

Background: Modulation of GABAA receptors trafficking is critical for controlling inhibitory neurotransmission.
Results: A point mutation or agonist application, both affecting the GABAA receptor extracellular domain, has an effect on receptor endocytosis.
Conclusion: Endocytosis of GABAA receptors is linked to agonist-induced conformational changes.
Significance: This represents one of the few reports demonstrating an influence of extracellular effectors on GABAA receptor trafficking.
Keywords: GABA Receptors, Neurobiology, Neurological Diseases, Receptor Endocytosis, Trafficking
Abstract
GABA-gated chloride channels (GABAARs) trafficking is involved in the regulation of fast inhibitory transmission. Here, we took advantage of a γ2(R43Q) subunit mutation linked to epilepsy in humans that considerably reduces the number of GABAARs on the cell surface to better understand the trafficking of GABAARs. Using recombinant expression in cultured rat hippocampal neurons and COS-7 cells, we showed that receptors containing γ2(R43Q) were addressed to the cell membrane but underwent clathrin-mediated dynamin-dependent endocytosis. The γ2(R43Q)-dependent endocytosis was reduced by GABAAR antagonists. These data, in addition to a new homology model, suggested that a conformational change in the extracellular domain of γ2(R43Q)-containing GABAARs increased their internalization. This led us to show that endogenous and recombinant wild-type GABAAR endocytosis in both cultured neurons and COS-7 cells can be amplified by their agonists. These findings revealed not only a direct relationship between endocytosis of GABAARs and a genetic neurological disorder but also that trafficking of these receptors can be modulated by their agonist.
Introduction
Inhibitory transmission relies greatly on ionotropic GABAA receptors (GABAARs)3 that are involved in many physiological functions and are the target of several drugs in wide clinical use (1). GABAAR trafficking is modulated by a number of different mechanisms, including surface targeting, mobility, and endocytosis. Moreover, studies in physiological and pathophysiological states have revealed that the number of GABAARs on the cell membrane have a profound influence on GABAergic neurotransmission (1–3). Inhibitory neurotransmission is also regulated by the exchange between surface and intracellular compartments via a constitutive clathrin-mediated dynamin-dependent endocytosis pathway (4–6). This constitutive internalization is modulated by intracellular mechanisms and is altered in pathological conditions (4, 6–9).
Epilepsies are complex syndromes with multiple causes and symptoms, but it is well established that alteration or modulation of GABA neurotransmission plays an important role in the disease and its treatment (10–15). Moreover, genetic evidence has revealed a direct link between epilepsy and GABAAR dysfunction, including trafficking alteration, supporting the hypothesis that defects in GABAARs lead to seizures (16–17). These mutations also offer an opportunity to obtain new insights into GABAAR structure and function as well as clues to the role of these receptors in neurological disorders (14). For example, an R43Q mutation located in the γ2 subunit N-terminal extracellular domain is linked to childhood absence epilepsy and febrile seizure (17); heterozygous mice harboring this mutation replicate the human clinical phenotype (16). Intensive research into this mutation (18–26) has led to controversial data on its effects on GABAergic physiology suggesting that γ2(R43Q) might modify the dynamics of subunit trafficking (27).
Here, we analyzed γ2(R43Q) trafficking in cultured hippocampal neurons and COS-7 cells and revealed that receptors containing the γ2(R43Q) subunit had a shorter residence time on the plasma membrane than their wild-type counterparts. We also showed that endocytosis of the mutated receptor was clathrin- and dynamin-dependent. However, it was surprising that a mutation in the extracellular domain (bearing binding sites for agonists and modulators) could have an influence on internalization, believed to be controlled through the intracellular domain. Moreover, endocytosis of GABAARs triggered by agonist exposure remains to be fully assessed (11, 28–31). Then, by using both imaging and biochemical methods, further experiments revealed that agonist exposure triggered an increase of wild-type GABAAR endocytosis, both on native and recombinant GABAARs.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies were raised in rabbits against the α1 GABAA subunit N terminus (Alomone Labs), the Myc tag (Upstate, Charlottesville, VA), membrin (1:1000; Synaptic Systems), calreticulin (1:500; Upstate Biotechnology, Inc.), or EEA1 (Sigma-Aldrich). Antibodies were raised in mice against the α1 intracellular loop (Neuromab) or the Myc tag (Roche Applied Science). Secondary antibodies were as follows: Alexa Fluor-568 goat anti-rabbit, Alexa Fluor-647 goat anti-rabbit, Alexa Fluor-488 goat anti-mouse (1:1000; Molecular Probes), and FITC-coupled anti-mouse antibody (1:200; Chemicon).
DNA Constructs
α1, β2, β3, and γ2S GABAAR subunits were subcloned in the pcDNA3 vector (Invitrogen), using constructs that were available from previous studies (32, 33). The Myc epitope (MEQKLISEEDLNE, repeated 6 times) was inserted between amino acids 4 and 5 of the γ2 subunit (22). As described previously, an insertion within this domain does not modify the functional properties of GABA- or glutamate-gated channels (32, 34–36). Point mutations were constructed using the QuikChange site-directed mutagenesis system (Stratagene). All constructs were verified by automatic dideoxy DNA sequencing (Genome Express, Meylan, France). Endoplasmic reticulum-Golgi intermediate compartment was revealed with ERGIC-GFP (37), kindly provided by Jochen Lang (Bordeaux, France).
Cell Culture and Transfection
COS-7 and HEK 293 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal calf serum (Eurobio). Embryonic hippocampal neurons were obtained from E18 rat embryos, as described earlier (22). COS-7 and HEK 293 cells were transfected using the FuGENE 6 reagent (Roche Applied Science), according to the manufacturer's specifications, with equal amounts of α1, β, and γ2 subunit cDNAs (and GFP for electrophysiology experiments on HEK 293 cells) (0.3 μg/well in 24-well plates). Cells were incubated with cDNAs for 24 h before analysis (22). Hippocampal neurons were transfected in vitro at 7–11 days, using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's specifications. The cells were analyzed 24–60 h after transfection (22).
Immunocytochemistry
For living cell surface labeling, COS-7 cells and hippocampal neurons were incubated with antibodies at room temperature for 20 min in Dulbecco's modified Eagle's medium or Neurobasal medium supplemented with 10 mm HEPES, respectively. Receptors on the surface were labeled with antibodies raised in rabbits against either the α1 GABAA subunit N-terminal domain or the Myc tag. Sera were diluted 1:500 (anti-α1) or 1:200 (anti-Myc) in medium. After incubation, cells were washed quickly by dipping coverslips in medium and fixed for 10 min in phosphate-buffered saline (PBS) containing 4% sucrose and 4% paraformaldehyde preheated to 37 °C, washed in PBS, and blocked in 0.3% bovine serum albumin and 50 mm glycine (in PBS) for 15 min. Cells were washed in PBS containing 0.3% bovine serum albumin. After cell permeabilization using 0.3% Triton X-100, intracellular tagged γ2 subunits were detected by incubating the cells with a mouse anti-Myc 9E10 antibody (1:1000; Roche Applied Science) for 2 h. Intracellular α1 subunits were detected with a mouse anti-α1 (1:1000). Polyclonal and monoclonal antibodies were detected using an Alexa Fluor-568-coupled anti-rabbit antibody and an Alexa Fluor-488-coupled anti-mouse antibody (1:1000), respectively. Endoreticulum and cis-Golgi staining were revealed with polyclonal anti-calreticulin (1:500) and anti-membrin (1:1000), respectively. The endoplasmic reticulum-Golgi intermediate compartment was revealed with ERGIC-GFP (37).
Internalization
Transfected neurons or COS cells were incubated in culture medium containing mouse monoclonal Myc antibody (1:200) at 37 °C for 30 min. The medium also contained GABAAR agonists or antagonists as required. In the case of neurons, a mixture of inhibitors (6-cyano-7-nitroquinoxalene-2,3-dione (10 μm), d-2-amino-5-phosphonovaleric acid (50 μm), and tetrodotoxin (1 μm)) was added, so as to prevent activity-dependent modulation of GABAAR trafficking (7). This mixture was added 5 min before the labeling experiments and was present throughout, both in control experiments and in GABAAR agonist/antagonist-treated neurons. Surface labeling was carried out at 20 °C using an anti-mouse secondary antibody coupled to Alexa Fluor-647 for 30 min (38); alternatively, labeled receptors remaining on the COS-7 cell surface were acid-washed for 2 min at 20 °C with culture medium adjusted at pH 2.5 (39, 40). Cells were then fixed using 4% paraformaldehyde in 4% sucrose and permeabilized in Triton X-100. Total protein content was assessed by incubating with a polyclonal antibody directed at the Myc tag. Internalized protein was then revealed by incubating with anti-mouse antibody coupled to Alexa Fluor-488 at room temperature for 1 h, whereas total protein was revealed using anti-rabbit antibody coupled to Alexa Fluor-568. For native α1 expressed in hippocampal neurons, cells were incubated in culture medium containing an antibody made in rabbits directed at the N-terminal domain (extracellular) of α1 (1:500) at 37 °C for 30 min. Surface labeling was carried as above using an anti-rabbit secondary antibody. Cells were then fixed and permeabilized as above. Total α1 subunit content was assessed by incubating with a mouse antibody directed at the Myc tag or at the native α1 at room temperature for 20 min. Internalized protein was then revealed by incubating cells with anti-mouse antibody coupled to Alexa Fluor-488 at room temperature for 1 h, whereas total protein was revealed using anti-rabbit antibody coupled to Alexa Fluor-568.
Quantitative Analysis of Fluorescence Signals
Fluorescence microscopy was performed using a Zeiss Axioplan 2 microscope, with a ×63, 1.4 numerical aperture oil immersion lens. Quantification of fluorescence signals and background subtraction were performed using ImageJ (National Institutes of Health). For each image acquired, background levels were determined using the surface and intracellular signals measured in neighboring non-transfected cells and subtracted from the values obtained in transfected cells. Numerical data are presented as mean ± S.E., and statistical significance was assessed using one-way analysis of variance (Origin, Originlab Corp.) (significance level, p < 0.05). Confocal microscopy was performed using an upright Leica DMR TCS SPZ AOBS, with a ×63, 1.4 numerical aperture Leica HPCL Fluotar oil objective. Colocalization was quantified using a plugin for ImageJ designed by F. Levet and C. Poujol (BIC (Bordeaux Imaging Center), Bordeaux, France). Briefly, two images, one containing GABAAR subunit labeling and one containing the labeling for a cellular compartment, were thresholded in the same way. The plugin calculates the percentage of pixels containing γ2 subunit labeling that also contain specific labeling for a cellular compartment. The percentage of colocalization was normalized for total γ2 and γ2(R43Q) immunoreactivity, respectively. Analyses were performed in parallel cultures, blind to experimental conditions.
Quantification of surface clusters or intracellular punctate labeling, blind to experimental conditions, was performed using ImageJ (National Institutes of Health). Threshold was applied to the images, and the number as well as the area of surface clusters or internalized particles were measured using the particle analyzer module of ImageJ. For COS-7 cells, the whole cell was counted. For neurons, an area of 10-μm length along a dendrite was counted. For all experiments, total protein expression was assessed by antibody labeling after permeabilization of the cells and was measured for the same area to allow normalization of the values. To calculate fluorescence ratios, a stack was created for each cell in ImageJ with the image corresponding to the surface and total labeling. This allows us to draw the outline of the cell and measure the average surface and total fluorescence for the same area.
Biotinylation Assays
Biotinylation experiments were performed essentially as described previously (36, 38). COS-7 cells were transfected in 6-well plates (2 wells/condition) and were incubated 24 h post-transfection. Cells were then washed two times with PBS, pH 8.0, incubated with 1 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Pierce) in PBS for 30 min at 4 °C, washed three times with PBS, and scraped in lysis buffer containing 25 mm HEPES, 150 mm NaCl, 1% Triton X-100, and a mix of protease inhibitors (Roche Applied Science). After centrifugation, the supernatant was immunoprecipitated with 50 μl of Immunopure immobilized streptavidin-beaded agarose overnight at 4 °C and washed extensively. Surface and total proteins were separated on SDS-PAGE and revealed by Western blotting using anti-Myc antibodies at a 1:1000 dilution. Quantification of Western blots was performed using ImageJ (National Institutes of Health) or with the Chemi Doc XRS+ under the control of the Image Lab software (Bio-Rad). To assess γ2 internalization, plates were returned to 37 °C for 30 min after biotinylation to allow endocytosis. Cells were then exposed to 50 mm MESNA, which cleaved biotin from proteins remaining on the surface. A sample was kept at 4 °C (instead of 37 °C) to control to ensure that the cleavage with MESNA was complete. Samples were analyzed by Western blot as above.
Electrophoretic and Western Blot Analyses
COS-7 cells were homogenized in buffer containing 20 mm HEPES, 0.15 mm EDTA, and 10 mm KCl, pH 8, supplemented with a mixture of protease inhibitors (Roche Applied Science). The buffer was then adjusted to 12% sucrose, and after four more strokes, the cells were centrifuged at 2000 rpm for 3 min to remove genomic DNA. The supernatant was centrifuged at 15,000 rpm for 30 min. The pellet was recovered, and cell membranes were solubilized with 15 strokes in a buffer containing 20 mm Tris-HCl, 0.15 mm EDTA, 150 mm NaCl, 2% Triton X-100, and 0.5% deoxycholate, pH 8, supplemented with a mixture of protease inhibitors, and then incubated for 45 min. The sample was centrifuged for 45 min at 15,000 rpm. The supernatant was supplemented with loading buffer and analyzed as described (22).
Electrophysiology
Brightly fluorescent isolated HEK 293 cells were selected for recording. Cells were bathed in a solution containing 150 mm NaCl, 2 mm KCl, 2 mm MgCl2, 2 mm CaCl2, 10 mm glucose, 10 mm HEPES, equilibrated to pH 7.4 with NaOH. Cells were recorded in whole cell mode and placed under the flow of a theta tube pulled to a final opening of ∼100 μm mounted on a piezoelectric translator (Physik Instrumente). Currents were evoked by applications of 100 μm GABA for 5 s every minute at −60 mV and recorded at a sampling frequency of 2 kHz by an EPC10 amplifier (HEKA). GABA was exchanged for gabazine (100 μm) and applied to the cell after 3 min, necessary for complete exchange. Thereafter, the medium was exchanged again for GABA, and we observed complete recovery of the response to the agonist. Data were analyzed with Igor 5 (Wavemetrics).
Model Building
The sequences of the human α1, β2, and γ2 GABA receptor subunits were retrieved from the Ligand Gated Ion Channel database (41). The model of the α1β2γ2 receptor was constructed by homology modeling using the structure of the glutamate-gated chloride channel as a template (Protein Data Bank code 3RIF) (42) and sequences alignments obtained with T-coffee (43). Homology modeling was performed with Modeler version 9.5 (44) using default settings. 100 models were prepared, and the best model according to the Discrete Optimized Protein Energy function (DOPE) was selected. Figures were prepared with PyMOL (77).
RESULTS
γ2(R43Q) Displays Increased Clathrin-mediated and Dynamin-dependent Endocytosis
To analyze the impact of the R43Q substitution on intracellular trafficking, the colocalization of wild-type or mutated γ2 with markers of intracellular compartments was assessed in transfected, cultured hippocampal neurons (Fig. 1, A and B). Increased retention in the endoplasmic reticulum (CalR colocalization: 38.3 ± 2.5% for γ2(R43Q)-containing subunits versus 26.7 ± 2.3% for wild-type receptors, n = 31 and 27, respectively; p < 0.005) confirmed previous findings in heterologous cells (HEK293 (24)). However, importantly, γ2(R43Q) colocalization with Golgi apparatus markers did not differ significantly from that of the wild-type subunit, showing that the mutated subunit was also present on the route to the cell membrane (membrin: 19.3 ± 2.4% for the wild-type versus 15.2 ± 1.6% for the γ2(R43Q)-containing receptor, n = 35 and 38, respectively; ERGIC: 18.6 ± 1.7% for the wild-type versus 13.4 ± 1.9% for the γ2(R43Q)-containing receptor, n = 28 and 34, respectively). We therefore checked whether the absence of surface labeling was due to fast endocytosis mechanisms, as proposed to explain the exclusion of sodium channels from somatic domains (45). When living neurons, expressing γ2(R43Q), were incubated at 37 °C with antibodies directed against the tagged extracellular N-terminal γ2-domain, intracellular punctate labeling was detected (Fig. 1C), suggesting receptor internalization. This was confirmed by the increased colocalization of γ2(R43Q) with EEA1, an early endosome marker (Fig. 1, D and E) (22.8 ± 4.7% for the wild-type versus 39.5 ± 4.1% for the γ2(R43Q)-containing receptor, n = 14 and 16, respectively, p < 0.005).
FIGURE 1.

GABAARs containing γ2(R43Q) are found in internalized compartments. A, cultured hippocampal neurons were transfected with γ2Myc or γ2Myc(R43Q) subunits and stained for Myc tag together with a cis-Golgi compartment marker (membrin) and analyzed by confocal microscopy. B, quantitative analyses of the colocalization of γ2 subunits with intracellular compartments (Ergic, endoplasmic reticulum-Golgi intermediate compartment; CalR, endoplasmic reticulum; membrin, cis-Golgi), normalized to Myc tag total immunoreactivity. C, live neurons expressing γ2Myc(R43Q) were incubated with a monoclonal Myc antibody and FITC-conjugated secondary antibodies (left) at 37 °C, and then the fixed, permeabilized neurons were labeled with a polyclonal antibody and Alexa Fluor-568-conjugated secondary antibodies (middle). Merged images revealed intracellular punctate labeling corresponding to internalized receptors within a transfected neuron (arrows). D, cultured hippocampal neurons were transfected with γ2Myc and γ2Myc(R43Q) subunits and stained for Myc tag together with an early endosome marker (EEA1). Arrowheads, labeled subunits not colocalized with EEA1; arrows, colocalized staining. E, quantitative analyses of the colocalization of γ2 subunits with EEA1, normalized to Myc tag total immunoreactivity. Scale bars, 40 μm (A and C) or 20 μm (D). **, p < 0.005. Error bars, S.E.
Because clathrin-mediated, dynamin-dependent endocytosis is the major neuronal GABAAR internalization mechanism (4, 6), we tested whether γ2(R43Q) internalization was driven by a similar pathway. When dynamin was inhibited by incubation with 80 μm dynasore (46), γ2(R43Q) was detected at the plasma membrane of transfected COS-7 cells (Fig. 2A). The ratio of γ2(R43Q) subunit labeled on the cell surface versus γ2(R43Q) labeled within intracellular compartments was increased from 0.42 ± 0.2 to 0.78 ± 0.18 (Fig. 2B). COS-7 cells were then transfected with a β2(LL/AA) mutant; this mutation within the β2 intracellular domain reduces the interaction of GABAAR with the AP2 complex (6). In this case, the signal ratio between surface and intracellular γ2(R43Q) increased from 0.18 ± 0.08 to 0.53 ± 0.1 (Fig. 2, C and D), showing that γ2(R43Q) was present on the cell surface when co-expressed with α1 and β2(LL/AA). Taken together, these data showed that the γ2 subunit containing the R43Q mutation increased endocytosis of GABAARs, hindering their detection on the cell surface.
FIGURE 2.

γ2(R43Q) internalization is clathrin- and dynamin-dependent. A, COS-7 cells were transiently co-transfected with α1-, β3-, and Myc-tagged γ2 subunits (1:1:1 ratio), either wild-type (γ2) or bearing the R43Q (γ2(R43Q)) substitution, as indicated at the top. Surface labeling was obtained after immunostaining living cells with a polyclonal antibody directed at the extracellular Myc tag and Alexa Fluor-568-conjugated secondary antibodies (surface), and intracellular staining of the same cells was revealed using a monoclonal antibody directed at the same tag and FITC-conjugated secondary antibodies (intra). Merged images show intracellular retention of the γ2(R43Q) subunit (control), whereas surface labeling was detected when cells were incubated with dynasore. Scale bar, 10 μm. B, the ratio of surface/total expression was measured in COS-7 cells expressing α1β3γ2 GABAARs, after incubation with dynasore (n = 13–22). C, cells were transiently co-transfected with α1-, β2-, or β2LL/AA and Myc-tagged γ2 subunit (1:1:1 ratio), either wild-type (γ2) or bearing the R43Q (γ2(R43Q)) substitution, as indicated at the top. Surface and intracellular labeling were obtained as above. Merged images show intracellular retention of the γ2(R43Q) subunit when cells were co-transfected with β2, whereas surface labeling was detected when cells were co-transfected with β2LL/AA. Scale bar, 10 μm. D, quantitative analysis of α1β2γ2 and α1β2(LL/AA)γ2 GABAARs surface expression in transfected COS-7 cells, (n = 48–65). ***, p < 0.001. Error bars, S.E.
γ2(R43Q) Endocytosis Is Inhibited by GABAAR Antagonists
This increased internalization of a ligand-gated channel, resulting from a mutation within the extracellular domain of the molecular complex, was surprising. This GABAAR domain contains binding sites for agonists and allosteric modulators, whereas the intracellular domain mediates interactions with trafficking factors (1, 4, 6, 47). We therefore tested whether GABAAR ligands interfered with endocytosis. Incubating transfected neurons (Fig. 3, A and B) or COS-7 cells (Fig. 3, C and D) with two different antagonists (i.e. picrotoxin and gabazine) for 1 h significantly increased γ2(R43Q) surface expression, whereas neither GABAAR agonist nor antagonists altered surface expression levels of receptors containing wild-type γ2 subunit. The area of clusters on the surface of neurons transfected with γ2 was 2.23 ± 0.23 μm2/10 μm in control conditions and 2.50 ± 0.19 or 2.36 ± 0.16 in the presence of gabazine (100 μm) or picrotoxin (100 μm), respectively (Fig. 3B). The area of clusters on the surface of neurons transfected with γ2(R43Q) was 0.08 ± 0.02 μm2/10 μm in control conditions and increased to 0.79 ± 0.15 and 0.79 ± 0.12 in the presence of gabazine (100 μm) or picrotoxin (100 μm), respectively (Fig. 3B). In COS-7 cells transfected with α1-, β2-, and γ2 subunits, the surface/intra ratio were 3.8 ± 0.5 in control conditions and 4 ± 0.9 or 3.3 ± 0.4 in the presence of gabazine (100 μm) or picrotoxin (100 μm), respectively (Fig. 3D). In COS-7 cells transfected with α1, β2, and γ2(R43Q) subunits, the surface/intra ratio increased from 0.3 ± 0.1 to 1.27 ± 0.2 for gabazine and 1.41 ± 0.23 for picrotoxin (Fig. 3D). Muscimol (10 μm), a GABAAR agonist, had no detectable effect, compared with the control experiments (Fig. 3, B and D). Cell surface biotinylation on α1-, β2-, and γ2-transfected COS-7 cells was not modified by 100 μm gabazine + picrotoxin treatment (107 ± 4%), whereas experiments on α1-, β2-, and γ2(R43Q)-transfected cells confirmed that antagonist treatment increased significantly the surface fraction of γ2(R43Q) (213 ± 29% of control, p < 0.05, three independent experiments; Fig. 3, E and F).
FIGURE 3.
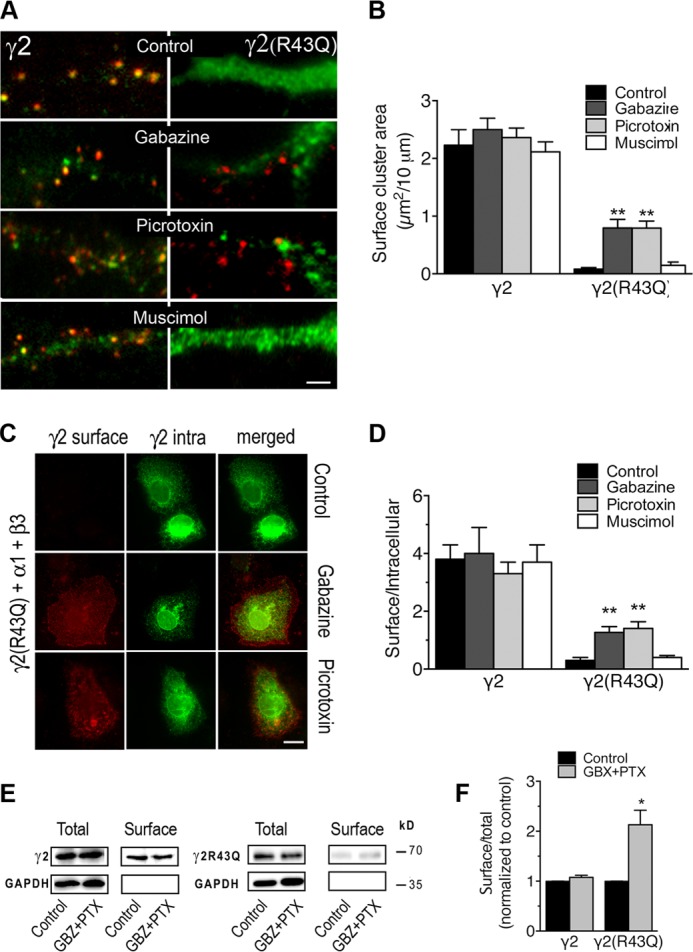
γ2(R43Q) internalization is inhibited by GABAAR antagonists. A and B, neurons were transfected with γ2Myc or γ2Myc(R43Q) subunits. Surface labeling was obtained after immunostaining live neurons at 20 °C with a polyclonal antibody directed at the extracellular Myc tag and Alexa Fluor-568-conjugated secondary antibodies. Intracellular staining of the same cells was obtained after permeabilization and labeling with a monoclonal antibody directed at the same tag and FITC-conjugated secondary antibodies. A, merged images revealed the γ2(R43Q) subunit on the surface of transfected neurons when cells were incubated with gabazine or picrotoxin at 37 °C for 1 h prior to immunostaining. Scale bar, 10 μm. B, quantitative analyses of the surface cluster area formed by γ2 constructs (n = 13–32). **, p < 0.005. C and D, COS-7 cells were transiently co-transfected with α1-, β3-, and Myc-tagged γ2 subunits, either wild-type (γ2) or bearing the R43Q (γ2R43Q) substitution. Treatment and labeling were as in A, and the surface/total expression ratio (D) was calculated as the ratio of the surface Alexa Fluor-568 fluorescence level on live cells to FITC fluorescence on permeabilized cells (n = 12–25). **, p < 0.005. E and F, COS-7 cells were transfected with α1-, β2-, and Myc-tagged γ2 or γ2(R43Q) subunits and incubated with or without picrotoxin and gabazine 30 min before surface biotinylation. E, representative Western blot shows Myc-tagged γ2 and γ2(R43Q) immunoreactivity in total or surface-biotinylated extracts from transfected cells. F, means of surface/total expression ratio from three independent experiments normalized to values of total γ2 or γ2(R43Q), as indicated, described in E show that treatment by picrotoxin and gabazine increases the surface-biotinylated fraction of cells expressing the γ2 subunit bearing the R43Q mutation. *, p < 0.01. Error bars, S.E.
The fact that picrotoxin and gabazine are both allosteric GABAAR antagonists (48–50) indicated that, compared with their wild-type counterparts, the equilibrium between states in γ2(R43Q)-containing GABAARs shifted away from the resting state toward the active or desensitized state. Our new homology model based on a glutamate-gated chloride channel (42) suggests that the Arg-43 residue of the γ2 subunit is connected to Tyr = 174 and Glu-178 from the loop B and to Asp-84 and Arg-86 of the β2 subunit via polar interactions (Fig. 4, B–D). In addition, this new model shows that γ2Arg-43 and β2Asp-84/Arg-86 are on loops (Allo1 and Allo2) identified as involved in the motion that opens the channel pore (51). This prompted us to test whether this alteration in the equilibrium between states favored an open channel conformation. We thus co-expressed wild-type or γ2(R43Q) constructs with α1 and β2 subunits in HEK cells. In α1-, β2-, and γ2(R43Q)-transfected cells, maximum GABA-gated currents were 16% (p < 0.01) of currents from cells expressing wild-type receptors (Fig. 4E), in agreement with previous findings (19, 23, 52). Gabazine, an allosteric antagonist, did not cause a significant change in the current base line during whole cell voltage clamp recordings (Fig. 4F) in HEK cells co-expressing α1-, β2-, and either wild-type or γ2(R43Q) subunits (n = 7 and 15, respectively), showing that γ2(R43Q)-containing receptors had no constitutive activity.
FIGURE 4.

The γ2(R43Q) mutation is not associated with an open channel conformation. A, schematic diagram of an α1β2γ2 GABAAR, which illustrates the five combined subunits that form the complex, the two GABA active binding sites at the β2 and α1 interfaces (gray circles), and the benzodiazepine (BDZ; blue circle) allosteric binding site at the α1 and γ2 interface. In current models, γ2Arg-43 is at the interface with β2. B–D, GABAAR model viewed from the outside (B), the top (C), and inside (D); only the γ2 and β2 subunits are shown for clarity (γ2 in green and β2 in blue). γ2 (Arg-43, Tyr-174, and Glu-178) and β2 (Asp-84 and Arg-86) residues are represented by sticks. These residues are within loops, shown in gray (γ2 subunit) or dark blue (β2), identified as being involved in the channel-opening motion. E, GABA application on HEK cells transfected with α1β2γ2 or α1β2γ2(R43Q) activated membrane currents, whereas gabazine application (F) did not change the holding current in either type of receptor.
Collectively, our data suggested that γ2(R43Q) mutation triggered GABAAR endocytosis through a structural change in the extracellular domain. Because agonist exposure triggers a long range conformational change (53, 54), our finding on mutated receptors opened the interesting possibility that endocytosis of wild-type GABAARs may be modulated by agonists. Our data did not yet indicate significant alteration of the surface level of wild-type γ2-containing receptors in the presence of agonists (Fig. 3). We thus decided to examine directly the impact of agonists on the internalized fraction of GABAARs.
GABAAR Agonist Enhances Receptor Endocytosis
To test a possible link between GABA-induced conformational change and endocytosis, suggested by our studies on γ2(R43Q), we directly quantified internalization by measuring the level of endogenous α1-containing receptors internalized during agonist application on cultured neurons (Fig. 5A). In control and muscimol experiments, excitatory activity was blocked with 6-cyano-7-nitroquinoxalene-2,3-dione, d-2-amino-5-phosphonovaleric acid, and tetrodotoxin in the medium. Neurons were incubated with an antibody directed at the extracellular N-terminal domain of α1 subunit for 30 min at 37 °C. Labeled receptors remaining at the cell surface were labeled with saturating concentration of Alexa Fluor-647 secondary antibodies, internalized receptors were labeled with Alexa Fluor-488 secondary antibodies (Fig. 5A, green), whereas total α1 subunits were labeled with a monoclonal antibody and Alexa Fluor-568 secondary antibodies (Fig. 5A, red). Quantification of the area of punctate labeling of internalized receptors showed a significant increase when neurons were incubated with 10 μm muscimol (the area of intracellular punctate labeling/10 μm increased to 3.14 ± 0.36 μm2 from 1.62 ± 0.17 in control condition). This increase was abolished when muscimol was co-applied with 100 μm gabazine or picrotoxin (1.68 ± 0.15 or 1.89 ± 018 μm2, respectively). Application of gabazine or picrotoxin alone had no effect (1.69 ± 0.19 or 1.61 ± 016 μm2, respectively) (Fig. 5B).
FIGURE 5.
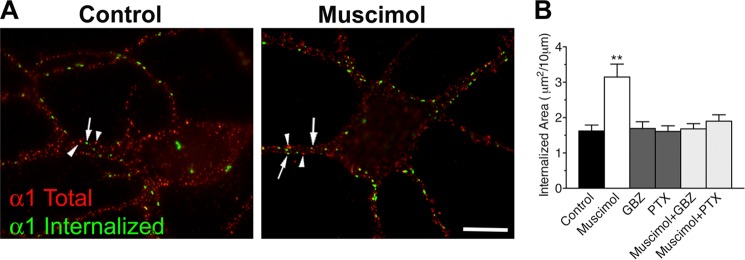
GABAAR internalization in neurons is increased by muscimol. A, internalized labeling was obtained after incubating live cultured hippocampal neurons with a polyclonal antibody directed at the extracellular domain of the native α1 subunit at 37 °C (Internalized). Total staining of the same neurons was performed after permeabilization and labeling with a monoclonal antibody directed at the intracellular domain of the native α1 subunit (total). Arrows, labeling of internalized receptors; arrowheads, labeling of the intracellular domain. Scale bar, 10 μm. B, quantitative analysis of the area of intracellular punctate labeling of internalized receptors formed by α1 subunits in dendrites of neurons incubated in culture medium (control), with muscimol, gabazine (gbz), or picrotoxin (ptx); a total of four experiments on independent neuron cultures where carried out, with the number of quantified neurons going from n = 32 to 40. **, p < 0.005. Error bars, S.E.
In another set of experiments, we analyzed γ2 subunit internalization in COS cells transfected with α1, β2, and tagged γ2 subunits (Fig. 6A). Average values ± S.E. from four independent experiments and 105 cells for each condition were plotted (Fig. 6B). These experiments showed that the number of internalized γ2 subunits increased during agonist treatment. The area of intracellular punctate labeling rose from 21.41 ± 3.28 μm2 in the control cells to 59.70 ± 6.89 μm2 when cells were treated with 100 μm GABA at 37 °C for 30 min. This increase was abolished when GABA was applied with gabazine, picrotoxin, or picrotoxin + gabazine (27.36 ± 3.29, 21.72 ± 2.82, or 21.41 ± 3.28 μm2, respectively). When compared with control, gabazine or picrotoxin had no significant effect on the area of intracellular punctate labeling (36.39 ± 5.42 or 28.89 ± 4.09, respectively).
FIGURE 6.

GABAAR internalization in COS-7 cells is increased by GABA. A, COS-7 cells were transiently co-transfected with α1-, β2-, and Myc-tagged γ2 subunits. Internalized labeling (red) was obtained after incubating live cells with polyclonal antibody directed at the extracellular Myc tag at 37 °C followed by stripping of antibodies remaining on the surface. Intracellular staining (green) of the same cells was obtained after permeabilization and labeling with a monoclonal antibody directed at the same tag (white frames detailed in lower panels). Scale bar, 10 μm. B, quantification of the area of intracellular punctate labeling of internalized receptors formed by γ2 subunit in cells incubated in culture medium (Ctrl) with GABA, gabazine (GBZ), or picrotoxin (PTX). ***, p < 0.001. C, COS-7 cells transfected as in A were labeled with biotin and returned to 37 °C in control medium, GABA, gabazine, or picrotoxin, as indicated. Biotin remaining on the surface after the incubation at 37 °C was removed by cleaving. Total and internalized (biotinylated) γ2 subunits were detected with an anti-Myc antibody. GAPDH staining shows that intracellular proteins were not biotinylated. D, means of internalized/total expression ratio normalized to total γ2 from experiments described in C. **, p < 0.005; *, p < 0.01. E, COS-7 cells transfected as in A and C and labeled with biotin. Representative Western blot shows Myc-tagged γ2 in total or surface-biotinylated extracts. F, means of surface/total expression ratio normalized to total γ2 from experiments described in E show that surface expression was unchanged by treatment with GABA. G, quantitative analysis of α1β2γ2 GABAARs surface expression in COS-7 cells transiently co-transfected with α1-, β2-, and Myc-tagged γ2 subunits and incubated with monensin with or without muscimol for 30 min before surface and total immunofluorescence labeling. A value of 1 for cells incubated without muscimol was used. **, p < 0.005. Error bars, S.E.
We also assessed γ2 internalization by performing a biotinylation assay on COS-7 cells transfected as above. After labeling with cleavable biotin, plates were returned to 37 °C for 30 min to allow endocytosis. Cells were then exposed to MESNA, which cleaved biotin from proteins remaining on the surface, allowing determination of intracellular biotinylated receptors. As shown in Fig. 6, C and D, application of GABA induced a significant increase of the internalization (212 ± 40% of control, n = 6, p < 0.05) that was suppressed when antagonists were co-applied with GABA (99.5 ± 27%).
We next examined whether agonist-induced internalization reduced the amount of receptors on the cell surface (Fig. 6E) by surface biotinylation experiments. Comparison of the relative surface/total ratio, in the absence or the presence of GABA (Fig. 6, E and F), showed that the ligand did not change significantly the amount of receptors on the surface. Expression is normalized to 1 for control and is 1.08 ± 0.11 for GABA-treated cells and 1.06 ± 0.12 for antagonist-treated cells; n = 3 independent experiments (Fig. 6F). These data implied that GABAAR endocytosis may be compensated for by exocytosis or reinsertion of GABAARs. Indeed, inhibition of receptor recycling with monensin (55, 56) (Fig. 6G) reduced significantly the surface expression of GABAARs following application of 10 μm muscimol. Expression is normalized to 1 for control and is 0.722 ± 0.034 for muscimol-treated cells (80 cells for each condition, five independent experiments), suggesting that removal and insertion of GABAARs on the cell surface is a use-dependent process that is tightly regulated.
DISCUSSION
Modulation of surface stability of GABAARs is essential for regulating the physiological properties of inhibitory neurotransmission. Modification of inhibitory signaling and altered receptor trafficking has been associated with several neurological diseases, including schizophrenia, substance abuse, pain, or epilepsy (2, 11, 57–61). It is also established that GABAARs endocytosis is regulated through intracellular signaling pathways (1, 2, 4). Here we show that a mutation in the N-terminal domain and ligand application have both an influence on receptor endocytosis. Thus, our data uncover a mechanism that links the extracellular domain of GABAARs to their stability on the cell surface. It must be noted that it was possible that we had detected the traffic of recombinant γ2 subunits in monomer form because it has been shown in some heterologous cells transfected with this subunit alone. However, we have previously shown that this is unlikely (22). Furthermore, in the present work, we found that the amount of γ2(R43Q) detected on the cell surface increased in the presence of a β2 subunit bearing the LL/AA substitution or following treatment with gabazine, a competitive antagonist of the GABA binding site. Because β2(LL/AA) should be associated with γ2(R43Q) to increase cell surface labeling and the gabazine binding site is within the interface between α and β subunits (48), these experiments strongly suggest that γ2(R43Q) detected on the cell surface is part of an oligomer also containing α and β subunits. The same holds true for wild type γ2 subunit endocytosis promoted by GABA (or muscimol), the binding site of which is at the α/β interface (Fig. 4A).
Analysis of γ2(R43Q) fate in neurons showed an increased retention in the endoplasmic reticulum, in agreement with previous findings in human embryonic kidney 293-T cells (24). Additionally, we provide evidence that γ2(R43Q) is not entirely retained within the reticulum and is transported via the Golgi apparatus to the cell membrane, where mutated receptors are highly internalized via a clathrin- and dynamin-dependent mechanism. Blockade of endocytosis leads to a major increase in γ2(R43Q) surface targeting in neurons and COS-7 cells, indicating that internalization is a major mechanism for down-regulating cell surface expression of γ2(R43Q)-containing receptors.
We also show that gabazine or picrotoxin increases dramatically the surface expression of γ2(R42Q) subunit. These two GABAAR antagonists are both negative allosteric modulators, acting at different sites (48–50, 62). The gabazine- and picrotoxin-sensitive internalization of γ2(R43Q) suggested that endocytosis could be linked to a constitutive activity of the mutated receptor. Electrophysiological recordings of γ2(R43Q)-expressing cells clearly showed that this mutated subunit did not give rise to constitutive currents. Therefore, the effect of antagonists on γ2(R43Q) endocytosis is probably related to another conformational state (e.g. the desensitized state). Interestingly, it has been shown that the γ2(R43Q) mutation favors desensitized states (52).
Consequently, our findings, showing that GABAAR antagonists prevent γ2(R43Q) endocytosis, suggest that internalization is driven by a global conformational change. Molecular models show that the γ2Arg-43 residue is at the γ2/β2 interface in the extracellular domain, on a loop positioned above the pocket, which is homologous to the GABA binding sites. Interestingly, many mutations in nicotinic receptors linked to diseases are at the interface between receptor subunits (63); they alter the gating allosterically (i.e. from a distance) (63–64). A model indicates that γ2Arg-43 and γ2Glu-178 are connected through a bifurcated salt bridge; this model has been used to study the γ2(R43Q) mutation (22, 26, 52). One of these studies has suggested that these positions have a long range allosteric effect (52). In our new GABAAR model derived from the glutamate-gated chloride channel (42), the Arg-43 residue of the γ2 subunit is connected to Tyr-174 and Glu-178 from the loop B and to the β2 subunit via polar interactions that should be sensitive to the R43Q substitution and positioned on a loop thought to be involved in the channel pore opening motion (51). Moreover, electrophysiological recordings and kinetic analyses have shown that the long distance effects of γ2(R43Q) substitution extend as far as the transmembrane domains (52). Therefore, γ2(R43Q) mutation might have an influence on receptor endocytosis in line with the current views on pentameric ligand-gated ion channels, describing a link between extracellular, transmembrane, and intracellular domains (53, 65, 66).
Because ligand binding in the Cys-loop receptor family is followed by a whole chain of interconnections, including the intracellular domain (67), it is of interest to assess whether GABA binding may influence GABAAR endocytosis. Although it is established that the number of surface GABAARs is regulated by constitutive endocytosis and neuromodulation through intracellular signaling (5–8, 68, 69), previous findings on ligand-independent or -dependent internalization are conflicting (28–31). Several studies have investigated GABAA receptor internalization following agonist application (29, 30). However, data were obtained by analyzing the amount of receptors remaining at the surface after agonist application. Here, we used a different approach (i.e. quantification of internalized receptors). Altogether, biochemical and immunocytochemical analyses of internalized receptor fraction, both in neurons and in COS-7, showed an increased number of internalized receptors during agonist application (Figs. 5B and 6, B and D), whereas the surface/intracellular ratio or surface labeling (Figs. 3, B and D, and 6E) remained unchanged. These data show that an overall counting of receptors on the cell membrane may overlook an increased endocytosis.
It has been suggested that internalization of GABAARs or increase in neuronal activity is accompanied by insertion of new receptors (4, 7, 70–72). Here, all of the experiments performed on neurons were conducted in the presence of tetrodotoxin and glutamate receptor inhibitors, suggesting that agonist binding endocytosis associated with the insertion of new receptors should instead represent an additional homeostasis mechanism. Knowing that internalized receptors are recycled back to the surface membrane or targeted for degradation and that endocytosis may be compensated for by surface targeting of distinct receptor subtypes, this balance between agonist-induced removal and insertion of receptors may regulate the number, but also the identity, of GABAARs on the cell surface. Because the functional and pharmacological properties of GABAARs depend on subunit composition, our findings showing that ligand stimulation increased endocytosis imply that this mechanism may be an important process for a fine tuning of GABAergic neurotransmission (4). It is of note that benzodiazepines (allosteric modulators of GABAARs) induce a subtype-specific change via enhanced degradation rather than alterations in receptor insertion or endocytosis, thus revealing another mechanism that might regulate GABAergic neurotransmission (73).
The γ2(R43Q) mutation is directly linked to epilepsy (16, 17). Thus, our findings, revealing a direct relationship between receptor endocytosis and a neurological disorder, are in line with the emerging concept that GABAAR trafficking deficiencies are key factors in initiating and maintaining several diseases, including epilepsy (4, 6, 15). For example, status epilepticus leads to enhanced GABAAR endocytosis (28–30). The K289M substitution in the γ2 subunit known to be responsible for generalized epilepsy with febrile seizures plus alters the membrane diffusion of GABAARs (61). It must be also noted that a shortened lifetime caused by the epilepsy mutation A322D on α1-containing GABAARs has been proposed (74) (but also see Ref. 75). Interestingly, experiments on γ2(R43Q) knock-in mice and transfected neurons or COS-7 cells have shown that α1 and α3 subunit surface expression was not reduced, despite a dramatic decrease in γ2(R43Q) surface labeling (16, 22). Earlier data, together with our present findings, show that the consequence of the mutation is a complex and dynamic process, suggesting that the defect is an active phenomenon instead of a more static retention of mutated receptors in the intracellular compartments and that γ2(R43Q)-containing receptor internalization is associated with a compensatory insertion of distinct GABAAR subtypes.
CONCLUSION
γ2(R43Q)-containing GABAARs are in a conformational state that promotes internalization, providing evidence for a direct link between GABAAR endocytosis and epilepsy. Furthermore, our data suggest that GABAAR endocytosis is use-dependent, consistent with a model in which ligand binding induces a conformation of the receptor that is a substrate for the biochemical events leading to endocytosis (70). Because GABA is the main inhibitory neurotransmitter in the brain and GABAARs are the target of many drugs, this property may have important functional and pathophysiological implications and should therefore be fully characterized (1, 4). Our data suggest that the γ2(R43Q) mutant is a useful model for this purpose. Our findings also illustrate the fact that mutations offer insights not only into diseases but also receptor physiology (61, 64, 74, 76). It would be also of interest to assess whether the different allosteric drugs acting on GABAARs have an influence on receptor trafficking.
Acknowledgments
We thank Dr. Daniel Choquet for helpful discussions, Christel Poujol for expertise with imaging, Erwin Sigel for human α1 cDNA, and Laura Cardoit and Frederique Masmejean for technical assistance.
This work was supported by CNRS, Université de Bordeaux, Conseil Régional Aquitaine, and Fédération de la Recherche sur le Cerveau.
- GABAAR
- GABAA receptor
- ERGIC
- endoplasmic reticulum-Golgi intermediate compartment
- MESNA
- sodium 2-mercaptoethanesulfonate.
REFERENCES
- 1. Luscher B., Fuchs T., Kilpatrick C. L. (2011) GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith K. R., Kittler J. T. (2010) The cell biology of synaptic inhibition in health and disease. Curr. Opin. Neurobiol. 20, 550–556 [DOI] [PubMed] [Google Scholar]
- 3. Triller A., Choquet D. (2008) New concepts in synaptic biology derived from single-molecule imaging. Neuron 59, 359–374 [DOI] [PubMed] [Google Scholar]
- 4. Jacob T. C., Moss S. J., Jurd R. (2008) GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kittler J. T., Delmas P., Jovanovic J. N., Brown D. A., Smart T. G., Moss S. J. (2000) Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J. Neurosci. 20, 7972–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leidenheimer N. J. (2008) Regulation of excitation by GABAA receptor internalization. Results Probl. Cell Differ. 44, 1–28 [DOI] [PubMed] [Google Scholar]
- 7. Bannai H., Lévi S., Schweizer C., Inoue T., Launey T., Racine V., Sibarita J. B., Mikoshiba K., Triller A. (2009) Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron 62, 670–682 [DOI] [PubMed] [Google Scholar]
- 8. Kittler J. T., Thomas P., Tretter V., Bogdanov Y. D., Haucke V., Smart T. G., Moss S. J. (2004) Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating γ-aminobutyric acid type A receptor membrane trafficking. Proc. Natl. Acad. Sci. U.S.A. 101, 12736–12741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith K. R., Muir J., Rao Y., Browarski M., Gruenig M. C., Sheehan D. F., Haucke V., Kittler J. T. (2012) Stabilization of GABAA receptors at endocytic zones is mediated by an AP2 binding motif within the GABAA receptor β3 subunit. J. Neurosci. 32, 2485–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cossart R., Bernard C., Ben-Ari Y. (2005) Multiple facets of GABAergic neurons and synapses. Multiple fates of GABA signalling in epilepsies. Trends Neurosci. 28, 108–115 [DOI] [PubMed] [Google Scholar]
- 11. Fritschy J. M. (2008) Epilepsy, E/I balance and GABAA receptor plasticity. Front. Mol. Neurosci. 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li H., Kraus A., Wu J., Huguenard J. R., Fisher R. S. (2006) Selective changes in thalamic and cortical GABAA receptor subunits in a model of acquired absence epilepsy in the rat. Neuropharmacology 51, 121–128 [DOI] [PubMed] [Google Scholar]
- 13. Meldrum B. S., Rogawski M. A. (2007) Molecular targets for antiepileptic drug development. Neurotherapeutics 4, 18–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noebels J. L. (2003) The biology of epilepsy genes. Annu. Rev. Neurosci. 26, 599–625 [DOI] [PubMed] [Google Scholar]
- 15. Ragozzino D., Palma E., Di Angelantonio S., Amici M., Mascia A., Arcella A., Giangaspero F., Cantore G., Di Gennaro G., Manfredi M., Esposito V., Quarato P. P., Miledi R., Eusebi F. (2005) Rundown of GABA type A receptors is a dysfunction associated with human drug-resistant mesial temporal lobe epilepsy. Proc. Natl. Acad. Sci. U.S.A. 102, 15219–15223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan H. O., Reid C. A., Single F. N., Davies P. J., Chiu C., Murphy S., Clarke A. L., Dibbens L., Krestel H., Mulley J. C., Jones M. V., Seeburg P. H., Sakmann B., Berkovic S. F., Sprengel R., Petrou S. (2007) Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc. Natl. Acad. Sci. U.S.A. 104, 17536–17541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wallace R. H., Marini C., Petrou S., Harkin L. A., Bowser D. N., Panchal R. G., Williams D. A., Sutherland G. R., Mulley J. C., Scheffer I. E., Berkovic S. F. (2001) Mutant GABAA receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 28, 49–52 [DOI] [PubMed] [Google Scholar]
- 18. Bailey M. E., Matthews D. A., Riley B. P., Albrecht B. E., Kostrzewa M., Hicks A. A., Harris R., Müller U., Darlison M. G., Johnson K. J. (1999) Genomic mapping and evolution of human GABAA receptor subunit gene clusters. Mamm. Genome 10, 839–843 [DOI] [PubMed] [Google Scholar]
- 19. Bianchi M. T., Song L., Zhang H., Macdonald R. L. (2002) Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J. Neurosci. 22, 5321–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bowser D. N., Wagner D. A., Czajkowski C., Cromer B. A., Parker M. W., Wallace R. H., Harkin L. A., Mulley J. C., Marini C., Berkovic S. F., Williams D. A., Jones M. V., Petrou S. (2002) Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation (γ 2(R43Q)) found in human epilepsy. Proc. Natl. Acad. Sci. U.S.A. 99, 15170–15175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eugène E., Depienne C., Baulac S., Baulac M., Fritschy J. M., Le Guern E., Miles R., Poncer J. C. (2007) GABAA receptor γ2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J. Neurosci. 27, 14108–14116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frugier G., Coussen F., Giraud M. F., Odessa M. F., Emerit M. B., Boué-Grabot E., Garret M. (2007) A γ2(R43Q) mutation, linked to epilepsy in humans, alters GABAA receptor assembly and modifies subunit composition on the cell surface. J. Biol. Chem. 282, 3819–3828 [DOI] [PubMed] [Google Scholar]
- 23. Hales T. G., Tang H., Bollan K. A., Johnson S. J., King D. P., McDonald N. A., Cheng A., Connolly C. N. (2005) The epilepsy mutation, γ2(R43Q) disrupts a highly conserved intersubunit contact site, perturbing the biogenesis of GABAA receptors. Mol. Cell Neurosci. 29, 120–127 [DOI] [PubMed] [Google Scholar]
- 24. Kang J., Macdonald R. L. (2004) The GABAA receptor γ2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of α1β2γ2S receptors in the endoplasmic reticulum. J. Neurosci. 24, 8672–8677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kang J. Q., Shen W., Macdonald R. L. (2006) Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J. Neurosci. 26, 2590–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sancar F., Czajkowski C. (2004) A GABAA receptor mutation linked to human epilepsy (γ2R43Q) impairs cell surface expression of αβγ receptors. J. Biol. Chem. 279, 47034–47039 [DOI] [PubMed] [Google Scholar]
- 27. Galanopoulou A. S. (2010) Mutations affecting GABAergic signaling in seizures and epilepsy. Pflugers Arch. 460, 505–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barnes E. M., Jr. (1996) Use-dependent regulation of GABAA receptors. Int. Rev. Neurobiol. 39, 53–76 [DOI] [PubMed] [Google Scholar]
- 29. Goodkin H. P., Joshi S., Mtchedlishvili Z., Brar J., Kapur J. (2008) Subunit-specific trafficking of GABAA receptors during status epilepticus. J. Neurosci. 28, 2527–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naylor D. E., Liu H., Wasterlain C. G. (2005) Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 25, 7724–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saito M., Toyoda H., Sato H., Ishii H., Kang Y. (2009) Rapid use-dependent down-regulation of γ-aminobutyric acid type A receptors in rat mesencephalic trigeminal neurons. J. Neurosci. Res. 87, 3120–3133 [DOI] [PubMed] [Google Scholar]
- 32. Boué-Grabot E., Emerit M. B., Toulmé E., Séguéla P., Garret M. (2004) Cross-talk and co-trafficking between rho1/GABA receptors and ATP-gated channels. J. Biol. Chem. 279, 6967–6975 [DOI] [PubMed] [Google Scholar]
- 33. Boué-Grabot E., Toulmé E., Emerit M. B., Garret M. (2004) Subunit-specific coupling between γ-aminobutyric acid type A and P2X2 receptor channels. J. Biol. Chem. 279, 52517–52525 [DOI] [PubMed] [Google Scholar]
- 34. Connolly C. N., Krishek B. J., McDonald B. J., Smart T. G., Moss S. J. (1996) Assembly and cell surface expression of heteromeric and homomeric γ-aminobutyric acid type A receptors. J. Biol. Chem. 271, 89–96 [DOI] [PubMed] [Google Scholar]
- 35. Coussen F., Normand E., Marchal C., Costet P., Choquet D., Lambert M., Mège R. M., Mulle C. (2002) Recruitment of the kainate receptor subunit glutamate receptor 6 by cadherin/catenin complexes. J. Neurosci. 22, 6426–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Toulmé E., Soto F., Garret M., Boué-Grabot E. (2006) Functional properties of internalization-deficient P2X4 receptors reveal a novel mechanism of ligand-gated channel facilitation by ivermectin. Mol. Pharmacol. 69, 576–587 [DOI] [PubMed] [Google Scholar]
- 37. Ben-Tekaya H., Miura K., Pepperkok R., Hauri H. P. (2005) Live imaging of bidirectional traffic from the ERGIC. J. Cell Sci. 118, 357–367 [DOI] [PubMed] [Google Scholar]
- 38. Huyghe D., Veran J., Labrousse V. F., Perrais D., Mulle C., Coussen F. (2011) Endocytosis of the glutamate receptor subunit GluK3 controls polarized trafficking. J. Neurosci. 31, 11645–11654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mondin M., Carta M., Normand E., Mulle C., Coussen F. (2010) Profilin II regulates the exocytosis of kainate glutamate receptors. J. Biol. Chem. 285, 40060–40071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petrini E. M., Lu J., Cognet L., Lounis B., Ehlers M. D., Choquet D. (2009) Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron 63, 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Le Novère N., Changeux J. P. (1999) The Ligand Gated Ion Channel Database. Nucleic Acids Res. 27, 340–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee. A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 44. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 45. Fache M. P., Moussif A., Fernandes F., Giraud P., Garrido J. J., Dargent B. (2004) Endocytotic elimination and domain-selective tethering constitute a potential mechanism of protein segregation at the axonal initial segment. J. Cell Biol. 166, 571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 47. Wang H., Bedford F. K., Brandon N. J., Moss S. J., Olsen R. W. (1999) GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature 397, 69–72 [DOI] [PubMed] [Google Scholar]
- 48. Boileau A. J., Newell J. G., Czajkowski C. (2002) GABAA receptor β2 Tyr97 and Leu99 line the GABA-binding site. Insights into mechanisms of agonist and antagonist actions. J. Biol. Chem. 277, 2931–2937 [DOI] [PubMed] [Google Scholar]
- 49. Ueno S., Bracamontes J., Zorumski C., Weiss D. S., Steinbach J. H. (1997) Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J. Neurosci. 17, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Torres V. I., Weiss D. S. (2002) Identification of a tyrosine in the agonist binding site of the homomeric ρ1 γ-aminobutyric acid (GABA) receptor that, when mutated, produces spontaneous opening. J. Biol. Chem. 277, 43741–43748 [DOI] [PubMed] [Google Scholar]
- 51. Taly A., Corringer P. J., Grutter T., Prado de Carvalho L., Karplus M., Changeux J. P. (2006) Implications of the quaternary twist allosteric model for the physiology and pathology of nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 16965–16970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goldschen-Ohm M. P., Wagner D. A., Petrou S., Jones M. V. (2010) An epilepsy-related region in the GABAA receptor mediates long-distance effects on GABA and benzodiazepine binding sites. Mol. Pharmacol. 77, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miller P. S., Smart T. G. (2010) Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol. Sci. 31, 161–174 [DOI] [PubMed] [Google Scholar]
- 54. Everitt A. B., Seymour V. A., Curmi J., Laver D. R., Gage P. W., Tierney M. L. (2009) Protein interactions involving the γ2 large cytoplasmic loop of GABAA receptors modulate conductance. FASEB J. 23, 4361–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roy S., Roy S. J., Pinard S., Taillefer L. D., Rached M., Parent J. L., Gallo-Payet N. (2011) Mechanisms of melanocortin-2 receptor (MC2R) internalization and recycling in human embryonic kidney (hek) cells. Identification of key Ser/Thr (S/T) amino acids. Mol. Endocrinol. 25, 1961–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stein B. S., Bensch K. G., Sussman H. H. (1984) Complete inhibition of transferrin recycling by monensin in K562 cells. J. Biol. Chem. 259, 14762–14772 [PubMed] [Google Scholar]
- 57. Charych E. I., Liu F., Moss S. J., Brandon N. J. (2009) GABAA receptors and their associated proteins. Implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology 57, 481–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Uusi-Oukari M., Korpi E. R. (2010) Regulation of GABAA receptor subunit expression by pharmacological agents. Pharmacol. Rev. 62, 97–135 [DOI] [PubMed] [Google Scholar]
- 59. Tan K. R., Brown M., Labouèbe G., Yvon C., Creton C., Fritschy J. M., Rudolph U., Lüscher C. (2010) Neural bases for addictive properties of benzodiazepines. Nature 463, 769–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Knabl J., Witschi R., Hösl K., Reinold H., Zeilhofer U. B., Ahmadi S., Brockhaus J., Sergejeva M., Hess A., Brune K., Fritschy J. M., Rudolph U., Möhler H., Zeilhofer H. U. (2008) Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 451, 330–334 [DOI] [PubMed] [Google Scholar]
- 61. Bouthour W., Leroy F., Emmanuelli C., Carnaud M., Dahan M., Poncer J. C., Lévi S. (2012) A Human Mutation in Gabrg2 Associated with Generalized Epilepsy Alters the Membrane Dynamics of GABAA Receptors. Cereb Cortex 22, 1542–1553 [DOI] [PubMed] [Google Scholar]
- 62. Krishek B. J., Moss S. J., Smart T. G. (1996) A functional comparison of the antagonists bicuculline and picrotoxin at recombinant GABAA receptors. Neuropharmacology 35, 1289–1298 [DOI] [PubMed] [Google Scholar]
- 63. Changeux J. P., Taly A. (2008) Nicotinic receptors, allosteric proteins and medicine. Trends Mol. Med. 14, 93–102 [DOI] [PubMed] [Google Scholar]
- 64. Sine S. M., Engel A. G. (2006) Recent advances in Cys-loop receptor structure and function. Nature 440, 448–455 [DOI] [PubMed] [Google Scholar]
- 65. Tierney M. L. (2011) Insights into the biophysical properties of GABAA ion channels. Modulation of ion permeation by drugs and protein interactions. Biochim. Biophys. Acta 1808, 667–673 [DOI] [PubMed] [Google Scholar]
- 66. Peters J. A., Cooper M. A., Carland J. E., Livesey M. R., Hales T. G., Lambert J. J. (2010) Novel structural determinants of single channel conductance and ion selectivity in 5-hydroxytryptamine type 3 and nicotinic acetylcholine receptors. J. Physiol. 588, 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsetlin V., Kuzmin D., Kasheverov I. (2011) Assembly of nicotinic and other Cys-loop receptors. J. Neurochem. 116, 734–741 [DOI] [PubMed] [Google Scholar]
- 68. Chen G., Kittler J. T., Moss S. J., Yan Z. (2006) Dopamine D3 receptors regulate GABAA receptor function through a phospho-dependent endocytosis mechanism in nucleus accumbens. J. Neurosci. 26, 2513–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kittler J. T., Chen G., Kukhtina V., Vahedi-Faridi A., Gu Z., Tretter V., Smith K. R., McAinsh K., Arancibia-Carcamo I. L., Saenger W., Haucke V., Yan Z., Moss S. J. (2008) Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor γ2 subunit. Proc. Natl. Acad. Sci. U.S.A. 105, 3616–3621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gonzalez C., Moss S. J., Olsen R. W. (2012) Ethanol promotes clathrin adaptor-mediated endocytosis via the intracellular domain of δ-containing GABAA receptors. J. Neurosci. 32, 17874–17881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liang J., Suryanarayanan A., Abriam A., Snyder B., Olsen R. W., Spigelman I. (2007) Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. J. Neurosci. 27, 12367–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saliba R. S., Kretschmannova K., Moss S. J. (2012) Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 31, 2937–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jacob T. C., Michels G., Silayeva L., Haydon J., Succol F., Moss S. J. (2012) Benzodiazepine treatment induces subtype-specific changes in GABAA receptor trafficking and decreases synaptic inhibition. Proc. Natl. Acad. Sci. U.S.A. 109, 18595–18600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bradley C. A., Taghibiglou C., Collingridge G. L., Wang Y. T. (2008) Mechanisms involved in the reduction of GABAA receptor α1-subunit expression caused by the epilepsy mutation A322D in the trafficking-competent receptor. J. Biol. Chem. 283, 22043–22050 [DOI] [PubMed] [Google Scholar]
- 75. Ding L., Feng H. J., Macdonald R. L., Botzolakis E. J., Hu N., Gallagher M. J. (2010) GABAA receptor α1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABAA receptors. J. Biol. Chem. 285, 26390–26405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Macdonald R. L., Kang J. Q., Gallagher M. J. (2010) Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 588, 1861–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrodinger, LLC, New York [Google Scholar]