

Background: Starved rat livers contain metabolites, which activate ChREBP and 14-3-3 interactions.
Results: βHB and AcAc in the extract stabilize the ChREBP·14-3-3 complex in cytosol and inhibit the ChREBP/importin interactions.
Conclusion: Ketone bodies are directly involved in the regulation of the nuclear/cytosol shuttle for ChREBP.
Significance: Under starvation, ketone bodies serve as a “low glucose” sensor to inhibit lipogenesis.
Keywords: Acetoacetate, Carbohydrate Metabolism, Lipogenesis, Signal Transduction, Transcription Factors
Abstract
The carbohydrate response element-binding protein (ChREBP) is a glucose-responsive transcription factor that plays a critical role in converting excess carbohydrate to storage fat in liver. In response to changing glucose levels, ChREBP activity is regulated by nucleo-cytoplasmic shuttling of ChREBP via interactions with 14-3-3 proteins and importins. The nuclear/cytosol trafficking is regulated partly by phosphorylation/dephosphorylation of serine 196 mediated by cAMP-dependent protein kinase and protein phosphatase. We show here that protein-free extracts of starved and high fat-fed livers contain metabolites that activate interaction of ChREBP·14-3-3 and inhibit the ChREBP/importin α interaction, resulting in cytosolic localization. These metabolites were identified as β-hydroxybutyrate and acetoacetate. Nuclear localization of GFP-ChREBP is rapidly inhibited in hepatocytes incubated in β-hydroxybutyrate or fatty acids, and the observed inhibition is closely correlated with the production of ketone bodies. These observations show that ketone bodies play an important role in the regulation of ChREBP activity by restricting ChREBP localization to the cytoplasm, thus inhibiting fat synthesis during periods of ketosis.
Introduction
The liver is the principal organ responsible for the conversion of excess dietary carbohydrates to fat. Carbohydrate response element-binding protein (ChREBP)2 is a transcription factor responsible for the coordinated metabolism of carbohydrate and fat synthesis, independent of insulin (1–3). ChREBP senses and responds to changing levels of glucose (“glucose signaling”) and activates transcription of genes encoding regulatory enzymes of glycolysis and lipogenesis. The action of ChREBP accounts for over half of the fat synthesis in liver (2).
ChREBP is a large transcription factor of 96 kDa containing several functional domains, including two nuclear export signals, NES1 and NES2, an extended bipartite nuclear import signal (NLS), and a DNA binding basic helix-loop-helix/Zip domain (Fig. 1). ChREBP function is regulated at two levels, nuclear translocation and DNA binding (1). The N-terminal region (residues 1–250) of ChREBP binds to 14-3-3 proteins and is responsible for regulating subcellular localization in response to glucose levels (4, 5). The C-terminal region of ChREBP is responsible for DNA binding and transcriptional activity by forming a heterodimer with Max-like protein (Mlx), which binds to E boxes in the promoter region of target genes (6, 7).
FIGURE 1.
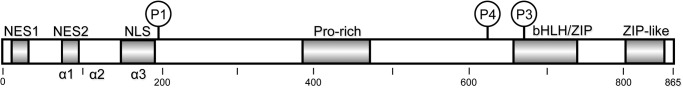
Domain organization of ChREBP. Residues 1–251 of the N-terminal regulatory region, including the predicted α1-, α2-, and α3-helices, the nuclear export signals (NES1 and NES2), and the NLS are shown. Amino acid residues 252–864 occur in the C-terminal transcriptional activity region and include the Pro-rich region, basic helix-loop-helix (bHLH)/ZIP, and a ZIP-like domain.
Phosphorylation/dephosphorylation is a major mechanism by which ChREBP activity is regulated, and it is mediated through multiple phosphorylation sites, including consensus sites for cAMP-dependent protein kinase (PKA) and AMP-dependent protein kinase (AMPK) (8, 9). Phosphorylation by either PKA or AMPK inhibits glycolysis and lipogenesis through inactivation of ChREBP. In addition to phosphorylation, acetylation and GlcNAc modification of ChREBP have also been reported (10–12). When glucose availability is low, the phosphorylated inactive pool of ChREBP (ChREBP-Ser(P)-196) binds 14-3-3 proteins and CRM-1, transits out of the nucleus, and localizes in the cytosol (5, 8). After ingestion of food, increased glucose levels lead to the conversion of inactive ChREBP(P) in the cytosol into active ChREBP, which results in ChREBP binding to import proteins and translocation into the nucleus to facilitate transcription of target genes encoding proteins important for glycolysis and lipogenesis (8, 13).
To begin to elucidate how ChREBP senses glucose, we have used a biochemical approach to determine the steps involved in ChREBP import and export (Fig. 2). We determined that the first step in nuclear localization of ChREBP is activation of the ChREBP(P)·14-3-3 complex by dephosphorylation of ChREBP in the cytosol, presumably by xylulose-5-P (Xu-5-P)-activated protein phosphatase (PP2A) (14). The activated dephospho-ChREBP next forms a complex with importin proteins, and the complex subsequently transits to the nucleus. Under conditions of low glucose, phosphorylation by PKA inactivates ChREBP, resulting in association of ChREBP(P) with 14-3-3 proteins and CRM-1, followed by translocation to the cytosol. Thus, the following functional domains can be assigned to the ChREBP/N-terminal region of ChREBP (residues 1–250), which binds 14-3-3, CRM-1, and/or importin α and is solely responsible for the import and export of ChREBP, and the C-terminal region (residues 251–846), which is responsible for transcriptional activity (5).
FIGURE 2.

Proposed nuclear-cytosol trafficking pathways of ChREBP. In response to high glucose, phosphorylated, inactive ChREBP is dephosphorylated by Xu-5-P-activated PP2A and translocates into the nucleus through interactions with importin α and β. ChREBP is further dephosphorylated in the nucleus and induces transcription of LPK and lipogenic genes. A decrease in glucose concentration results in inactivation of ChREBP by phosphorylation by PKA and complex formation with 14-3-3 and CRM1 followed by translocation to the cytosol.
Despite extensive investigation, the biochemical mechanisms responsible for glucose sensing and for induction of ChREBP activity remain unclear. No approaches have yet identified a specific metabolite or allosteric ligand that is responsive to circulating blood glucose levels, thus serving as a glucose-signaling second messenger. It has been suggested that glucose-6-P instead, or in addition to Xu-5-P, functions as a glucose-signaling compound (15, 16). Xu-5-P activates only the dephosphorylation reaction of ChREBP(P) catalyzed by PP2ABδC, the first step in ChREBP activation. We hypothesize that other metabolites might control nuclear import of ChREBP, the second step in the activation process.
EXPERIMENTAL PROCEDURES
Chemicals and Vectors
All chemicals were purchased from Sigma unless otherwise indicated. Bacterial expression vectors for GST-tagged importin α were gifts from Dr. Y. Yoneda (Osaka University, Osaka, Japan), and those for the human 14-3-3 proteins were gifts from Dr. Steven L. McKnight (University of Texas Southwestern Medical Center, Dallas, TX).
Proteins and Peptides
His-tagged human 14-3-3β was transformed into Escherichia coli strain BL21. Expression of the 14-3-3β proteins was facilitated by the addition of isopropyl 1-thio-β-d-galactopyranoside (0.1 mm) followed by incubation at 20 °C with shaking at 200 rpm for 16 h. GST fusion proteins were purified using glutathione-Sepharose (GE Healthcare) according to the manufacturer's instructions. His-tagged proteins were purified by affinity chromatography using nickel-nitrilotriacetic acid. The peptides containing α-helix (residues 117–149) of ChREBP phosphorylated at Ser-140, designated as “α2-Ser-140p,” were synthesized by the Peptide Synthesis Group at the University of Texas Southwestern. The peptide containing the α3-helix regions and the phosphorylated Ser-196 site of ChREBP (residues 170–198), designated as “α3-Ser-196p” was also synthesized by the Peptide Synthesis group at the University of Texas Southwestern.
Animals
The Institutional Animal Care and Use Research Advisory Committee of the Veterans Affairs Medical Center approved all animal studies. Male rats 6–10 weeks of age were housed in temperature-controlled facilities with 12-h light/dark cycles and maintained on standard rodent chow (“NIH”) (Harlan-Teklad Mouse/Rat Diet 7002; Harlan-Teklad Premier Laboratory Diets). The high sucrose (“HS”) and high fat (“HF”) diets were described as before (17).
Interaction of ChREBP with 14-3-3 Proteins
The following two assay methods were used.
Assay A
Immunoblotting method (“pulldown”) was carried out as described before (5). Briefly, FLAG-tagged wild type (WT)-ChREBP was expressed in HEK293 cells and then purified from cell lysate by binding to anti-FLAG antibody beads in the reaction mixture containing 20 mm HEPES-KOH, pH 7.3, 0.5 mm EGTA, 60 mm KAc, 2 mm MgAc, 5 mm NaAc, 1% Nonidet P-40 and incubated for 1 h at 4 °C with gentle rocking. Pure exogenous 14-3-3β and/or GST-tagged importin α were added to the beads bound with ChREBP and the 14-3-3 and/or importin α proteins and incubated with shaking for 1.5 h at 4 °C. Bound FLAG-ChREBP and the 14-3-3 and importin α proteins were then eluted with SDS-PAGE sample buffer and subjected to gel electrophoresis followed by immunoblotting.
Assay B, Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were carried out using a VP-ITC microcalorimeter (MicroCal) as described before (5). Briefly, His-tagged 14-3-3 proteins were dialyzed overnight into 50 mm Tris-HCl, pH 7.4, containing 0.1 mm EDTA. From these samples, 25 μm solutions of monomeric subunits were placed in the cell chamber of the calorimeter (cell volume 1.8 ml). For most ITC measurements, human 14-3-3β was used. Binding isotherms were measured as α2-Ser-140p or α3-Ser-196p and added to the chamber from a syringe with an initial peptide concentration of 250 μm. All titration experiments were performed at 20 °C in 50 mm Tris-HCl, pH 7.4, containing 0.1 mm EDTA. The ChREBP peptide was injected into the calorimetric cell in 10-μl increments. The binding isotherm derived from heat changes was used to calculate the standard free energy of binding (ΔG0) according to the equation ΔG0 = −RTlnKa, where R is the gas constant; T is the absolute temperature, and Ka is the association constant. From the binding isotherm, the number of binding sites (n) was obtained. Curve fitting and the derivation of thermodynamic parameters were carried out with the ORIGIN version 7.0 software package (MicroCal). The concentrations of 14-3-3 proteins and the ChREBP peptides, α2-Ser-140p (residues 117–149) and α3-Ser-196p (residues 170–198), were determined using extinction coefficients at 280 nm of 25.7 and 13.98 mm−1 cm−1, respectively.
Preparation of Protein-free Extracts
For metabolite isolation, protein-free extracts of livers were prepared according to the extraction methods described by Newton et al. (19) with slight modifications. Freeze-clamped rat liver (1 g) was homogenized in 3 ml of ice-cold 0.6 n HClO4 using a Polytron homogenizer at 0–4 °C, and the homogenate was centrifuged at 15,000 × g for 10 min. The supernatant solution was neutralized with cold 1 n KOH. The precipitate was removed by centrifugation at 14,000 × g for 10 min at 4 °C. The supernatant solution was either used directly for enzymatic assays for ketone bodies or for isolation of metabolites by HPLC.
HPLC Analysis of Protein-free Extracts
HPLC analysis of protein-free extracts was carried out as follows. Metabolite extracts (0.2 ml) were applied on a CarboPac PA1 column (4 mm × 250 mm) (Dionex) and eluted with a linear gradient of ammonium acetate (0–1 m, pH 5.0) at a flow rate of 1 ml/min. Eluted fractions were collected, concentrated by lyophilization, and assayed for an activity to increase the ChREBP·14-3-3β interaction using the “pulldown assay.”
HPLC analysis was also carried out using a Synergi Fusion column (4.6 mm × 250 mm) (Phenomenex) and eluted with a linear gradient from 0 to 50% saturation of methanol for the initial 5 min, followed by a linear gradient of 50–75% from 5 to 35 min. Eluates were concentrated by lyophilization and assayed.
RESULTS AND DISCUSSION
Metabolites in Starved Rat Liver Activated ChREBP and 14-3-3 Interaction
The early steps in the nuclear/cytosol translocation of ChREBP are direct protein/protein interactions between active dephospho-ChREBP with importin α in the nuclear import step, and inactive phosphorylated ChREBP(P) with the 14-3-3 proteins and CRM-1 in the export step (5). Because ChREBP plays a critical role in coordinating carbohydrate metabolism and lipogenesis, independent of insulin, we hypothesize the existence of specific metabolites that might activate nuclear import of ChREBP in response to increased glucose uptake after eating. These proposed metabolites along with others might trigger nuclear export of ChREBP upon starvation. To this end, we searched first for a metabolite(s) in starved rat liver that might promote ChREBP binding to 14-3-3β. Using a ChREBP·14-3-3β pulldown assay, we identified a crude protein-free extract in the livers from starved rats containing a metabolite that promoted interaction between these proteins. This metabolite was highest in livers from starved (ST) animals, lowest in livers from HS animals (Fig. 3A), and intermediate in livers from animals fed lab chow (NIH) and HF diets. To isolate and identify the metabolite, ST liver extracts were subjected to HPLC separation, and the resolved eluates were assayed in a pulldown reaction containing FLAG-ChREBP and 14-3-3β. The results (Fig. 3B) indicated that only one fraction (fraction 10) contained a metabolite(s) that activated the ChREBP(P)/14-3-3 interaction. The active metabolites in the fraction were subsequently identified as the ketone bodies βHB and AcAc by enzymatic assay using β-hydroxybutyrate dehydrogenase and GC-mass spectrometry analysis. Both βHB and AcAc standards elute within the same HPLC fraction, confirming the identities of the activator metabolites as ketone bodies.
FIGURE 3.

Effects of metabolites and ketone bodies on ChREBP·14-3-3 interactions. FLAG-ChREBP (full-length) was expressed in HEK293 cells, which were incubated for 48 h. FLAG-ChREBP in cell extracts was bound to anti-FLAG beads, which were mixed with metabolites. A, reaction mixtures containing FLAG-ChREBP beads and pure 14-3-3β were mixed with crude protein-free metabolite extracts of livers of rats fed different diets. After 1.5 h, the beads were washed, and ChREBP-bound 14-3-3β was eluted and subjected to PAGE. Bound 14-3-3β were measured by immunoblotting (top panel). The values presented are the means ± S.D. of at least three sets of experiments (bottom panel). **, p < 0.01;***, p < 0.001. B, to identify the metabolite activator of the ChREBP·14-3-3β interaction, starved liver extract was subjected to HPLC analysis using a CarboPac PA1 column (4 × 250 mm). The eluted fractions were assayed using the pulldown assay for ChREBP·14-3-3β interaction (top panel). The elution of UV-absorbing compounds (A = 254 nm) and elution positions of standard βHB and AcAc as determined with LC/MS are shown (bottom panel). C, total ketone bodies (βHB + AcAc) in the crude metabolite extracts of the freeze-clamped livers were determined with LC/MS. These extracts of three livers each from the rats fed different diets were measured. * = <0.05; *** = <0.001.
To confirm that βHB and AcAc were responsible for promoting ChREBP(P)/14-3-3 interaction in vivo, we measured the concentrations of total ketone bodies in the crude liver extracts of rats fed different diets (Fig. 3C). The concentration of βHB and AcAc was highest in the starved livers (2.2 μmol/g liver) and 5.2-fold higher than that in livers from the NIH-diet fed animals (0.43 μmol/g), which compares favorably to the extent of activation (4.5-fold) of ChREBP(P)/14-3-3 interaction (Fig. 3A). Similarly, the total ketone concentration in the livers from the high fat-fed animals (0.8 μmol/g) was 1.9-fold higher than that in the livers from animals fed the NIH diet, and the level of ChREBP(P)/14-3-3 activation by these extracts was ∼1.9-fold. The ketone concentration in livers from animals fed the HS diet (0.41 μmol/g) was similar to that in livers from the NIH diet-fed animals, but the activation was lower (0.8-fold). These observations indicate that activation of the ChREBP(P)·14-3-3 complex formation correlates very closely with the total ketone body concentrations in liver, further supporting a regulatory role for βHB and AcAc in promoting cytosolic localization of ChREBP during starvation.
Ketone bodies are synthesized from β-hydroxy-β-methylglutaryl-CoA in cytoplasm generated by fatty acid oxidation in liver tissue exposed to starvation or a high fat diet (17). Ketone bodies cannot be metabolized in liver because the tissue lacks 3-oxoacid CoA transferase (20), and they are therefore exported out of the liver to other tissues where they are used as a source of energy. βHB and AcAc increase between 1 and 2 mm in blood and liver of rats when fasting as the liver switches to fatty acid oxidation (21), increasing to as much as 6–8 mm in liver during prolonged starvation (22). In contrast, most major metabolites of glycolysis, the pentose-phosphate shunt, and glycogenolysis are decreased at least 2–4-fold under these conditions. Thus, it is plausible that βHB and AcAc activate and stabilize the ChREBP(P)·14-3-3 complex to ensure that ChREBP remains in the cytoplasm during starvation where it is incapable of activating lipogenesis.
How Do Ketone Bodies Activate ChREBP·14-3-3 Interaction?
To determine how ketones promote ChREBP(P)/14-3-3 heterodimer formation, we measured the effect of βHB and AcAc on ChREBP affinity for 14-3-3 using a pulldown assay. Both ketone bodies promote ChREBP·14-3-3 heterodimer interactions with similar affinity (Ka = 0.5 mm) (Fig. 4A). The results also demonstrate that the affinity (Ka) of ChREBP for 14-3-3 increased ∼2-fold in the presence of 1 mm βHB compared with its absence (Fig. 4B), which is within the physiological concentration ranges of ketone bodies in livers of starved animals (>2 mm) and fed livers (<0.4 mm) (23).
FIGURE 4.
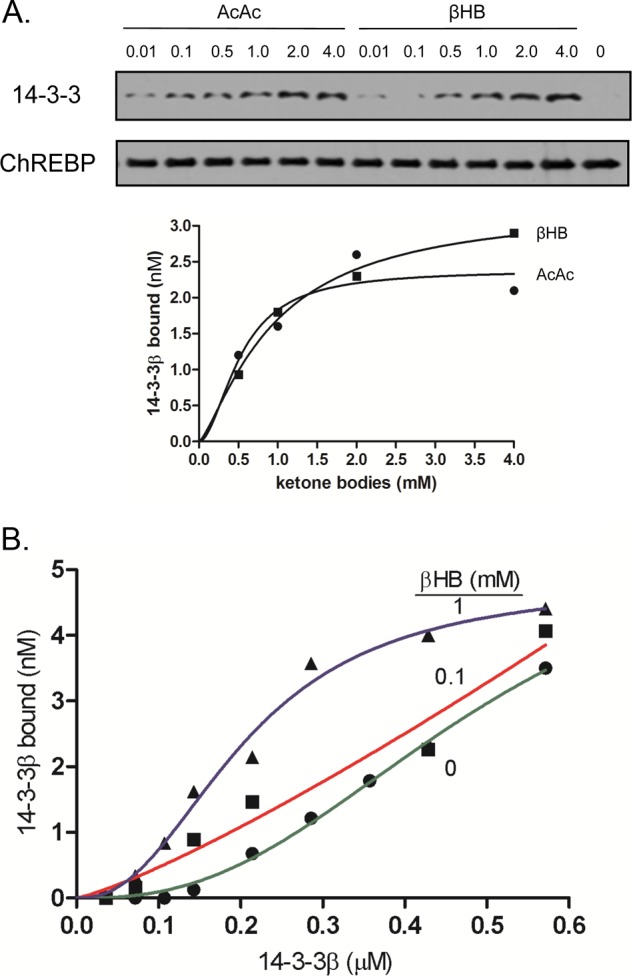
Activation of ChREBP and 14-3-3 interactions with βHB and AcAc. A, activation of FLAG-ChREBP·14-3-3β by varying concentrations of standard βHB and AcAc was determined using the pulldown assay as described in Fig. 1A. Representative images showing the 14-3-3 bound to ChREBP in varying βHB and AcAc are shown. B, βHB increased affinity of 14-3-3β binding to ChREBP. Varying concentrations of 14-3-3β to FLAG-ChREBP were added to the reaction mixture in the presence of 0, 0.1, or 1 mm βHB, and 14-3-3β bound to FLAG-ChREBP beads was subjected to the pulldown assay.
To further investigate the mechanism by which ketone bodies promote ChREBP(P)/14-3-3 interactions, ITC was used to measure the effect of βHB on the interaction between 14-3-3β protein and two distinct ChREBP peptides corresponding to two 14-3-3-binding sites (amino acids 117–149, ”α2-Ser-140p,” and amino acids 170–198, “α3-Ser-196p”) (5, 13). β-HB failed to bind to either the ChREBP peptides or the 14-3-3 proteins individually, but they did bind when both 14-3-3β and either of the ChREBP peptides were present (Table 1 and Fig. 5A). The interaction between the peptides and the 14-3-3β protein increased nearly 2-fold (KD decreased from 1.5 × 10−6 to 0.6 × 10−6 m) in the presence of 10 μm and 1 mm β-HB, respectively, and the binding of 14-3-3β protein to α3-Ser-196p peptide, which is a weaker binding site, was 30% lower than the same concentrations of the ketone body (KD decreased from 0.83 × 10−6 to 0.59 × 10−6 m). AcAc had little effect on the peptides binding under these conditions (Table 2 and Fig. 5B). These results suggest that ketone bodies directly promote ChREBP·14-3-3β protein interactions, possibly by inducing conformational changes in ChREBP and thereby stabilizing the ChREBP(P)·14-3-3 protein complex. Thus, we conclude based on the ITC data that ketone bodies represent a physiological signal produced by the liver in response to low blood glucose levels during starvation that inhibit fat synthesis by promoting ChREBP(P)·14-3-3 complex formation in the cytosol.
TABLE 1.
Thermodynamic parameters for the interactions of human 14-3-3 proteins with human ChREBP peptides α2-Ser-140p and α3-Ser-196p in the presence of βHB as determined by isothermal titration calorimetry
Data represent at least two ITC measurements.
| Peptides and β-hydroxybutyrate | Kd |
|---|---|
| m | |
| α2-Ser-140p | 1.1 × 10−6 |
| α2-Ser-140p + 10 μm βΗΒ | 1.5 × 10−6 |
| α2-Ser-140p + 100 μm βΗΒ | 0.71 × 10−6 |
| α2-Ser-140p + 1000 μm βΗΒ | 0.60 × 10−6 |
| α3-Ser-196p | 0.78 × 10−6 |
| α3-Ser-196p + 10 μm βHB | 0.83 × 10−6 |
| α3-Ser-196p + 100 μm βΗΒ | 0.63 × 10−6 |
| α3-Ser-196p + 1000 μm βΗΒ | 0.59 × 10−6 |
FIGURE 5.

ITC titrations of 14-3-3β with ChREBP peptide in presence of βHB. ITC experiments were carried out using a VP-ITC microcalorimeter (MicroCal) as described above. Binding isotherms were measured as phosphorylated peptides were added to the chamber from a syringe with an initial peptide concentration of 250 μm. All titration experiments were performed at 20 °C in 50 mm Tris-HCl, pH 7.4, containing 0.1 mm EDTA. Curve fitting and the derivation of thermodynamic parameters were carried out with the ORIGIN version 7.0 software package (MicroCal). The concentrations of 14-3-3β proteins and the ChREBP peptide, α2-Ser-140p (residues 117–149) were determined as mentioned previously. A and B contained 10 and 1000 μm βHB, respectively.
TABLE 2.
Thermodynamic parameters for the interactions of human 14-3-3 proteins with human ChREBP peptides α2-Ser-140p and α3-Ser-196p in the presence of acetoacetate as determined by isothermal titration calorimetry
Data represent at least two ITC measurements.
| Peptides and acetoacetate | Kd |
|---|---|
| m | |
| α2-Ser-140p | 1.1 × 10−6 |
| α2-Ser-140p + 10 μm acetoacetate | 1.2 × 10−6 |
| α2-Ser-140p + 100 μm acetoacetate | 1.3 × 10−6 |
| α2-Ser-140p + 1000 μm acetoacetate | 1.1 × 10−6 |
| α3-Ser-196p | 0.51 × 10−6 |
| α3-Ser-196p + 10 μm acetoacetate | 0.62 × 10−6 |
| α3-Ser-196p + 100 μm acetoacetate | 0.64 × 10−6 |
| α3-Ser-196p + 1000 μm acetoacetate | 0.65 × 10−6 |
The discrepancy in the AcAc binding to ChREBP·14-3-3 could be because the ITC measurements were performed using the ChREBP peptides, whereas the pulldown assays were carried out with full-length FLAG-ChREBP protein, which would be closer to physiological conditions.
Effects of Ketones on Nuclear Localization and Transcriptional Activities of ChREBP in Hepatocytes
To determine whether our biochemical findings with respect to ketone body promotion of ChREBP(P)/14-3-3 interactions have physiological relevance, we investigated whether ketone bodies affect nuclear localization and transcriptional activity of ChREBP. Ketone bodies are transported into hepatocytes by a monocarboxylate transporter (24). Primary hepatocytes expressing GFP-ChREBP were incubated in high glucose in the presence of βHB or AcAc, and GFP-ChREBP nuclear export was determined. Cells were incubated in high glucose (27.5 mm) along with βHB and AcAc to minimize ketone bodies that were otherwise produced by hepatocytes if plated under low glucose conditions. Under high glucose conditions, nuclear localization of GFP-ChREBP was rapidly inhibited by βHB and AcAc, reaching a maximum level within 2–3 h (Fig. 6A). Cytoplasmic localization of GFP-ChREBP in the presence of ketones was comparable with that present under low glucose (5.5 mm) conditions. In the high glucose control cells incubated in the absence of ketone bodies, GFP-ChREBP remained in the nucleus.
FIGURE 6.

βHB and AcAc inhibit nuclear localization of ChREBP. A, primary cultures of rat hepatocytes were transfected with GFP-ChREBP proteins in DMEM containing low (5.5 mm) glucose for 4 h and then the medium was changed to DMEM containing high (27.5 mm) glucose for 12 h and incubated in low (5.5 mm) or high (27.5 mm) glucose in the presence or absence of βHB (2 mm) or AcAc (2 mm). At the indicated time, the cells were fixed with 4% paraformaldehyde, and subcellular localization of GFP-ChREBP in the nucleus and cytoplasm was determined with using a confocal microscopy as described previously (5). The values presented are the mean ± S.D. of three sets of about 100 fluorescent cells. *, p < 0.05; **, p < 0.01 cf. high Glc. B, transcriptional activity in the hepatocytes incubated in βHB or AcAc were measured after 6 h using the luciferase reporter system as described (22) *, p < 0.05. C, fatty acids inhibit nuclear localization of ChREBP. Rat hepatocytes transfected with GFP-ChREBP were cultured in high glucose for 12 h, and the cells were incubated in the medium-containing stearate, palmitate, oleate, and linoleate, and cellular localization of GFP-ChREBP in nucleus and cytoplasm was determined at different time periods as above. The control cells were incubated in either low or high glucose. *, p < 0.05; **, p < 0.01; ***, p < 0.0001 cf. high Glc. D, βHB and AcAc produced by the cells incubated in the stearate and oleate were measured by removing the cells rapidly by centrifugation, and the packed cells were quickly frozen in liquid nitrogen. Acid extracts of the cells were prepared, and ketone bodies in the extracts were determined with GC-MS.
We next investigated whether ketone bodies would affect ChREBP signaling. The transcriptional activity of ChREBP, which was assayed using a reporter containing the ChREBP-responsive liver pyruvate kinase promoter, decreased ∼20% when assayed 6 h after addition of ketone bodies (Fig. 6B). However, the transcriptional activity increased slowly after 6 h and recovered completely from the ketone inhibition after 12 h, suggesting that βHB and AcAc were either exported out of the cells or degraded or both. The concentrations of ketones in hepatocytes plated using high glucose conditions could not be measured accurately because the cells were not only incubated in high βHB concentrations but the hepatocytes also released ketone bodies rapidly upon washing. However, ketone levels in control hepatocytes incubated under low glucose conditions increased, whereas the concentrations of ketone bodies remained constant under high glucose conditions for at least 6 h.
Effect of Ketones Produced by Fatty Acid Metabolism on the Nuclear Localization of ChREBP in Hepatocytes
Because ketone bodies are also produced by the metabolism of fatty acid in liver, we investigated the effect of the addition of saturated fatty acids, stearate and palmitate, and unsaturated fatty acids, oleate and linoleate, on the nuclear/cytosol localization of GFP-ChREBP in hepatocytes. The results demonstrated that fatty acid metabolism also inhibited ChREBP nuclear localization with similar kinetics as those during starvation, reaching maximum inhibition within 3 h following addition of the saturated fatty acids (Fig. 6C), but the unsaturated fatty acids continued to inhibit the nuclear localization of ChREBP beyond 3 h. The rates and the amounts of βHB and AcAc produced by palmitate and oleate oxidation in hepatocytes were the same and correlated well with inhibition of nuclear localization of ChREBP (Fig. 6D). The differences in the extent of inhibition of the ChREBP activity observed between the saturated and unsaturated fatty acids suggest that the latter fatty acids had additional inhibitory effects on ChREBP, possibly involving denaturation of proteins and membranes.
Because the rates of ChREBP nuclear export and ketone production by fatty acid oxidation correlate closely, we conclude that ketone bodies are responsible for the inhibition of ChREBP nuclear import. These in vivo and in vitro results are in total support of a role for βHB and AcAc in the inhibition of ChREBP nuclear localization in hepatocytes and suggest that this is a critical mechanism by which ChREBP is maintained as an inactive ChREBP(P)·14-3-3 complex in cytosol under low glucose conditions induced by starvation or induced by a high fat diet.
Effects of Metabolites and 14-3-3 on ChREBP and Importin α Interaction
We have demonstrated previously that 14-3-3 inhibits importin α binding to ChREBP directly at a “secondary or regulatory” site that overlaps a long bipartite nuclear localization signal (NLS) (13). It is possible that ketone bodies provide an additional stabilizing effect for ChREBP(P)/14-3-3 interactions while at the same time decreasing access of importin α to ChREBP, the first step in nuclear localization. To investigate this possibility, we used a pulldown assay to compare the effects of crude liver metabolite extracts from starved rats or rats fed different diets on importin α in the presence and the absence of 14-3-3β binding to ChREBP. The results shown in Fig. 7A (left panel) demonstrate that 14-3-3β binding to the secondary binding site of ChREBP (NLS-containing site) was completely dependent on the metabolite extract of the ST liver, partially activated by liver extracts prepared from the HF chow-fed rats, but completely inhibited by liver extracts prepared from the HS chow or NIH standard chow-fed rats. However, in the absence of 14-3-3β, the importin α binding was not inhibited significantly by the ST extract or the HF extract (Fig. 7A, right panel). These results strongly suggest that ketone bodies promote 14-3-3 binding to the regulatory site of ChREBP and promote ChREBP(P)/14-3-3 heterodimer formation. More importantly, there was an opposite effect of dietary conditions on the ChREBP/importin α interaction compared with ChREBP·14-3-3 complex formation. Importin α binding was activated by the HS and the NIH extracts and was inhibited almost completely by the ST extract and partially by the HF extract when both 14-3-3β and importin α were present. Thus, ChREBP·importin α and ChREBP·14-3-3 complex formation is inversely regulated by different diets, and these differences can be accounted for by differences in ketone body production in the liver.
FIGURE 7.

Ketone bodies activate ChREBP·14-3-3β interaction and inhibit ChREBP/importin α interaction. A, FLAG-ChREBP (α2 mutant, “A129P/Y131P”) (13) bound to anti-FLAG beads were incubated in the reaction mixture containing a mixture of pure 14-3-3β (2 μm) and pure importin α (0.12 μm) in the presence of crude liver extracts of rats fed different diet, as in Fig. 1A. After incubation for 1.5 h, the beads were washed, and ChREBP-bound 14-3-3β and importin α were eluted and subjected to PAGE. Bound 14-3-3β and importin α were measured by immunoblotting. B, starved rat liver extract was subjected to HPLC analysis using a Synergi Fusion column (4 μm, 4.6 × 250 mm) and eluted in a tributylamine buffer, pH 5.0, with two linear gradients of methanol, as described under “Experimental Procedures.” The eluted fractions 11 and 12 contained βHB and AcAc, respectively, as shown by the enzymatic assay, and the standard βHB and AcAc were eluted at the same respective times. These fractions containing the ketone bodies activated the ChREBP/14-3-3β interaction but inhibited the ChREBP/importin α interaction.
To confirm that ketone bodies in ST liver extracts are responsible for the reciprocal effects on ChREBP·importin α and ChREBP·14-3-3 complex formation, ST extract was subjected to additional HPLC analysis on a Synergi column (Fig. 7B), and the eluates were assayed for effects on both the ChREBP(P)/14-3-3 and ChREBP/importin α interactions. The results demonstrated that only two fractions, which contain βHB and AcAc, conferred the reciprocal effects (Fig. 7B). The ketone bodies in these fractions were identified as βHB in fraction 11 and AcAc in fraction 12 by the enzymatic assay using βHB dehydrogenase. In addition, standard βHB and AcAc were eluted in the same fractions under the identical conditions. This is further evidence that ketone bodies inhibit nuclear import by blocking importin α binding to the NLS of ChREBP as well as by promoting ChREBP(P)/14-3-3 interactions, thus resulting in cytosolic localization of ChREBP.
Recently, we solved the crystal structure of 14-3-3β bound to its primary binding site on ChREBP, the α2-helix (amino acid residues 117–137), which occurs in a novel phosphate-independent manner. 14-3-3 also binds to the secondary binding site of ChREBP consisting of α3-helix (170–190), especially when a nearby residue Ser-196 is phosphorylated by PKA (8). This secondary binding site of 14-3-3 overlaps with NLS resulting in a competitive binding between 14-3-3 and importin α, and it serves as a regulatory site for nuclear/cytosol localization of ChREBP in response to glucose (25). In low glucose, 14-3-3 binds to both the primary and the secondary sites containing the nearby phosphorylated Ser(P)-196 and blocks importin α from binding to the NLS, thus promoting export, whereas under high glucose Ser-196 is dephosphorylated, which facilitates importin α binding and activates nuclear import of ChREBP.
We suggest that the molecular mechanism for promotion of ChREBP(P)·14-3-3 complex formation is an allosteric activation induced by βHB binding at the interface between these two proteins, which further increases the affinity of ChREBP(P) and 14-3-3. ChREBP lacks conformation in isolation when in solution and is stabilized only by binding to the more rigid 14-3-3 proteins, which can be considered an example of “allosteric stabilization” (18). The crystal structure of the ChREBP(P)·14-3-3β complex revealed that the ChREBP α2-helix adopts an α-helical conformation and binds 14-3-3 in a unique phosphorylation-independent manner (25). The interaction involves both electrostatic and van der Waals interaction, and interestingly, the binding is also strengthened by anions such as free sulfate or phosphate present in the molecular structure of the crystals. Thus, small ligands such as β-HB may allosterically enhance the interaction between ChREBP and 14-3-3, which is supported by ITC measurements (Table 1). It is possible that monocarboxylate ions such as βHB and AcAc can bind to the same anion- binding site on the interface to enhance the binding and stabilization of the ChREBP(P)/14-3-3 heterodimer. Structural studies will be needed to determine precisely how βHB is bound to the ChREBP(P)·14-3-3 complex.
In summary, we demonstrate that metabolites generated in animals fed specific diets or subjected to starvation alter gene expression patterns by directly affecting an unexpectedly simple signal transduction pathway. Ketone bodies produced during fasting or in animals fed a high fat diet inhibit lipogenesis by sequestering inactive ChREBP(P)/14-3-3 heterodimer in the cytosol and, at the same time, by inhibiting nuclear import by preventing importin α binding to ChREBP. Thus, ketone bodies are proposed to represent a “low glucose signal” that reciprocally regulates ChREBP signaling mediated by phosphorylation/dephosphorylation events induced by a high glucose signal.
Acknowledgments
We thank Drs. Steven L. McKnight, Richard Bruick, and Joseph Garcia for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant DK06394804. This work was also supported by Veterans Affairs Merit Review and a Welch Grant.
This article was selected as a Paper of the Week.
- ChREBP
- carbohydrate response element-binding protein
- AcAc
- acetoacetate
- βHB
- β-hydroxybutyrate
- HS
- high sucrose
- HF
- high fat
- NLS
- nuclear localization signal
- Xu-5-P
- xylulose-5-P
- ITC
- isothermal titration calorimetry
- ST
- starved.
REFERENCES
- 1. Uyeda K., Repa J. J. (2006) Carbohydrate-response element-binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 4, 107–110 [DOI] [PubMed] [Google Scholar]
- 2. Iizuka K., Bruick R. K., Liang G., Horton J. D., Uyeda K. (2004) Deficiency of carbohydrate-response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. U.S.A. 101, 7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamashita H., Takenoshita M., Sakurai M., Bruick R. K., Henzel W. J., Shillinglaw W., Arnot D., Uyeda K. (2001) A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. U.S.A. 98, 9116–9121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merla G., Howald C., Antonarakis S. E., Reymond A. (2004) The subcellular localization of the ChoRE-binding protein, encoded by the Williams-Beuren syndrome critical region gene 14, is regulated by 14-3-3. Hum. Mol. Genet. 13, 1505–1514 [DOI] [PubMed] [Google Scholar]
- 5. Sakiyama H., Wynn R. M., Lee W. R., Fukasawa M., Mizuguchi H., Gardner K. H., Repa J. J., Uyeda K. (2008) Regulation of nuclear import/export of carbohydrate response element-binding protein (ChREBP): interaction of an α-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 283, 24899–24908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cairo S., Merla G., Urbinati F., Ballabio A., Reymond A. (2001) WBSCR14, a gene mapping to the Williams-Beuren syndrome deleted region, is a new member of the Mlx transcription factor network. Hum. Mol. Genet. 10, 617–627 [DOI] [PubMed] [Google Scholar]
- 7. Ma L., Tsatsos N. G., Towle H. C. (2005) Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J. Biol. Chem. 280, 12019–12027 [DOI] [PubMed] [Google Scholar]
- 8. Kawaguchi T., Takenoshita M., Kabashima T., Uyeda K. (2001) Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. U.S.A. 98, 13710–13715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawaguchi T., Osatomi K., Yamashita H., Kabashima T., Uyeda K. (2002) Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 277, 3829–3835 [DOI] [PubMed] [Google Scholar]
- 10. Bricambert J., Miranda J., Benhamed F., Girard J., Postic C., Dentin R. (2010) Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Invest. 120, 4316–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakiyama H., Fujiwara N., Noguchi T., Eguchi H., Yoshihara D., Uyeda K., Suzuki K. (2010) The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem. Biophys. Res. Commun. 402, 784–789 [DOI] [PubMed] [Google Scholar]
- 12. Guinez C., Filhoulaud G., Rayah-Benhamed F., Marmier S., Dubuquoy C., Dentin R., Moldes M., Burnol A. F., Yang X., Lefebvre T., Girard J., Postic C. (2011) O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60, 1399–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ge Q., Nakagawa T., Wynn R. M., Chook Y. M., Miller B. C., Uyeda K. (2011) Importin-α protein binding to a nuclear localization signal of carbohydrate response element-binding protein (ChREBP). J. Biol. Chem. 286, 28119–28127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kabashima T., Kawaguchi T., Wadzinski B. E., Uyeda K. (2003) Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. U.S.A. 100, 5107–5112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dentin R., Tomas-Cobos L., Foufelle F., Leopold J., Girard J., Postic C., Ferré P. (2012) Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 56, 199–209 [DOI] [PubMed] [Google Scholar]
- 16. Li M. V., Chen W., Harmancey R. N., Nuotio-Antar A. M., Imamura M., Saha P., Taegtmeyer H., Chan L. (2010) Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 395, 395–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iizuka K., Miller B., Uyeda K. (2006) Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 291, E358–E364 [DOI] [PubMed] [Google Scholar]
- 18. Gardino A. K., Smerdon S. J., Yaffe M. B. (2006) Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the x-ray crystal structures of all human 14-3-3 isoforms. Semin. Cancer Biol. 16, 173–182 [DOI] [PubMed] [Google Scholar]
- 19. Newton R. P., Salih S. G., Salvage B. J., Kingston E. E. (1984) Extraction, purification and identification of cytidine 3′,5′-cyclic monophosphate from rat tissues. Biochem. J. 221, 665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Orii K. E., Fukao T., Song X. Q., Mitchell G. A., Kondo N. (2008) Liver-specific silencing of the human gene encoding succinyl-CoA:3-ketoacid CoA transferase. Tohoku J. Exp. Med. 215, 227–236 [DOI] [PubMed] [Google Scholar]
- 21. Williamson D. H., Lund P., Krebs H. A. (1967) The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 103, 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lawson J. W., Guynn R. W., Cornell N. W., Veech R. L. (1976) in Gluconeogenesis: Its Regulation in Mammalian Species (Hansen R. W., Mehlman M. A., eds) John Wiley and Sons, Inc., New York [Google Scholar]
- 23. Lawson J. W., Uyeda K. (1987) Effects of insulin and work on fructose 2,6-bisphosphate content and phosphofructokinase activity in perfused rat hearts. J. Biol. Chem. 262, 3165–3173 [PubMed] [Google Scholar]
- 24. Koehler-Stec E. M., Simpson I. A., Vannucci S. J., Landschulz K. T., Landschulz W. H. (1998) Monocarboxylate transporter expression in mouse brain. Am. J. Physiol. 275, E516–E524 [DOI] [PubMed] [Google Scholar]
- 25. Ge Q., Huang N., Wynn R. M., Li Y., Du X., Miller B., Zhang H., Uyeda K. (2012) Structural characterization of a unique interface between carbohydrate response element-binding protein (ChREBP) and 14-3-3β protein. J. Biol. Chem. 287, 41914–41921 [DOI] [PMC free article] [PubMed] [Google Scholar]