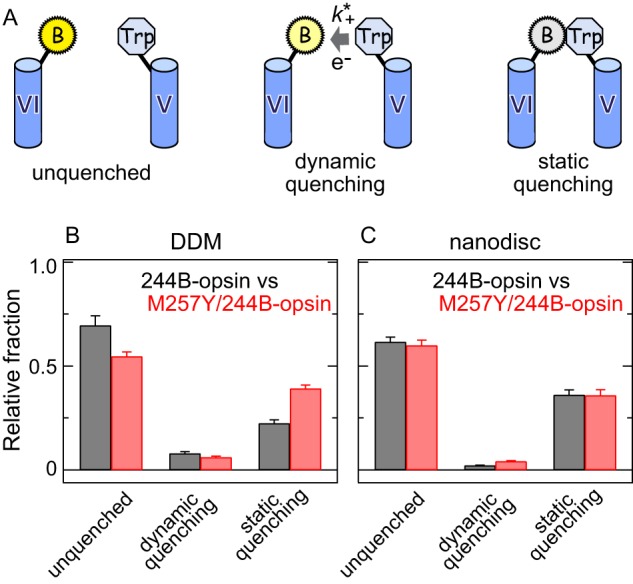
The M257Y CAM shifts the population of receptor conformations toward active ones, as indicated by the changes in the amount of static and dynamic quenching of the bimane at site 2446.27 by the Trp at site 2315.66. A, schematic scheme of the different quenching species that can be detected by TrIQ analysis; unquenched, dynamic quenching, and static quenching. B and C, steady-state fluorescence data in Fig. 5 with the fluorescence lifetime data allows calculation of the relative fraction of unquenched, dynamically quenched, and statically quenched in 244B-opsin (black) and M257Y/244B-opsin (red) in DDM micelles (B) and in nanodiscs (C). The error bars represent the S.E. for three independent experiments. The calculation of different quenching fractions is described under “Experimental Procedures,” using the Fw/Fo values shown in Fig. 5, and the following ratio of average fluorescence lifetimes (〈τo〉/〈τw〉) obtained from three exponential fitting of the decay data: 244B in DDM (1.09 ± 0.02), 244B in nanodiscs (1.04 ± 0.01), M257Y/244B in DDM (1.13 ± 0.01), M257Y/244B in nanodiscs (1.07 ± 0.01). The experiments were carried out at 10 °C.
FIGURE 7.
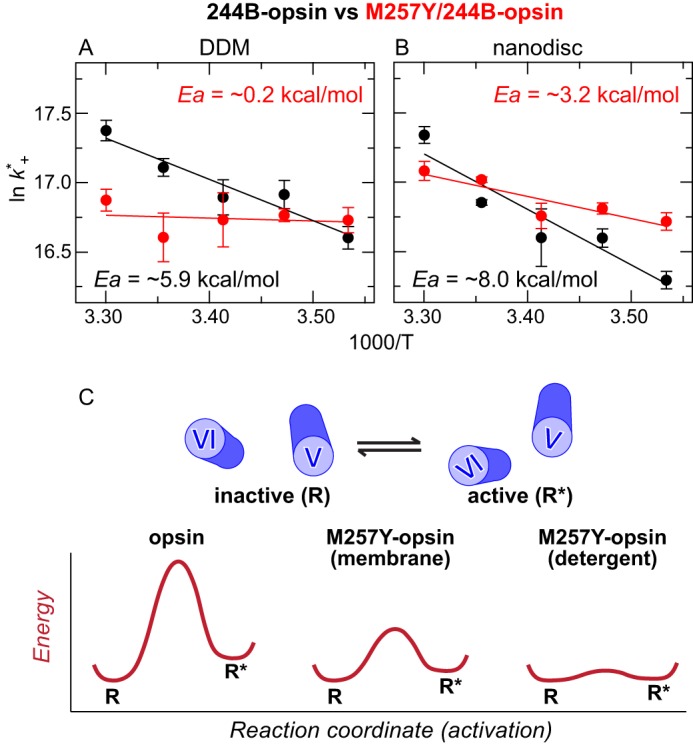
The M257Y CAM appears to lower the activation energy for TM5/TM6 movement. A and B, Arrhenius plots of the dynamic quenching constants (k+*) of the Trp at position 2315.66 and the bimane probe at position 2446.27 in opsins as a function of temperature. Calculation of k+* values were obtained using the single lifetime values obtained from analyzing the decay using a stretched exponential function (I(t) = αexp(−t/τ)β) (29). The τw and τo values used are reported in supplemental Table S1. Black and red circles indicate 244B-opsin and M257Y/244B-opsin, respectively, in DDM (A) and nanodiscs (B). The calculated Ea values of 244B-opsin (black) and M257Y/244B-opsin (red) are provided. The error bars represent the S.E. for three independent experiments. Further details are provided under “Experimental Procedures.” C, simplified two-dimensional energy landscape of opsin with or without M257Y CAM. This schematic shows the energy of the receptor conformations is plotted along the activation pathway (2). Note that virtually the same conclusion was obtained from Arrhenius plots based on k+* values that were calculated using either averaged lifetime values (〈τ〉), or the long-lifetime component obtained from a bimodal Lorentzian distribution.
The dynamic quenching rate constant, k+*, used in the Arrhenius analysis (see Fig. 7 and supplemental Table S1) was determined as k+* = (1/τw − 1/τo). For these calculations, the τw and τo lifetime values were obtained by fitting the decay data to a single lifetime using a stretched exponential function, I(t) = αexp(−t/τ)β (29). The τw and τo values used in Fig. 7 are shown in supplemental Table S1.
RESULTS
Structural changes accompanying GPCR activation can be studied by placing spectroscopic probes at site 2446.27 on TM6 (the superscript gives the Ballesteros-Weinstein numbering (18)) (Fig. 1). In rhodopsin, activation causes a spin label at position 2446.27 to show increased mobility (3). Similarly, when the fluorescent label bimane is attached to the same site, activation causes a red-shifted fluorescence emission λmax, both in rhodopsin (12) and in the β-adrenergic receptor (βAR, labeled at position 2656.27) (30). These spectroscopic changes are consistent with crystal structures of dark and meta-II states of rhodopsin (10, 11, 31, 32), which show the residue at position 2446.27 moves from the interior of the protein to outside the protein in meta-II state (Figs. 1A and 3A). We therefore placed a bimane label at this site in opsin to monitor conformational changes.
FIGURE 3.

Fluorescence spectra of opsin, 11-cis-retinal bound rhodopsin, and the photoactivated form (meta-II) of 244B and M257Y/244B in DDM and nanodiscs. A, schematic model indicating the different relative orientations of TM5 and TM6 between 11-cis-retinal bound rhodopsin (black) and the photoactivated meta-II (red). B, schematic model indicating where meta-II interacts with the Gα C terminus peptide. C, the interaction site is absent in the inactive rhodopsin. D, comparison of fluorescence emission spectra of the retinal-free 244B-opsin (blue, λmax = 451 nm), 11-cis-retinal-bound 244B-rhodopsin (black, λmax = 451 nm), and the photoactivated 244B-meta-II (red, λmax = 463 nm) in DDM. Inset, the spectra normalized by maximal fluorescence intensity as 1.0. E, fluorescence spectra of 244B-meta-II in the absence (red, λmax = 463 nm) and presence (purple, λmax = 470 nm) of Gα-peptide in DDM. F, fluorescence spectra of 244B-rhodopsin in the absence (black, λmax = 451 nm) and presence (purple, λmax = 451 nm) of Gα-peptide in DDM. G, comparison of fluorescence spectra of M257Y/244B-opsin (blue, λmax = 456 nm), M257Y/244B-rhoodpsin (black, λmax = 452 nm), and M257Y/244B-meta-II (red, λmax = 462 nm) in DDM. Inset, the spectra normalized by maximal fluorescence intensity as 1.0. H, fluorescence spectra of M257Y/244B-meta-II in the absence (red, λmax = 462 nm) and presence (purple, λmax = 469 nm) of Gα-peptide in DDM. I, fluorescence spectra of M257Y/244B-rhodopsin in the absence (black, λmax = 452 nm) and presence (purple, λmax = 452 nm) of Gα-peptide in DDM. J, comparison of fluorescence spectra of 244B-opsin (blue, λmax = 454 nm), 244B-rhodopsin (black, λmax = 455 nm), and 244B-meta-II (red, λmax = 464 nm) in nanodiscs. Inset, the spectra normalized by maximal fluorescence intensity as 1.0. K, fluorescence spectra of 244B-meta-II in the absence (red, λmax = 464 nm) and presence (purple, λmax = 468 nm) of Gα-peptide in nanodiscs. L, fluorescence spectra of 244B-rhodpsin in the absence (black, λmax = 455 nm) and presence (purple, λmax = 455 nm) of Gα-peptide in nanodiscs. M, comparison of fluorescence spectra of M257Y/244B-opsin (blue, λmax = 460 nm), M257Y/244B-rhodopsin (black, λmax = 460 nm) and M257Y/244B-meta-II (red, λmax = 464 nm) in nanodiscs. Inset, the spectra normalized by maximal fluorescence intensity as 1.0. N, fluorescence spectra of M257Y/244B-meta-II in the absence (red, λmax = 464 nm) and presence (purple, λmax = 469 nm) of Gα-peptide in nanodiscs. O, fluorescence spectra of M257Y/244B-rhodopsin in the absence (black, λmax = 460 nm) and presence (purple, λmax = 460 nm) of Gα-peptide in nanodiscs. To enable direct comparison, all spectra are normalized to the spectrum of free bimane released from the protein by TCEP reduction of the attaching disulfide (see Fig. 2, D–F, and “Experimental Procedures”). Concentrations of labeled opsin and Gα-peptide are 500 nm and 100 μm, respectively. The experiments were carried out at 10 °C.
The Fluorescent Probe PDT-bimane Can Be Specifically Attached on TM6 at Position 2446.27 in Opsin without Perturbing the Receptor Structure
Our SDFL studies required us to uniquely and specifically attach a fluorescent probe on TM6 of opsin. To achieve this, we introduced mutation Q244C into a non-reactive background opsin mutant called “θ” (see “Experimental Procedures”), and we also introduced two cysteine substitutions, N2C and D282C, which Oprian and colleagues (33) have shown to form an intra-molecular disulfide bond that makes opsin highly stable in detergent. The resulting opsin mutant Q244C/N2C/D282C/θ, was expressed, purified, and labeled with PDT-bimane (see Fig. 1A).
PDT-bimane was used because it attaches to the protein with a disulfide linkage that can be cleaved with a reducing agent TCEP (Fig. 2D). This feature makes possible quantitative comparison between samples (34). As shown in Fig. 2, E and F, to enable comparison, we thus normalized the fluorescence intensity data for the different labeled opsin to their fluorescence intensity after release of probe by TCEP reduction.
The PDT-bimane-labeled 244C/2C/282C/θ opsin mutant, hereafter referred to as 244B-opsin, showed two absorption peaks, a protein peak at ∼280 nm and a bimane peak at ∼380 nm (Fig. 2A). In contrast, the labeled “background” mutant, 2C/282C/θ opsin, showed only a single absorption peak at ∼280 nm (Fig. 2B), and negligible fluorescence (Fig. 2C). These results indicate specific attachment of the bimane to Cys-244 on 244B-opsin, and no significant background labeling of 2C/282C/θ. In a similar fashion, we also prepared the PDT-bimane-labeled opsin containing the CAM M257Y (24), hereafter referred to as M257Y/244B-opsin.
The 244B-opsin and M257Y/244B-opsin were prepared both in DDM micelles and incorporated into small lipid particle nanodiscs (25). The samples showed normal ability to bind 11-cis-retinal to form 244B-rhodopsin and M257Y/244B-rhoodpsin, and they form the meta-II state (244B-meta-II and M257Y/244B-meta-II) upon light irradiation (indicated by the decrease in 500 nm and increase in 380-nm absorbance, data not shown).
The Bimane Probe on TM6 Detects Differences between the Inactive and Active Receptor Forms, in Both Detergent and Lipids
We first assessed TM6 conformations by comparing the fluorescence emission λmax of the labeled opsins. In general, when a bimane fluorophore is brought into a more polar environment, the λmax of the fluorescence spectrum is red-shifted and the fluorescence intensity decreases (30, 34). However, for the rhodopsin samples, we had to first take into account the fact that binding of 11-cis-retinal to 244B-opsin and M257Y/244B-opsin caused a decrease in fluorescence intensity, and photoactivation to meta-II caused an increase in intensity (Fig. 3, D, G, J, and M). We have previously established that these dramatic changes in fluorescence intensity are primarily due to varying amounts of fluorescence energy transfer from the bimane to the retinal in the different rhodopsin forms (6, 7, 12). In dark-state rhodopsin, the fluorescence of bimane overlaps substantially with the retinal absorbance, resulting in significant energy transfer, whereas in the meta-II form, this overlap is diminished (but not completely abolished), resulting in the partial increase in bimane fluorescence (see Fig. 3, D, G, J, and M). Thus, because of this additional complication, when we studied retinal containing samples, we did not compare intensities, but rather assessed the environment around the bimane label by comparing shifts in the λmax of emission, which are not affected by the amount of energy transfer but are representative of local environmental polarity (34). We compared fluorescence intensities only for opsin samples that contained no retinal.
In DDM micelles, the emission λmax value of 244B-rhodopsin was almost identical to that of (retinal-free) 244B-opsin (inset of Fig. 3D). However, 244B-meta-II showed a substantial red shift in the bimane emission λmax (inset of Fig. 3D), indicating a change in the environment around the bimane label, as we have seen previously (12). Interestingly, the bimane emission λmax for M257Y/244B-opsin was between those of M257Y/244B-rhodopsin and M257Y/244B-meta-II (inset of Fig. 3G), suggesting that the M257Y CAM produces an intermediate state and/or a mixture of inactive and active conformations.
In nanodiscs, the bimane emission λmax for 244B-opsin was similar to that of 244B-rhodopsin, although the latter possessed a broader spectral shape (inset of Fig. 3J). Interestingly, in nanodiscs, M257Y/244B-opsin had a similar λmax to the M257Y/244B-rhodopsin (inset of Fig. 3M), suggesting that in lipids, conformational differences between ligand-free and 11-cis-retinal bound states of M257Y/244B are less than in detergent. 244B-meta-II and M257Y/244B-meta-II showed a red-shifted λmax (see insets of Fig. 3, J and M), although the light-dependent shifts were smaller in nanodiscs than in DDM.
As shown in Fig. 4, these results are even clearer when opsin (non-retinal containing) fluorescence spectra are compared. The spectra show that the M257Y mutation moves the bimane at position 2446.27 on TM6 into a more polar environment, as indicated by the red-shifted λmax and a decreased intensity of M257Y/244B-opsin compared with 244B-opsin, in both DDM and nanodiscs (Fig. 4, A and D).
FIGURE 4.
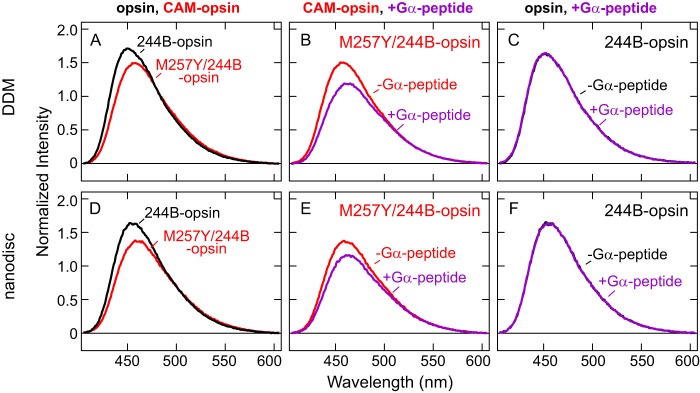
Comparison of fluorescence spectra of 244B-opsin and M257Y/244B-opsin in DDM and nanodiscs. A, fluorescence emission spectra of 244B-opsin (black, λmax = 451 nm) and M257Y/244B-opsin (red, λmax = 456 nm) in DDM. B, fluorescence spectra of M257Y/244B-opsin in the absence (red, λmax = 456-nm) and presence (purple, λmax = 462-nm) of Gα-peptide in DDM. C, fluorescence spectra of 244B-opsin in the absence (black, λmax = 451 nm) and presence (purple, λmax = 451 nm) of Gα-peptide in DDM. D, fluorescence spectra of 244B-opsin (black, λmax = 454 nm) and M257Y/244B-opsin (red, λmax = 460 nm) in nanodiscs. E, fluorescence spectra of M257Y/244B-opsin in the absence (red, λmax = 460 nm) and presence (purple, λmax = 463 nm) of Gα-peptide in nanodiscs. F, fluorescence spectra of 244B-opsin in the absence (black, λmax = 454 nm) and presence (purple, λmax = 454 nm) of Gα-peptide in nanodiscs. The experiments were carried out at 10 °C.
We next investigated spectral changes in the bimane-labeled opsins caused by binding of Gα-peptide, a high-affinity G protein mimetic of the transducin Gα C terminus (9, 28). The Gα-peptide binds to a hydrophobic patch on the inner face of TM6 that is exposed upon meta-II formation (9–11) (see Fig. 3B). We anticipated that interaction of opsin with the peptide could induce (or stabilize) conformational changes around position 2446.27 on TM6. In both DDM and nanodiscs, no shifts were seen when the Gα-peptide was added to (inactive) 244B-rhodopsin or M257Y/244B-rhodopsin (Fig. 3, F, I, L, and O), but shifts were observed when it was added to 244B-meta-II and M257Y/244B-meta-II (Fig. 3, E, H, K, and N).
Similarly, addition of the Gα-peptide caused no effect to fluorescence emission of 244B-opsin (Fig. 4, C and F). In contrast, the Gα-peptide caused a red-shifted emission λmax and decrease in intensity for the M257Y/244B-opsin (Fig. 4, B and E), indicating the bimane probe had moved into a more polar environment. These shifts also show the Gα-peptide can bind to M257Y/244B-opsin, but not 244B-opsin, consistent with the extremely low inherent G protein activity (20) of opsin and the dramatically elevated activity previously observed for the CAM M257Y (24).
Taken together, these data demonstrate the ligand-free 244B-opsin, as well as 11-cis-retinal bound 244B-rhodopsin and M257Y/244B-rhodopsin are highly constrained to an inactive conformation (Fig. 3C), whereas the ligand-free M257Y/244B-opsin as well as photoactivated (all-trans-retinal bound) 244B-meta-II and M257Y/244B-meta-II can interact with the Gα-peptide, resulting in a stabilization of the active conformation.
Tryptophan-induced Quenching (TrIQ) Studies Show Different Amounts of TM6 Movement for the CAM M257Y Opsin in Detergent and Lipids
Figs. 3 and 4 suggest that CAM M257Y causes some conformational changes of TM6 in opsin. To better resolve this phenomena, we next studied TM6 movement using the TrIQ method, which we have extensively calibrated (34–36), and used to study various rhodopsins (12) and the βAR (14).
Briefly, the TrIQ method measures how efficiently a Trp can quench the fluorescence of a nearby bimane probe. TrIQ only occurs over short distances (within ∼15 Å), and is distance-dependent. To assess how the M257Y CAM causes TM6 to move relative to TM5, we introduced a Trp at position 2315.66 (231W) on TM5 for both 244B-opsin and M257Y/244B-opsin. The Cα-Cα distance between these sites goes from 12.6 Å in the inactive rhodopsin crystal structure (31) to 8.5 Å in the active meta-II structure (10) (Figs. 1A and 5A). Thus, more TrIQ should occur between a Trp at site 2315.66 and a bimane at site 2446.27 in the active receptor conformation, and be observed as lower fluorescence intensity.
We first confirmed that the introduction of Trp-231 does not alter the 11-cis-retinal binding or photobleaching properties for the regenerated rhodopsin mutants (data not shown). In DDM, the bimane fluorescence was quenched by Trp-231 more efficiently in M257Y/244B-opsin than in 244B-opsin (Fig. 5, B and C), but in nanodiscs, the quenching efficiency is almost the same for both opsins (Fig. 5, D and E), suggesting that in the detergent micelles, the M257Y mutation induces a significant rearrangement of TM5 and TM6 that is not observed in lipids.
The M257Y CAM Increases the Relative Population of Active Receptor Conformation in Detergent, as Indicated by the Amount of Static and Dynamic Fluorescence Quenching
We next more precisely assessed the proximity between residues at positions 2315.66 and 2446.27 by quantifying the amount of dynamic and static quenching caused by the Trp residue on the bimane probe. Dynamic quenching occurs when the bimane interacts with the Trp quencher after excitation of the fluorophore, whereas static quenching occurs when the two form a complex before light excitation (35) (see Fig. 6A). Thus, static quenching is especially useful in TrIQ studies as it identifies species where the fluorophore and quencher are within contact distance during the time of light excitation (35).
Quantifying the amount of dynamic and static quenching requires measuring the fluorescence lifetimes of the samples and comparing this data with the steady-state fluorescence data. Thus, the fluorescence lifetimes of the samples were measured using an instrument with an excellent time resolution (∼60 ps). These data were then used to calculate the relative fraction of static quenching species, defined as (1 − (Fw/Fo)(〈τo〉/〈τw〉)) (see “Experimental Procedures”) (35), where Fw and 〈τw〉 represent the fluorescence intensity and average lifetime in the presence of the quenching tryptophan, respectively, and Fo and 〈τo〉 represent the corresponding values in the absence of the quenching tryptophan.
Our analysis finds that the fraction of static quenching species increased for M257Y/244B-opsin in DDM (Fig. 6B) but not in nanodiscs (Fig. 6C), indicating that at any given moment, positions 2315.66 and 2446.27 are closer together in the M257Y mutation when in detergent rather than lipids. The results suggest that the M257Y mutation induced significant rearrangement of TM5 and TM6 in detergent reflecting a shift of the relative population of “active” and “inactive” conformations (see “Discussion”), but the CAM had much less effect for opsin in lipid membranes, at least at 10 °C.
Arrhenius Analysis of the TrIQ Data Suggests M257Y CAM Lowers the Activation Energy for TM5/TM6 Movement
We next assessed how the CAM affected the energetic barrier to TM6 movement in opsins. To do this, we measured the rate constant for the formation of a dynamic quenching complex between the bimane on TM6 (position 2446.27) by the Trp residue on TM5 (position 2315.66) as a function of different temperatures, and analyzed the results using an Arrhenius plot (Fig. 7, A and B) (37). Specifically, we determined the dynamic quenching constant k+* (defined as ((1/τw) − (1/τo))) (35) from fluorescence lifetime measurements made at different temperatures, and plotted this on the y axis and the inverse temperature on the x axis. For these calculations, the τ values were determined using a stretched exponential function, to enable calculation of single k+* values for the Arrhenius analysis (29).
We stress that the k+* values do not directly represent the formation of an active, stable receptor conformation, R*, nor tell how long R* would remain in that state. The k+* values only report how often the Trp residue on TM5 collides with the probe on TM6. Because each movement does not necessarily produce R*, neither the amount of R* formation nor its stability can be determined from absolute values of k+*. However, the temperature-dependent changes of k+* values do provide a unique way to assess the underlying energetic barrier (Ea) to TM6 movement that must be overcome so that this key, initial step in R* formation can occur.
Interestingly, these analyses show that for both DDM (Fig. 7A) and nanodiscs (Fig. 7B), the M257Y mutation decreases the slopes of the plot, indicating a lower activation energy for the dynamic quenching complex formation. The same effect was observed in our other Arrhenius plots calculated using dynamic quenching k+* values obtained from different fitting approaches for the lifetime data (see “Experimental Procedures”).4 Because positions 2315.66 and 2446.27 are closer in an active conformation (see Fig. 5A), the lower activation energy strongly suggests that the M257Y CAM lowers the energy barrier for the TM5/TM6 movement that must occur before an inactive receptor can be converted into an active conformation. A possible mechanism underlying the CAM-induced changes on the conformational dynamics is discussed below.
DISCUSSION
In this study, we probed the mechanism and energetics underlying dynamic transitions involved in GPCR activation, by using SDFL to compare the dynamic features of opsin with or without a CAM in both solution and lipid membranes. As discussed below, our data reveal differences in how a CAM can affect receptor dynamics in detergent compared with lipids, and provide new insights into how a CAM can change a ligand-free GPCR structure.
Opsin Conformation in Detergent Micelles and Lipid Bilayer
Previous infrared spectroscopic (21) and biochemical studies (19, 20) have concluded that opsin has an inactive conformation under physiological conditions, yet surprisingly, the recent crystal structures of opsin possess many features of an active conformation (22) (see Fig. 1). In our present study, we find that features of ligand-free opsin are more similar to an inactive form rather than to an active form in both detergent and lipid environments, at least regarding the environment of a probe on TM6. The probe at position 2446.27 in opsin showed a very similar λmax to the 11-cis-retinal bound (inactive) state (insets of Fig. 3, D and J), and both 244B-rhodopsin and 244B-opsin were insensitive to Gα-peptide (Figs. 3, F and L, and 4, C and F). In contrast, the light-activated 244B-meta-II showed a red-shifted λmax from 244B-rhodopsin (insets of Fig. 3, D and J), and a further red shift was induced by the Gα-peptide (Fig. 3, E and K). Taken together, these results suggest that opsin is highly constrained to an inactive conformation in both detergent and lipid environments.
Why then do the crystal structures of opsin possess features of an active conformation, with a movement of TM6 (22)? One possibility is that the solubilized opsin used for the crystallization possesses a more flexible structure than membrane-embedded opsin, and the active conformation is stabilized in the crystals. This idea is consistent with the result from Hubbell and co-workers (38) that found the lipid environment reduces flexibility of rhodopsin conformations. The higher flexibility of opsin in detergent is more prominent in the presence of the CAM M257Y, resulting in larger conformational changes in TM 5/6 (Figs. 5 and 6, see below).
Effect of a Constitutively Activating Mutation on Receptor Dynamic
Our results indicate that the M257Y CAM converts opsin into a species with enough active-state features to bind the Gα C terminus. Opsin containing this mutation shows a red shift and decrease in intensity for a bimane probe at site 2446.27 on TM6 (compare M257Y/244B-opsin with 244B-opsin in Fig. 4, A and D). These changes are further enhanced upon addition and binding of Gα-peptide (Fig. 4, B and E). Interestingly, studies of βAR find that a bimane probe on the equivalent site exhibits a similar red shift and decrease in intensity upon G protein binding, even when no CAM is present in the receptor (30). In contrast, opsin requires the M257Y CAM to show Gα-peptide-dependent fluorescence changes (compare Fig. 4, C and F, with B and E). These differences are consistent with observations that, like M257Y opsin (24), wild-type βAR possess a significant basal G protein activity (1), whereas wild-type opsin does not (20).
How does the CAM exert its effect on the conformational dynamics of opsin? One way to address this question is through the use of a simplified two-dimensional energy landscape (Fig. 7C), where the energy of the receptor conformations is plotted along the activation pathway (2). As described by Deupi and Kobilka (2), this representation of the receptor energy landscape has the following characteristics. 1) The energy minima correspond to inactive (R) and active (R*) conformations that are separated by a region of higher energy (energy barrier). 2) The rate of transition between R and R* is determined by the height of the barrier. 3) The relative population of R and R* is determined by their relative difference in energy levels (2).
For normal opsin, the R and R* states are drawn separated by an energy barrier (Fig. 7C, left). Note that the energy level of R is also drawn as significantly lower than R*, to reflect our results and previous studies (19–21) that find opsin is constrained to the inactive R state. The combination of these two factors would mean transition from R to R* is very unlikely.
This model can be used to explain the effect of M257Y CAM on opsin dynamics and the constitutive activity. We propose the M257Y CAM works by decreasing the energy barrier between R and R* (Fig. 7C, middle and right), resulting in more frequent productive transitions between R and R*. If the difference in energy levels of R and R* is also sufficiently decreased, the result will be an increase in the relative population of R*. Such a change in receptor dynamics was observed in a recent NMR and simulation study for agonist-induced activation of βAR in detergent solution (17).
Interestingly, the effect of the M257Y CAM is more dramatic in detergent than in lipids. For M257Y opsin in detergent, the apparent energy barrier to TM5/TM6 movement is substantially decreased (Fig. 7A), and significant movement is observed (Figs. 5 and 6). Together, these two factors, the decrease the energy barrier and the difference in energy level between R and R*, result in an increase of the R* population.
In contrast, in nanodiscs, although the CAM does lower the energy barrier, the effect is less than in detergent (Fig. 7, B and C), and little CAM-induced movement of TM5/TM6 is observed (less than detection limit of our TrIQ method) (Figs. 5D and 6C). Together, these data suggest that in lipids, M257Y CAM significantly affects opsin dynamics, but does not significantly alter the relative stable population of R and R*.
How then is it possible for the M257Y CAM opsin to still be constitutively active in lipids? The lower energy barrier for TM6 movement for M257Y opsin in lipids would still result in an increase in the transition rate between R and R*, thus resulting in the adoption of enough constitutively active form (R*) to enable binding of the Gα-peptide and the shift of the fluorescence emission spectra we observed for M257Y/244B-opsin (Fig. 4, B and E). It is important to not overgeneralize about these observations. Without further studies, we cannot rule out the possibility that the M257Y CAM effect is specific to opsin in this combination of lipids in nanodiscs. Certainly the magnitude of the effect could vary with different lipid combinations, as well as different CAMs, and different GPCRs. However, we feel the results noted here are generally relevant for rhodopsin, because rhodopsin under these exact same conditions shows optimal arrestin binding (26) and photoactivation processes like native rod outer segment membranes (27). Thus, it is unlikely that the effect seen here for M257Y is due to some distortion of opsin structural features caused only by the specific combination of lipids and nanodiscs used in the present study.
Why does the M257Y mutation affect the energetics and dynamics of opsin conformations? Photoactivation of rhodopsin involves relocation of water molecules inside the proteins and rearrangement of a hydrogen-bonding network around TM6 (11, 39, 40), leading Deupi et al. (40) to propose “mutations of Met-257 thus likely reduces the energy barrier between the active and inactive states by disturbing the hydrophobic barrier between the intramolecular water-mediated hydrogen bond network and the cytoplasmic side that prevents helix movement.” Our experimental data obtained on ligand-free M257Y opsin are in full agreement with this concept.
Insight into CAMs in Other GPCRs
A number of CAMs have been identified in different GPCRs, and some are known to be involved in genetic diseases (23). Our results here provide new insights into how some CAMs can result in G protein activation. They show that it may not be necessary for a CAM to induce dramatic new changes in the static conformation of a receptor. Rather, under some conditions, a CAM can exert its effect by altering the energetics and dynamics of receptor conformations, for example, by decreasing the activation energy barrier in conversion of the inactive to active receptor conformations. As seen in our detergent experiments, such a primary effect of a CAM on receptor dynamics can lead to shifting the relative population of active and inactive receptor conformations.
Finally, we note that the novel approach described here should prove applicable to the study of other proteins. The use of static crystallographic structures to guide fluorescence TrIQ studies can provide a unique, powerful way to gain mechanistic insights into the role of dynamics and energetics underlying protein function.
Acknowledgement
We thank Mark DeWitt for preparing MSP proteins.
This work was supported, in whole or in part, by National Institutes of Health Grant EY015436 (to D. L. F.).

This article contains supplemental Table S1.
During the preparation of this manuscript, we fit the fluorescence decay data using three different approaches (stretched exponential function (Fig. 7, A and B), three exponential fit and bimodal Lorentzian distribution, see “Experimental Procedures”), and used these values to calculate k+*. Although the different fitting procedures produced slightly different k+* values, the same trend of temperature-dependent changes in k+* values and Arrhenius values were observed in all cases (data not shown).
- GPCR
- G protein-coupled receptor
- βAR
- β-adrenergic receptor
- CAM
- constitutively activating mutation
- DDM
- n-dodecyl-β-d-maltoside
- Gα-peptide
- peptide derived from C terminus of G protein α-subunit
- k+*
- dynamic quenching constant
- meta-II
- metarhodopsin II
- SDFL
- site-directed fluorescence labeling
- TM
- transmembrane helix
- TrIQ
- tryptophan-induced quenching
- PDT-bimane
- (2-pyridyl)dithiobimane
- TCEP
- tris(2-carboxyethyl)phosphine.
REFERENCES
- 1. Kobilka B. K. (2007) G protein-coupled receptor structure and activation. Biochim. Biophys. Acta 1768, 794–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deupi X., Kobilka B. K. (2010) Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology 25, 293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Altenbach C., Yang K., Farrens D. L., Farahbakhsh Z. T., Khorana H. G., Hubbell W. L. (1996) Structural features and light-dependent changes in the cytoplasmic interhelical E-F loop region of rhodopsin. A site-directed spin-labeling study. Biochemistry 35, 12470–12478 [DOI] [PubMed] [Google Scholar]
- 4. Farrens D. L., Altenbach C., Yang K., Hubbell W. L., Khorana H. G. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274, 768–770 [DOI] [PubMed] [Google Scholar]
- 5. Sheikh S. P., Zvyaga T. A., Lichtarge O., Sakmar T. P., Bourne H. R. (1996) Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature 383, 347–350 [DOI] [PubMed] [Google Scholar]
- 6. Dunham T. D., Farrens D. L. (1999) Conformational changes in rhodopsin. Movement of helix f detected by site-specific chemical labeling and fluorescence spectroscopy. J. Biol. Chem. 274, 1683–1690 [DOI] [PubMed] [Google Scholar]
- 7. Farrens D. L. (2010) What site-directed labeling studies tell us about the mechanism of rhodopsin activation and G-protein binding. Photochem. Photobiol. Sci. 9, 1466–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knierim B., Hofmann K. P., Ernst O. P., Hubbell W. L. (2007) Sequence of late molecular events in the activation of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 104, 20290–20295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janz J. M., Farrens D. L. (2004) Rhodopsin activation exposes a key hydrophobic binding site for the transducin α-subunit C terminus. J. Biol. Chem. 279, 29767–29773 [DOI] [PubMed] [Google Scholar]
- 10. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 11. Standfuss J., Edwards P. C., D'Antona A., Fransen M., Xie G., Oprian D. D., Schertler G. F. (2011) The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsukamoto H., Farrens D. L., Koyanagi M., Terakita A. (2009) The magnitude of the light-induced conformational change in different rhodopsins correlates with their ability to activate G proteins. J. Biol. Chem. 284, 20676–20683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gether U., Lin S., Ghanouni P., Ballesteros J. A., Weinstein H., Kobilka B. K. (1997) Agonists induce conformational changes in transmembrane domains III and VI of the β2 adrenoceptor. EMBO J. 16, 6737–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yao X., Parnot C., Deupi X., Ratnala V. R., Swaminath G., Farrens D., Kobilka B. (2006) Coupling ligand structure to specific conformational switches in the β2-adrenoceptor. Nat. Chem. Biol. 2, 417–422 [DOI] [PubMed] [Google Scholar]
- 15. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 16. Ye S., Zaitseva E., Caltabiano G., Schertler G. F., Sakmar T. P., Deupi X., Vogel R. (2010) Tracking G-protein-coupled receptor activation using genetically encoded infrared probes. Nature 464, 1386–1389 [DOI] [PubMed] [Google Scholar]
- 17. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K. (2013) The dynamic process of β2-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 19. Cohen G. B., Yang T., Robinson P. R., Oprian D. D. (1993) Constitutive activation of opsin. Influence of charge at position 134 and size at position 296. Biochemistry 32, 6111–6115 [DOI] [PubMed] [Google Scholar]
- 20. Melia T. J., Jr., Cowan C. W., Angleson J. K., Wensel T. G. (1997) A comparison of the efficiency of G protein activation by ligand-free and light-activated forms of rhodopsin. Biophys. J. 73, 3182–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vogel R., Siebert F. (2001) Conformations of the active and inactive states of opsin. J. Biol. Chem. 276, 38487–38493 [DOI] [PubMed] [Google Scholar]
- 22. Park J. H., Scheerer P., Hofmann K. P., Choe H. W., Ernst O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 23. Thompson M. D., Percy M. E., McIntyre Burnham W., Cole D. E. (2008) G protein-coupled receptors disrupted in human genetic disease. Methods Mol. Biol. 448, 109–137 [DOI] [PubMed] [Google Scholar]
- 24. Han M., Smith S. O., Sakmar T. P. (1998) Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry 37, 8253–8261 [DOI] [PubMed] [Google Scholar]
- 25. Bayburt T. H., Sligar S. G. (2010) Membrane protein assembly into Nanodiscs. FEBS Lett. 584, 1721–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsukamoto H., Sinha A., DeWitt M., Farrens D. L. (2010) Monomeric rhodopsin is the minimal functional unit required for arrestin binding. J. Mol. Biol. 399, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsukamoto H., Szundi I., Lewis J. W., Farrens D. L., Kliger D. S. (2011) Rhodopsin in nanodiscs has native membrane-like photointermediates. Biochemistry 50, 5086–5091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin E. L., Rens-Domiano S., Schatz P. J., Hamm H. E. (1996) Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J. Biol. Chem. 271, 361–366 [DOI] [PubMed] [Google Scholar]
- 29. Shi X., Duft D., Parks J. H. (2008) Fluorescence quenching induced by conformational fluctuations in unsolvated polypeptides. J. Phys. Chem. B 112, 12801–12815 [DOI] [PubMed] [Google Scholar]
- 30. Yao X. J., Vélez Ruiz G., Whorton M. R., Rasmussen S. G., DeVree B. T., Deupi X., Sunahara R. K., Kobilka B. (2009) The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc. Natl. Acad. Sci. U.S.A. 106, 9501–9506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li J., Edwards P. C., Burghammer M., Villa C., Schertler G. F. (2004) Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 343, 1409–1438 [DOI] [PubMed] [Google Scholar]
- 32. Okada T., Sugihara M., Bondar A. N., Elstner M., Entel P., Buss V. (2004) The retinal conformation and its environment in rhodopsin in light of a new 2.2-Å crystal structure. J. Mol. Biol. 342, 571–583 [DOI] [PubMed] [Google Scholar]
- 33. Xie G., Gross A. K., Oprian D. D. (2003) An opsin mutant with increased thermal stability. Biochemistry 42, 1995–2001 [DOI] [PubMed] [Google Scholar]
- 34. Mansoor S. E., Farrens D. L. (2004) High-throughput protein structural analysis using site-directed fluorescence labeling and the bimane derivative (2-pyridyl)dithiobimane. Biochemistry 43, 9426–9438 [DOI] [PubMed] [Google Scholar]
- 35. Mansoor S. E., Dewitt M. A., Farrens D. L. (2010) Distance mapping in proteins using fluorescence spectroscopy. The tryptophan-induced quenching (TrIQ) method. Biochemistry 49, 9722–9731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mansoor S. E., McHaourab H. S., Farrens D. L. (2002) Mapping proximity within proteins using fluorescence spectroscopy. A study of T4 lysozyme showing that tryptophan residues quench bimane fluorescence. Biochemistry 41, 2475–2784 [DOI] [PubMed] [Google Scholar]
- 37. Spencer R. D. (1970) Fluorescence Lifetimes: Theory, Instrumentation, and Application of Nanosecond Fluorometry. Ph.D. thesis, University of Illinois at Urbana-Champaign [Google Scholar]
- 38. Kusnetzow A. K., Altenbach C., Hubbell W. L. (2006) Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry 45, 5538–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Angel T. E., Chance M. R., Palczewski K. (2009) Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 106, 8555–8560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deupi X., Edwards P., Singhal A., Nickle B., Oprian D., Schertler G., Standfuss J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. U.S.A. 109, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]