

Background: Copper amine oxidases activate O2 either at the copper center or aminoquinol cofactor.
Results: Catalytic intermediates from the oxidative half-reaction are structurally and spectroscopically characterized.
Conclusion: The mechanism of O2 activation may depend on accessibility dictated by two conformers of the quinone cofactor.
Significance: Structural changes that inform on catalytic mechanism have been revealed in the ubiquitous copper amine oxidases.
Keywords: Copper, Enzyme Mechanisms, Oxidation-Reduction, Oxygen Binding, Quinones, Spectroscopy, X-ray Crystallography
Abstract
The mechanism of molecular oxygen activation is the subject of controversy in the copper amine oxidase family. At their active sites, copper amine oxidases contain both a mononuclear copper ion and a protein-derived quinone cofactor. Proposals have been made for the activation of molecular oxygen via both a Cu(II)-aminoquinol catalytic intermediate and a Cu(I)-semiquinone intermediate. Using protein crystallographic freeze-trapping methods under low oxygen conditions combined with single-crystal microspectrophotometry, we have determined structures corresponding to the iminoquinone and semiquinone forms of the enzyme. Methylamine reduction at acidic or neutral pH has revealed protonated and deprotonated forms of the iminoquinone that are accompanied by a bound oxygen species that is likely hydrogen peroxide. However, methylamine reduction at pH 8.5 has revealed a copper-ligated cofactor proposed to be the semiquinone form. A copper-ligated orientation, be it the sole identity of the semiquinone or not, blocks the oxygen-binding site, suggesting that accessibility of Cu(I) may be the basis of partitioning O2 activation between the aminoquinol and Cu(I).
Introduction
The activation and reduction of molecular oxygen to hydrogen peroxide occur during catalysis in copper amine oxidases (CAOs).2 In addition to a mononuclear copper ion, the active site contains a quinone cofactor, 2,4,5-trihydroxyphenylalanine quinone, derived from a conserved tyrosine in the CAO polypeptide. Catalysis in CAOs proceeds via a Ping Pong Bi Bi reaction mechanism expressed as two half-reactions: reductive and oxidative (see Fig. 1). The oxidative half-reaction, in which molecular oxygen is reduced to hydrogen peroxide via cofactor reoxidation, follows a reductive half-reaction consisting of the oxidation of a primary amine to an aldehyde.
FIGURE 1.

Catalysis performed by CAOs including reductive and oxidative half-reactions. P represents the rest of the polypeptide chain. R represents the moiety of the substrate amine, which varies from a hydrogen atom to a polypeptide. TPQ, 2,4,5-trihydroxyphenylalanine quinone.
CAOs can be grouped based upon their catalytic properties and source organisms. These include plant, bacterial, and non-plant eukaryotic CAOs. Unlike plant CAOs, which show the highest kcat values, fast electron transfer rates, and considerable amounts of semiquinone formation upon reduction (up to 80%), non-plant eukaryotic CAOs exhibit the lowest kcat values, slower electron transfer rates, and the formation of little, if any, semiquinone (see Fig. 1) (1–4). Bacterial enzymes fall in the middle of these two extremes (5). These differences in enzymatic properties have caused some researchers to propose that enzymes from difference sources utilize different mechanisms for the activation of molecular oxygen (6, 7).
Studies of CAOs from divergent organisms have led to mechanistic proposals for the catalytic cycle (8, 9). The information gained from these studies has led to a consensus mechanism for the amine oxidation to aldehyde (10). However, the mechanism of molecular oxygen activation remains the subject of controversy (11–16). Kinetic isotope effects, steady-state kinetics, viscosogen, and stopped-flow experiments have shown that the reduction of molecular oxygen contributes 29% to the overall kcat of catalysis in Hansenula polymorpha amine oxidase 1 (HPAO-1)3 (17, 18). This is unexpected, as the reduction of O2 by Cu(I) to give superoxide is likely to be extremely fast, with low temperatures (183 K) required to measure the kinetics in synthetic model systems (19). As a result, the utilization of copper as a redox center in HPAO-1 has been called into question. At least three pieces of evidence exist in support of this proposal. First, the enzyme is active at pH values <7.0, where the amount of measurable Cu(I)-semiquinone formed is negligible (20). Second, Co(II)-substituted HPAO-1 has a kcat almost identical to that of copper-containing HPAO-1 at pH 7 under O2-saturating conditions (18, 21). Because the reduction potential for the Co(II)/Co(I) couple is so negative, Co(I) makes for an unlikely intermediate in catalysis (22), although a Co(III)/Co(II) couple has been recently proposed (15). Finally, model compounds of the reduced aminoquinol form of the cofactor can consume molecular oxygen in the absence of a metal ion (21). Based on these results, a mechanism has been proposed for non-plant eukaryotic enzymes that invokes an off-metal molecular oxygen-binding pocket for the first electron transfer to O2 directly from the reduced aminoquinol cofactor (23). Site-directed mutagenesis of Met-634, adjacent to the copper, found a positive correlation between side chain size and kcat/Km(O2). This resulted in the description of a “hydrophobic pocket” at which O2 might undergo the initial one-electron reduction required for activation (24). Subsequent to activation, the superoxide species is proposed to move and become a ligand of the Cu(II) ion.
Proponents for a redox role for copper suggest that under conditions in which the enzyme is active and the amount of Cu(I)-semiquinone is unobservable, there is a minimal but sufficient amount of Cu(I) to activate O2. Temperature jump relaxation studies in Arthrobacter globiformis amine oxidase (AGAO) have confirmed that the electron transfer step to form the Cu(I)-semiquinone intermediate from the Cu(II)-aminoquinol would not be rate-limiting (5). Stopped-flow kinetics of the oxidative half-reaction in AGAO (7) and kinetic competitive oxygen kinetic isotope effects (KIE) and density functional theory calculations in pea seedling CAO (16) confirmed inner-sphere electron transfer from Cu(I) to be the likely mechanism of O2 activation in these enzymes. This makes sense in light of their faster electron transfer rates, elevated formation of Cu(I)-semiquinone upon anaerobic substrate reduction, and higher kcat values (2).
HPAO-1 is a member of the non-plant eukaryotic CAO family that has been kinetically characterized in great detail (18, 21, 23, 25–27). At pH ∼7.0 the kcat of HPAO-1 has been measured to be ∼2.0 s−1 concomitant with levels of Cu(I)-semiquinone insufficient for accurate measurement (20). Although low levels are typical in HPAO-1 at neutral pH values, the propensity to form Cu(I)-semiquinone has been shown to be increased under basic conditions in the pH range of 7.5–8.5 (20). At pH ∼8.6, ∼15% of the reduced cofactor is in the semiquinone form in the anaerobically methylamine-reduced enzyme. The kcat/Km(O2) value has also been shown to be pH-dependent with increased bimolecular rate constants under basic conditions (20). A comparison of the kinetic parameters for both kcat and kcat/Km(O2) shows that HPAO-1 activates oxygen optimally between pH 8.0 and 8.5. This link between enzymatic efficiency and the presence of semiquinone has been weakened by experimental results indicating that 18O KIE fail to impact theoretical kcat/Km(O2) values for the reaction of Cu(I)-semiquinone with 18O2 (20). That said, observations of oxidative half-reaction structural intermediates in this pH regime still have the potential of revealing whether increased kinetic performance of the enzyme and the presence of elevated semiquinone are coincidental or causative at basic pH values.
To date, the best structural information for intermediates in the oxidative pathway has come from work performed in Escherichia coli amine oxidase (ECAO) (28) and through the characterization of synthetic models (29, 30). In the crystal, anaerobically substrate-reduced ECAO gave the aminoquinol intermediate, which is the starting structure of the oxidative half-reaction. The aerobic steady-state intermediate of ECAO in crystalline form also revealed the reoxidized iminoquinone (see Fig. 1) cofactor in combination with a bound oxygen species present at the axial position of the copper. One important omission, however, in the structures of oxidative half-reaction intermediates is a structure of the Cu(I)-semiquinone state of the enzyme.
Through a combination of single-crystal UV-visible microspectrophotometry, x-ray crystallography, and freeze trapping, reaction intermediates from the oxidative half-reaction have been observed in crystals of HPAO-1. By performing substrate reduction in a limiting oxygen atmosphere and at varying pH values, we were able to trap three spectroscopically and structurally distinct oxidative half-reaction intermediates in HPAO-1. In particular, the trapping of the semiquinone form of the cofactor in a copper-ligated orientation reveals new mechanistic possibilities for the activation of molecular oxygen in CAOs.
EXPERIMENTAL PROCEDURES
HPAO-1 Purification
HPAO-1 for crystallization was heterologously expressed in Saccharomyces cerevisiae and purified as described previously (27, 31) with specific modifications recently described (32). The protein was buffer-exchanged into 50 mm HEPES (pH 7.0) and concentrated to 13 mg/ml prior to crystallization.
HPAO-1 Crystallization
Crystallization was performed as reported previously (32) with 13 mg/ml HPAO-1 set up in hanging drop at a 1:1 ratio with 8.0–9.5% (w/v) PEG 8000 and 0.28–0.30 m phosphate (pH 6.0) and incubated at 293 K.
Intermediate Trapping
In addition to crystals grown at pH 6.0, two sets of oxidized HPAO-1 crystals were equilibrated against new mother liquor prepared with phosphate buffer at pH 7.0 and 8.5 by moving the caps of crystal trays, which contained hanging drops, to new wells containing the target pH to reduce osmotic shock upon subsequent transfer to the target pH mother liquor. This step prevented crystal cracking and loss of diffraction, and an equilibration time of 3 days was allowed. One-step soaks were then performed in a low oxygen glove box (10–20 ppm) with 5 mm methylamine prepared by adding concentrated stock (0.5 μl of 100 mm) to a drop containing equilibration buffer of the desired pH and 25% (v/v) glycerol required for cryoprotection (final volume of 10 μl). Crystals were soaked for ∼20 s in this condition before being flash-frozen in liquid nitrogen.
Single-crystal UV-visible Microspectrophotometry
Single-crystal spectra were collected at 100 K using a microspectrophotometer (4DX Systems AB) equipped with an MS125TM 1/8-m spectrograph (Thermo Oriel), a CCD detector (Andor Technology), and a xenon lamp (Zeiss) emitting from 250 to 800 nm. Spectra were generated from the integration of 10 19-ms exposures (33). Optimal diffraction data were collected from crystals too small for accurate spectral measurement by the microspectrophotometer. Instead, measurements were taken from larger crystals prepared under conditions identical to those specified for each structure.
To check that the reaction was turning over in the crystals at the three pH values, a quartz capillary flow cell was constructed as described previously (34, 35). Briefly, HPAO crystals were immobilized in a quartz capillary in Sephadex G-25 superfine resin equilibrated with artificial mother liquor composed of 8.5% (w/v) PEG 8000 and 0.28 m phosphate (pH 6.0, 7.0, or 8.5) by pumping the solution through the capillary at ∼1 ml/h using a syringe pump (Thermo Orion). 160 μl of 100 mm methylamine in artificial mother liquor was introduced to the crystal through a loop containing ∼160 μl of substrate solution. Spectra were recorded every 30 s through to steady state. As the substrate solution was depleted, crystal spectra were collected every 60 s.
Structure Determination and Refinement
Each x-ray data-set was collected from single crystals at 100 K at the Advanced Photon Source of Argonne National Laboratory (beamline 19-ID, Structural Biology Center Collaborative Access Team). High resolution diffraction data were measured at λ = 0.979 Å. Data were processed with HKL2000 and SCALEPACK (36). Structure solution for data collected from crystals harvested at pH 6.0 and 7.0 was by direct Fourier synthesis using the CCP4 suite (37) and the previously deposited HPAO-1 model (Protein Data Bank code 2OOV) (32). For structure solution of data collected from a crystal harvested at pH 8.5, molecular replacement was performed using Phaser (37) and a protein monomer from the previously deposited HPAO-1 model (code 2OOV) (32). Model building was performed using Coot (38). Refinement of the models was performed using REFMAC5 (39). No non-crystallographic symmetry restraints were used during the refinement.
RESULTS
X-ray structural information was obtained for reactive intermediates in the oxidative half-reaction by soaking substrate into crystals under oxygen-limiting conditions. This was performed with methylamine at pH 6.0, at which the crystals grew, and also at pH 7.0 and 8.5. Diffraction data collection, processing, and model refinement statistics are reported in Table 1. The P21 crystal form of HPAO-1 has a large asymmetric unit containing three dimers, and thus, six total active sites were built and refined independently. The C2221 crystal form of HPAO-1 contains 1.5 physiological dimers in the asymmetric unit, and thus, a total of three active sites were built and refined independently. Despite the different crystal forms, the HPAO-1 structures are essentially identical, with differences limited to the active site. Differences were observed between active sites within the same crystal (Table 1).
TABLE 1.
Data collection, processing, and refinement statistics
| Methylamine-reduced at pH 6.0 | Methylamine-reduced at pH 7.0 | Methylamine-reduced at pH 8.5 | |
|---|---|---|---|
| Data collection | |||
| Space group | P21 | P21 | C2221 |
| Unit cell parameters | |||
| a, b, c (Å) | 103.8, 222.8, 103.6 | 103.8, 222.8, 103.6 | 139.1, 153.3, 223.2 |
| α, β, γ | 90.0°, 95.9°, 90.0° | 90.0°, 95.8°, 90.0° | 90.0°, 90.0°, 90.0° |
| Resolution range (Å) | 50.0–1.70 (1.76–1.70) | 50.0–2.10 (2.14–2.10) | 50.0–2.15 (2.19–2.15) |
| Rmerge | 0.09 (0.59) | 0.10 (0.41) | 0.11 (0.56) |
| I/σI | 14.1 (1.7) | 12.3 (1.83) | 15.8 (3.19) |
| Completeness (%) | 99.7 (97.3) | 98.6 (89.4) | 100.0 (100.0) |
| Redundancy | 4.7 (3.3) | 3.6 (2.4) | 6.8 (6.5) |
| Refinement statistics | |||
| Resolution (Å) | 46.8-1.70 | 49.0-2.10 | 38.6-2.15 |
| No. of reflections (work/test) | 487,365/25,742 | 252,434/13,290 | 122,479/6492 |
| Rwork/Rfree | 13.4/16.4 | 12.9/18.0 | 15.1/19.1 |
| No. of atoms | |||
| Protein | 31,433 | 31,442 | 15,662 |
| Copper | 6 | 6 | 3 |
| Water | 4737 | 4380 | 1260 |
| All other | 335 | 337 | 83 |
| r.m.s.d.a | |||
| Bond lengths (Å) | 0.021 | 0.019 | 0.019 |
| Bond angles | 1.99° | 1.88° | 1.96° |
| Average B-factor (Å2) | 16.1 | 22.8 | 21.2 |
| Relative proportion of cofactor conformers for each protein chain in asymmetric unit (%) | Off-Cu, short cofactor N5/Asp-319 interaction:off-Cu, weak or H2O-mediated cofactor N5/Asp-319 interaction:on-Cu | ||
| A | 100:0:0 | 100:0:0 | 0:35:65 |
| B | 100:0:0 | 100:0:0 | 0:30:70 |
| C | 50:50:0 | 0:100:0 | 0:30:70 |
| D | 100:0:0 | 0:100:0 | NA |
| E | 100:0:0 | 0:100:0 | NA |
| F | 50:50:0 | 0:100:0 | NA |
| Protein Data Bank code | 4KFD | 4KFE | 4KFF |
a r.m.s.d., root mean square deviation; NA, not applicable.
To confirm that the crystals were undergoing turnover at the three pH values, UV-visible spectra of HPAO-1 crystals treated with substrate and tracked over time clearly show that the level of 2,4,5-trihydroxyphenylalanine quinone cofactor was regenerated to at least ∼70% at each pH following substrate depletion (Fig. 2).
FIGURE 2.
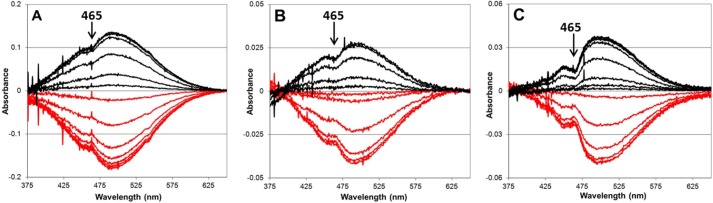
Aerobic methylamine turnover by HPAO-1 crystals at 298 K and at pH 6.0 (A), 7.0 (B), and 8.5 (C). The initial oxidized HPAO-1 UV-visible spectrum was subtracted from spectra obtained every 30 s until no further changes were observed (red traces). The steady-state spectrum was subtracted from spectra obtained every 60 s during regeneration of the HPAO-1 resting state (black traces). The increasing amount of semiquinone can be clearly seen at 465 nm as the pH is increased.
Diffraction data were recorded to 2.10 Å for HPAO-1 crystals methylamine-reduced at pH 7.0, and the crystals were observed to contain predominately deprotonated iminoquinone on the basis of their spectra (Fig. 3B). Crystals reduced at this pH contain a λmax at 450 nm, and in spectral work with another amine oxidase, bovine serum amine oxidase, this feature was previously assigned to a deprotonated iminoquinone (25). In this structure, two of the six active sites (chains C and F) show the cofactor to be in an identical position to the off-copper cofactor in oxidized HPAO-1 (Protein Data Bank code 2OOV) (32). This orientation of the cofactor is hydrogen-bonded directly to the conserved Tyr-305 at O4 and indirectly to the catalytic base Asp-319 at N5 through an intervening solvent molecule (Fig. 4B). The remaining four active sites of the asymmetric unit (chains A, B, D, and E) contain cofactors that are directly hydrogen-bonded to Asp-319 at a close distance (∼2.5 Å; chains A and B) or a weak interaction at a longer distance (∼3.5 Å; chains D and E) without an intervening water. Thus, it appears that the deprotonated iminoquinone interaction with Asp-319 can be either mediated by water or direct. The O2 atoms of all cofactors in the asymmetric unit are hydrogen-bonded to an oxygen species present at the axial position of the copper. This oxygen species has been assigned to the product hydrogen peroxide, as the presence of the iminoquinone suggests that the second substrate, molecular oxygen, has already been activated. In addition to hydrogen bonding with O2 of the cofactor, hydrogen bonding is also observed with an ordered solvent molecule (commonly referred to as W2 in amine oxidase literature) that bridges the axial copper ligand position and the hydroxyl of the conserved Tyr-305 that helps stabilize the cofactor in the off-copper conformation (Fig. 4D). The copper of this structure also has electron density for an equatorially bound water molecule (hidden behind the copper in Fig. 4), making the copper ion five-coordinate.
FIGURE 3.
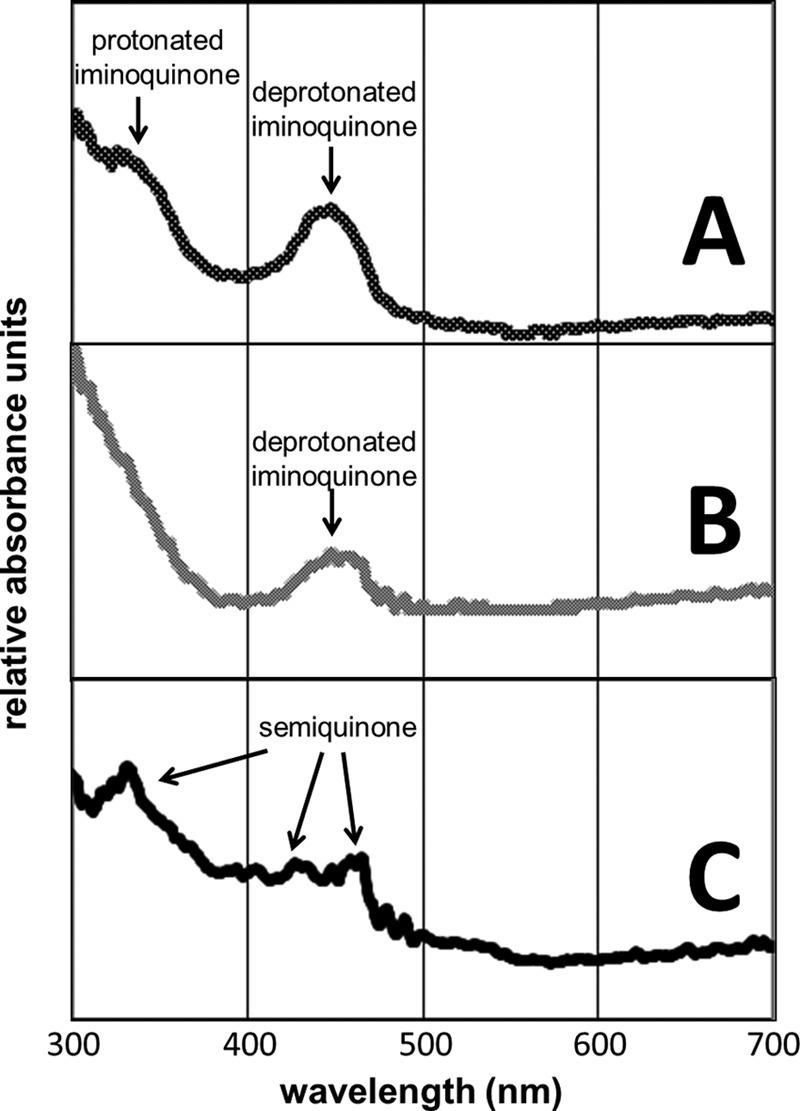
UV-visible spectra at 100 K taken from HPAO-1 crystals exposed to methylamine in a limiting oxygen environment at pH 6. 0 (A), 7.0 (B), and 8.5 (C).
FIGURE 4.

Active site stereo images of methylamine-reduced structures in a low oxygen environment at pH 6.0 (A), 7.0 (B), and 8.5 (C) and the oxidized structure (1.7 Å resolution; Protein Data Bank code 2OOV) in the same space group as the pH 6.0 and 7.0 structures reported here (D). Residues and bound peroxide are shown as sticks, copper is shown as a large brown sphere, and waters (Wa) are shown as small red spheres. Dashed lines indicate hydrogen bonding patterns. Blue mesh represents the 2Fo − Fc map at a value of 1σ. TPQ, 2,4,5-trihydroxyphenylalanine quinone.
Diffraction data were recorded to 1.70 Å for HPAO-1 crystals methylamine-reduced at pH 6.0, and the crystals were observed to contain a mixture of deprotonated and protonated iminoquinone on the basis of their spectra (Fig. 3A). Crystals of methylamine-reduced HPAO-1 at pH 6.0 contain a λmax at 350 nm in addition to the λmax at 450 nm also observed in the pH 7.0 structure. The new feature at 350 nm is assigned to a protonated iminoquinone form of the cofactor based upon spectral work performed with model compounds (40). In this structure, four of the six active sites (chains A, B, D, and E) show the cofactor to be in an orientation in which N5 directly interacts with the carboxylate of Asp-319 (Fig. 4A). Chains C and F contain partial occupancy for the cofactor in a similar conformation but also contain partial occupancy for a cofactor orientation like that found in the pH 7.0 reduced enzyme. Also similar to the deprotonated iminoquinone structure at pH 7.0, there is an oxygen species present at the axial position of the copper ion and a similar hydrogen bonding pattern involving the oxygen attached to C2 of the cofactor and the solvent molecule W2. As the UV-visible spectra of these crystals indicate only the presence of iminoquinone species, the product hydrogen peroxide is again presumed. Also identical to the structure at pH 7.0 is the presence of water equatorially bound to the copper, making this copper again five-coordinate.
The UV-visible crystal absorbance spectrum measured for methylamine-reduced HPAO-1 at pH 8.5 contains peaks at 350, 435, and 465 nm characteristic of the semiquinone, as well as features associated with the iminoquinone (Fig. 3C). Diffraction data recorded to 2.15 Å reveal structural features different from those at pH 6.0 and 7.0. In this structure, all three active sites of the asymmetric unit show the cofactor in two distinct orientations, one with ∼30–35% occupancy (off-copper) and the other with ∼65–70% occupancy (on-copper) (Fig. 4C). The occupancies were determined by selecting a partition between the species that resulted in similar B-factors for conformationally ordered atoms adjacent to the cofactor within the structure following refinement and gave Fo − Fc electron density at the noise level of the map. The off-copper orientation of the cofactor is similar to that of the predominant orientation observed at pH 7.0, where N5 of the cofactor interacts with the carboxylate of Asp-319 at long range through an intervening solvent molecule that is assigned to the deprotonated iminoquinone. The second orientation is an on-copper conformation with O4 of the cofactor directly ligated to the copper ion. In this orientation, the cofactor fully occupies the axial position of the copper. Absent in this structure are both the axially bound oxygen species and the equatorially bound water, making the copper four-coordinate.
DISCUSSION
Methylamine reduction of HPAO-1 in an oxygen-limiting atmosphere at pH 6.0, 7.0, and 8.5 has allowed the successful trapping of three distinct species relevant to the oxidative half-reaction. Two of these intermediates correspond to the iminoquinone form of the cofactor, as protonated and deprotonated species. The third corresponds to a semiquinone form of the cofactor, mixed with the deprotonated iminoquinone. Single-crystal UV-visible spectra confirm the identity of each of these species due to their distinctive λmax (Fig. 3).
A significant feature of the iminoquinone structures at pH 6.0 and 7.0 is that all of the cofactors in the asymmetric unit (of which there are six) are in the off-copper conformation. This is an important observation because the oxidized structure solved in the same space group indicates that four of the six cofactors are in mixed populations of on-copper and off-copper orientations (32). In the past, those orientations of the cofactor in which O4 is directly bound to the copper have been described as nonproductive “inactive” conformers of the cofactor (41, 42). However, despite this starting orientation, those cofactors in the on-copper orientation are obviously in equilibrium with the productive off-copper conformer, allowing all active sites to be reactive with amine substrate. The pH 6.0 and 7.0 structures also show that deprotonated and protonated forms of the iminoquinone have different orientations and interactions within the active site. The protonated form of the cofactor, which is positively charged at N5, directly interacts with the carboxylate of Asp-319 (Fig. 4A). Charge stabilization between the negative carboxylate and positive imine of the protonated iminoquinone explains the subtle orientation difference observed versus the deprotonated iminoquinone that lacks a positive charge at N5 (Fig. 4B). A kinetic and spectroscopic study with AGAO demonstrated that both forms of the iminoquinone could undergo hydrolysis, albeit with different rates, to regenerate 2,4,5-trihydroxyphenylalanine quinone (7).
A second significant feature of the iminoquinone structures is associated with the copper. Both structures display the presence of an axially bound oxygen species that has been assigned to the product hydrogen peroxide. The axially bound hydrogen peroxide of these structures shows striking similarity to that seen previously in the bacterial enzyme ECAO (Fig. 5) and anaerobically ethylamine-reduced HPAO-1, where its presence with aminoquinol was proposed to be a consequence of residual O2 in the crystallization drop leading to some initial turnover (28, 43). Additionally, although H2O2 is axially bound to the copper, an equatorially bound water is present at high occupancy, creating a five-coordinate copper ligation sphere. This is different from the resting oxidized state, where discernible electron density for the equatorial water is present in only two of the six active sites of the asymmetric unit of the isomorphous crystal form. That said, the five-coordinate ligation sphere preference for Cu(II) in CAOs has been observed through extended x-ray absorption fine structure (12) and supports the proposal that the oxygen species present in these structures is not a Cu(I)-bound species.
FIGURE 5.
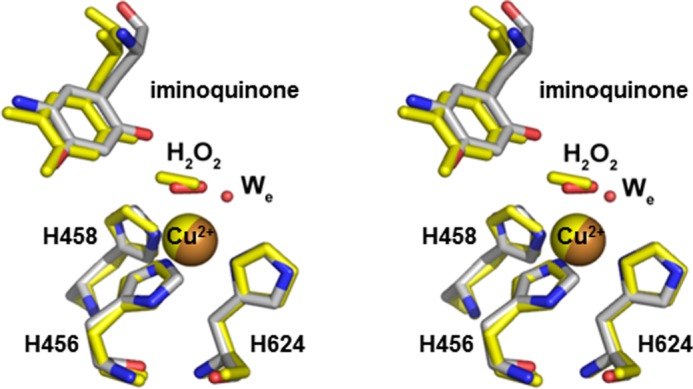
Stereoview of the aerobic ECAO steady-state structure (Protein Data Bank code 1D6Z) overlaid with the methylamine-exposed HPAO-1 structure at pH 7.0. Residues of ECAO are shown in yellow, and residues of HPAO-1 are colored by atom. Copper ions are shown as spheres.
The final oxidative half-reaction intermediate characterized structurally is that of the semiquinone measured at pH 8.5. In addition to an off-copper iminoquinone orientation similar to that observed at pH 7.0, up to 70% of the cofactor is directly ligated to the copper through the axial position. Despite the presence of partial occupancy iminoquinone in this structure, the contribution of the axially bound cofactor makes the presence of H2O2 indeterminate, although positive peaks in the Fo − Fc map indicate the presence of an axial copper ligand in all three active sites. Additionally, the equatorial position, which contained a bound water in the pH 6.0 and 7.0 iminoquinone structures, now lacks any evidence of electron density. As a four-coordinate tetrahedral environment is more consistent with Cu(I) than with Cu(II), this supports the on-copper species being assigned to Cu(I)-semiquinone. These observations pose a key mechanistic question: is the Cu(I)-semiquinone form of the enzyme a copper-ligated species?
These three structural observations allow an expansion of the possible equilibria that exist on the mechanistic pathway of the oxidative half-reaction (Fig. 6). These equilibria assist in describing routes of highest probability with regard to the pH of the enzymatic environment. Substrate reduction at slightly acidic pH values favors a reaction pathway lacking reduction of Cu(II) to Cu(I) and would proceed through a protonated iminoquinone pathway (Fig. 6A). Substrate reduction at slightly basic pH values would favor a reaction pathway that includes formation of the Cu(I)-semiquinone, which may or may not be the O2-activating species, and proceeds through a deprotonated iminoquinone pathway (Fig. 6, B and C).
FIGURE 6.

Possible reaction pathways for the oxidative half-reaction of CAOs. A, the proposed pathway utilized in the absence of the Cu(I)-semiquinone intermediate for the activation of molecular oxygen (acidic pH). B, the proposed pathway utilized in the presence of an off-copper Cu(I)-semiquinone (neutral pH). C, the proposed pathway utilized in the presence of a copper-bound Cu(I)-semiquinone intermediate (basic pH). Gray boxes represent intermediates for which there are crystal structures from this work and elsewhere (28). PCET, proton-coupled electron transfer; TPQ, 2,4,5-trihydroxyphenylalanine quinone.
Although we have visualized the semiquinone as a copper-bound species, the possibility also exists that the semiquinone, like the oxidized cofactor, can have two orientations, including both copper-ligated and copper-dissociated. A three-coordinate Cu(I) has been observed by x-ray absorption spectroscopy in dithionite-reduced CAOs, although this is in the absence of semiquinone (12). This scenario has mechanistic implications. If the semiquinone is not directly ligated to the copper, then the axial position of the copper remains open for molecular oxygen reduction to form a Cu(II)-superoxo intermediate (Fig. 6B). From this intermediate, the second electron reduction can take place, forming the bound hydrogen peroxide product previously observed in ECAO and now here in HPAO-1. This mechanistic possibility finds favor in the observation that the Cu(I)-bound semiquinone builds up in the crystal to a higher occupancy than that observed in solution (20). The buildup of any Cu(I) is counterintuitive to the highly reactive catalytic properties suggested by synthetic model complexes (44), and furthermore, intermediates that normally build up in crystals are usually kinetic dead ends or rate-limiting steps. Therefore, in this scenario, the presence of a Cu(I)-bound semiquinone could be viewed as off the reaction pathway and part of an inactive complex that cannot react with the low levels of O2 present in the experiment. By process of elimination, only that proportion of semiquinone that is off-copper would be able to react with O2.
Although this study offers no evidence that Cu(I)-semiquinone is exclusively copper-ligated, the observation that it can be copper-ligated offers further intriguing mechanistic possibilities. If CAOs utilize Cu(I)-mediated molecular oxygen activation, then a copper-bound cofactor would require that O2 use an equatorial site (Fig. 6C). This possibility exists, as the equatorial site is vacant in this structure, and there is chemical precedent in the literature, with both synthetic model compound (44) and enzymatic examples of Cu(I)-phenolate/tyrosinate reactivity with molecular oxygen (45). However, as suggested by product complexes both in ECAO and here, the activated oxygen species would at some point need to migrate from the equatorial to the axial position before dissociating from the enzyme (28). Rate-limiting steps in HPAO-1 include almost equal contributions from aldehyde release, oxidation of the reduced enzyme, and release of the axially bound hydrogen peroxide at pH 7.0 (17). It is interesting to note that the rate-limiting step of at least one plant enzyme is contained entirely within the reductive half-reaction (46). If hydrogen peroxide release does not control the rate of catalysis in plant enzymes, then its more rapid dissociation may be facilitated by another reductive half-reaction and subsequent formation of copper-bound semiquinone. This effect would manifest itself more readily in the plant enzymes because they have a higher propensity for Cu(I)-semiquinone formation. It would be of interest to determine whether and how the rate-limiting steps of HPAO-1 change when the kinetics are measured at basic pH values (pH >8). It is already known that O2 utilization approaches maximum efficiency at pH ≥8.5 (18).
The strongest evidence against Cu(I)-required O2 activation in HPAO-1 (Fig. 6, B and C) still lies in the observation that Co(II)-substituted HPAO-1 has nearly identical kcat values to the native form (18). In contrast to HPAO-1, Co(II)-substituted AGAO, pea seedling CAO, and ECAO all have very low catalytic activities (15, 47–49). The catalytic rates are similar to wild-type copper-containing HPAO-1, HPAO-2, and bovine serum amine oxidase, which are also posited to employ an outer-sphere electron transfer mechanism from aminoquinol to activate O2 (24, 50, 51). Therefore, it was suggested that the Co(II)-AGAO, pea seedling CAO, and ECAO enzymes use the less efficient mechanism of outer-sphere electron transfer for O2 activation. However, the existence of an outer-sphere electron transfer mechanism in either copper- or cobalt-containing CAOs has been recently challenged in pea seedling CAO and AGAO based on 18O KIE studies and density functional theory (15, 16), coupled with the smaller than expected 18O KIE observed in HPAO-1 and bovine serum amine oxidase that are more in line with inner-sphere electron transfer (18, 24). However, it should be noted that a larger 18O KIE than observed in CAO is seen in the Cu(I) inner-sphere electron transfer activation of O2 in galactose oxidase, indicating caution is warranted when linking 18O KIE magnitude to the O2 activation mechanism (52).
The possibility of a Co(III)/Co(II) couple being in operation rather than the unlikely Co(II)/Co(I) couple cannot be discounted, and recent competitive 18O KIE and deuterium KIE studies in AGAO coupled with density functional theory are consistent with an identical inner-sphere oxidative half-reaction mechanism for both the copper and cobalt forms of the enzyme (15). In the case of HPAO-1, the experimental data are not definitive. The kcat and 18O KIE are within experimental error for the copper and cobalt forms of the enzyme, so the probability that the presence of the protein would tune the parameters such that they were the same for the two transition metals seems unlikely (18).
Previous studies have suggested that the identity of second-sphere residues can control both the propensity for the formation of semiquinone and the subsequent activity of CAOs toward molecular oxygen by charge stabilization/destabilization of Cu(II) and the resultant effect on the pKa of an axially bound water (20). The results obtained here come to a similar conclusion by visualizing how perturbation of the solution pH affects the structure of the active site, suggesting that at more basic pH values, there is a higher propensity for the cofactor to collapse onto the copper and form the semiquinone (Fig. 6, top to bottom). Thus, the pKa values of the cofactor and axial water likely have the effect of modulating the equilibrium for the aminoquinol-semiquinone. As a result, residue differences across species are responsible not only for the catalytic properties of the enzymes but potentially the mechanistic route available to the enzyme at a given pH. Therefore, we propose that this finely balanced interplay explains why plant, bacterial, and non-plant eukaryotic enzymes would seem to utilize different mechanisms. Ultimately, the reason why Cu(I)-semiquinone CAO may activate O2 in some species rather than in others may simply be a consequence of whether it is predominantly on-copper, blocking O2 binding to the copper, or off-copper, allowing O2 to access the Cu(I) axial coordination site.
Acknowledgments
We thank Teresa De la Mora-Rey for help during x-ray data collection; Arwen Pearson, Richard Welford, and Elena Kovaleva for discussions; and the staff of the Structural Biology Center Collaborative Access Team of the Advanced Photon Source, especially Steve Ginell. We thank Ed Hoeffner for Kahlert Structural Biology Laboratory support at the University of Minnesota.
This work was supported, in whole or in part, by National Institutes of Health Training Grant GM008700 (to B. J. J.) and Grants GM66569 (to C. M. W.) and GM25765 (to J. P. K.). This work was also supported by Minnesota Medical Foundation Grant 3714-9221-06 (to C. M. W.). Final X-ray diffraction data were collected at the Structural Biology Center of the Advanced Photon Source at Argonne National Laboratory. Argonne National Laboratory is operated by UChicago Argonne, LLC, for the United States Department of Energy, Office of Biological and Environmental Research, under Contract DE-AC02-06CH11357. Computer resources were provided by the Basic Sciences Computing Laboratory of the Minnesota Supercomputing Institute.
The atomic coordinates and structure factors (codes 4KFD, 4KFE, and 4KFF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
H. polymorpha has been reclassified as Pichia angusta. The names H. polymorpha and HPAO-1 are used to correspond with past literature.
- CAO
- copper amine oxidase
- HPAO-1
- H. polymorpha amine oxidase 1
- AGAO
- A. globiformis amine oxidase
- KIE
- kinetic isotope effect(s)
- ECAO
- E. coli amine oxidase.
REFERENCES
- 1. Dooley D. M., Brown D. E. (1996) Intramolecular electron transfer in the oxidation of amines by methylamine oxidase from Arthrobacter P1. J. Biol. Inorg. Chem. 1, 205–209 [Google Scholar]
- 2. Turowski P. N., McGuirl M. A., Dooley D. M. (1993) Intramolecular electron transfer rate between active-site copper and topa quinone in pea seedling amine oxidase. J. Biol. Chem. 268, 17680–17682 [PubMed] [Google Scholar]
- 3. Dooley D. M., McGuirl M. A., Brown D. E., Turowski P. N., McIntire W. S., Knowles P. F. (1991) A Cu(I)-semiquinone state in substrate-reduced amine oxidases. Nature 349, 262–264 [DOI] [PubMed] [Google Scholar]
- 4. Medda R., Padiglia A., Bellelli A., Pedersen J. Z., Agrò A. F., Floris G. (1999) CuI-semiquinone radical species in plant copper-amine oxidases. FEBS Lett. 453, 1–5 [DOI] [PubMed] [Google Scholar]
- 5. Shepard E. M., Dooley D. M. (2006) Intramolecular electron transfer rate between active-site copper and TPQ in Arthrobacter globiformis amine oxidase. J. Biol. Inorg. Chem. 11, 1039–1048 [DOI] [PubMed] [Google Scholar]
- 6. Pietrangeli P., Nocera S., Mondovi B., Morpurgo L. (2003) Is the catalytic mechanism of bacteria, plant, and mammal copper-TPQ amine oxidases identical? Biochim. Biophys. Acta 1647, 152–156 [DOI] [PubMed] [Google Scholar]
- 7. Shepard E. M., Okonski K. M., Dooley D. M. (2008) Kinetics and spectroscopic evidence that the Cu(I)-semiquinone intermediate reduces molecular oxygen in the oxidative half-reaction of Arthrobacter globiformis amine oxidase. Biochemistry 47, 13907–13920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mure M., Mills S. A., Klinman J. P. (2002) Catalytic mechanism of the topa quinone containing copper amine oxidases. Biochemistry 41, 9269–9278 [DOI] [PubMed] [Google Scholar]
- 9. Dooley D. M. (1999) Structure and biogenesis of topaquinone and related cofactors. J. Biol. Inorg. Chem. 4, 1–11 [DOI] [PubMed] [Google Scholar]
- 10. Klinman J. P. (1996) Mechanisms whereby mononuclear copper proteins functionalize organic substrates. Chem. Rev. 96, 2541–2562 [DOI] [PubMed] [Google Scholar]
- 11. Dove J. E., Klinman J. P. (2001) Trihydroxyphenylalanine quinone (TPQ) from copper amine oxidases and lysyl tyrosylquinone (LTQ) from lysyl oxidase. Adv. Protein Chem. 58, 141–174 [DOI] [PubMed] [Google Scholar]
- 12. Dooley D. M., Scott R. A., Knowles P. F., Colangelo C. M., McGuirl M. A., Brown D. E. (1998) Structures of the Cu(I) and Cu(II) forms of amine oxidases from x-ray absorption spectroscopy. J. Am. Chem. Soc. 120, 2599–2605 [Google Scholar]
- 13. Hirota S., Iwamoto T., Kishishita S., Okajima T., Yamauchi O., Tanizawa K. (2001) Spectroscopic observation of intermediates formed during the oxidative half-reaction of copper/topa quinone-containing phenylethylamine oxidase. Biochemistry 40, 15789–15796 [DOI] [PubMed] [Google Scholar]
- 14. Padiglia A., Medda R., Lorrai A., Paci M., Pedersen J. Z., Boffi A., Bellelli A., Agrò A. F., Floris G. (2001) Irreversible inhibition of pig kidney copper-containing amine oxidase by sodium and lithium ions. Eur. J. Biochem. 268, 4686–4697 [DOI] [PubMed] [Google Scholar]
- 15. Liu Y., Mukherjee A., Nahumi N., Ozbil M., Brown D., Angeles-Boza A. M., Dooley D. M., Prabhakar R., Roth J. P. (2013) Experimental and computational evidence of metal-O2 activation and rate-limiting proton-coupled electron transfer in a copper amine oxidase. J. Phys. Chem. B 117, 218–229 [DOI] [PubMed] [Google Scholar]
- 16. Mukherjee A., Smirnov V. V., Lanci M. P., Brown D. E., Shepard E. M., Dooley D. M., Roth J. P. (2008) Inner-sphere mechanism for molecular oxygen reduction catalyzed by copper amine oxidases. J. Am. Chem. Soc. 130, 9459–9473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takahashi K., Klinman J. P. (2006) Relationship of stopped flow to steady state parameters in the dimeric copper amine oxidase from Hansenula polymorpha and the role of zinc in inhibiting activity at alternate copper-containing subunits. Biochemistry 45, 4683–4694 [DOI] [PubMed] [Google Scholar]
- 18. Mills S. A., Goto Y., Su Q., Plastino J., Klinman J. P. (2002) Mechanistic comparison of the cobalt-substituted and wild-type copper amine oxidase from Hansenula polymorpha. Biochemistry 41, 10577–10584 [DOI] [PubMed] [Google Scholar]
- 19. Lewis E. A., Tolman W. B. (2004) Reactivity of dioxygen-copper systems. Chem. Rev. 104, 1047–1076 [DOI] [PubMed] [Google Scholar]
- 20. Welford R. W. D., Lam A., Mirica L. M., Klinman J. P. (2007) Partial conversion of Hansenula polymorpha amine oxidase into a “plant” amine oxidase: implications for copper chemistry and mechanism. Biochemistry 46, 10817–10827 [DOI] [PubMed] [Google Scholar]
- 21. Mills S. A., Klinman J. P. (2000) Evidence against reduction of Cu2+ to Cu+ during dioxygen activation in a copper amine oxidase from yeast. J. Am. Chem. Soc. 122, 9897–9904 [Google Scholar]
- 22. Drummond J. T., Matthews R. G. (1994) Nitrous oxide degradation by cobalamin-dependent methionine synthase: characterization of the reactants and products in the inactivation reaction. Biochemistry 33, 3732–3741 [DOI] [PubMed] [Google Scholar]
- 23. Goto Y., Klinman J. P. (2002) Binding of dioxygen to non-metal sites in proteins: exploration of the importance of binding site size versus hydrophobicity in the copper amine oxidase from Hansenula polymorpha. Biochemistry 41, 13637–13643 [DOI] [PubMed] [Google Scholar]
- 24. Su Q., Klinman J. P. (1998) Probing the mechanism of proton coupled electron transfer to dioxygen: the oxidative half-reaction of bovine serum amine oxidase. Biochemistry 37, 12513–12525 [DOI] [PubMed] [Google Scholar]
- 25. Plastino J., Green E. L., Sanders-Loehr J., Klinman J. P. (1999) An unexpected role for the active site base in cofactor orientation and flexibility in the copper amine oxidase from Hansenula polymorpha. Biochemistry 38, 8204–8216 [DOI] [PubMed] [Google Scholar]
- 26. Schwartz B., Olgin A. K., Klinman J. P. (2001) The role of copper in topa quinone biogenesis and catalysis, as probed by azide inhibition of a copper amine oxidase from yeast. Biochemistry 40, 2954–2963 [DOI] [PubMed] [Google Scholar]
- 27. Cai D., Klinman J. P. (1994) Copper amine oxidase: heterologous expression, purification, and characterization of an active enzyme in Saccharomyces cerevisiae. Biochemistry 33, 7647–7653 [DOI] [PubMed] [Google Scholar]
- 28. Wilmot C. M., Hajdu J., McPherson M. J., Knowles P. F., Phillips S. E. (1999) Visualization of dioxygen bound to copper during enzyme catalysis. Science 286, 1724–1728 [DOI] [PubMed] [Google Scholar]
- 29. Mure M., Klinman J. P. (1993) Synthesis and spectroscopic characterization of model compounds for the active site cofactor in copper amine oxidases. J. Am. Chem. Soc. 115, 7117–7127 [Google Scholar]
- 30. Mure M., Klinman J. P. (1995) Model studies of topa quinone: synthesis and characterization of topa quinone derivatives. Methods Enzymol. 258, 39–52 [DOI] [PubMed] [Google Scholar]
- 31. DuBois J. L., Klinman J. P. (2004) Methods for characterizing TPQ-containing proteins. Methods Enzymol. 378, 17–31 [DOI] [PubMed] [Google Scholar]
- 32. Johnson B. J., Cohen J., Welford R. W., Pearson A. R., Schulten K., Klinman J. P., Wilmot C. M. (2007) Exploring molecular oxygen pathways in Hansenula polymorpha copper-containing amine oxidase. J. Biol. Chem. 282, 17767–17776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hadfield A., Hajdu J. (1993) A fast and portable microspectrophotometer for protein crystallography. J. Appl. Crystallogr. 26, 839–842 [Google Scholar]
- 34. Pearson A. R., Wilmot C. M. (2003) Catching catalysis in the act: using single crystal kinetics to trap methylamine dehydrogenase reaction intermediates. Biochim. Biophys. Acta 1647, 381–389 [DOI] [PubMed] [Google Scholar]
- 35. Wilmot C. M., Sjögren T., Carlsson G. H., Berglund G. I., Hajdu J. (2002) Defining redox state of x-ray crystal structures by single-crystal ultraviolet-visible microspectrophotometry. Methods Enzymol. 353, 301–318 [DOI] [PubMed] [Google Scholar]
- 36. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 37. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 40. Mure M., Klinman J. P. (1995) Model studies of topaquinone-dependent amine oxidases. 2. characterization of reaction intermediates and mechanism. J. Am. Chem. Soc. 117, 8707–8718 [Google Scholar]
- 41. Airenne T. T., Nymalm Y., Kidron H., Smith D. J., Pihlavisto M., Salmi M., Jalkanen S., Johnson M. S., Salminen T. A. (2005) Crystal structure of the human vascular adhesion protein-1: unique structural features with functional implications. Protein Sci. 14, 1964–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Duff A. P., Cohen A. E., Ellis P. J., Kuchar J. A., Langley D. B., Shepard E. M., Dooley D. M., Freeman H. C., Guss J. M. (2003) The crystal structure of Pichia pastoris lysyl oxidase. Biochemistry 42, 15148–15157 [DOI] [PubMed] [Google Scholar]
- 43. Klema V. J., Solheid C. J., Klinman J. P., Wilmot C. M. (2013) Structural analysis of aliphatic versus aromatic substrate specificity in a copper amine oxidase from Hansenula polymorpha. Biochemistry 52, 2291–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jazdzewski B. A., Reynolds A. M., Holland P. L., Young V. G., Jr., Kaderli S., Zuberbühler A. D., Tolman W. B. (2003) Copper(I)-phenolate complexes as models of the reduced active site of galactose oxidase: synthesis, characterization, and O2 reactivity. J. Biol. Inorg. Chem. 8, 381–393 [DOI] [PubMed] [Google Scholar]
- 45. Whittaker J. W. (2005) The radical chemistry of galactose oxidase. Arch. Biochem. Biophys. 433, 227–239 [DOI] [PubMed] [Google Scholar]
- 46. Padiglia A., Medda R., Bellelli A., Agostinelli E., Morpurgo L., Mondovi B., Agrò A. F., Floris G. (2001) The reductive and oxidative half-reactions and the role of copper ions in plant and mammalian copper-amine oxidases. Eur. J. Inorg. Chem. 2001, 35–42 [Google Scholar]
- 47. Kishishita S., Okajima T., Kim M., Yamaguchi H., Hirota S., Suzuki S., Kuroda S., Tanizawa K., Mure M. (2003) Role of copper ion in bacterial copper amine oxidase: spectroscopic and crystallographic studies of metal-substituted enzymes. J. Am. Chem. Soc. 125, 1041–1055 [DOI] [PubMed] [Google Scholar]
- 48. Mills S. A., Brown D. E., Dang K., Sommer D., Bitsimis A., Nguyen J., Dooley D. M. (2012) Cobalt substitution supports an inner-sphere electron transfer mechanism for oxygen reduction in pea seedling amine oxidase. J. Biol. Inorg. Chem. 17, 507–515 [DOI] [PubMed] [Google Scholar]
- 49. Smith M. A., Pirrat P., Pearson A. R., Kurtis C. R. P., Trinh C. H., Gaule T. G., Knowles P. F., Phillips S. E. V., McPherson M. J. (2010) Exploring the roles of the metal ions in Escherichia coli copper amine oxidase. Biochemistry 49, 1268–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bellelli A., Morpurgo L., Mondovì B., Agostinelli E. (2000) The oxidation and reduction reactions of bovine serum amine oxidase–a kinetic study. Eur. J. Biochem. 267, 3264–3269 [DOI] [PubMed] [Google Scholar]
- 51. Chang C. M., Klema V. J., Johnson B. J., Mure M., Klinman J. P., Wilmot C. M. (2010) Kinetic and structural analysis of substrate specificity in two copper amine oxidases from Hansenula polymorpha. Biochemistry 49, 2540–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Humphreys K. J., Mirica L. M., Wang Y., Klinman J. P. (2009) Galactose oxidase as a model for reactivity at a copper superoxide center. J Am. Chem. Soc. 131, 4657–4663 [DOI] [PMC free article] [PubMed] [Google Scholar]