

Background: Shape-function studies are necessary to design better therapeutic alternatives of the plasma gelsolin.
Results: N-terminal fragment 30–161 is the smallest segment with F-actin depolymerization potential, and G1-G3 can function independent of Ca2+ ions or low pH.
Conclusion: The g2-g3 linker plays a role in imparting pH/Ca2+ insensitivity to G1-G3.
Significance: We provide the first evidence that g2-g3 linker regulates mobility of the G1 domain.
Keywords: Actin, Molecular Modeling, Protein Engineering, Sepsis, X-ray Scattering, ab Initio Modeling, Minimal Gelsolin, Small Angle X-ray Scattering
Abstract
Because of its ability to rapidly depolymerize F-actin, plasma gelsolin has emerged as a therapeutic molecule in different disease conditions. High amounts of exogenous gelsolin are, however, required to treat animal models of different diseases. Knowing that the F-actin depolymerizing property of gelsolin resides in its N terminus, we made several truncated versions of plasma gelsolin. The smaller versions, particularly the one composed of the first 28–161 residues, depolymerized the F-actin much faster than the native gelsolin and other truncates at the same molar ratios. Although G1-G3 loses its dependence on Ca2+ or low pH for the actin depolymerization function, interestingly, G1-G2 and its smaller versions were found to regain this requirement. Small angle x-ray scattering-based shape reconstructions revealed that G1-G3 adopts an open shape in both the presence and the absence of Ca2+ as well as low pH, whereas G1-G2 and residues 28–161 prefer collapsed states in Ca2+-free conditions at pH 8. The mutations in the g2-g3 linker resulted in the calcium sensitivity of the mutant G1-G3 for F-actin depolymerization activity, although the F-actin-binding sites remained exposed in the mutant G1-G3 as well as in the smaller truncates even in the Ca2+-free conditions at pH 8. Furthermore, unlike wild type G1-G3, calcium-sensitive mutants of G1-G3 acquired closed shapes in the absence of free calcium, implying a role of g2-g3 linker in determining the open F-actin depolymerizing-competent shape of G1-G3 in this condition. We demonstrate that the mobility of the G1 domain, essential for F-actin depolymerization, is indirectly regulated by the gelsolin-like sequence of g2-g3 linker.
Introduction
Gelsolin, a hexa-domain actin-severing, nucleating, and capping protein, exists as a cytosolic as well as a secreted plasma isoform in the body (1). These isoforms exhibit a compact globular structure under Ca2+-free conditions at the physiological pH and attain an open functionally active structure in the presence of 1 mm Ca2+ or at lower pH (2–4). Radiolytic footprinting and small angle x-ray scattering (SAXS)4 studies have elucidated a three-stage opening of the gelsolin molecule upon sequential binding of free Ca2+. The first transition from the inactive molecule to the capping and severing-competent intermediate state is achieved primarily by the opening of the tail latch at a Ca2+ concentration of ∼5 μm, although further opening occurs at 100 μm Ca2+ by the unwinding of the g3-g4 linker (3, 5). The structural and functional data from the last few decades have suggested that the actin filament-depolymerization activity of gelsolin resides in its N-terminal region, and the N-terminal half (G1-G3) of the molecule can depolymerize filamentous actin as well as cap the barbed end in a calcium-independent but PIP2-sensitive manner (6–10). Despite the lack of absolute control of the depolymerizing activity of G1-G3 by Ca2+, these divalent ions do regulate the structure of the N-terminal half of gelsolin as evident from dynamic light scattering and fluorescence studies and analysis of its crystal structure (7, 11, 12). The fluorescence-based kinetic studies have also shown that the Ca2+ binding-triggered conformational changes in G1-G3 enhance its interaction with F-actin, as well as increase its F-actin depolymerizing efficiency. The quantitative measurement of the F-actin depolymerization activity of G1-G3 showed that the F-actin depolymerization by G1-G3 is a cooperative process requiring the binding of two G1-G3 molecules in close proximity and suggested an active participation of G4-G6 in the F-actin depolymerization process by the intact gelsolin (13). However, the C-terminal half of the molecule is a Ca2+-dependent regulatory region, which senses the Ca2+ level and maintains the protein either in active or inactive conformation (14).
Deletion mutagenesis of the N-terminal region of gelsolin has revealed that the F-actin depolymerization activity of gelsolin resides within its first 160 residues, with a critical dependence on the sequence 150–160 in the g1-g2 linker (15). This 1–160-residue fragment depolymerized the F-actin roughly as well as the full-length gelsolin in a calcium- and PIP2-sensitive manner (15). The importance of the g1-g2 linker in the depolymerization mechanism was further elucidated by the gain of function studies on CapG, a gelsolin family protein that caps but does not depolymerize actin filaments (16). This study showed that the two stretches of amino acid sequences, the residues 148–152 (GFKHV) in the g1-g2 linker and the residues 108–114 (LDDYLNG) in the G1 domain, as well as the exact length of the g1-g2 linker are critical for the F-actin depolymerization function of the gelsolin (16, 17). The structure of the F-actin depolymerization-competent 25–160-residue fragment of gelsolin in complex with the actin supported the hypothesis that the partial G2 domain binds along the side of the actin filament in such a way that it targets the G1 domain to intercalate between the actin subunit bound to G2 and the adjacent actin subunit and to rupture the noncovalent interactions holding them together (18). The opening of the G1 domain from rest of the molecule might thus be a prerequisite for the F-actin depolymerization function of gelsolin as has also been supported by the recent studies (3, 4).
Plasma gelsolin (pGSN) is the fourth most abundant protein in human plasma and is primarily involved in the clearance of actin filaments released into the circulation upon cell necrosis (19, 20). The pGSN levels decrease by 20–50% in a wide variety of disease conditions, although a minimum threshold level of pGSN (≥25%) is essential for maintaining the normal physiology of the body (21, 22). At the time of admission of patients to an intensive care unit, the pGSN levels below the critical threshold have been associated with the adverse outcomes, although an improvement in the pGSN levels has been documented in the patients recovering from the illnesses. In addition, a repletion with the exogenous recombinant pGSN increases the survivability and counters the adverse outcomes in various rodent models (23–25) advocating a need for the “gelsolin replacement therapy.” However, the optimum dosage of exogenous gelsolin found to be effective in mice (∼8 mg of protein per animal) is high, and the translation of this dosage to human body weight projects the need of a very large amount (∼28 g for 70 kg or 150 pounds of body mass) of recombinant protein per administration. This shows the necessity of a minimized yet efficient version of gelsolin that can be used as an alternative in replacement therapy.
Essentially, although a lot of progress has been made in the structural and the functional aspects of the gelsolin, the exact mechanism and the regulation of F-actin depolymerization by the N-terminal region still remain elusive. Each domain of the gelsolin contains a site for calcium binding, and the whole molecule as well as its smaller truncates are dependent on Ca2+ binding for their activation. It is, however, unclear what imparts the calcium insensitivity to G1-G3 and some of the G1-G2 fragments for their F-actin depolymerizing activities. Furthermore, the therapeutic use of gelsolin would benefit by the substitution of the full-length protein with its minimal versions that retain full actin-filament depolymerization activity. In this investigation, we have made various truncated versions of the full-length gelsolin by deletion mutagenesis and determined their ability to depolymerize the F-actin. Our in vitro and in vivo data brought forth that the 28–161-residue fragment of gelsolin is a fully potent minimal gelsolin in the F-actin depolymerization assay in a calcium/low pH-dependent manner. The SAXS-based shape reconstruction revealed that the depolymerization-competent G1-G3 adopts an open shape, whereas G1-G2 and 28–161 prefer collapsed states in Ca2+-free conditions at pH 8, suggesting the freely mobile G1 domain to be a key requirement for the depolymerization activity. Also, we show that the g2-g3 linker region plays an important role in determining the calcium or pH insensitivity of the N-terminal half of gelsolin for the F-actin depolymerization function.
MATERIALS AND METHODS
Cloning and Purification of Recombinant Gelsolin Mutants
Cloning of the full-length gelsolin (GSN) has been described earlier (4). The plasmids harboring the genes for recombinant gelsolin mutants G1-G3 (residues 1–371), G1-G2 (residues 1–241), G2-G6 (residues 158–755), G2-G3 (residues 158–371), G4-G6 (residues 412–755), dT-GSN (residues 1–731), 1–161, 25–161, 28–161, 30–161, 32–161, 34–161, 36–161, 40–161, 42–161, 56–161, 25–158 and 25–156) were created by PCR amplification of the corresponding fragments using specific primers and pET303/gelsolin as template. The amplified fragments were digested with XbaI and XhoI and subcloned into the vector backbone of pET303/CT-His (Invitrogen). In the reverse primers, no stop codon was inserted so as to include the His tag at the C terminus. The sequences of subcloned gelsolin mutant's DNA were verified by automated DNA sequencing (Applied Biosystems). The plasmids thus created were used to transform Escherichia coli (BL21 (DE3)) for the heterologous protein expression. Expression and purification of GSN, dT-GSN, and G1-G3 was performed as described previously (4). Smaller gelsolin mutants (G1-G3, G1-G2, G2-G6, G4-G6, 1–161, 25–161, 28–161, 30–161, 32–161, 36–161, 40–161, G2-G3, 25–158, and 25–156) were affinity-purified using nickel-nitrilotriacetic acid resin. The proteins bound to resin were eluted using 250 mm imidazole in elution buffer. Imidazole was then removed by extensive dialysis against buffer A (25 mm Tris-HCl, pH 8, 45 mm NaCl, 1 mm EGTA). The deletion mutants 42–161 and 56–161 were affinity-purified under denaturing conditions (buffers containing 8 m urea). After purification, these mutants were refolded by the gradual removal of urea by stepwise dialysis in buffers containing reduced concentrations of urea followed by final dialysis in buffer A containing no denaturant. The refolded proteins were analyzed by circular dichroism spectroscopy to estimate and confirm the presence of comparable levels of secondary structural content as seen in crystal structures (Jasco 810). All the proteins were finally subjected to an S200 gel filtration step using an FPLC system (AKTA Prime, GE Healthcare). The purity of the proteins was ascertained by 10–12% SDS-PAGE containing molecular mass markers (Fermentas, India). Identities of the proteins were confirmed from their intact mass values from MALDI-TOF (Voyager). Whenever required, proteins were concentrated using membrane concentrators (Millipore, Ireland). The concentration of purified proteins was ascertained by measuring absorption at 280 nm followed by calculations using their molar absorption coefficients (U-2900 Spectrophotometer, Hitachi, Japan).
Site-directed Mutagenesis
Mutations in the g2-g3 linker region of the construct G1-G3 were carried out sequentially using the primers containing the desired mutations and following QuikChange mutagenesis protocol. The primers used for the mutagenesis were as follows: Mut1, 5′-cggctctgcctgcaaaggaggatgcggc-3′; Mut2, 5′-gtgctgggcggcaagggggctctggctgcaaagcatgatgcggcc-3′; Mut3, 5′-gcaaagcatgatgcggcccccaccaagctggccaagctcta-3′; and Mut4, 5′-gatgcggcccccaccaagtcggagaagctctacaaggtctc-3′.
Actin Extraction from Chicken Muscle
Actin was extracted from chicken breast muscle following a protocol reported by Spudich and co-workers (26) with slight modifications. Briefly, actin was solubilized by suspending the acetone muscle powder in pre-chilled G-buffer (0.2 mm Tris, 0.2 mm CaCl2, 0.2 mm ATP, 0.5 mm 2-mercaptoethanol, pH 8) at 4 °C. Polymerization of the actin was induced by the addition of 50 mm KCl and 2 mm MgCl2 at room temperature for 2 h followed by addition of solid KCl with continuous stirring at 4 °C to the final concentration of 0.8 m. The polymerized actin was collected by ultracentrifugation at 40,000 × g for 2.5 h at 4 °C in Beckman rotor 50.2, and the pellet was thoroughly washed and softened by overnight incubation in G-buffer. The softened pellet was homogenized thoroughly and kept for extensive dialysis against G-buffer for 3 days with buffer exchange at every 24 h and monitored by dynamic light scattering (Beckman Coulter) for gradual decrement in the diffusion coefficient values of actin. Finally, the G-actin was obtained by centrifugation at 40,000 × g for 2 h, and the concentration was determined by measuring the absorbance at 290 nm (A290 ∼0.62, 1 mg/ml). The purified G-actin has been characterized recently in ATP/ADP/AMP-NP state by SAXS analyses (27). The G-actin was labeled with a 7-fold molar excess of pyrenyl-iodoacetamide as described previously (28) and stored at −80 °C for further use.
Fluorescence Assays of Actin Filament Depolymerization
The ability of gelsolin to depolymerize actin filaments was determined by its effect on the rate and the extent of decrement in the fluorescence of pyrene-labeled F-actin. The pyrene-labeled G-actin (14% labeling) was converted to F-actin by adding 3 mm MgCl2 and 100 mm KCl in the presence of full-length GSN at a molar ratio of 1:500 (GSN/actin) followed by overnight incubation at 20 °C. For the measurement of depolymerization effects of different versions of gelsolin, F-actin (4 μm) was diluted to 100 nm in F-buffer (G-buffer containing 3 mm MgCl2 and 100 mm KCl, pH 8) with or without gelsolin variants at different molar ratios in the presence of either 1 mm calcium or 1 mm EGTA, and the fluorescence measurements were started with in 15–25 s (time taken for the mixing of reaction components). This time was recorded for each sample (done in triplicates) and was taken into account for data plotting and the rate of depolymerization calculations. For the calcium titration experiments, PHEM buffer (60 mm Pipes, 20 mm Hepes, 10 mm EGTA, 2 mm MgCl2, 0.5 mm ATP, 0.2 mm DTT, 150 mm KCl), pH 8, was used, and different amounts of CaCl2 were added based on Web-Maxc version 2.1. To study the effect of pH on the F-actin depolymerizing activities of the gelsolin mutants, PHEM buffers of pH 8, 7, 6, and 5 were used for the dilution of the gelsolin mutants and the pyrene-labeled F-actin. The pH of the PHEM buffers ranging from 8 to 5 was obtained by titrating with HCl. For the kinetic studies, fluorescence intensities were measured at 407 nm (excitation at 365 nm) as a function of the time using a PTI fluorimeter, and for end point assays fluorescence intensity was measured using Synergy 4 multimode detection microplate reader after 5–10 min.
To compare the actin depolymerization activities of different gelsolin mutants, the depolymerization curves were subdivided into three time frames, 0–30, 31–45, and 46–60 s, post-addition of the reaction components, and the rates of decrement in the fluorescence units were determined by calculating the slopes of the curves.
Direct Observation of Filamentous Actin Depolymerization by Confocal Microscopy
The G-actin was labeled with the fluorescent dye, BODIPY FL iodoacetamide, as described previously (29). 200 μm of the BODIPY FL-C1-IA (Molecular Probes, Eugene) was added slowly to 48 μm of F-actin while stirring, and the reaction mixture was incubated for 2 h at room temperature. The reaction was terminated by the addition of 5 mm dithiothreitol, and the labeled actin was collected after centrifugation at 40,000 × g for 1 h at 4 °C. The pellet was dissolved in G-buffer, and the homogenized actin was dialyzed against G-buffer for 2 days at 4 °C with 2–3 times of buffer change. After centrifugation at 40,000 × g for 1 h, BODIPY FL-labeled G-actin concentration in the supernatant was determined by subtracting 0.029 × A505 from the A290 value. The degree of labeling was calculated to be 50%. BODIPY FL-labeled G-actin was mixed with unlabeled G-actin in a 1:3 ratio and co-polymerized by adding 3 mm MgCl2 and 100 mm KCl in the presence of full-length GSN at a molar ratio of 1:500 by overnight incubation at 20 °C. Glass coverslips (thickness 0.08–0.12 mm, size 22 × 30 mm) were coated with poly-l-lysine (0.1%, Sigma), rinsed with water, and air-dried. F-actin (1 μm, 16% labeled) was incubated on the coated coverslips with either a gelsolin variant (0.2 μm) in F-buffer or buffer alone in the presence of 1 mm CaCl2, mounted on the glass slide at room temperature, and observed within 5 min using Nikon A1R confocal microscope. A 488 nm laser light was used for excitation, and emission was captured at 512 nm. The images were captured using ×100 objective and numeric aperture = 1.3 and processed using Nikon's NIS-elements imaging software.
Determination of the Critical Concentration (Cc) of Actin
To determine the effect of gelsolin variants on the Cc of actin for actin polymerization, full-length GSN or different gelsolin mutants (G1-G3, G4-G6, G1-G2, 1–161, 28–161, and 42–161) (0.1 μm) were added to increasing concentrations (0.1–0.8 μm) of pyrene-labeled G-actin (14% labeled) in F-buffer in the presence of either 0.1 mm CaCl2 or 5 mm EGTA, and the actin was allowed to polymerize at room temperature. The pyrene fluorescence was measured (excitation at 365 nm and emission at 407 nm) using Synergy 4 multimode detection microplate reader after 18 h.
Inhibition of Actin Filament Elongation Assay
Inhibition assay of the elongation of actin filaments at the barbed end was carried out as described previously (30, 31). Monomeric actin (10 μm) was polymerized by the addition of 100 mm KCl and 3 mm MgCl2 at RT for 30 min and added at final concentration of 1 μm to the wells containing 0.5 μm pyrene-labeled (15%) monomeric actin and increasing concentrations (0.25-4 nm) of the gelsolin mutants in the presence of 2 mm calcium. Fluorescence intensity (excitation 365 nm and emission 407 nm) was measured for 2 h at 10-min intervals. The change in the fluorescence units in the absence of the mutants was taken as 100% polymerization, and the relative inhibition of polymerization in the presence of each mutant was calculated.
F-actin Binding Assay
F-actin co-sedimentation assay was performed by adding different gelsolin mutants (5 μm) to F-actin (5 μm) in the presence of either 2 mm EGTA or 2 mm CaCl2 and incubating the samples for 1 h at room temperature. After incubation, the samples were ultracentrifuged at 46,000 × g for 30 min at 20 °C using Beckman TLA 100.2 rotor, and the pellets were resuspended in equal volumes and subjected to SDS-PAGE followed by staining with Coomassie Brilliant Blue R-250. The gels were scanned using gel doc system, and the band intensities were measured using AlphaEaseFC image analysis software. To determine the cosedimentation of different variants with F-actin, the actin-independent sedimentation of individual proteins was subtracted from their experimental data in the presence of actin.
Generation of Polyclonal Anti-gelsolin Antibodies
The antibodies against recombinant human pGSN were raised in rabbit. Briefly, the rabbit was immunized with 250 μg of human recombinant pGSN emulsified in Freund's complete adjuvant, followed by two booster doses in incomplete Freund's adjuvant at 3-week intervals. The titer and specificity of anti-gelsolin antibodies in serum were determined by ELISA and Western blotting.
Lipopolysaccharide (LPS)-induced Murine Sepsis Model
Male C57BL/6 mice, 6–8 weeks old (obtained from animal house, CSIR-IMTECH), each weighing 18–20 g, were used for the following experiments. The animals were housed under normal conditions and had free access to standard diet and water. All the protocols on mice were performed according to the National Regulatory Guidelines issued by Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Environment and Forests (Government of India). The mice (four per group) were injected intraperitoneally with 25 mg/kg LPS (LPS, E. coli O55:B5, Sigma) followed by subcutaneous administration of different amounts of recombinant human plasma GSN containing 1 mm calcium in phosphate-buffered saline (PBS). The control group of mice was given saline without LPS challenge. Blood was collected by retro-orbital bleeding into EDTA containing tubes at 24 h after LPS treatment to measure the plasma gelsolin levels.
In a different experiment, mice were divided into six groups (each containing eight mice), and the blood was collected by retro-orbital bleeding into EDTA-containing tubes at 24 h prior to LPS treatment. These mice were injected subcutaneously with 0.5 ml (2 mg) of GSN, G1-G3, G2-G6, G4-G6, or 28–161 containing 1 mm calcium in PBS just after the LPS administration and at 24 h after the LPS treatment. Another control group of mice was given saline without LPS challenge. The blood was collected into EDTA-containing tubes for checking the plasma gelsolin levels and the cytokine expression profile by sacrificing four mice at 24 h and the remaining mice from each group at 48 h. Similarly, other groups of mice were injected subcutaneously with 2 mg of GSN, G1-G3, G2-G6, G4-G6, or 28–161 as above, just after the LPS administration and at 24 and 48 h after the LPS treatment, and the survival of mice in each group was monitored for 6 days. The survival experiments were repeated twice. Surviving mice were sacrificed in all the experiments after 7 days. The blood collected in the above experiments was centrifuged at 2000 × g for 5 min to collect the plasma and stored at −80 °C. Plasma gelsolin and cytokines levels were assayed by Western blotting and ELISA, respectively.
Gelsolin Level Measurements by Immunoblotting
The plasma gelsolin levels were determined in diluted plasma samples (1:2.5) using quantitative Western blot analysis. 10 μl of the sample was subjected to 10% SDS-PAGE and transferred onto Immobilon-P membrane (Millipore, Bedford, MA). The respective purified recombinant gelsolin variant was loaded as standard in at least five lanes of each gel in a concentration range comparable with the gelsolin concentrations in the samples to quantify GSN, G1-G3, G4-G6, G2-G6, and 28–161. After the transfer, the membranes were blocked with 5% (w/v) nonfat dry milk powder followed by probing either with polyclonal rabbit anti-human gelsolin (for exogenous and endogenous GSN, G2-G6, G4-G6, and G1-G3) or mouse anti-His monoclonal antibodies (Pierce) (28–161-residue fragment, as this could not be detected with polyclonal anti-human gelsolin) overnight at 4 °C. Membranes washed thoroughly with PBS, 0.1% Tween (PBS-T) were then incubated with peroxidase-conjugated secondary antibody (Sigma) for 1 h. After washing, chemiluminescence of horseradish peroxidase was developed with Luminata-Forte substrate (Millipore) and exposed to x-ray films. Developed x-ray films were scanned using a phosphorimager (Fujifilm FLA-900), and the integrated intensity of each band on Western blot, minus the adjacent background signal, was quantified using MultiGauge version 3.1 images analysis software. The concentration of gelsolin variants in unknown samples was calculated from the standard curve constructed by known protein concentrations of the respective purified variant. Because both the endogenous and exogenous gelsolin (seen as a single band in Western blots) were detected together by anti-gelsolin polyclonal antibodies, these values have been reported here as the combined values. Total albumin levels in the plasma samples were measured as control using the BCG albumin assay kit (BioAssay Systems, Hayward, CA) according to the manufacturer's instructions.
Determination of Plasma Cytokine Levels
Plasma interleukin (IL)-6, IL-10, interferon (INF)-γ, and tumor necrosis factor (TNF)-α levels were quantified using respective ELISA kits according to the manufacturer's instructions (BioLegend, Inc., San Diego). The resulting absorbance in each well was measured at 450 nm with a microplate ELISA reader (Lab Tech, India). The fold change in the cytokine levels was calculated as the level in the test group divided by the level in the control group.
Synchrotron SAXS Experiments
Samples of different gelsolin truncates (volume of 60 μl) were prepared as follows: free and total calcium levels were estimated using WEB-MAXC version 2.1, and the required amounts of CaCl2 to obtain the desired free Ca2+ concentration (1 mm) were dried at the bottom of Eppendorf tubes. These tubes were prepared in duplicates. Just prior to the SAXS experiments, diluted gelsolin stocks (GSN, G1-G3, G1-G2, 28–161, G2-G3, and mutants of G1-G3) were added to the tubes containing Ca2+ and mixed. Matched buffers (the S200 elution buffer) for each sample were prepared in a similar way. Lack of aggregation in the samples during storage and buffer conditions was confirmed through dynamic light scattering experiments using Beckman Coulter back-scattering instrument (DelsaNano). To check the pH-induced conformational changes, the gelsolin mutants (G1-G3, G1-G2, G2-G3, 28–161, and mutants of G1-G3) in EGTA containing Tris buffer at pH 8 were dialyzed sequentially against the buffers of different pH values from pH 8 to pH 5 for ∼45 min (three changes: 10-, 15-, and 20-min intervals) as described earlier (4) before the SAXS experiments. Post-data collections, the samples exposed to x-rays were collected for analysis by SDS-PAGE (except for G2-G3). In all the cases, the migration pattern was comparable with the protein samples that had not left our laboratory, supporting that no degradation had occurred in the samples during travel and data collection.
X-ray scattering data at low angles were collected at X9 beam line at National Synchrotron Light Source (Brookhaven National Laboratory, New York) and analyzed as described previously (4). Each dataset (solutions and buffers) was an average of three independent runs. All the scattering profiles were normalized to the transmission counts registered at the pin-diode during data collection of EGTA gelsolin (pH 8) (except for G2-G3, because this dataset was acquired under different conditions). Guinier analyses of the datasets to estimate the radius of gyration (RG) and the radius of cross-section (RC) were done using PRIMUS software (32) presuming a globular and rod-like scattering shape of molecules, respectively. Long dimension of the molecules, L, was estimated using Equation 1,
Using GNOM45 program (33), indirect Fourier transformation of the scattering datasets computed a pairwise distribution function of interatomic vectors, P(r) (Equation 2).
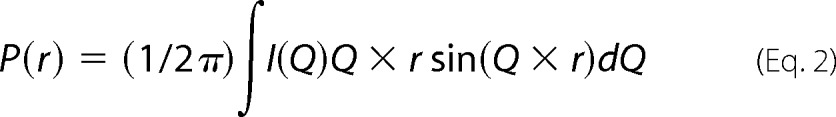 |
P(r) is a histogram of the frequency of vector lengths connecting small volume elements within the entire volume of the scattering particle. During the indirect Fourier transformation, P(r) was considered to be zero for vector lengths equal to 0 and Dmax (Dmax is the maximum linear dimension of the protein or longest possible vector in the scattering shape). P(r) analysis also provided RG and I0 for each SAXS dataset. Because I0 values are directly proportional to the mass of the scattering species, the I0 values estimated from P(r) analysis were utilized to estimate the concentration of the protein samples relative to the I0 value estimated from a dilution series of pre-characterized lysozyme solutions, except for the G2-G3, for which the data were acquired under different conditions (Table 1 and supplemental Table S1).
TABLE 1.
Structural parameters of various gelsolin mutants in different buffer conditions as deduced from indirect Fourier transformation of the SAXS data acquired from the samples of the proteins (Dmax and RG values are in Å)
For the parameters deduced from Guinier analysis, please refer to supplemental Table S1.
| Protein (mass kDa) concentration (mg/ml) | EGTA, pH 8 |
1 mm free Ca2+, pH 8 |
EGTA, pH 5 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Dmax | RG | I0 | Dmax | RG | I0 | Dmax | RG | I0 | |
| Gelsolin (82.9) | |||||||||
| 5 | 100 | 30.9 | 92 | 160 | 37.5 | 90 | 125 | 34.1 | 93 |
| 2.5 | 100 | 31.0 | 46 | 160 | 37.5 | 43 | 125 | 34.0 | 44 |
| G1-G3 (42.7) | |||||||||
| 3.7 | 110 | 33.7 | 36 | 105 | 31.6 | 35 | 110 | 33.0 | 34 |
| 2.3 | 110 | 33.6 | 23 | 105 | 31.6 | 22 | 110 | 33.1 | 24 |
| G1-G2 (28.6) | |||||||||
| 4.2 | 68 | 20.6 | 28 | 105 | 29.4 | 25 | 103 | 29.3 | 23 |
| 3.3 | 67 | 20.5 | 23 | 105 | 29.3 | 22 | 107 | 29.4 | 20 |
| 28–161 (16.8) | |||||||||
| 4.0 | 55 | 16.5 | 16 | 65 | 18.4 | 15 | 65 | 18.4 | 17 |
| 3.4 | 55 | 16.4 | 12 | 65 | 18.4 | 13 | 65 | 18.3 | 14 |
| G2-G3 (24.5) | |||||||||
| 2.7a | 60 | 19.5 | 11.4a | 63 | 19.9 | 10.2a | 62 | 19.6 | 11.2a |
| Mut1-G1-G3 (42.15) | |||||||||
| 3.1 | 110 | 32.3 | 29 | 105 | 30.8 | 30 | 108 | 32.2 | 31 |
| 2.6 | 110 | 32.1 | 23 | 105 | 30.9 | 24 | 107 | 32.2 | 24 |
| Mut2-G1-G3 (42.06) | |||||||||
| 2.7 | 88 | 25.7 | 29 | 130 | 38.7 | 26 | 130 | 38.6 | 28 |
| 1.9 | 87 | 25.7 | 24 | 130 | 38.7 | 24 | 130 | 38.6 | 22 |
| Mut3-G1-G3 (41.98) | |||||||||
| 2.5 | 85 | 25.6 | 23 | 125 | 37.9 | 24 | 120 | 37.4 | 23 |
| 1.7 | 85 | 25.7 | 15 | 125 | 37.8 | 16 | 120 | 37.4 | 14 |
| Mut4-G1-G3 (42.02) | |||||||||
| 2.2 | 88 | 25.6 | 21 | 130 | 38.5 | 20 | 125 | 38.0 | 23 |
| 1.4 | 87 | 25.6 | 13 | 130 | 38.5 | 14 | 125 | 37.9 | 15 |
a SAXS data for G2-G3 was acquired in a different trial, and the protein concentration is based on absorbance at 280 nm.
Structure Reconstruction within SAXS Profiles
To visualize the Ca2+ ion and pH-induced changes in the global structure of gelsolin mutants, models of the proteins were reconstructed within the shape constraints provided by SAXS data using chain-ensemble protocol (GASBOR22IQ program (34)). The models were generated for each dataset considering no shape and symmetry bias using 380 dummy residues for G1-G3 and its mutants, 250 for G1-G2, 133 for 28–161, and 250 for G2-G3, respectively (supplemental Table S2). Different mutants of G1-G3 were modeled using 380 dummy residues. For all the reconstructions, a Fibonacci grid order of 15 was used to model the hydration layer around the chain ensemble. Each calculation was repeated 10 times. Of the 10 models constructed, the structure with the χ2 close to 1 between computed I(Q) for the modeled structure versus the experimental data was used for structure interpretation (supplemental Table S3). Using SUPCOMB20 program, the inertial axes of the resultant low resolution shapes for the proteins and known crystal structures from x-ray diffraction were superimposed (35). Open source programs PyMOL and SPDB viewer were used for graphical analysis, manual alignment of models, and figure generation. Dummy models have been presented in this work in surface mode. The van der Waals radii of the dummy ASP residues have been set to 1.9 to correspond to the volume of 27 Å3. Data plotting was done using OriginLab software.
Statistical Analysis
The results are expressed as the mean ± S.D. The statistical significance of the differences between the rates of depolymerization was evaluated by the unpaired t tests, although those of the plasma gelsolin levels and cytokine levels were evaluated by using one-way analysis of variance followed by the post hoc Tukey-Kramer test. The animal survival data are presented as Kaplan-Meier curves, and the impact of treatment on the animal survival was analyzed by the log-rank test. The differences were considered significant if the probability value was ≤0.05.
RESULTS
Redefining Minimal Gelsolin Capable of F-actin Depolymerization
To decipher the minimal region of plasma gelsolin required for F-actin depolymerization, we generated different truncated versions of GSN, retaining F-actin binding and depolymerization segments, by deletion mutagenesis (Fig. 1). Each truncated mutant was purified and examined for its F-actin depolymerization ability by measuring the decrement in relative fluorescence of the pyrene-labeled F-actin in the presence of free Ca2+ (Fig. 2). Noticeably, the F-actin in the presence of buffer alone also exhibited a substantial depolymerization effect (Fig. 2A), which was repeatedly observed in the absence of any gelsolin variant and could be due to the dilution of F-actin from 4 μm to 100 nm concentration just prior to the fluorescence measurements (as described under “Materials and Methods”). Because the G1 domain and the native g1-g2 linker are essential for the F-actin depolymerizing ability of GSN (3, 4, 17), as expected, the addition of G2-G6 and G4-G6 (data not shown) to labeled F-actin in the buffer containing 1 mm of free Ca2+ at pH 8 did not lower the relative fluorescence (Fig. 2A). Maintaining a molar ratio of 1:10 between (all) gelsolin variants and actin, a substantial decrement in fluorescence was observed upon the addition of GSN, G1-G3, G1-G2, 1–161, and 25–161 to labeled actin. Interestingly, under identical conditions, the 36–161-residue fragment showed an intermediate F-actin depolymerizing potency, and the deletion mutant 42–161 (and its smaller versions) completely lost the F-actin depolymerizing ability (profiles similar to F-actin alone, and thus not shown in Fig. 2A). Evidently, even though the fraction of pyrene-labeled F-actin added to each sample was identical, the fluorescence curves did not converge to the same fluorescence value at the origin in the presence of gelsolin variants. To our best knowledge and as has been seen earlier (9), this could be attributed to the quasi-instantaneous nature of the F-actin depolymerization, which could not be measured during the first 15–25 s taken for the mixing of the samples.
FIGURE 1.

A schematic representation of different gelsolin variants generated in this work. Plasma GSN contains six domains represented as G1-G6. Domain organization shown is as described earlier (2). Small black and white ovals represent the segments 108–114 and 148–152, respectively, which are essential for actin binding and depolymerizing activities of gelsolin.
FIGURE 2.
Redefinition of minimal gelsolin capable of depolymerizing F-actin. A, kinetics of pyrene- labeled F-actin (100 nm) depolymerization were studied by incubating with either buffer alone or different gelsolin variants (10 nm) in the presence of 1 mm CaCl2 and measuring the fluorescence at 407 nm for 150 s. B and C, rates of depolymerization (decrement in fluorescence/s) of labeled F-actin by the addition of different gelsolin variants (at 1:10 gelsolin/actin ratio) in the presence of CaCl2 at different time intervals post-addition are shown. The difference in the depolymerization rate was considered significant if p ≤ 0.5 as compared with the rate of depolymerization by GSN. p values <0.01 and <0.05 are shown as ** and *, respectively.
Upon comparing the rates of decrement in fluorescence units of pyrene-labeled F-actin in the presence of different gelsolin truncates, it was observed that GSN, dT-GSN, G1-G3, and 25–161 depolymerize F-actin much faster in the time frame of 0–30 s post-addition of the sample components than G1-G2 and 1–161 (Fig. 2B). At the same time, G2-G6, G4-G6, and 42–161 showed almost no depolymerizing activities, whereas residues 36–161 showed very slow and reduced depolymerizing ability. Conversely, G1-G2 and 1–161 showed higher depolymerizing rates in the time frame of 31–45 s post-addition compared with those of GSN, dT-GSN, and G1-G3, suggesting a delayed action in these proteins. The smallest of this group, i.e. 25–161-residue fragment, not only exhibited fast rates initially in the time frame of 0–30 s comparable with GSN, but also exhibited a prolonged action, thus showing maximum overall F-actin depolymerizing potency. To evaluate if any further reduction in the size of the 25–161-fragment would affect its F-actin depolymerizing property, some more constructs 28–161, 30–161, 32–161, 34–161, 25–158 and 25–156 were prepared. The latter two proteins were designed to explore the role of the C-terminal residues of 25–161 in F-actin depolymerization function. Interestingly, the removal of three residues from the N terminus of 25–161 led to a substantial increase in the actin depolymerizing ability in Ca2+ (Fig. 2C). But further reduction of the N terminus led to a gradual loss in actin depolymerizing ability. However, the removal of just three residues from the C terminus of 25–161 led to a sudden decrease in the depolymerizing activity of the resultant protein fragment, which further disappeared in residues 25–156. Earlier, residues 150–160 have been demonstrated to be essential for the F-actin depolymerization activity of gelsolin (15). Furthermore, because the rates of decrement in fluorescence in different time frames did not correlate among different gelsolin truncates, it seems plausible that the F-actin depolymerization capabilities of different gelsolin variants are qualitatively different. The depolymerization activities of different gelsolin variants were also visualized by confocal microscopy using fluorescent (BODIPY FL)-labeled F-actin. As seen in Fig. 3, incubation of the labeled F-actin with GSN, G1-G3, 1–161, or 28–161 at a molar ratio of 1:5 (gelsolin variant/actin) resulted in a shortening of the actin filaments or their disappearance in the presence of Ca2+ ions, whereas the incubation with G4-G6 or buffer alone did not modulate the filamentous actin morphology. Overall, our data shows that the fragment 30–161 is the smallest N-terminal region of gelsolin that can depolymerize F-actin in the presence of Ca2+.
FIGURE 3.

Visualization of F-actin depolymerization activities of gelsolin variants by confocal microscopy. BODIPY-labeled F-actin was incubated with either buffer alone (control) or a gelsolin variant at a molar ratio of 1:5 (gelsolin/actin) in F-buffer in the presence of 1 mm CaCl2, and the depolymerization activity was directly observed by confocal microscopy at ×100 magnification. Representative snapshot images are shown here. The scale bar in each image corresponds to 10 μm.
Regulation of F-actin Depolymerizing Activity of Truncated Gelsolins by Ca2+ Ions and pH
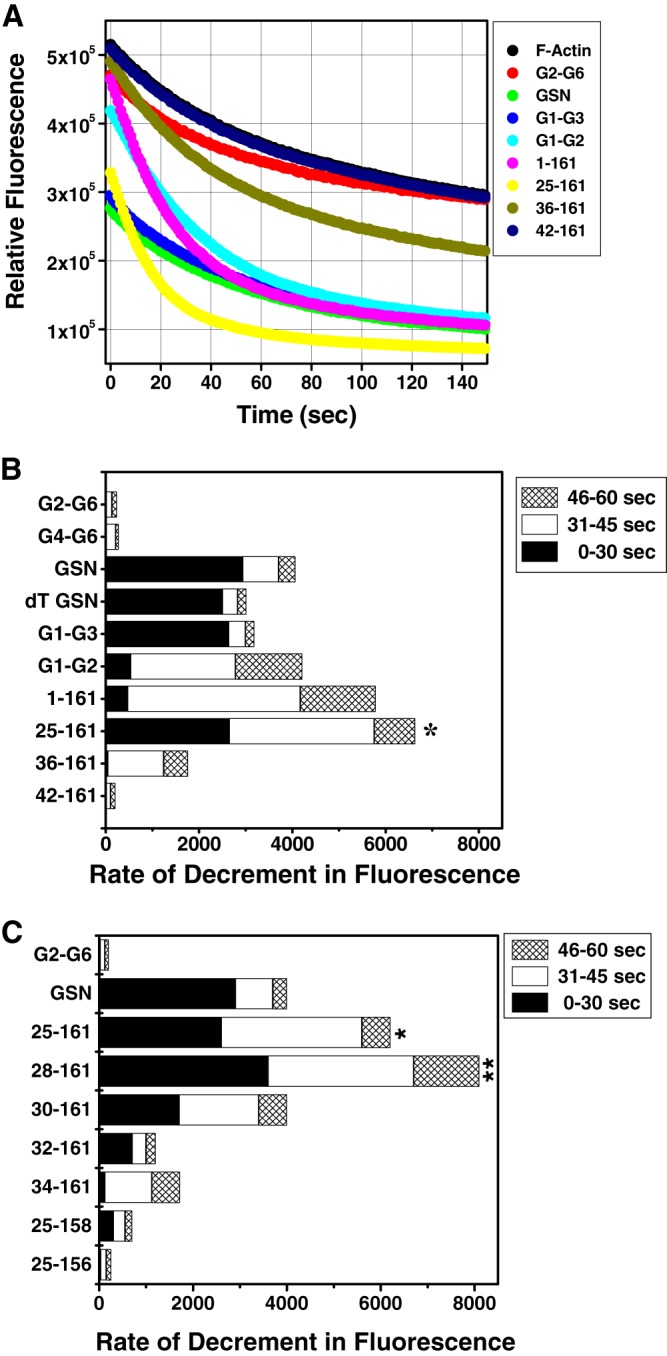
The F-actin depolymerizing activities of different deletion mutants of gelsolin were further examined for their dependence on the presence of free calcium in the buffer. As seen in Fig. 4, A and B, the full-length gelsolin as well as different truncated versions of gelsolin were F-actin depolymerization-competent only in the presence of 1 mm free Ca2+ with the exception of G1-G3 and tail-less gelsolin, which have earlier been found to depolymerize F-actin independently of free Ca2+ (9, 14). In our experiments, whereas dT-GSN showed partial activity in EGTA conditions, G1-G3 showed almost no dependence on calcium for actin depolymerizing activity. As expected, G2-G6 and G4-G6 (data not shown) did not show any F-actin depolymerizing activity and acted as negative control. At similar (1:10) protein/actin molar ratios, 36–161 and 42–161 showed very little actin depolymerizing potencies even in the presence of Ca2+ ions (Fig. 4A), but there was some gain in the depolymerizing activities of these proteins at higher (1:2–1:1) protein/actin molar ratios (supplemental Fig. S1). F-actin depolymerizing activities of different gelsolin variants were further found to increase with increasing molar ratios of the gelsolin truncates/actin in a calcium-dependent manner as above (supplemental Fig. S1). Weak F-actin depolymerizing activity exhibited by G2-G6 at higher molar ratios is in accordance with the earlier reports (10). Each of the six domains of gelsolin has one Ca2+-binding site; thus, we speculated that smaller versions of gelsolin would require lesser amounts of free Ca2+ in the buffer to initiate the F-actin depolymerization. However, upon comparing the pyrene fluorescence units in the samples varying in [Ca2+]/[protein], we found that all three of the full-length gelsolins, G1-G2 and 28–161, required 1–2 Ca2+ ions per protein molecule to show the sudden increment in their actin depolymerizing potency (Fig. 4C). As expected, the tail-less gelsolin (dT-GSN) and G1-G3 required much less and relatively no free Ca2+ ions, respectively, in the buffer to execute F-actin depolymerizing activities. This was an interesting experiment, because it indirectly indicated that the Ca2+ binding induced primary structural change that imparts the F-actin depolymerizing ability in full-length gelsolin, G1-G2 and 28–161 are common in all of these proteins.
FIGURE 4.
Regulation of F-actin depolymerization activities of different gelsolin variants by free Ca2+ levels. A and B, percent decrement in the fluorescence of pyrene-labeled F-actin (100 nm) by different gelsolin variants (10 nm) in the presence of either CaCl2 (1 mm) or EGTA (1 mm) as measured after 5 min is plotted here. C, plot showing the dependence of different gelsolin versions on free Ca2+ levels to depolymerize the pyrene-labeled F-actin. End point (after 60 s from mixing) fluorescence values are plotted here as a function of the molar ratio of free Ca2+ in buffer to the protein used.
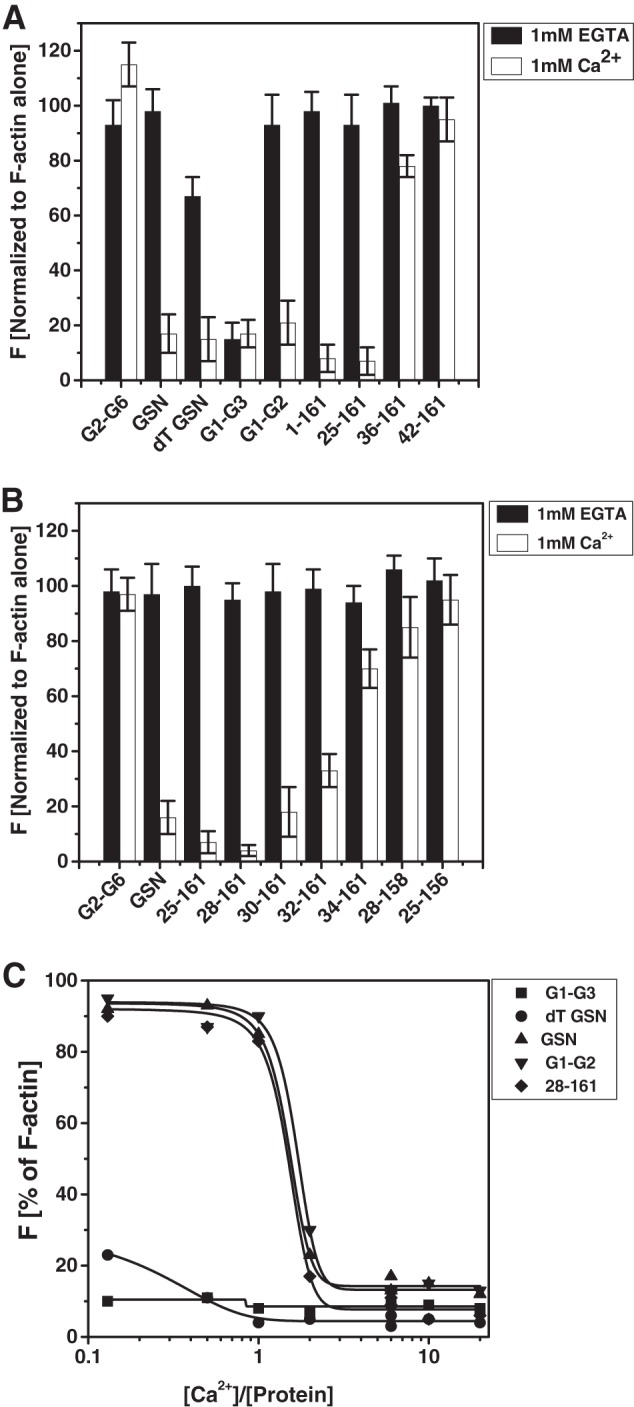
Because it is known that the low pH overrides the need for Ca2+ ions by gelsolin to acquire the F-actin depolymerizing ability (4, 36), we also investigated the effect of the buffer pH on the depolymerizing activities of gelsolin mutants by incubating these with pyrene-labeled F-actin at a 1:10 (gelsolin/actin) molar ratio under calcium-free conditions (10 mm EGTA). The results in Fig. 5 show that under EGTA conditions, the full-length GSN as well as the gelsolin truncates (except dT-GSN and G1-G3) did not change the fluorescence of pyrene-labeled F-actin at pH 7. A decrease in the pH value to 6 resulted in partial F-actin depolymerizing capabilities of GSN, G1-G2, and 1–161, whereas at pH 5, GSN, G1-G2, 1–161, 25–161, and 36–161 became depolymerizing competent, although G2-G6 and 42–161 remained depolymerizing incompetent throughout (Fig. 5A). In addition, other N-terminal truncated fragments of gelsolin, 28–161, 30–161, 32–161, and 34–161, also showed low pH activation in the absence of free calcium (Fig. 5B). Interestingly, dT-GSN and G1-G3 showed the ability to depolymerize F-actin at pH 7 also in the absence of Ca2+ ions. Gelsolin lacking Ca2+-sensitive C-tail latch (dT-GSN) has been shown to depolymerize F-actin in a calcium-independent manner, and low pH is also capable of opening this molecule to dimensions similar to GSN in the buffer containing 1 mm free Ca2+ ions (4). Drawing analogy with dT-GSN, the Ca2+-independent F-actin depolymerizing ability of G1-G3 can be explained, but the restoration of dependence on the Ca2+ ions or low pH for the depolymerization function upon further truncation of the C-terminal portion in G1-G3 remained puzzling to us.
FIGURE 5.
Regulation of the F-actin depolymerization activities of different gelsolin variants by buffer pH. A and B, effect of pH on the pyrene-labeled filamentous actin depolymerizing activities of different gelsolin mutants at 1:10 gelsolin/actin ratio by measuring the fluorescence intensities after 60 s is plotted as percent decrement in fluorescence.
Barbed End Capping Activity of Truncated Gelsolin Variants
To determine the actin filament capping activity of different gelsolin truncates, we investigated their effect on the Cc of actin required for its polymerization. As described earlier, Cc of actin (defined as the concentration of unpolymerized actin at steady-state in a solution containing F-actin) at the barbed end is 0.1 μm; however, it is 0.5–0.6 μm at the pointed end (37–39). Thus, if capped at the barbed end, the Cc of actin shifts toward that of the pointed end, which is seen as a drastic change in the slope of the plots near the Cc. As seen in Fig. 6, A and B, in the presence of Ca2+ ions, the addition of GSN, G1-G3, G1-G2, and G4-G6 substantially shifted the Cc of actin (∼2 mol of actin/mol of mutant), whereas the addition of fragments 1–161, 28–161, and 42–161 shifted the Cc of actin only weakly (0.8–1.2 mol of actin/mol of mutant). However, in the absence of free calcium ions, only G1-G3 was capable of bringing this effect (Fig. 6C).
FIGURE 6.

Capping activity of different gelsolin variants. Effect of different gelsolin mutants on the critical concentration of actin for polymerization in the presence of either CaCl2 (A and B) or EGTA (C) in F-buffer at room temperature was determined by measuring pyrene fluorescence after 18 h. Relative fluorescence intensity is plotted here as a function of increasing concentrations of actin (0.1–0.8 μm). D, graph represents the percent inhibition of the actin filaments elongation at the barbed end by the increasing concentrations of the gelsolin mutants (0.25-4 nm). To 1 μm of F-actin seeds, 0.5 μm monomeric G-actin was added with the increasing concentrations of gelsolin mutants in the presence of 2 mm calcium. The change in fluorescence in the absence of gelsolin mutants was taken as 100% polymerization, and the relative inhibition of polymerization in the presence of different mutants was calculated.
To further examine whether the observed shift in Cc of actin for its polymerization in the presence of gelsolin mutants was indeed due to the barbed end capping activity of the gelsolin truncates or due to mere nonpolymerizing sequestration of G-actin by the gelsolin truncates, we further measured the inhibition of actin filament elongations at the barbed ends at the concentrations where the fraction of sequestered monomeric actin is insignificant. For this, the gelsolin truncates were added at the increasing concentrations (0.25–4 nm) to the actin filaments (1 μm) along with pyrene-labeled G-actin (0.5 μm). At this actin concentration, the rate of the polymerization is a linear function of the number of free barbed ends and of the available polymerizable G-actin (30). As seen in Fig. 6D, a significant inhibition of the elongation of actin filaments was observed in the presence of increasing concentrations of GSN and G1-G3 with 50% inhibition occurring at a concentration of 0.98 and 2.8 nm, respectively. However, only a weak inhibition or no inhibition was observed in the presence of the gelsolin truncates, 1–161, 28–161, and 42–161, suggesting a weak capping ability in these mutants. These results are in corroboration with the previous reports that the fragment 1–150 caps the actin filaments with a Kcap of 10 nm, although 1–161 does not cap the filament stably (30, 40). Additionally, the residues 161–172 have been shown to be critical for the capping activity of gelsolin fragments (40). Thus, these results further supported that the shift in Cc of actin in the presence of full-length GSN, G1-G3, G4-G6 and G1-G2 might be due to the barbed end capping of the actin filaments by these gelsolin truncates. However, because both the F-actin depolymerization-efficient (1–161 and 28–161) as well as depolymerization-deficient (42–161) mutants of G1 domain were incapable of preventing the actin polymerization even at 4 nm concentration (molar ratio of 1:250 (gelsolin mutant/actin)), the small shift in the Cc of actin observed in the presence of these mutants at 1:1 to 1:8 (gelsolin mutant/actin) ratios could be due to the nonpolymerizing sequestration of the monomeric actin by these fragments (41). Likewise, at these higher molar ratios (1:1–1:8) at least a partial contribution from the G-actin sequestration in the observed Cc shift by the capping efficient gelsolin truncates also cannot be ruled out.
Structural Insights into the Solution Shapes of Minimal Gelsolins
Intrigued by the differential abilities of minimized forms of gelsolin to depolymerize F-actin as a function of free Ca2+ levels in buffer and the pH of buffer, we attempted to gain visual insight into the predominant solution shapes of these proteins from their solution SAXS intensity profiles. Data acquired from solutions of G1-G3, G1-G2, 28–161, and G2-G3 in buffers containing either EGTA or 1 mm free Ca2+ ions at pH 8, and EGTA at pH 5 were analyzed, and the shapes were constructed. Guinier analysis presuming globular and rod-like shape of the scattering species provided RG and RC values of the protein molecules, respectively, and Equation 1 was employed to estimate the long dimension, L, of the molecules (supplemental Table S1). For the Guinier analysis, only low Q data (Q·RG or Q· RC ≤1.3) were used. Employing a wider Q range (0.008–0.5 Å−1), indirect Fourier transformation of the datasets using the GNOM45 program provided the Dmax and RG of the protein molecules under different conditions studied (Table 1 and supplemental Fig. S2). For each protein (except G2-G3), two different protein concentrations were studied. Importantly, the full-length protein (GSN) showed the parameters in agreement with those published earlier (3, 4). We earlier showed that GSN adopts a tight collapsed shape of maximum linear dimension (Dmax) of ∼100 Å under calcium-free conditions at pH 8, which opens up to ∼160 Å in the presence of 1 mm Ca2+ (pH 8) or partially opens to ∼125 Å in Ca2+-free buffer having pH 5. Interestingly, in the buffers containing ± 1 mm Ca2+ ions at pH 8 or the Ca2+-free buffer having pH 5, G1-G2 and 28–161 also showed an increment in Dmax upon sensing Ca2+ ions or low pH. At the same time, G1-G3 and G2-G3 showed no increment in the dimensions under these conditions. It is pertinent to highlight here that agreement of the I0 values estimated from the measured SAXS datasets with the expected values based on the mass and the concentration of proteins provided confidence in the shape parameters estimated by indirect Fourier transformation.
To gain visual insight into the predominant shapes of the protein molecules, we calculated models of the proteins using the chain-ensemble modeling approach (Fig. 7A and supplemental Fig. S3). Statistics of the modeling work are summarized in supplemental Tables S2 and S3, and comparison of the calculated I(Q) profiles with the experimental data are presented in supplemental Fig. S2. Although the normalized spatial density of the 10 models restored for each protein/condition indicated values about 0.7 to 1.2 suggesting variation among the individual models solved from the same dataset, the theoretical SAXS profile of the models was well within the error in the measured data used for structure restoration (Q range 0.008–0.5 Å−1) (supplemental Fig. S2). The SAXS profiles of the models selected for shape change interpretation were also compared with the computed SAXS profile from the crystal structures, which showed good correlation for some cases (supplemental Table S3). Importantly, the open versus closed shapes were in agreement with the changes seen in the shape parameters deduced from Guinier and P(r) analysis for proteins in buffers with EGTA, pH 8 and 5, and buffer having 1 mm free Ca2+, pH 8.
FIGURE 7.

A, SAXS data based chain-ensemble models computed for different proteins in three different conditions have been presented here. The dummy atom models have been shown in surface mode, and the overlaid structures from crystallography have been presented in ribbon format (and colored for the secondary structural content). B, schematic showing a preferential binding of the G3 domain to the G2 domain leaving the G1 domain free in G1-G3 and making it F-actin depolymerization-capable. In the absence of the G3 domain, the G1 domain interacts with the G2 domain acquiring a closed conformation that is depolymerization-inefficient and requires either free calcium or low pH conditions to attain an open structure.
The models solved for the scattering shapes of G1-G3, G1-G2, 28–161, and G2-G3 showed that the three-domain protein, G1-G3, and the two-domain protein, G2-G3, adopt an open three-lobed and a closed shape, respectively, in all the conditions without any major changes. The other two proteins, i.e. G1-G2 and 28–161, however, appeared to prefer a collapsed state in Ca2+-free conditions at pH 8, although addition of Ca2+ ions or lowering of buffer pH induced an opening of the shapes of these two proteins. Although two clear domains could be seen for G1-G2 in the presence of 1 mm Ca2+ ions or pH 5, a tail-like feature appeared in the 28–161 protein under these conditions. Knowing the sequence of the 28–161 construct, very likely the tail-like feature is the g1-g2 linker present in the molecule. The collapsed models of G1-G2 and 28–161, and the open model of G1-G3 under EGTA conditions in pH 8 can be explained based on the hypothesis that there is some kind of competitive affinity of G1 and G3 domains for the same or overlapping epitopes in the G2 domain, and the G3 domain supersedes the G1 domain (Fig. 7B). Support for the conclusion that the G1 domain extends away from the G2 (and G3) domain could be seen from the SAXS-based models for the G2-G3 protein that did not undergo any shape change either in the presence of Ca2+ ions or the low pH in buffer. Additionally, all the SAXS data-based dummy models were compared with the spatial positioning of the domains and linker as seen in the crystal structures of gelsolin in EGTA and the Ca2+-activated G-actin-bound form by attempting automated superimposition of their inertial axes (Fig. 7A and supplemental Fig. S3). Besides visual confirmation of the comparable nature of volumes of the domains, calculated χ2 values provided confidence about the similarities in the shapes in solution and crystal structures (supplemental Table S3). Overall, our modeling results brought forth that when the G3 domain is not present, the G1 domain binds to the G2 domain resulting in a collapsed state. This contact is broken by Ca2+ binding or lowering of the buffer pH leading to an opened-up shape. Because the actin-binding residues residing in the g1-g2 linker and possibly a freely mobile G1 domain are the requisites, the actin depolymerizing ability of G1-G2 and 28–161 is thus triggered by Ca2+ ions or low pH. The same explanation extends to Ca2+- and pH-independent F-actin-depolymerizing functions of G1-G3 protein. Interestingly, a recent publication also showed that G1-G3 can depolymerize actin in vitro and in cultured cells in Ca2+-independent manner (42).
Role of g2-g3 Linker in Ca2+/pH-independent F-actin Depolymerizing Activity of N-terminal Half of Gelsolin
One corollary of our hypothesis assuming a role of the G3 domain in keeping the g1-g2 linker open in the N-terminal half of gelsolin, i.e. G1-G3 is that the g2-g3 linker might indirectly regulate the Ca2+/pH-independent activity of G1-G3. As observed by F-actin depolymerization assays, G1-G3 (residues 1–371), but not G1-G2 (residues 1–241), was capable of depolymerizing F-actin independently of free calcium, and it implied that the additional region in G1-G3 comprising the g2-g3 linker and the G3 domain might be imparting calcium insensitivity to G1-G3. Interestingly, earlier a mutant consisting of residues 1–266 had also been found to depolymerize F-actin in a calcium-independent manner (43), which further narrowed down the region to 242–266 residues (spanning g2-g3 linker i.e. residues 247–271) that might be playing a role in determining the differential calcium sensitivities of these truncated gelsolins. Besides this, severin, a three-domain homolog of gelsolin, is dependent on free Ca2+ for its F-actin depolymerizing activity (44). Upon comparing the amino acid sequences of both the proteins, we found some interesting differences in the g2-g3 linker (Fig. 8A). Not only the g2-g3 linker of severin is six residues shorter in comparison with that of gelsolin but also few of the proline residues present in the gelsolin linker are substituted by glycines and alanine in this linker region of severin. To examine whether this linker plays any role in the determination of the calcium sensitivities of truncated gelsolins, we generated mutants of G1-G3 containing mutations in its g2-g3 linker to make it similar to that of severin (Fig. 8A). Determination of the F-actin depolymerizing activities by the purified mutant G1-G3 proteins showed that although Mut1-G1-G3 (with truncated g2-g3 linker) could still depolymerize F-actin independently of Ca2+ ions or low pH, further mutations suddenly made the resultant proteins dependent on Ca2+ ions or low pH for their depolymerizing function (Fig. 8, B and C). The SAXS data analyses showed that although Mut1-G1-G3 adopted the solution shape similar to G1-G3 in the presence or absence of Ca2+ ions and low pH, other mutants adopted a smaller dimension under Ca2+-free conditions at pH 8, which opened substantially (125–130 Å) in the presence of Ca2+ ions or at pH 5 (Table 1). Interestingly, these open dimensions of the Ca2+/pH-dependent F-actin depolymerization-competent mutants were even larger than that of G1-G3 by almost 20 Å. Structure reconstructions of these mutants revealed that although the three domains of Mut1-G1-G3 adopt an open shape similar to G1-G3 under EGTA conditions at pH 8, Mut2 through Mut4-G1-G3 preferred a collapsed state under these conditions (Fig. 9). A comparison of their open shapes in the presence of Ca2+ ions or at pH 5 suggested that the mutations in the g2-g3 linker region altered the native-like relative disposition of G2-G3 domains, possibly moving G3 away from its interaction site in G2 in Ca2+-free conditions at pH 8, thus allowing the G1 domain to interact with the G2 domain.
FIGURE 8.

Role of g2-g3 linker in calcium sensitivity of G1-G3 actin depolymerizing activity. A, sequence comparison of gelsolin and severin in the g2-g3 linker region is shown. Lowercase letters in Mut1-Mut4 of G1-G3 indicate the amino acids changed from wild type gelsolin to that of severin. B, percent decrement in the fluorescence of pyrene-labeled F-actin (100 nm) by either wild type or mutant G1-G3 (10 nm) in the presence of either CaCl2 (1 mm) or EGTA (1 mm) as measured after 5 min is plotted here. C, effect of the pH on the pyrene-labeled filamentous actin depolymerizing activity of G1-G3 and its mutants at a 1:10 gelsolin/actin ratio by measuring the fluorescence intensity after 60 s is shown.
FIGURE 9.

Chain-ensemble models restored for mutated G1-G3 molecules aided us in visualizing that although Mut1-G1-G3 acquires an open conformation similar to the wild type G1-G3 (as in Fig. 7) in all three conditions, other mutants Mut2 to Mut4-G1-G3 attain a closed conformation in EGTA conditions at pH 8. Low pH or the presence of free calcium at pH 8 results in the opening up of the global shapes of Mut2 to Mut4-G1-G3 thus making these proteins F-actin depolymerization-competent.
Furthermore, to investigate whether the F-actin-binding sites in closed conformations of G1-G2 and Mut4-G1-G3 remain exposed, binding of these mutants to F-actin was examined in the presence or absence of free calcium. As seen in Fig. 10A, Mut4-G1-G3, G1-G2, and the truncate 42–161 showed binding to F-actin even in the absence of free calcium similar to G1-G3, whereas GSN showed no binding to F-actin in the absence of free calcium. G4-G6 (lacking F-actin-binding site) did not bind to F-actin in any of these conditions as expected. Lower amounts of actin were found to sediment when the F-actin was mixed with GSN (+CaCl2) or G1-G3 (±CaCl2) compared with that in the F-actin alone sample (Fig. 10B), which could be attributed to the depolymerizing activities of these proteins in respective conditions. These results suggested that the F-actin-binding site remains exposed in Mut4-G1-G3, G1-G2, and smaller truncates even in the closed shape acquired in the absence of free calcium. The lack of F-actin depolymerizing activity exhibited by these gelsolin truncates could thus be a result of improper conformation of these fragments in the absence of free calcium. Overall, we show that the freely mobile G1 domain is another key requirement for filamentous actin depolymerizing ability of gelsolin truncates, and this feature is indirectly regulated by the positioning of G3 domain in the proteins augmented by gelsolin-like sequence of the g2-g3 linker.
FIGURE 10.
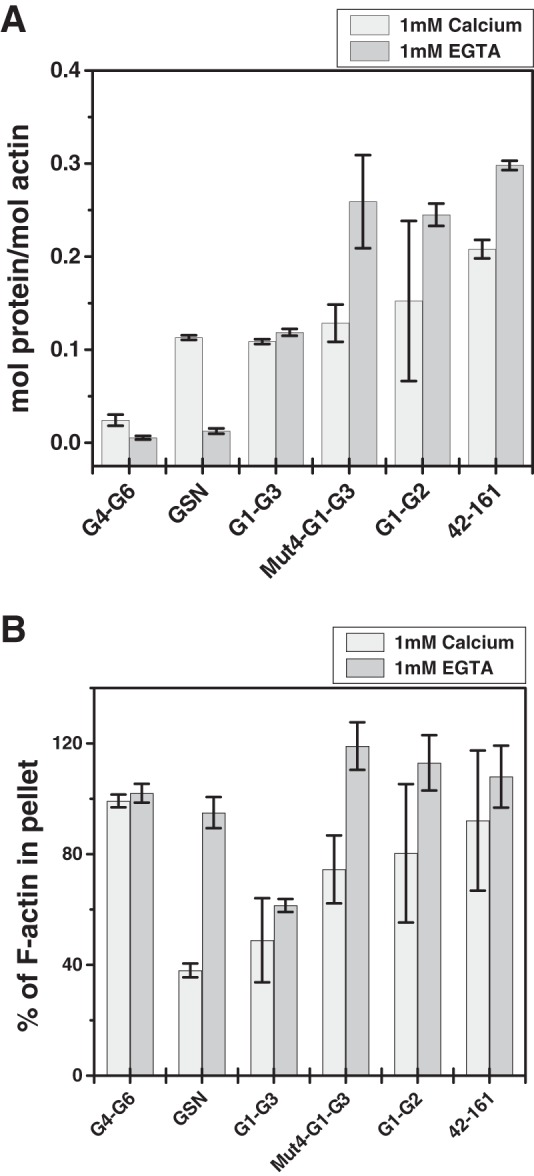
F-actin binding of different gelsolin variants. Cosedimentation of different gelsolin mutants (GSN, G1-G3, Mut4-G1-G3, G1-G2, 42–161, and G4-G6) mixed with equimolar concentrations of F-actin (5 μm) in the presence or absence of free calcium was performed. A, molar ratio of different gelsolin variants to the actin in the pellet fraction is plotted here. Actin-independent sedimentation of individual proteins was subtracted from the experimental data in the presence of F-actin. B, percentage of F-actin in the pellets of different samples was calculated using densitometry analysis taking the intensity in the F-actin-alone sample as 100%. Values are shown as mean ± S.D. of two independent experiments.
In Vivo Activity of Minimal Gelsolin in Septic Mice
The current literature concludes that intraperitoneal administration of LPS induces sepsis in mice, and repletion with exogenous recombinant full-length gelsolin improves the survivability in the experimental animal models (23–25). The effective dose of exogenous recombinant human gelsolin leading to the improvement in the animal survival in the LPS-induced sepsis mouse model was 8 mg, whereas it was 10 mg in the rat model of cecal ligation and double puncture-induced sepsis (23, 25). Because a mouse with an average weight of 20 g and a rat with an average weight of 300 g would theoretically have about 0.32 and 4.8 mg of endogenous plasma gelsolin, respectively, earlier studies have used ∼25× and 2× amounts of exogenous gelsolin per dose in the mouse and rat models, respectively. We first asked the question regarding much exogenous protein would be required to bring the depleted plasma gelsolin levels in the septic mouse back to normal levels by administering different amounts (0.5–8 mg) of the recombinant human GSN (93% similar to mouse plasma gelsolin) in LPS-induced septic mice. Placebo-treated mice had about 45% of the plasma gelsolin level as measured by Western blotting compared with the unchallenged control mice after 24 h, confirming the induction of sepsis upon LPS administration (data not shown). The plasma gelsolin levels in septic mice started to rise upon administration of exogenous gelsolin reaching approximately 78% of the control level upon administration of 2 mg of exogenous gelsolin. The plasma gelsolin levels rose to the levels same or above the control levels upon administration of 4 mg or higher amounts of exogenous gelsolin (data not shown). Importantly, plasma albumin levels remained constant in all groups of mice (data not shown).
To investigate the F-actin depolymerization potential (measured in terms of therapeutic effect) of truncated gelsolins in the LPS-induced mouse sepsis model, we used a dose of 2 mg of recombinant GSN per administration (which is ∼6.25× the amount of endogenous plasma gelsolin level in normal mice) because the molar quantities of minimal gelsolins are 2–5 times higher compared with full-length gelsolin in 2 mg of protein. The therapeutic potential of G1-G3, 28–161, G2-G6, and G4-G6 was explored and compared with that of GSN in septic mice. G2-G6 was chosen as an LPS binding but nondepolymerizing control, although G4-G6 served as both a non-LPS binding as well as nondepolymerizing control (Fig. 2B) (45). The plasma gelsolin levels in LPS-challenged and the exogenous recombinant truncated gelsolin-treated mice were measured over a 48-h time period by Western blotting. Administration of 2 mg (dose/mouse) of exogenous recombinant truncated gelsolins, G1-G3 and 28–161, significantly increased the plasma gelsolin levels to above normal levels compared with full-length gelsolin after 24 h (256 and 231 μg/ml in the G1-G3- and 28–161-treated groups, respectively, compared with 152 μg/ml in the GSN-treated group) (Fig. 11A). Higher plasma levels in the case of G1-G3 and 28–161 administered mice compared with those in GSN could be due to the better absorption rate of the truncated gelsolins than that of full-length gelsolin upon subcutaneous administration (Fig. 11A).
FIGURE 11.
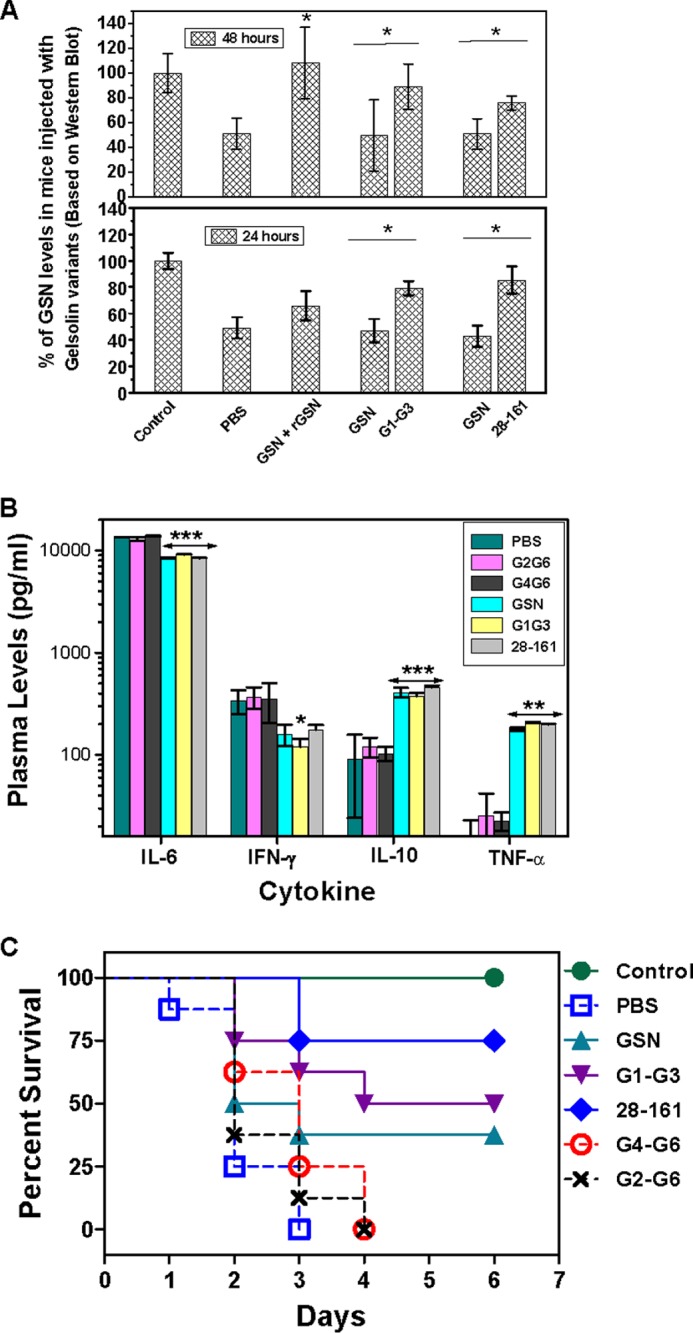
Effect of administration of different gelsolin variants in lipopolysaccharide-challenged mice. A, plasma gelsolin and recombinant truncated gelsolin levels were measured at 24- and 48-h time points after subcutaneous administration in LPS-challenged mice. GSN bars show endogenous plasma gelsolin levels, whereas rGSN, G1-G3, and 28–161 bars represent the levels of exogenous administered recombinant full-length plasma gelsolin and its truncated versions (G1-G3, 28–161). B, cytokine profiles of the mice in different groups compared with unchallenged control (nondetectable levels) are shown. Data are shown as mean ± S.D. C, percent survival of mice in different groups observed for 6 days after LPS challenge is shown. The p values <0.001, <0.01, and <0.05 compared with PBS-treated control values are shown as ***, **, and *, respectively.
To examine the effect of LPS treatment and truncated gelsolin administration on the cytokine profiles of the septic mice, we measured the levels of four plasma cytokines, IL-6, IFN-γ, and TNF-α, which are among the strong markers of the tissue damage/inflammation, and IL-10. The results summarized in Fig. 11B showed that the levels of all four cytokines increased steeply in LPS-treated mice as compared with the control group (without any LPS treatment) in which all these cytokines were undetectable, further confirming the induction of sepsis in our LPS-treated mice. The treatment with GSN, G1-G3, and 28–161 significantly reduced the levels of IL-6 (to ∼0.6-fold of those in the PBS control group) and IFN-γ (to ∼0.5-fold of those in the PBS control group) after 24 h of LPS challenge. In contrast, IL-10 and TNF-α levels were increased significantly to ∼5- and ∼12-fold, respectively, upon treatment with GSN, G1-G3, and 28–161 in septic mice compared with those levels in the PBS-treated control group. Essentially, these cytokine profiles suggested a shift in the immune responses from pro-inflammatory to anti-inflammatory mode. Interestingly, no change in the plasma cytokine levels was observed in G2-G6- and G4-G6-treated groups compared with the PBS-treated groups even though these proteins were able to raise the plasma gelsolin levels to normal levels (data not shown).
Finally, replenishment with a 2 mg dose of G1-G3 (50% survival, p < 0.007 versus PBS control group) and 28–161 (75% survival, p < 0.001 versus PBS control group) protected the LPS-challenged mice from mortality significantly better than GSN (33% survival, p = 0.07 versus PBS control group) (Fig. 11C), whereas all the mice in the PBS, G2-G6, and G4-G6 administered control groups died within 4 days of LPS administration. Thus, although the administration of a 2-mg dose of GSN could significantly modulate the cytokine profiles of septic mice from pro-inflammatory to anti-inflammatory mode, this dose provided only a marginal protection to these mice from mortality. In addition, F-actin depolymerization-incompetent gelsolin truncates G2-G6 and G4-G6 were incapable of providing any protection to septic mice from mortality. Overall, the therapeutic potential of truncated gelsolin fragments, G1-G3 and 28–161, as measured in LPS-induced septic mice demonstrated the filamentous actin depolymerizing potency of these proteins under in vivo conditions also.
DISCUSSION
Kwiatkowski et al. (15) had earlier shown that the first 1–160 N-terminal residues of gelsolin expressed in COS cell lines and secreted into media are sufficient for filamentous actin depolymerization, roughly as well as the full-length protein. Further deletion of the C-terminal residues to 149 significantly abolished the F-actin depolymerizing activity of the fragment (15, 43), whereas truncation of the N-terminal region to 25–160 residues has been found to retain its depolymerizing activity (36). We now have further dissected the first 1–161 residues of gelsolin and have revealed that fragment 28–161 is the smallest truncate capable of depolymerizing F-actin even more potently than the full-length gelsolin, whereas the smaller fragment 30–161 has depolymerizing activity roughly similar to that of the full-length gelsolin. Even smaller fragments, 32–161, 34–161, 36–161, and 42–161, were also able to depolymerize F-actin but with much less efficiency, revealing that the residues in the region 28–31 are also critically important for F-actin depolymerizing activity in addition to the two key regions reported previously (17). Moreover, because the residues 40–46 form a β-strand based on the gelsolin crystal structures (46), deletions in this region might lead to a structural collapse of the protein, which could be the reason for the loss of F-actin depolymerization activity of these proteins. Furthermore, the depolymerizing activity of 1–160-residue fragment has been shown to be quantitatively weaker than that of GSN in some studies, potentially due to the lack of capping activity, and at the same time it has also been proposed that in the absence of capping, actin depolymerization can take place from both the ends (47). In our studies, we have observed that although fragment 1–161 is much slower than GSN in depolymerizing F-actin in first 30 s upon addition to F-actin, it becomes faster than GSN in next 30 s. Furthermore, fragments 1–161, 28–161, and 42–161 were found to have much weaker capping ability as compared with the full-length GSN, G1-G3, and G1-G2. Interestingly, G4-G6 also exhibited capping activity during actin polymerization as has been suggested earlier (14). The differences between the capping abilities of different gelsolin variants might also lead to the qualitative changes observed in the depolymerizing activities of different variants and also raises a possibility of depolymerization of filaments from both the ends. Overall, it implies that possibly fragment 1–161 takes a longer time to bind to the F-actin compared with the full-length protein, but after that lag period, it results in rapid actin depolymerization probably from both the ends because of inefficient capping of the barbed ends. Surprisingly, further smaller fragments 25–161 and 28–161 are equally efficient as GSN in depolymerizing F-actin also in the first 30 s post-addition of the reaction components and are even faster than GSN in the second phase, which is indicative of some hindrance posed by residues 1–24 in the depolymerization action of the protein. It also indicates that the cytosolic gelsolin might be more potent than the plasma gelsolin in F-actin depolymerizing function. The faster rate of F-actin depolymerization of smaller fragments than that of the full-length protein and other truncates capable of capping could also be due to the engagement of the capping efficient versions with the depolymerized filaments, thus reducing the available gelsolin molecules to carry out further depolymerization after an initial faster phase of depolymerization as in the full-length protein and G1-G3. Overall, the capping-inefficient smaller fragments were more potent than the capping-efficient bigger truncates, including the full-length protein, until the critically important region for F-actin depolymerization (residues 28–31) was present in them.
Additionally, all the truncated versions of the N-terminal half of gelsolin in this study were dependent on the presence of Ca2+ for their F-actin depolymerizing activities just like the full-length protein, except for G1-G3. Although GSN is unable to bind F-actin in the absence of calcium, smaller variants (1–161 and 25–161) have been shown to bind F-actin in the absence of calcium, although the positioning/orientation of G1 is not optimal for actin depolymerization (18). The binding to calcium then enhances the affinity of G1 for actin by 1000-fold and facilitates its depolymerizing function (41). Thus, the calcium requirement of 1–161 and smaller fragments, for the F-actin depolymerization function, seems to be for aiding their optimal binding to actin. If that was the case, then G1-G2, which contains a proper F-actin- binding site (residues for this site), should have been calcium-insensitive for depolymerizing activity, but in this study it was found otherwise. In addition, our observations that G1-G2 and smaller variants are able to bind F-actin even in the absence of free calcium support the presence of cryptic calcium regulation in smaller constructs as reported earlier (15). To gain some understanding regarding this, in this study we have mutated the g2-g3 linker region of G1-G3 to make it similar with that in severin. The mutations in the g2-g3 linker region were found to result in regaining the calcium sensitivity of G1-G3 (otherwise calcium-insensitive) for F-actin depolymerization activity. Nevertheless, calcium-sensitive mutants of G1-G3 were capable of binding actin filaments in the absence of free calcium. Structural insights into the solution shapes of truncated gelsolins based on the SAXS studies revealed that although G1-G3 adopts an open shape in the presence or absence of free calcium, the smaller truncates G1-G2 and 28–161 adopt a closed shape in the calcium-free conditions. Furthermore, mutations in the g2-g3 linker result in the G1-G3 molecule adopting a collapsed shape in the absence of free calcium. These results led us to hypothesize that both the G1 and G3 domains have affinity for the same or overlapping regions in the G2 domain, and this affinity of G3 domain is higher than that of the G1 domain. Thus, in the presence of the G3 domain, the G1 domain remains free, and the shape of G1-G3 becomes F-actin depolymerization-competent. However, in the absence of G3 domain, the G1 domain interacts with the G2 domain acquiring a closed shape and becomes F-actin depolymerization-incompetent requiring Ca2+ binding or lowering of the buffer pH to break this interaction. Our hypothesis gets substantiated by the earlier reports that in G1-G3, G1 translates away from G2 by displacement of the 137–141-residue fragment (A strand) from the edge of the core β-sheet of G2, resulting in the extension of the g1-g2 linker (7). This β-sheet edge thus gets exposed and forms an interface with the G3 domain. In addition, Asp-259 (in g2-g3 linker) has been shown to initiate a helix formation in coordination with Thr-260 and condense the otherwise disordered linker to allow close packing of G2 and G3 (7, 48). Interestingly, an Asp-260 and Thr-261 are also present at similar positions in this linker region of severin, but the smaller length of this linker or the other specific residues like proline might result in the calcium sensitivity of severin.
Furthermore, the treatment of LPS-challenged mice with GSN and F-actin depolymerization-competent truncated gelsolins, G1-G3 and 28–161, resulted in a significant decrease in IL-6 and IFN-γ levels and a significant increase in IL-10 and TNF-α levels compared with the saline-treated mice shifting the immune response from the pro-inflammatory mode to anti-inflammatory mode as reported earlier (23). IL-6 and IFN-γ are among major proinflammatory cytokines, whereas IL-10 is an immune modulator cytokine that reduces the expression of other pro-inflammatory cytokines by the heme oxygenase-1-mediated pathway (49). Although higher TNF-α levels in the depolymerization-competent gelsolin variant-treated septic mice remained puzzling, it is pertinent to mention here that because IFN-γ has been shown to up-regulate the expression of the receptors for TNF-α (50), a reduction in INF-γ levels might mitigate the effects of TNF-α and may also result in higher TNF-α levels in the circulation. The therapeutic potential of G1-G3 and minimal gelsolin (residues 28–161) was also evident by the fact that these F-actin depolymerization-competent fragments of gelsolin were capable of protecting septic mice from mortality as compared with the nondepolymerizing fragment G4-G6. Moreover, the LPS-binding fragment, G2-G6, was also incapable of protecting mice from mortality, suggesting that the depolymerizing activity of gelsolin is required for this protection instead of the LPS sequestration as has also been suggested earlier (23). These results demonstrate for the first time that depolymerization-competent truncated gelsolins can be used at lower doses (4-fold lesser amount in this study) for the gelsolin replacement therapy in critical illnesses. Finally, it might be argued that the capping deficiency in the smaller fragment would reduce its potential in clearing the actin filaments from the circulation, but the presence of the Gc globulin that sequesters monomeric actin with higher affinity than full-length gelsolin would help the F-actin depolymerization to go forward (51). Also, because the level of Gc globulin in plasma can be replenished by increased synthesis in liver as an acute-phase response, whereas plasma gelsolin synthesis remains uninduced (24), the administration of exogenous minimal gelsolin would definitely provide a therapeutic effect in case of disease conditions accompanying cell necrosis. We thus propose here that small F-actin depolymerization-competent fragments of gelsolin hold great potential as therapeutic molecules in various disease conditions, and eventual gelsolin replacement therapy in the future could consist of formulations of some of these small molecules instead of the full-length gelsolin.
Acknowledgments
We acknowledge the help from S. Maiti (Indian Institute of Science Education and Research, Kolkata, India) in pyrene labeling of actin, Bhupinder Singh (IMTECH, Chandigarh, India) in animal experiments, and Dr. Vito Graziano, X9 Beam Line National Synchrotron Light Source, Brookhaven National Laboratory, for data collection on our behalf. We also thank Pradip Sen (IMTECH, Chandigarh, India) for critical reading of the manuscript. We acknowledge the consistent support from faculty and staff of IMTECH.
This work was supported in part by Projects UNSEEN NWP, OLP-0056, and Sip-10. This is IMTech Communication Number 65/2012.

This article contains supplemental Figs. S1–S3 and Table S1–S3.
- SAXS
- small angle x-ray scattering
- pGSN
- plasma gelsolin
- GSN
- gelsolin
- Cc
- critical concentration.
REFERENCES
- 1. Sun H. Q., Yamamoto M., Mejillano M., Yin H. L. (1999) Gelsolin, a multifunctional actin regulatory protein. J. Biol. Chem. 274, 33179–33182 [DOI] [PubMed] [Google Scholar]
- 2. Burtnick L. D., Koepf E. K., Grimes J., Jones E. Y., Stuart D. I., McLaughlin P. J., Robinson R. C. (1997) The crystal structure of plasma gelsolin: implications for actin severing, capping, and nucleation. Cell 90, 661–670 [DOI] [PubMed] [Google Scholar]
- 3. Ashish, Paine M. S., Perryman P. B., Yang L., Yin H. L., Krueger J. K. (2007) Global structure changes associated with Ca2+ activation of full-length human plasma gelsolin. J. Biol. Chem. 282, 25884–25892 [DOI] [PubMed] [Google Scholar]
- 4. Garg R., Peddada N., Sagar A., Nihalani D., Ashish (2011) Visual insight into how low pH alone can induce actin-severing ability in gelsolin under calcium-free conditions. J. Biol. Chem. 286, 20387–20397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kiselar J. G., Janmey P. A., Almo S. C., Chance M. R. (2003) Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proc. Natl. Acad. Sci. U.S.A. 100, 3942–3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kothakota S., Azuma T., Reinhard C., Klippel A., Tang J., Chu K., McGarry T. J., Kirschner M. W., Koths K., Kwiatkowski D. J., Williams L. T. (1997) Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 278, 294–298 [DOI] [PubMed] [Google Scholar]
- 7. Burtnick L. D., Urosev D., Irobi E., Narayan K., Robinson R. C. (2004) Structure of the N-terminal half of gelsolin bound to actin: roles in severing, apoptosis and FAF. EMBO J. 23, 2713–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kwiatkowski D. J., Janmey P. A., Mole J. E., Yin H. L. (1985) Isolation and properties of two actin-binding domains in gelsolin. J. Biol. Chem. 260, 15232–15238 [PubMed] [Google Scholar]
- 9. Chaponnier C., Janmey P. A., Yin H. L. (1986) The actin filament-severing domain of plasma gelsolin. J. Cell Biol. 103, 1473–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Way M., Gooch J., Pope B., Weeds A. G. (1989) Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J. Cell Biol. 109, 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roustan C., Ferjani I., Maciver S. K., Fattoum A., Rebière B., Benyamin Y. (2007) Calcium-induced conformational changes in the amino-terminal half of gelsolin. FEBS Lett. 581, 681–686 [DOI] [PubMed] [Google Scholar]
- 12. Pope B. J., Gooch J. T., Weeds A. G. (1997) Probing the effects of calcium on gelsolin. Biochemistry 36, 15848–15855 [DOI] [PubMed] [Google Scholar]
- 13. Selden L. A., Kinosian H. J., Newman J., Lincoln B., Hurwitz C., Gershman L. C., Estes J. E. (1998) Severing of F-actin by the amino-terminal half of gelsolin suggests internal cooperativity in gelsolin. Biophys. J. 75, 3092–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin K. M., Mejillano M., Yin H. L. (2000) Ca2+ regulation of gelsolin by its C-terminal tail. J. Biol. Chem. 275, 27746–27752 [DOI] [PubMed] [Google Scholar]
- 15. Kwiatkowski D. J., Janmey P. A., Yin H. L. (1989) Identification of critical functional and regulatory domains in gelsolin. J. Cell Biol. 108, 1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Southwick F. S. (1995) Gain-of-function mutations conferring actin-severing activity to human macrophage cap G. J. Biol. Chem. 270, 45–48 [DOI] [PubMed] [Google Scholar]
- 17. Zhang Y., Vorobiev S. M., Gibson B. G., Hao B., Sidhu G. S., Mishra V. S., Yarmola E. G., Bubb M. R., Almo S. C., Southwick F. S. (2006) A CapG gain-of-function mutant reveals critical structural and functional determinants for actin filament severing. EMBO J. 25, 4458–4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Irobi E., Burtnick L. D., Urosev D., Narayan K., Robinson R. C. (2003) From the first to the second domain of gelsolin: a common path on the surface of actin? FEBS Lett. 552, 86–90 [DOI] [PubMed] [Google Scholar]
- 19. Lee W. M., Galbraith R. M. (1992) The extracellular actin-scavenger system and actin toxicity. N. Engl. J. Med. 326, 1335–1341 [DOI] [PubMed] [Google Scholar]
- 20. Haddad J. G., Harper K. D., Guoth M., Pietra G. G., Sanger J. W. (1990) Angiopathic consequences of saturating the plasma scavenger system for actin. Proc. Natl. Acad. Sci. U.S.A. 87, 1381–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee P. S., Drager L. R., Stossel T. P., Moore F. D., Rogers S. O. (2006) Relationship of plasma gelsolin levels to outcomes in critically ill surgical patients. Ann. Surg. 243, 399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peddada N., Sagar A., Ashish, Garg R. (2012) Plasma gelsolin: a general prognostic marker of health. Med. Hypotheses 78, 203–210 [DOI] [PubMed] [Google Scholar]
- 23. Lee P. S., Waxman A. B., Cotich K. L., Chung S. W., Perrella M. A., Stossel T. P. (2007) Plasma gelsolin is a marker and therapeutic agent in animal sepsis. Crit. Care Med. 35, 849–855 [DOI] [PubMed] [Google Scholar]
- 24. Rothenbach P. A., Dahl B., Schwartz J. J., O'Keefe G. E., Yamamoto M., Lee W. M., Horton J. W., Yin H. L., Turnage R. H. (2004) Recombinant plasma gelsolin infusion attenuates burn-induced pulmonary microvascular dysfunction. J. Appl. Physiol. 96, 25–31 [DOI] [PubMed] [Google Scholar]
- 25. Cohen T. S., Bucki R., Byfield F. J., Ciccarelli N. J., Rosenberg B., DiNubile M. J., Janmey P. A., Margulies S. S. (2011) Therapeutic potential of plasma gelsolin administration in a rat model of sepsis. Cytokine 54, 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pardee J. D., Spudich J. A. (1982) Purification of muscle action. Methods Enzymol. 85, 164–181 [DOI] [PubMed] [Google Scholar]
- 27. Sagar A., Peddada N., Solanki A. k., Choudhary V., Garg R., Ashish (2013) Visualizing the elusive open shape of G-actin in solution by SAXS data analysis. Biochem. Biophys. Res. Commun. 435, 740–744 [DOI] [PubMed] [Google Scholar]
- 28. Kouyama T., Mihashi K. (1981) Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 114, 33–38 [PubMed] [Google Scholar]
- 29. Terada N., Shimozawa T., Ishiwata S., Funatsu T. (2007) Size distribution of linear and helical polymers in actin solution analyzed by photon counting histogram. Biophys. J. 92, 2162–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weber A., Pring M., Lin S. L., Bryan J. (1991) Role of the N- and C-terminal actin-binding domains of gelsolin in barbed filament end capping. Biochemistry 30, 9327–9334 [DOI] [PubMed] [Google Scholar]
- 31. Selve N., Wegner A. (1986) Rate constant for capping of the barbed ends of actin filaments by the gelsolin-actin complex. Eur. J. Biochem. 155, 397–401 [DOI] [PubMed] [Google Scholar]
- 32. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) A Windows-based system for small-angle scattering data analysis J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 33. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 34. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozin M. B., Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 36. Lamb J. A., Allen P. G., Tuan B. Y., Janmey P. A. (1993) Modulation of gelsolin function. Activation at low pH overrides Ca2+ requirement. J. Biol. Chem. 268, 8999–9004 [PubMed] [Google Scholar]
- 37. Klaavuniemi T., Yamashiro S., Ono S. (2008) Caenorhabditis elegans gelsolin-like protein 1 is a novel actin filament-severing protein with four gelsolin-like repeats. J. Biol. Chem. 283, 26071–26080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Z., Klaavuniemi T., Ono S. (2010) Distinct roles of four gelsolin-like domains of Caenorhabditis elegans gelsolin-like protein-1 in action filament severing, barbed end capping, and phosphoinositide binding. Biochemistry 49, 4349–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pollard T. D. (1983) Measurement of rate constants for actin filament elongation in solution. Anal. Biochem. 134, 406–412 [DOI] [PubMed] [Google Scholar]
- 40. Sun H. Q., Wooten D. C., Janmey P. A., Yin H. L. (1994) The actin side-binding domain of gelsolin also caps actin filaments. Implications for actin filament severing. J. Biol. Chem. 269, 9473–9479 [PubMed] [Google Scholar]
- 41. Way M., Pope B., Gooch J., Hawkins M., Weeds A. G. (1990) Identification of a region in segment 1 of gelsolin critical for actin binding. EMBO J. 9, 4103–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mazumdar B., Meyer K., Ray R. (2012) N-terminal region of gelsolin induces apoptosis of activated hepatic stellate cells by a caspase-dependent mechanism. PLoS One 7, e44461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Way M., Pope B., Weeds A. G. (1992) Evidence for functional homology in the F-actin binding domains of gelsolin and α-actinin: implications for the requirements of severing and capping. J. Cell Biol. 119, 835–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eichinger L., Schleicher M. (1992) Characterization of actin- and lipid-binding domains in severin, a Ca2+-dependent F-actin fragmenting protein. Biochemistry 31, 4779–4787 [DOI] [PubMed] [Google Scholar]
- 45. Bucki R., Georges P. C., Espinassous Q., Funaki M., Pastore J. J., Chaby R., Janmey P. A. (2005) Inactivation of endotoxin by human plasma gelsolin. Biochemistry 44, 9590–9597 [DOI] [PubMed] [Google Scholar]
- 46. McLaughlin P. J., Gooch J. T., Mannherz H. G., Weeds A. G. (1993) Structure of gelsolin segment 1-actin complex and the mechanism of filament severing. Nature 364, 685–692 [DOI] [PubMed] [Google Scholar]
- 47. Weeds A. G., Gooch J., Hawkins M., Pope B., Way M. (1991) Role of actin-binding proteins in cytoskeletal dynamics. Biochem. Soc. Trans. 19, 1016–1020 [DOI] [PubMed] [Google Scholar]
- 48. Nag S., Ma Q., Wang H., Chumnarnsilpa S., Lee W. L., Larsson M., Kannan B., Hernandez-Valladares M., Burtnick L. D., Robinson R. C. (2009) Ca2+ binding by domain 2 plays a critical role in the activation and stabilization of gelsolin. Proc. Natl. Acad. Sci. U.S.A. 106, 13713–13718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee T. S., Chau L. Y. (2002) Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 8, 240–246 [DOI] [PubMed] [Google Scholar]
- 50. Pandita R., Pocsik E., Aggarwal B. B. (1992) Interferon-γ induces cell surface expression for both types of tumor necrosis factor receptors. FEBS Lett. 312, 87–90 [DOI] [PubMed] [Google Scholar]
- 51. Meier U., Gressner O., Lammert F., Gressner A. M. (2006) Gc-globulin: roles in response to injury. Clin. Chem. 52, 1247–1253 [DOI] [PubMed] [Google Scholar]