

Background: Microsomal prostaglandin E (PGE) synthase-1 (mPGES-1) is an inducible enzyme with unknown properties in adipose homeostasis.
Results: mPGES-1 is necessary for pre-adipocyte differentiation into beige/brite adipocytes through functional interaction with peroxisome proliferator-activated receptor γ (PPARγ).
Conclusion: A coordinate interaction between mPGES-1 and PPARγ is required for white-to-brown fat conversion.
Significance: Increases in the number of beige cells in fat exerts beneficial metabolic actions.
Keywords: Adipose Tissue, Cyclooxygenase (COX) Pathway, Eicosanoid, Metabolic Diseases, Prostaglandins, PPARγ, Beige Adipocytes, mPGES-1, Pre-adipocytes
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated nuclear receptor and a master regulator of adipogenesis. Microsomal prostaglandin E (PGE) synthase-1 (mPGES-1) is an inducible enzyme that couples with cyclooxygenase-2 for the biosynthesis of PGE2. In this study we demonstrate the existence of a coordinate functional interaction between PPARγ and mPGES-1 in controlling the process of pre-adipocyte differentiation in white adipose tissue (WAT). Adipocyte-specific PPARγ knock-out mice carrying an aP2 promoter-driven Cre recombinase transgene showed a blunted response to the adipogenic effects of a high fat diet. Pre-adipocytes from these knock-out mice showed loss of PPARγ and were resistant to rosiglitazone-induced WAT differentiation. In parallel, WAT from these mice showed increased expression of uncoupling protein 1, a mitochondrial enzyme that dissipates chemical energy as heat. Adipose tissue from mice lacking PPARγ also showed mPGES-1 up-regulation and increased PGE2 levels. In turn, PGE2 suppressed PPARγ expression and blocked rosiglitazone-induced pre-adipocyte differentiation toward white adipocytes while directly elevating uncoupling protein 1 expression and pre-adipocyte differentiation into mature beige/brite adipocytes. Consistently, pharmacological mPGES-1 inhibition directed pre-adipocyte differentiation toward white adipocytes while suppressing differentiation into beige/brite adipocytes. This browning effect was reproduced in knockdown experiments using a siRNA directed against mPGES-1. The effects of PGE2 on pre-adipocyte differentiation were not seen in mice lacking PPARγ in adipose tissue and were not mirrored by other eicosanoids (i.e. leukotriene B4). Taken together, these findings identify PGE2 as a key regulator of white-to-brown adipogenesis and suggest the existence of a coordinate regulation of adipogenesis between PPARγ and mPGES-1.
Introduction
The formation of new adipocytes from precursor cells is a crucial aspect in controlling normal adipose tissue function (1, 2). During the adipogenic process, adipocytes differentiated from mesenchymal stem cells give rise to two main types of adipose tissue: WAT,3 characterized by the presence of adipocytes containing large unilocular lipid droplets, and BAT, composed by multiloculated adipocytes containing large numbers of mithocondria (1, 2). WAT is the predominant fat type in human adults and is widely distributed through the body; its main function is to store excess energy as triglycerides (1, 2). WAT is not only important for energy storage but also as an endocrine organ because it regulates whole body homeostasis by secreting adipokines (cytokines, chemokines, and growth factors) and biologically active lipid mediators (3, 4). By contrast, BAT is located in discrete pockets and is specialized to generate heat by dissipating chemical energy, counteracting hypothermia, obesity, and diabetes (1, 5). Apart from “classical” brown adipocytes that reside in BAT depots, another type of brown adipocytes, called beige or brite (brown-in-white) adipocytes, are sporadically found in WAT upon prolonged cold exposure or in response to β-adrenergic or thiazolidinedione exposure (1, 5, 6). Importantly, increases in the number of beige adipocytes in WAT are associated with protection against diet-induced obesity and metabolic diseases (7–9).
Adipocyte differentiation requires expression of peroxisome proliferator-activated receptor γ (PPARγ), a member of the nuclear receptor superfamily that functions as a ligand-activated transcription factor regulating the genes implicated in adipogenesis (10). Although PPARγ functions as a master regulator of adipocyte differentiation and is absolutely necessary for both white and brown fat cell development (11, 12), the mechanisms underlying terminal differentiation of pre-adipocytes into white or brown adipocytes up-stream and/or downstream from this transcription factor have not been completely elucidated.
Microsomal prostaglandin E (PGE) synthase-1 (mPGES-1) is an inducible enzyme that cooperates with cyclooxygenase-2 (COX-2) in the biosynthesis of PGE2, one of the most ubiquitous and biologically active endogenous lipid mediators (13). Several studies have linked this lipid mediator to the adipogenic program, although their results have yielded controversial views. For example, PGE2 is recognized as a negative regulator of WAT lipolysis and adipocyte development (14, 15), and pre-adipocytes stably transfected with either COX-1 or COX-2 show lower PPARγ expression (16). Consistent with this view, mice genetically deficient for mPGES-1 show basal elevations in PPARγ expression and transcriptional activity (17). However, more recent studies have shown that transgenic mice overexpressing COX-2 do not exhibit changes in PPARγ expression in adipose tissues (18), although PPARγ expression has been shown to be significantly reduced in adipose tissue from COX-2-deficient mice (19). The existence of this undefined role of the COX-2/mPGES-1/PGE2 pathway in WAT physiology led us to investigate the functional interaction between PPARγ and mPGES-1 in the process of adipogenesis. Our results demonstrate that removing PPARγ specifically in fat cells triggers mPGES-1 expression and PGE2 biosynthesis, uncovering an important role in the browning process of WAT pre-adipocytes.
EXPERIMENTAL PROCEDURES
Reagents
Rosiglitazone, prostaglandin E2 (PGE2) and leukotriene B4 (LTB4) and PGE2 EIA kits were from Cayman Chemicals (Ann Arbor, MI). 15-Deoxy-Δ12,14-PGJ2 (15d-PGJ2) EIA was from Assay Designs (Ann Harbor, MI). The mPGES-1 inhibitor benzo[g]indol-3-carboxylate was synthesized and pharmacologically characterized by Dr. Oliver Werz (University of Jena, Jena, Germany). Krebs-Ringer bicarbonate buffer, DMEM, and fatty acid-free BSA were from Sigma. Nylon mesh filters (100 μm) were from BD Biosciences. TRIzol was from Invitrogen, and l-glutamine was from Biological Industries (Kibbutz Beit Haemek, Israel). FBS and Dulbecco's PBS with (+/+) and without (−/−) calcium and magnesium were from Lonza (Vervieres, Belgium). Lab-TeK Chamber Slides were from Nalge Nunc International (Rochester, NY). TaqMan Gene Expression Assays were from Applied Biosystems (Foster City, CA). Anti-Dlk/Pref-1 Antibody was from MBL (Woburn, MA).
Mice
Mice carrying the loxP-targeted PPARγ gene (PPARγf/f) were crossed with transgenic mice expressing Cre recombinase under the control of the aP2 promoter to yield adipocyte-specific PPARγ KO mice (Δadip) (20). aP2 is an adipose-specific gene that is also expressed in undifferentiated adipogenic progenitors (21, 22). The control group included wild-type (WT) mice carrying the loxP-targeted PPARγ gene. Mice were housed on wood-chip bedding cages at a ratio of 4 mice per cage with a humidity level of 50–60% and a 12-h light-dark cycle and were given free access to food and water. Genomic DNA from mouse ear was isolated using the Omni-Pure Tissue Genomic DNA Purification System (Gene Link, Hawthorne, NY) following the manufacturer's protocol. Mice were genotyped by a multiplex PCR, which was performed in a total of 20 μl containing 1 μl of DNA, 2 μl of 10× reaction buffer (2.5 mm MgCl2), 3.2 μl of 1.25 mm dNTPs, 0.5 units of PRIME HotMaster TaqDNA Polymerase (5 PRIME, Hamburg, Germany), and 0.2–0.75 μm 6-carboxyfluorescein and NED (2′-chloro-5′-fluoro-7′,8′-fused phenyl-1.4-dichloro-6-carboxyfluorescein) fluorescent-labeled primers (supplemental Table 1). PCR samples were amplified at 94 °C (30 s), 60 °C (30 s), and 71.5 °C (90 s) for 40 cycles in a GeneAmp PCR System 9700, and PCR products were analyzed by capillary electrophoresis in a 3130 Genetic Analyzer (Applied Biosystems). Genotyping was also performed by conventional PCR in a total volume of 20 μl containing 1 μl of DNA, 2 μl of 10× reaction buffer (2.5 mm MgCl2), 3.2 μl of 1.25 mm dNTPs, 1 μl of unlabeled primers, and 0.5 units of PRIME HotMaster TaqDNA polymerase. PCR samples were amplified as described above, and products were analyzed by electrophoresis in 2.5% LM Sieve-agarose gels and visualized by GelRedTM Nucleic Acid Gel Stain (Biotium, Hayward, CA) using a 100-bp DNA ladder marker (Invitrogen).
The mice used in the study were F5 or subsequent generations of age-matched littermates. To induce obesity, male WT and Δadip mice were fed a high fat diet (HFD) (60% Kcal from fat; Research Diets Inc., New Brunswick, Canada) for 12 weeks starting at week 6 of age. The chow group received a standard pelleted chow diet (13% kcal from fat) for 12 weeks. At the end of the study mice were euthanized under ketamine/xylazine (4:1) anesthesia via intraperitoneal injection (Ketolar, Parke Davis-Pfizer, Dublin, Ireland; Rompun, Bayer Leverkusen, Germany). Epididymal WAT (eWAT) and interscapular BAT (iBAT) were collected and snap-frozen in liquid nitrogen for further analysis. All animal studies were conducted in accordance with the criteria of the Investigation and Ethics Committee of Hospital Clinic and the European Community laws governing the use of experimental animals.
Ex Vivo Experiments in eWAT Explants
Fat pads were collected under sterile conditions and placed in P60 plates in prewarmed (37 °C) DPBS+/+ containing penicillin (100 units/ml) and streptomycin (100 mg/ml). Connective tissue and blood vessels were removed by dissection before cutting the tissue into small pieces. Explants were washed with DPBS at 37 °C by centrifugation for 1 min at 400 × g to remove blood cells and pieces of tissue containing insufficient adipocytes to float as described in Refs. 23 and 24. Thereafter, explants were cultured in 12-well plates (40 mg/well) with DMEM, l-glutamine (2 mm), penicillin (50 units/ml), streptomycin (50 mg/ml), and 2% fatty acid-free BSA and incubated with vehicle (0.04% ethanol), PGE2 (1 μm), the mPGES-1 inhibitor benzo[g]indol-3-carboxylate (1, 5, and 10 μm), the selective COX-1 inhibitor SC-560 (1 and 5 μm), the selective COX-2 inhibitors SC-58635 (5 μm) and SC-236 (1 μm), and the selective 5-lipoxygenase (5-LO) inhibitor CJ-016 (1 μm) for 12 h. Supernatants were collected and kept at −80 °C.
Analysis of Eicosanoids by Enzyme Immunoassay (EIA) and LC-Electrospray Ionization-MS/MS
LTB4, PGE2 and 15d-PGJ2 levels were extracted from eWAT samples (200 mg) from WT and Δadip mice. Each sample was individually homogenized in a Pellet pestle, Cordless Motor (Sigma) in 400 μl of cold MeOH and kept at −80 °C overnight. Subsequently, homogenates were centrifuged at 2000 rpm for 10 min at 4 °C. Supernatants were collected and brought to a final volume of 10 ml with distilled water, transferred into syringes, acidified to pH 3.5, and loaded onto Sep-Pak C18 Cartridges (Waters, Milford, MA). Materials were eluted with methyl formate, evaporated, and resuspended in EIA buffer. Confirmation of PGE2 and 15d-PGJ2 levels and analysis of 6-keto-PGF1α, PGF2α, PGD2, PGJ2, and thromboxane B2 concentrations were performed by LC-electrospray ionization-MS/MS analysis. Briefly, 50 mg of frozen adipose tissue were extracted on solid phase columns before injection into an Agilent 1200 HPLC system coupled with an Agilent 6460 Triplequad mass spectrometer with electrospray ionization source. Analysis of lipid mediators was performed with MRM in negative mode.
Analysis of Gene Expression
Total RNA was isolated using the TRIzol reagent. RNA concentration was assessed in a NanoDrop-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE), and its integrity was tested with a RNA 6000 Nano Assay in a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). cDNA synthesis from 0.5–1.0 μg of total RNA was performed using the High Capacity cDNA Archive kit (Applied Biosystems). Quantitative analysis of gene expression was performed by real-time PCR in an ABI Prism 7900 Sequence Detection System in Fast Real Time System. Optimal and pre-designed TaqMan Gene Expression Assays were used (TNF-α (ID Mm00443258_m1), PPARγ (ID Mm00440945_m1), 5-LO (ID Mm001182747_m1), 5-LO-activating protein FLAP (ID Mm00802100_m1), COX-2 (ID Mm00478374_m1), mPGES-1 (ID Mm00460181_m1), PGD2 (ID Mm01330613_m1), 15-prostaglandin dehydrogenase (15-PGDH) (ID Mm0051521_m1), PPARγ co-activator-1 α (PGC-1α; ID Mm01208835_m1), PPARγ isoform2 (ID Mm00440940_m1), mPGES-2 (ID Mm00460181_m1), Cidea (ID Mm00432554_m1), uncoupling protein 1 (UCP1; ID Mm01244861_m1), PRD1-BF-1-RIZ1 homologous domain containing protein-16 (PRDM16; ID Mm00712556_m1)) and custom TaqMan Assay PPARγ isoform1 (NCBI NM_001127330.1) as previously described (20). β-Actin (Actb; ID Mm00607939_s1) was used as an endogenous control. PCR results were analyzed with the Sequence Detector Software Version 2.1 (Applied Biosystems). Relative quantification of gene expression was performed using the comparative Ct method. The amount of target gene normalized to β-actin and relative to a calibrator was determined by the arithmetic equation 2−ΔΔCt described in the comparative Ct method.
Isolation of Pre-adipocytes from the eWAT Stromal Vascular Cell (SVC) Fraction
eWAT was excised, weighed, rinsed twice in cold carbogen-gassed Krebs-Ringer supplemented with 1% fatty acid-free BSA and 2 mm EDTA and centrifuged at 500 × g for 5 min at 4 °C to remove free erythrocytes and leukocytes. Tissue suspensions (300–600 mg) were placed in 5 ml of digestion buffer containing Krebs-Ringer supplemented with 1% fatty acid-free BSA and 1 mg/ml collagenase A (Roche Applied Science) and incubated at 37 °C for 30 min as described (23). Cell pellets corresponding to the SVC were incubated with erythrocyte lysis buffer (0.15 m NH4Cl, 10 mm KHCO3, and 0.1 mm EDTA) for 5 min and centrifuged at 500 × g for 5 min. SVC with a predominant population of pre-adipocytes (∼72%) were further enriched using the magnetic labeling system Lineage Cell Depletion kit (Miltenyi Biotech, Auburn, CA), which depletes mature hematopoietic cells, such as monocytes/macrophages, T cells, B cells, granulocytes, and erythrocytes and their committed precursors in the SVC fraction. Pre-adipocyte purity was tested by immunocytochemistry of Pref-1, which is expressed specifically in pre-adipocytes but not in mature adipocytes.
Pref-1 Immunocytochemistry
After cytospin at 500 × g for 5 min, cells on coverslips were fixed with acetone for 10 min at −20 °C followed by incubation with peroxidase blocking solution (S2023, DAKO, Glostrup, Denmark) for 15 min at room temperature to block endogenous peroxidase activity. After washing with DPBS, cells were incubated with blocking serum (Vectastin ABC Kit, Vector Laboratories, Burlingame, CA). Cells were then incubated overnight at 4 °C with the primary rat anti-mouse Pref-1 antibody (1/250) followed by incubation for 30 min at room temperature with a biotinylated rabbit anti-rat IgG secondary antibody and incubation with ABC for 30 min at room temperature as described (25). Color was developed using the diaminobenzidine substrate (Roche Applied Science), and cells were counterstained with hematoxylin and mounted with aqueous solution. Cells were visualized at magnification ×200 in a Nikon Eclipse E600 microscope (Kawasaki, Japan).
Pre-adipocyte Differentiation into White Adipocytes
Freshly isolated pre-adipocytes were seeded onto 96-well plates (40,000 cells/well) with DMEM supplemented with 10% FBS and 10 ng/ml basic FGF (R&D Systems, Minneapolis, MN) and maintained in a 5% CO2 atmosphere. Cells were allowed to grow to confluence for 2 days and then exposed to the white adipocyte differentiation mixture (25) (1 μg/ml insulin, 0.25 μg/ml dexamethasone, 0.5 mm isobutylmethylxanthine, and 1 μm rosiglitazone) with 100 units/ml penicillin/streptomycin and 2 mm l-glutamine in the presence of vehicle (0.01% ethanol), PGE2 (0.1 or 1 μm), SC-560 (3 μm), SC-58635 (3 μm), and benzo[g]indol-3-carboxylate (3 μm). After 72 h, cells were grown in fresh DMEM with 10% FBS until day 12 for harvesting. Mature adipocytes were fixed in 4% paraformaldehyde for 1 h and then with 60% isopropyl alcohol before incubation with 0.2% Oil Red-O for 30 min at room temperature. To quantify the amount of Oil Red-O retained by the cells, cells were incubated with isopropyl alcohol for 30 min with shaking to elute the stain, and the optical density was measured at 500 nm in a FluoStar Optima microplate reader (BMG Labtech, Offenburg, Germany). For visualization, cells were counterstained with Gill's hematoxylin, washed with distilled water (4%), and mounted with aqueous solution. Cells were visualized at a magnification of ×200 in a Nikon Eclipse E600 microscope.
Pre-adipocyte Differentiation into Beige/Brite Adipocytes
Freshly isolated eWAT pre-adipocytes were cultured and grown to confluence as indicated before and then exposed to the beige adipogenic mixture (26, 27) containing insulin (5 μg/ml), dexamethasone (0.5 μm), isobutylmethylxanthine (0.5 mm), 3,5,3′-triiodothyronine (T3) (1 nm), rosiglitazone (1 μm), penicillin/streptomycin (100 units/ml), and l-glutamine (2 mm). Cells were incubated with vehicle (0.01% ethanol), PGE2 (0.1 μm), SC-560 (3 μm), SC-58635 (3 μm), and benzo[g]indol-3-carboxylate (3 μm) for 48 h. The induction medium was replaced with fresh DMEM containing 10% FBS, 1 μg/ml insulin, and 1 nm T3 for 2 additional days. Thereafter, cells were incubated in DMEM with 10% FBS until day 10. Differentiation of pre-adipocytes into beige/brite adipocytes was evaluated by assessing the mitochondrial distribution using MitoTracker Red CMXRos kit (Invitrogen). The MitoTracker Red CMXRos reagent was diluted in DMSO at 100 nm and incubated for 30 min at 37 °C. The adipocytes were washed with DPBS+/+ and fixed with 4% (w/v) formaldehyde for 15 min before mounting with Vectashield medium containing DAPI (Molecular Probes, Eugene, OR). Mitochondrial distribution was examined under a fluorescence microscope (Olympus BX51, Hamburg, Germany).
Differentiation of 3T3-L1 Adipocytes
Mouse 3T3-L1 cells were maintained in DMEM supplemented with 10% (v/v) FBS and 100 units/ml penicillin/streptomycin. Cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C and differentiated into white or beige adipocytes as described before.
siRNA Transfection
Synthetic mPGES-1 siRNA and Universal Scrambled Negative Control (siCON) oligonucleotides were designed and synthesized by OriGene (Rockville, MD). 3T3-L1 cells were transfected at 70% confluence with 10 nm siRNA using Metafectene PRO (Biontex, Martinsried, Germany) at a 1:3 (w/v) in 24-well plates according to the manufacturer's instructions. To assess siRNA efficiency, RNA and protein samples were obtained after 48 h. Three different siRNAs targeting mPGES-1 were screened (data not shown), and the sequence of the most efficient was used. The effects of mPGES-1 siRNA on adipocyte differentiation were assessed 4 days after the addition of the differentiation mixture.
Western Blotting
Total protein from adipose tissue was extracted in lysis buffer containing 50 mm HEPES, 20 mm β-glycerophosphate, 2 mm EDTA, 1% Igepal, 10% glycerol, 1 mm MgCl2, 1 mm CaCl2, and 150 mm NaCl, supplemented with protease inhibitor (Complete Mini) and phosphatase inhibitor (PhosSTOP) cocktails. For protein isolation from cell cultures, cells were scraped into ice-cold DPBS and resuspended in 150 μl of lysis buffer. Homogenates from tissues were incubated on ice for 15 min and centrifuged at 16,000 × g for 20 min at 4 °C. Homogenates from cells were incubated on ice for 10 min and centrifuged at 1000 rpm for 2 min. Supernatants were collected, and COX-1, COX-2, and mPGES-1 protein expression was analyzed by Western blot. A total of 60 μg of protein from tissues and 30 μg of protein from cells were resuspended in SDS-containing Laemmli sample buffer, heated for 5 min at 95 °C, and resolved on 8% (tissue) or 15% (cells) SDS-PAGE. Proteins were electroblotted for 60–90 min at 100 V at 4 °C onto polyvinyl difluoride membranes. Transfer was performed by the iBlot Dry Blotting System (Invitrogen) at 20 V in 7 min. The transfer was visualized by Ponceau S solution. Membranes were then soaked for 1 h at room temperature in TBS (20 mm Tris/HCl, pH 7.4, and 0.5 m NaCl) containing 0.1% (v/v) Tween 20 (0.1% TBS-T) and 5% (w/v) nonfat dry milk. Blots were washed 3 times for 5 min each with 0.1% TBS-T and subsequently incubated overnight at 4 °C with primary mouse anti-mouse COX-1 (dilution 1:300), rabbit anti-mouse COX-2 (dilution 1:1000), and rabbit anti-mouse mPGES-1 (dilution 1:150) antibodies (Cayman Chemicals) and primary anti-mouse GAPDH (dilution 1:1000) (Abcam, Cambridge, UK) in 0.1% TBS-T containing 5% BSA. After washing the blots 3 times for 5 min each with 0.1% TBS-T, membranes were incubated for 1 h at room temperature with an HRP-linked donkey anti-rabbit secondary (dilution 1:2000) antibody (Biolegend, San Diego, CA) in 0.1% TBS-T (for COX-2 and GAPDH) and HRP-linked anti-mouse IgG (dilution 1:2000) (Cell Signaling, Danvers, MA) in 0.1% TBS-T for COX-1. Bands were visualized using the EZ-ECL chemiluminescence detection kit (Biological Industries) in a LAS 4000 imaging system (GE Healthcare) and quantified using Image GE ImageQuant TL analysis software.
RESULTS
Mice lacking PPARγ specifically in adipocytes were generated by introducing transgenic Cre recombinase under the control of the aP2 promoter into mice homozygous for the loxP-flanked (floxed) allele of PPARγ. The presence of respective cell-specific Cre recombinase transgenes was confirmed by DNA genotyping, which showed either a 210-bp band corresponding to WT mice carrying the loxP-targeted PPARγ gene or 210- and 524-bp bands corresponding to the presence of both floxed alleles and aP2-Cre recombinase (supplemental Fig. 1). Consistent with earlier studies showing that ∼30% of cells in adipose tissue are nonfat cells (21, 28), expression of PPARγ isoforms 1 and 2 was reduced in base-line conditions by 47–61% in eWAT from adipocyte-specific PPARγ knock-out (Δadip) mice (Fig. 1A). Because PPARγ is a master regulator of adipocyte differentiation, we used the HFD model to induce obesity in WT and Δadip mice. As shown in Fig. 1B, ablation of PPARγ isoforms in Δadip mice under HFD feeding ranged between 63 and 65%. HFD feeding by itself produced a significant reduction in PPARγ1 but not in PPARγ2 in both WT and Δadip mice (supplemental Fig. 2). Compared with WT mice, Δadip mice showed a lesser body weight gain (Fig. 1C) and reduced eWAT weight (Fig. 1D) in response to HFD feeding. Under the chow diet, Δadip mice showed similar body weight gain and slightly reduced eWAT weight (Fig. 1, C and D).
FIGURE 1.

Effects of targeted deletion of PPARγ in adipocytes. A, adipose mRNA expression for PPARγ isoforms 1 and 2 assessed by real-time PCR in WT (n = 10) and aP2-Cre driven PPARγ KO (Δadip) (n = 9) mice under chow conditions. B, expression for PPARγ isoforms 1 and 2 in eWAT from WT (n = 6) and Δadip (n = 6) mice after 12 weeks of HFD (60% kcal from fat) feeding. C, body weight in WT (n = 5) and Δadip (n = 4) mice receiving a chow diet and in WT (n = 13) and Δadip (n = 11) mice during the obesity-induced model of HFD feeding for 12 weeks. D, eWAT weight in WT (n = 18) and Δadip (n = 15) mice under Chow and HFD conditions. E, representative photomicrographs of SVC fraction and pre-adipocyte cultures stained with Diff-quick or immunostained with Pref-1 specific antibody (×200 magnification). F, PPARγ expression in pre-adipocytes from WT (n = 3) and Δadip (n = 3) mice. G, representative bright field images of pre-adipocytes from WT and Δadip mice incubated with rosiglitazone (Rosi, 1 μm) or vehicle (Veh). Absorbance values from Oil Red-O staining of rosiglitazone-induced pre-adipocyte differentiation toward white adipocytes are shown on the right. H, mitochondrial UCP1 expression in eWAT and iBAT from WT (n = 17) and Δadip (n = 11) mice. I, UCP1 and PGC-1α expression (fold change versus eWAT for each condition and gene studied) in eWAT and iBAT from WT mice under Chow and HFD conditions. Results are expressed as the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus WT mice. #, p < 0.05 versus Veh. a, p < 0.05 versus chow.
To assess the effects of PPARγ deletion on adipogenesis, we compared pre-adipocyte differentiation in SVC fractions from Δadip mice with those from WT mice. After depletion of mature hematopoietic cells, ∼90–95% pre-adipocyte purity was confirmed by Pref-1 immunocytochemistry (Fig. 1E). Purified pre-adipocytes from Δadip mice showed a greater loss of PPARγ (>72%) (Fig. 1F) compared with that seen in whole adipose tissue (Fig. 1A). Of note, pre-adipocytes from Δadip mice were resistant to rosiglitazone-induced differentiation toward white adipocytes (Fig. 1G). Interestingly, the expression of the mitochondrial marker UCP1 was significantly up-regulated in eWAT from Δadip mice, whereas this brown marker was down-regulated in iBAT from these mice (Fig. 1H). As expected, UCP1 and PGC-1α were predominantly expressed in iBAT (Fig. 1I). UCP1 expression was significantly reduced by HFD (Fig. 1I).
PGs are key regulatory components of adipogenesis. The status of the PGE2 biosynthetic pathway in adipose tissue in the absence of PPARγ has not been investigated. Consequently, we next assessed the expression of the key enzymes involved in adipose PGE2 biosynthesis in WT and Δadip mice. COX-2 was found up-regulated at the protein level in adipose tissue from Δadip mice under HFD but not in chow diet (Fig. 2A). No changes in COX-2 expression were detected at the mRNA level in both conditions (data not shown). In contrast, COX-1 was up-regulated in Δadip mice under the chow diet but not in HFD (Fig. 2B). Consistently, increased PGE2 levels were detected in fat tissue from these KO mice either under chow or HFD conditions (Fig. 2C). Further evidence on the relationship between PGE2 and PPARγ was obtained in ex vivo experiments in fat pads. First, fat explants from Δadip mice displayed a remarkable induction in COX-2 expression (Fig. 2D). Second, the addition of exogenous PGE2 up-regulated COX-2 in fat explants from both WT and Δadip mice (Fig. 2E), suggesting the existence of a PPARγ-independent positive regulatory feedback. Third, fat explants from Δadip mice also displayed mPGES-1 up-regulation (Fig. 2F). Last, the addition of exogenous PGE2 also up-regulated mPGES-1 in WT and Δadip fat explants (Fig. 2G). Changes in other members of the PG cascade and biosynthetic pathways were also detected in Δadip mice. Indeed, increased 15d-PGJ2 levels were elevated in these mice under chow conditions but not during HFD (Fig. 2H). This was consistent with increased prostaglandin D synthase (PGDS) expression in Δadip mice only under Chow diet (Fig. 2I). Moreover, Δadip mice showed reduced mPGES-2 expression (Fig. 2K) without changes in 15-prostaglandin dehydrogenase, the enzyme that catalyzes the dehydrogenation of active PGE2 (Fig. 2K). Another COX-derived product analyzed by LC- electrospray ionization-MS/MS and found increased in Δadip mice was PGF2α (supplemental Table 2).
FIGURE 2.

COX-2 and mPGES-1 up-regulation and increased adipose PGE2 levels in aP2-Cre driven PPARγ KO (Δadip) mice. A, adipose tissue protein expression for COX-2 in WT (n = 8) and Δadip (n = 8) mice under Chow and HFD conditions. B, adipose tissue protein expression for COX-1 in WT (n = 9) and Δadip (n = 7) mice under chow and HFD conditions. C, PGE2 levels in adipose tissue from WT (n = 6) and Δadip (n = 6) mice under chow and HFD conditions. D, expression of COX-2 in fat explants from WT and Δadip mice incubated ex vivo. E, expression of COX-2 in WT and Δadip fat explants incubated with vehicle (Veh, 0.04% ethanol) and PGE2 (1 μm) for 12 h. F, expression of mPGES-1 in fat explants from WT and Δadip mice incubated ex vivo. G, expression of mPGES-1 in WT and Δadip fat explants incubated with vehicle (Veh, 0.04% ethanol) and PGE2 (1 μm) for 12 h. H, levels of 15d-PGJ2 in adipose tissue from WT (n = 6) and Δadip (n = 6) mice under chow and HFD conditions. I, expression of prostaglandin D synthase (PGDS) in these mice. J, adipose tissue mPGES-2 expression. K, adipose tissue 15-prostaglandin dehydrogenase (15-PGDH) expression. Results are expressed as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus WT mice. a, p < 0.05; b, p < 0.01 versus vehicle.
To elucidate the ability of COX-derived products to regulate the adipogenic program, we next assessed the effects of PGE2 on pre-adipocyte differentiation. Pre-adipocytes isolated from eWAT were primed with the differentiation-inducing mixture containing insulin, dexamethasone, isobutylmethylxanthine, and rosiglitazone in the presence of PGE2 or vehicle, and the differentiation toward white adipocytes was monitored by Oil Red-O staining. As shown in Fig. 3A, PGE2 significantly reduced rosiglitazone-induced white adipocyte differentiation. PGE2 did not modify the Oil Red-O signal in the absence of rosiglitazone in the differentiation mixture (Fig. 3A, inset). Conversely, PGE2 stimulated pre-adipocytes incubated with the brown adipocyte differentiation mixture containing insulin, dexamethasone, isobutylmethylxanthine, T3, and rosiglitazone to induce the development of beige/brite cells, as monitored by MitoTracker Red CMXRos (Fig. 3, B–D). Images from these experiments are shown at a lower magnification in supplemental Fig. 3A. Interestingly, a direct stimulatory effect of PGE2 on the brown marker UCP1 was observed in fat pads from WT mice (Fig. 3E). In contrast, PGE2 was unable to stimulate the browning of pre-adipocytes from Δadip mice lacking PPARγ (Fig. 3E). A suppression of both PPARγ 1 and 2 isoforms was observed in WT adipose tissue explants incubated with PGE2, suggesting a negative regulation of PPARγ by this eicosanoid (Fig. 3F).
FIGURE 3.

Effects of PGE2 on pre-adipocyte differentiation. A, Oil Red-O staining of primary eWAT pre-adipocytes at day 12 of differentiation incubated with rosiglitazone (Rosi, 1 μm) in the absence or presence of PGE2 (0.1 μm) for 72 h. The bottom graph shows the quantification of the Oil Red-O content. Inset, pre-adipocytes incubated with increasing concentrations of PGE2 in the absence of rosiglitazone. B, pre-adipocytes from eWAT induced to brown differentiation in the absence or presence of PGE2 (0.1 μm) for 72 h. Cells were labeled with Mito Tracker and co-stained with DAPI to identify nuclei (middle panel) (×200 magnification). C, morphometric quantification of the area stained with MitoTracker. D, corresponding bright-field images (×200 magnification). E, UCP1 expression in pre-adipocytes from WT (n = 4) and aP2-Cre driven PPARγ KO (Δadip) mice incubated with PGE2 (1 μm) or vehicle (Veh). F, mRNA expression for PPARγ isoforms 1 and 2 assessed by real-time PCR in fat eWAT explants from WT mice incubated with PGE2 (1 μm) or vehicle for 12 h. Results are expressed as the mean ± S.E. *, p < 0.05; **, p < 0.001 versus vehicle. a, p < 0.05 versus rosiglitazone.
Consistent with the stimulatory role of exogenous PGE2 on pre-adipocyte differentiation toward beige/brite adipocytes, inhibition of endogenous PGE2 synthesis was associated with pre-adipocyte commitment toward the WAT phenotype (Fig. 4A). The capability of inhibitors of PGE2 synthesis to induce the WAT program was more evident in the presence of rosiglitazone in the differentiation mixture (Fig. 4A). In these experiments, SC-560, a selective COX-1 inhibitor, produced similar effects to those of SC-58635, a selective COX-2 inhibitor (Fig. 4A). By contrast, in the absence of rosiglitazone, the benzo[g]indol-3-carboxylate compound, which is a potent inhibitor of mPGES-1 without significant activity on COX-1/COX-2 (29), exerted a more efficacious stimulatory effect on WAT development (Fig. 4A). Importantly, the mPGES-1 inhibitor significantly decreased pre-adipocyte differentiation into beige/brite cells induced by the differentiation mixture (Fig. 4B). Images from these experiments are shown at a lower magnification in supplemental Fig. 3B. To discard potential off-target actions of the mPGES-1 inhibitor, we performed knockdown experiments using a siRNA directed against mPGES-1. Because primary pre-adipocytes are difficult to transfect, knockdown experiments were performed in the 3T3-L1 adipogenic cell line, the most studied cell line used for adipocyte differentiation (30). As shown in Fig. 4C, both mRNA and protein expression for mPGES-1 as measured by real-time PCR and Western blot, respectively, were significantly reduced 48 h after transfection. Interestingly, mPGES-1 gene silencing reduced the expression of markers of pre-adipocyte browning (i.e. UCP1, Cidea, and PGC-1α) to a similar extent as that of pharmacological mPGES-1 inhibition (Fig. 4, D–G). Of note, genetic or pharmacological inhibition of mPGES-1 reduced the expression of the brown adipose determination factor PRDM16 (Fig. 4E). Consistent with findings described in Fig. 4B, pharmacological inhibition of either COX-1 or COX-2 did not translate into changes in adipocyte browning (Fig. 4H).
FIGURE 4.

Effects of PGE2 inhibition on pre-adipocyte differentiation. A, quantification of Oil Red-O staining of primary eWAT pre-adipocytes at day 12 of differentiation incubated with 3 μm selective COX-1 (SC-560), COX-2 (SC-58635), and mPGES-1 (benzo[g]indol-3-carboxylate) inhibitors (Inh) for 72 h in the absence or presence of rosiglitazone (1 μm). Veh, vehicle. B, representative microphotographs of pre-adipocytes from eWAT induced to brown differentiation in the absence or presence of COX-1, COX-2, or mPGES-1 inhibitors at 3 μm. Cells were labeled with MitoTracker and co-stained with DAPI to identify nuclei (×200 magnification). The morphometric assessment of the area stained with MitoTracker is shown on the right. C, mRNA expression for mPGES-1 48 h after transfection of 3T3-L1 cells with a mPGES-1 siRNA or an universal scrambled negative control (siCON). Inset, protein levels for mPGES-1 and GAPDH as measured by Western blot. D, UCP1 expression in 3T3-L1 cells transfected with mPGES-1 siRNA and siCON or incubated with the selective mPGES-1 inhibitor benzo[g]indol-3-carboxylate (3 μm). E, expression of PRDM16 in 3T3-L1 after genetic or pharmacological inhibition of mPGES-1. F, changes in Cidea expression in these experiments. G, PGC-1α expression. H, UCP1, Cidea, and PGC-1α expression in primary pre-adipocytes incubated with vehicle (0.04% ethanol) or selective COX-1 (SC-560) and COX-2 (SC-58635) inhibitors (3 μm). Results are expressed as the mean ± S.E. of three individual experiments in duplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle or siCON.
Consistent with previous publications, the mPGES-1 inhibitor benzo[g]indol-3-carboxylate blocked PGE2 formation by ∼55% (Fig. 5A) (29). In addition, confirming the existence of a negative regulatory loop between PGE2 and PPARγ, fat explants incubated with this mPGES-1 inhibitor displayed enhanced PPARγ expression (Fig. 5B). Given that changes in pre-adipocyte browning were only seen with the mPGES-1 inhibitor despite similar suppression of PGE2 levels with selective COX-1 and COX-2 inhibitors, we next explored the pharmacological properties of this compound on other eicosanoid-generating pathways. In this regard, this benzo[g]indol-3-carboxylate compound has been shown to suppress leukotriene biosynthesis by inhibiting 5-LO (31). Accordingly, we assessed the effects of this compound on LTB4 formation by adipose tissue. As shown in Fig. 5, C and D, this compound inhibited LTB4 levels and 5-LO expression in a concentration-dependent manner in fat explants from WT mice. Noteworthy, Δadip mice exhibited increased adipose LTB4 levels accompanied by reduced 5-LO expression (Fig. 5E). The presence of augmented LTB4 levels in Δadip mice was likely related to a parallel increase in FLAP expression (Fig. 5E). When tested on Δadip mice, the benzo[g]indol-3-carboxylate compound also produced concentration-dependent inhibitory actions on LTB4 levels and 5-LO expression (Fig. 5, F and G). The participation of LTB4 in the browning process was excluded by demonstrating the lack of effect of this 5-LO product on the expression of UCP1, Cidea and PGC-1α in WAT pre-adipocytes (Fig. 5H).
FIGURE 5.
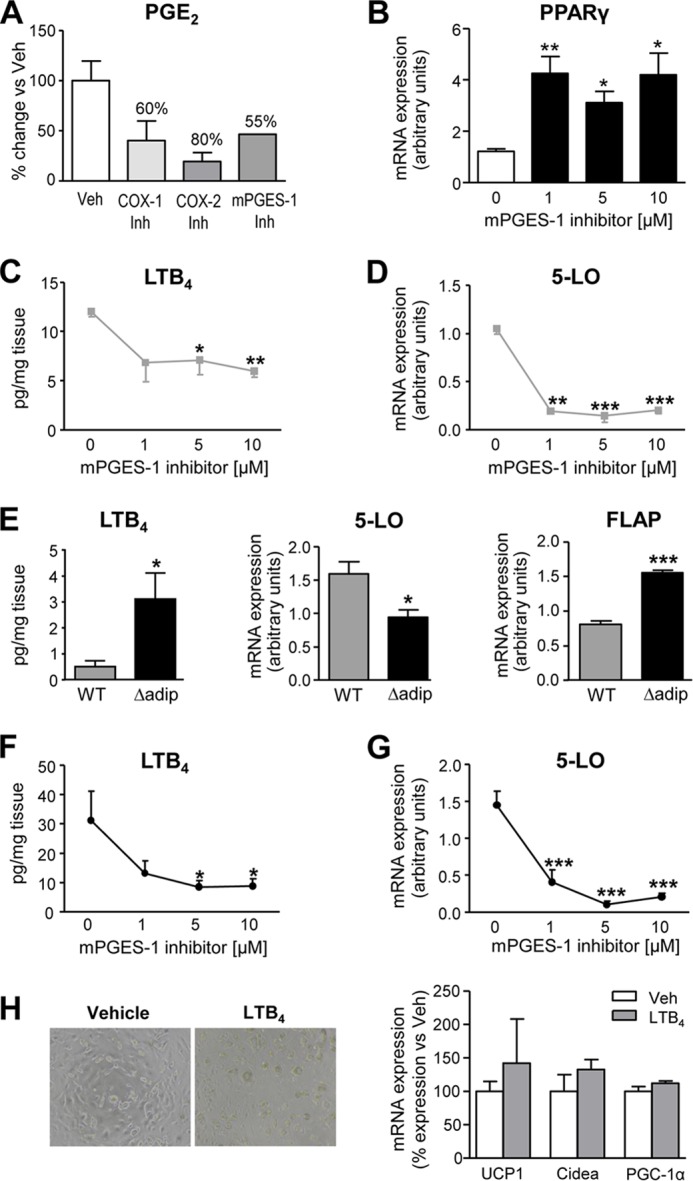
Lack of effect of the 5-LO product LTB4 on pre-adipocyte differentiation. A, percent (%) inhibition of PGE2 synthesis as determined by enzyme immunoassay (EIA) in WT fat explant supernatants incubated ex vivo at 5 μm concentration with selective COX-1 (SC-560), COX-2 (SC-58635), and mPGES-1 (benzo[g]indol-3-carboxylate) inhibitors (Inh). Veh, vehicle. B, mRNA expression for PPARγ assessed by real-time PCR in fat eWAT explants from WT mice incubated with increasing concentrations of the mPGES-1 inhibitor for 12 h. C, effects of the mPGES-1 inhibitor on LTB4 production by eWAT explants from WT mice. D, effects of the mPGES-1 inhibitor on eWAT 5-LO expression. E, LTB4 levels and 5-LO and 5-LO-activating protein (FLAP) expression in eWAT from WT (n = 10) and aP2-Cre driven PPARγ KO (Δadip) (n = 9) mice under HFD conditions. F and G, effects of increasing concentrations of the mPGES-1 inhibitor on LTB4 production and 5-LO expression in eWAT explants from Δadip mice. H, representative bright-field images and UCP1, Cidea, and PGC-1α expression in primary eWAT pre-adipocytes incubated with LTB4 (0.1 μm) or vehicle for 48 h. Data are expressed as the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 with respect to vehicle or WT mice.
Because in addition to adipogenesis, PPARγ also plays a role in regulating inflammatory response, we finally assessed the consequences of lacking PPARγ on TNFα expression in fat explants. As shown in Fig. 6A, Δadip mice exhibited increased expression of TNFα as compared with tissue explants from WT mice. This result was consistent with the observation that rosiglitazone was able to down-regulate the expression of this adipokine (Fig. 6B). Given that mPGES-1 products are well known lipid mediators participating in inflammatory response (13), we also assessed the consequences of mPGES-1 inhibition on adipose TNFα expression. In fat pads from both WT and Δadip mice, TNFα was down-regulated in a concentration-dependent manner by the pharmacological mPGES-1 inhibitor (Fig. 6C). This finding was reproduced in knockdown experiments using a siRNA directed against mPGES-1 (Fig. 6C, inset). Similar to TNFα, Δadip mice exhibited increased expression of IL-6 and MCP-1 as compared with tissue explants from WT mice (Fig. 6, D and E). These two inflammatory adipokines were also blocked by different selective COX and 5-LO inhibitors (Fig. 6F).
FIGURE 6.

Modulation of pro-inflammatory adipokines by PPARγ and mPGES-1. A, TNFα expression in eWAT explants from WT (n = 6) and aP2-Cre driven PPARγ KO (Δadip) (n = 6) mice. B, TNFα expression in eWAT explants from WT mice incubated in the absence or presence of rosiglitazone (Rosi, 10 μm). Veh, vehicle. C, effects of increasing concentrations of the mPGES-1 inhibitor (benzo[g]indol-3-carboxylate, 3 μm) on TNFα expression in eWAT explants from WT and Δadip mice. Inset, mRNA expression for TNFα 48 h after transfection of 3T3-L1 cells with a mPGES-1 siRNA or an universal scrambled negative control (siCON). D and E, IL-6 and MCP-1 expression in eWAT explants from WT and Δadip mice. F, IL-6 and MCP-1 expression in WT fat explants incubated with vehicle (0.2% DMSO), SC-236 (COX-2 inhibitor), SC-260 (COX-1 inhibitor), CJ-016 (5-LO inhibitor), and a combination of SC-236 and CJ-016. Results are the mean ± S.E. *, p < 0.05; **, p < 0.001 with respect to vehicle or WT mice. a, p < 0.05 versus siCON.
DISCUSSION
PPARγ plays a unique regulatory role in adipose tissue homeostasis by exerting strict control of the adipogenic process. Here we present evidence that mice lacking PPARγ specifically in the adipose tissue are resistant to HFD-induced eWAT formation by mechanisms linked to PGE2 biosynthesis. Indeed, our study demonstrates the induction of mPGES-1 expression and augmented PGE2 levels in eWAT from aP2-Cre PPARγ-deficient mice. This mPGES-1-derived product is able to divert pre-adipocyte differentiation in WAT from white adipocytes to beige/brite mature adipocytes accompanied by up-regulation of UCP1. A proof of concept of the mPGES-1 role was obtained by inhibiting either the expression or the activity of this terminal enzyme. In particular, the addition of a selective pharmacological mPGES-1 inhibitor as well as a siRNA directed against mPGES-1 to adipogenic precursors resulted in the reduction of browning markers (i.e. UCP1, Cidea, and PGC-1α) and browning determination factors (i.e. PRDM16). Together, these findings are relevant in terms of energy homeostasis because the engagement of beige/brite adipocytes and the induction of a thermogenic program in WAT depots are able to waste the surplus of energy through increased heat production, which ultimately exerts protection against obesity and obesity-related co-morbidities.
Previous studies indicate that beige/brite cells emerging in WAT depots come from a completely different cell lineage than those in the classical BAT depots (6). To date, the beige/brite cells in WAT depots have been described upon chronic cold exposure or in response to β-adrenergic or PPARγ agonists (1, 5, 6). Our study is the first investigation providing evidence that exposure of pre-adipocytes of WAT origin to the eicosanoid PGE2 results in a browning effect during the adipocyte differentiation process. These findings add value to the previous observation that overexpression of COX-2, the first upstream enzyme in the PG biosynthetic cascade, was associated with de novo recruitment of brown adipocytes in WAT (18). Similarly, a previous study using COX-2-deficient mice reported that induction of UCP1 expression in WAT is dependent on COX activity (32). However, these studies left unanswered the relative role of PGs in beige fat development and whether WAT beige cells come from progenitor cells pushed to develop into beige/brown fat by PGs or whether white adipocytes could directly convert into beige cells. Moreover, these previous studies pose the limitation that in addition to PGE2, COX-2 activity gives rise to other PGs, including PGI2 and specially PGD2. PGD2 undergoes spontaneous dehydration to a number of derivatives including 15d-PGJ2, which is a potent ligand for the activation of PPARγ, although its function in adipogenesis is still unproven (33). Our study obviates these limitations by providing evidence of the direct involvement of a COX-2 downstream terminal synthase responsible for the biosynthesis of PGE2 (i.e. mPGES-1), which drives beige/brite fat development in WAT by direct stimulation of the conversion of pre-adipocytes into beige/brite adipocytes.
An interesting aspect of our study is that we provide evidence of the existence of a coordinate functional regulation of brown-in-white adipogenesis by mPGES-1 and PPARγ. Indeed, our study presents a number of findings supporting a negative regulation between the nuclear receptor PPARγ and the PGE2 biosynthetic pathway (Fig. 7). On one hand, mice deficient in PPARγ specifically in adipose tissue showed increased expression of COX-2 and mPGES-1 and augmented PGE2 levels. We noted that elevations in COX-2 expression were more evident in ex vivo experiments (fat explants cultured in the absence of circulatory factors) than in vivo. This is probably related to the existence of complex mechanisms regulating COX-2 in vivo that may mask the effects of PPARγ deletion on adipose tissue COX-2 expression (34). Indeed, a number of circulating factors has been reported to exert opposite and counter-regulatory roles on adipose tissue COX-2 expression, including angiotensin II and glucocorticoids (down-regulating) and TNFα and IL-1β (up-regulating) (35, 36). On the other hand, exogenous PGE2 was able to suppress PPARγ expression, whereas opposite effects were seen after inhibition of endogenous PGE2 biosynthesis in adipose tissue from WT mice. The existence of this negative regulatory loop is consistent with previous studies showing that pre-adipocytes stably transfected with either COX-1 or COX-2 had lower PPARγ expression (16) and that mice genetically deficient for mPGES-1 had basal elevations in PPARγ expression and transcriptional activity (17). Moreover, our results are suggestive of the existence of a positive feedback loop between PGE2 and local COX-2 and mPGES-1 expression in adipose tissue (Fig. 7). This finding is consistent with a previously described positive feedback loop between PGE2 and mPGES-1 in the porcine endometrium (37) and between PGE2 and COX-2 in abdominal WAT (38).
FIGURE 7.
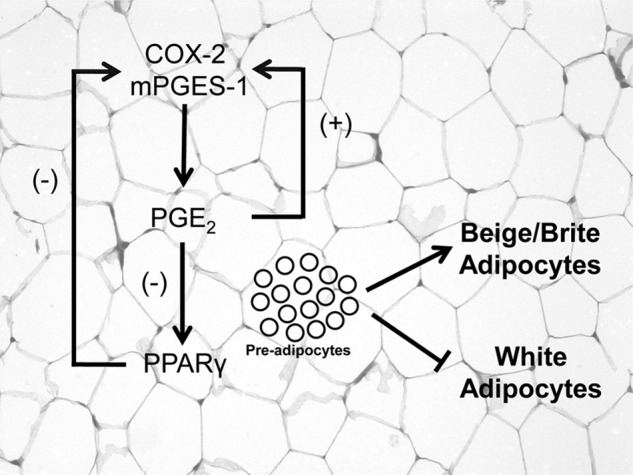
Schematic diagram of the proposed coordinated functional regulation of mPGES-1 and PPARγ in beige/brite adipogenesis. PPARγ deletion in WAT uncovers a negative regulation of COX-2 and mPGES-1 expression by this nuclear receptor. In addition, PGE2 is able to down-regulate PPARγ expression in WAT. Furthermore, in this tissue the interaction between PGE2 and PPARγ has the ability to divert pre-adipocyte differentiation from white into beige/brite adipocytes. Interestingly, PGE2 exerts a PPARγ-independent positive feedback loop with local COX-2 and mPGES-1 expression in adipose tissue.
Our findings indicate that the end result of the coordinate functional regulation between COX-2/mPGES-1/PGE2 and the nuclear receptor PPARγ is the blockage of differentiation of pre-adipocytes into mature white adipocytes and the concomitant rise of beige/brite adipocytes. The mechanisms by which the COX-2/mPGES-1/PGE2 axis and the nuclear receptor PPARγ interact during the process of adipogenesis are not completely delineated but might be related to the ability of PGE2 to increase intracellular cAMP, which is a well known mediator of the induction of “brown fat-like” cells residing in the WAT (7, 8). Another mechanism potentially implicated is the stabilization of several dominant transcriptional regulators of white-to-brown adipocyte development and function, including PRDM16 (9, 11, 38). In fact, our results demonstrate a reduction in PRDM16 in adipocytes incubated with an inhibitor of mPGES-1 activity or transfected with a siRNA that induce gene silencing of this terminal PG synthase. In any event, our findings indicate that this process requires the integrity of both systems because PGE2 is not able to inhibit white adipocyte differentiation in the absence of a PPARγ agonist. Moreover, PGE2 is able to directly induce the browning of WAT (i.e. UCP1) in WT mice but not in mice lacking PPARγ specifically in adipocytes. Finally, the existence of a loop between PGE2 and PPARγ in the regulation of adipocyte browning appears not to be a generalized event to all fat depots. In this regard, Δadip mice showed more UCP1 expression in eWAT and less expression in iBAT than WT animals. Although the mechanisms underlying this observation are unknown, a remarkable reduction of UCP1 expression in interscapular, but not in perigonadal adipose tissue has been previously described in Δadip mice (39).
In addition to regulating adipogenesis, PGs are well known lipid mediators that reproduce the cardinal signs of inflammation (40). PGs are formed through the sequential actions of COXs and a variety of terminal PG synthases, which are expressed with some tissue specificity (41). Among the different PGs, PGE2 is the most abundant and closely associated with inflammatory conditions and is synthesized by the concerted action of COXs and PGE synthases. In particular, mPGES-1 is preferentially linked with inducible COX-2, which is then coupled with inflammatory conditions in a variety of cells and tissues (13). On the other hand, PPARγ is a ligand-activated transcription factor implicated in the regulation of inflammatory responses and a nuclear factor assisting the resolution of inflammation (42). Therefore, it was pertinent in our study to assess the inflammatory status in adipose tissue from mice specifically lacking PPARγ in this tissue. Our findings showing increased proinflammatory adipokine expression (i.e. TNF-α, IL-6, and MCP-1) in fat from PPARγ knock-out mice further support the previously described anti-inflammatory role of this nuclear receptor (42). Our findings also show that either pharmacological or genetic inhibition of mPGES-1 results in a reduction of TNF-α expression in adipose tissue from WT mice, a response that was not significantly different from that seen in PPARγ KO mice. This observation suggests that contrary to the adipogenic process, there is no coordinate functional regulation between mPGES-1 and PPARγ in the control of adipose tissue inflammation. Considering the duality of PGE2 in WAT where it promotes the browning and heat dissipating function of adipocytes in parallel with a causative responsibility in adipose inflammation, further studies are needed to fully elucidate the overall role of the COX-2/mPGES-1/PGE2 pathway on the insulin axis and the development or prevention of obesity-related co-morbidities.
In conclusion, the present study demonstrates the existence of a functional interaction between PPARγ and mPGES-1 in the process of adipogenesis, especially in the formation of beige/brite adipocytes from WAT pre-adipocytes. The PGE2-mediated actions on pre-adipocyte differentiation are not mirrored by other lipid mediators such as LTB4. Finally, our findings suggest that the coordinate functional interaction between PPARγ and mPGES-1 does not appear to contribute to the regulation of the unbalanced inflammatory tone associated with obesity.
Acknowledgments
CIBERehd is funded by the Instituto de Salud Carlos III. Our laboratory is a Consolidated Research Group recognized by the Generalitat de Catalunya (Grant 2009SGR1484).
This work was supported by Ministerio de Ciencia e Innovación Grants SAF09/08767 and SAF12/32789.

This article contains supplemental Tables 1 and 2 and Figs. 1–3.
- WAT
- white adipose tissue
- eWAT
- epididymal WAT
- Δadip
- adipocyte-specific PPARγ-deficient mice
- aP2
- adipocyte fatty acid-binding protein 4
- BAT
- brown adipose tissue
- iBAT
- interscapular BAT
- HFD
- high fat diet
- PGE
- prostaglandin E
- mPGES-1
- microsomal PGE synthase-1
- PPARγ
- peroxisome proliferator-activated receptor γ
- PGC-1α
- PPARγ co-activator-1 α
- PRDM16
- PRD1-BF-1-RIZ1 homologous domain containing protein-16
- UCP1
- uncoupling protein 1
- 15d-PGJ2
- 15-deoxy-Δ12,14-PGJ2
- DPBS
- Dulbecco's PBS
- 5-LO
- 5-lipoxygenase
- SVC
- stromal vascular cell
- T3
- 3,5,3′-triiodothyronine
- LTB4
- leukotriene B4.
REFERENCES
- 1. Cristancho A. G., Lazar M. A. (2011) Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 12, 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosen E. D., Spiegelman B. M., (2006) Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444, 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galic S., Oakhill J. S., Steinberg G. R. (2010) Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 316, 129–139 [DOI] [PubMed] [Google Scholar]
- 4. Trayhurn P. (2005) Endocrine and signalling role of adipose tissue. New perspectives on fat. Acta Physiol. Scand. 184, 285–293 [DOI] [PubMed] [Google Scholar]
- 5. Kajimura S., Seale P., Spiegelman B. M. (2010) Transcriptional control of brown fat development. Cell Metab. 11, 257–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu J., Boström P., Sparks L. M., Ye L., Choi J. H., Giang A. H., Khandekar M., Virtanen K. A., Nuutila P., Schaart G., Huang K., Tu H., van Marken Lichtenbelt W. D., Hoeks J., Enerbäck S., Schrauwen P., Spiegelman B. M. (2012) Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cederberg A., Grønning L. M., Ahrén B., Taskén K., Carlsson P., Enerbäck S. (2001) FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 106, 563–573 [DOI] [PubMed] [Google Scholar]
- 8. Leonardsson G., Steel J. H., Christian M., Pocock V., Milligan S., Bell J., So P. W., Medina-Gomez G., Vidal-Puig A., White R., Parker M. G. (2004) Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc. Natl. Acad. Sci. U.S.A. 101, 8437–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seale P., Conroe H. M., Estall J., Kajimura S., Frontini A., Ishibashi J., Cohen P., Cinti S., Spiegelman B. M. (2011) Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Invest. 121, 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barak Y., Nelson M. C., Ong E. S., Jones Y. Z., Ruiz-Lozano P., Chien K. R., Koder A., Evans R. M. (1999) PPAR γ is required for placental, cardiac, and adipose tissue development. Mol. Cell 4, 585–595 [DOI] [PubMed] [Google Scholar]
- 11. Koppen A., Kalkhoven E. (2010) Brown vs white adipocytes. the PPARγ coregulator story. FEBS Lett. 584, 3250–3259 [DOI] [PubMed] [Google Scholar]
- 12. Ohno H., Shinoda K., Spiegelman B. M., Kajimura S. (2012) PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 15, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jakobsson P. J., Thorén S., Morgenstern R., Samuelsson B. (1999) Identification of human prostaglandin E synthase. A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. U.S.A. 96, 7220–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chatzipanteli K., Rudolph S., Axelrod L. (1992) Coordinate control of lipolysis by prostaglandin E2 and prostacyclin in rat adipose tissue. Diabetes 41, 927–935 [DOI] [PubMed] [Google Scholar]
- 15. Tsuboi H., Sugimoto Y., Kainoh T., Ichikawa A. (2004) Prostanoid EP4 receptor is involved in suppression of 3T3-L1 adipocyte differentiation. Biochem. Biophys. Res. Commun. 322, 1066–1072 [DOI] [PubMed] [Google Scholar]
- 16. Chu X., Xu L., Nishimura K., Jisaka M., Nagaya T., Shono F., Yokota K. (2009) Suppression of adipogenesis program in cultured preadipocytes transfected stably with cyclooxygenase isoforms. Biochim. Biophys. Acta 1791, 273–280 [DOI] [PubMed] [Google Scholar]
- 17. Kapoor M., Kojima F., Qian M., Yang L., Crofford L. J. (2007) Microsomal prostaglandin E synthase-1 deficiency is associated with elevated peroxisome proliferator-activated receptor γ. Regulation by prostaglandin E2 via the phosphatidylinositol 3-kinase and Akt pathway. J. Biol. Chem. 282, 5356–5366 [DOI] [PubMed] [Google Scholar]
- 18. Vegiopoulos A., Müller-Decker K., Strzoda D., Schmitt I., Chichelnitskiy E., Ostertag A., Berriel Diaz M., Rozman J., Hrabe de Angelis M., Nüsing R. M., Meyer C. W., Wahli W., Klingenspor M., Herzig S. (2010) Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 328, 1158–1161 [DOI] [PubMed] [Google Scholar]
- 19. Ghoshal S., Trivedi D. B., Graf G. A., Loftin C. D. (2011) Cyclooxygenase-2 deficiency attenuates adipose tissue differentiation and inflammation in mice. J. Biol. Chem. 286, 889–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morán-Salvador E., López-Parra M., García-Alonso V., Titos E., Martínez-Clemente M., González-Périz A., López-Vicario C., Barak Y., Arroyo V., Clària J. (2011) Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 25, 2538–2550 [DOI] [PubMed] [Google Scholar]
- 21. He W., Barak Y., Hevener A., Olson P., Liao D., Le J., Nelson M., Ong E., Olefsky J. M., Evans R. M. (2003) Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. U.S.A. 100, 15712–15717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shan T., Liu W., Kuang S. (2013) Fatty acid binding protein 4 expression marks a population of adipocyte progenitors in white and brown adipose tissues. FASEB J. 27, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Titos E., Rius B., González-Périz A., López-Vicario C., Morán-Salvador E., Martínez-Clemente M., Arroyo V., Clària J. (2011) Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J. Immunol. 187, 5408–5418 [DOI] [PubMed] [Google Scholar]
- 24. Horrillo R., González-Périz A., Martínez-Clemente M., López-Parra M., Ferré N., Titos E., Morán-Salvador E., Deulofeu R., Arroyo V., Clària J. (2010) 5-Lipoxygenase-activating protein signals adipose tissue inflammation and lipid dysfunction in experimental obesity. J. Immunol. 184, 3978–3987 [DOI] [PubMed] [Google Scholar]
- 25. Rodeheffer M. S., Birsoy K., Friedman J. M. (2008) Identification of white adipocyte progenitor cells in vivo. Cell 135, 240–249 [DOI] [PubMed] [Google Scholar]
- 26. Fisher F. M., Kleiner S., Douris N., Fox E. C., Mepani R. J., Verdeguer F., Wu J., Kharitonenkov A., Flier J. S., Maratos-Flier E., Spiegelman B. M. (2012) FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun L., Xie H., Mori M. A., Alexander R., Yuan B., Hattangadi S. M., Liu Q., Kahn C. R., Lodish H. F. (2011) Mir193b-365 is essential for brown fat differentiation. Nat. Cell Biol. 13, 958–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Brien S. N., Mantzke K. A., Kilgore M. W., Price T. M. (1996) Relationship between adipose stromal-vascular cells and adipocytes in human adipose tissue. Anal. Quant. Cytol. Histol. 18, 137–143 [PubMed] [Google Scholar]
- 29. Koeberle A., Haberl E. M., Rossi A., Pergola C., Dehm F., Northoff H., Troschuetz R., Sautebin L., Werz O. (2009) Discovery of benzo[g]indol-3-carboxylates as potent inhibitors of microsomal prostaglandin E(2) synthase-1. Bioorg. Med. Chem. 17, 7924–7932 [DOI] [PubMed] [Google Scholar]
- 30. Scott M. A., Nguyen V. T., Levi B., James A. W. (2011) Current methods of adipogenic differentiation of mesenchymal stem cells. Stem. Cells Dev. 20, 1793–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karg E. M., Luderer S., Pergola C., Bühring U., Rossi A., Northoff H., Sautebin L., Troschütz R., Werz O. (2009) Structural optimization and biological evaluation of 2-substituted 5-hydroxyindole-3-carboxylates as potent inhibitors of human 5-lipoxygenase. J. Med. Chem. 52, 3474–3483 [DOI] [PubMed] [Google Scholar]
- 32. Madsen L., Pedersen L. M., Lillefosse H. H., Fjaere E., Bronstad I., Hao Q., Petersen R. K., Hallenborg P., Ma T., De Matteis R., Araujo P., Mercader J., Bonet M. L., Hansen J. B., Cannon B., Nedergaard J., Wang J., Cinti S., Voshol P., Døskeland S. O., Kristiansen K. (2010) UCP1 induction during recruitment of brown adipocytes in white adipose tissue is dependent on cyclooxygenase activity. PLoS ONE 5, e11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bell-Parikh L. C., Ide T., Lawson J. A., McNamara P., Reilly M., FitzGerald G. A. (2003) Biosynthesis of 15-deoxy-Δ12,14-PGJ2 and the ligation of PPARγ. J. Clin. Invest. 112, 945–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith W. L., DeWitt D. L., Garavito R.M. (2000) Cyclooxygenases. Structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182 [DOI] [PubMed] [Google Scholar]
- 35. Santos S. H., Fernandes L. R., Pereira C. S., Guimarães A. L., de Paula A. M., Campagnole-Santos M. J., Alvarez-Leite J. I., Bader M., Santos R. A. (2012) Increased circulating angiotensin-(1–7) protects white adipose tissue against development of a proinflammatory state stimulated by a high-fat diet. Regul. Pept. 178, 64–70 [DOI] [PubMed] [Google Scholar]
- 36. Huang Z. F., Massey J. B., Via D. P. (2000) Differential regulation of cyclooxygenase-2 (COX-2) mRNA stability by interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) in human in vitro differentiated macrophages. Biochem. Pharmacol. 59, 187–194 [DOI] [PubMed] [Google Scholar]
- 37. Waclawik A., Jabbour H. N., Blitek A., Ziecik A. J. (2009) Estradiol-17β, prostaglandin E2 (PGE2), and the PGE2 receptor are involved in PGE2 positive feedback loop in the porcine endometrium. Endocrinology 150, 3823–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahfeldt T., Schinzel R. T., Lee Y. K., Hendrickson D., Kaplan A., Lum D. H., Camahort R., Xia F., Shay J., Rhee E. P., Clish C. B., Deo R. C., Shen T., Lau F. H., Cowley A., Mowrer G., Al-Siddiqi H., Nahrendorf M., Musunuru K., Gerszten R. E., Rinn J. L., Cowan C. A. (2012) Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 14, 209–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones J. R., Barrick C., Kim K. A., Lindner J., Blondeau B., Fujimoto Y., Shiota M., Kesterson R. A., Kahn B. B., Magnuson M. A. (2005) Deletion of PPARγ in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 102, 6207–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Willoughby D. A., Colville-Nash P. R., Seed M. P. (1993) Inflammation, prostaglandins, and loss of function. J. Lipid. Mediat. 6, 287–293 [PubMed] [Google Scholar]
- 41. Romano M., Claria J. (2003) Cyclooxygenase-2 and 5-lipoxygenase converging functions on cell proliferation and tumor angiogenesis. Implications for cancer therapy. FASEB J. 17, 1986–1995 [DOI] [PubMed] [Google Scholar]
- 42. Ricote M., Li A. C., Willson T. M., Kelly C. J., Glass C. K., (1998) The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 391, 79–82 [DOI] [PubMed] [Google Scholar]