

Background: The oligomerization of GRASP65 and GRASP55 is required to tether Golgi membranes.
Results: The crystal structures reveal two types of intermolecular interactions, and biochemical and cellular assays confirm these observations.
Conclusion: Two relatively weak interactions in combination are needed for GRASP-mediated Golgi stacking.
Significance: These data suggest a novel mode of Golgi membrane stacking by the GRASP proteins.
Keywords: Golgi, Membrane, Protein Assembly, Protein Structure, Protein-Protein Interactions, PDZ Domain, Membrane Stacking
Abstract
The stacking of Golgi cisternae involves GRASP65 and GRASP55. The oligomerization of the N-terminal GRASP domain of these proteins, which consists of two tandem PDZ domains, is required to tether the Golgi membranes. However, the molecular basis for GRASP assembly is unclear. Here, we determined the crystal structures of the GRASP domain of GRASP65 and GRASP55. The structures reveal similar homotypic interactions: the GRASP domain forms a dimer in which the peptide-binding pockets of the two neighboring PDZ2 domains face each other, and the dimers are further connected by the C-terminal tail of one GRASP domain inserting into the binding pocket of the PDZ1 domain in another dimer. Biochemical analysis suggests that both types of contacts are relatively weak but are needed in combination for GRASP-mediated Golgi stacking. Our results unveil a novel mode of membrane tethering by GRASP proteins and provide insight into the mechanism of Golgi stacking.
Introduction
In eukaryotic cells, the Golgi apparatus is composed of apposed and flattened membrane cisternae (1). Each cisterna compartmentalizes a distinctive set of enzymes that catalyze sequential steps of protein glycosylation and maturation (2). The stacking of these cisternae ensures that membrane proteins and secretory proteins are processed directionally and efficiently through the Golgi complex. Electron microscopy has shown that Golgi cisternae are linked by proteinaceous bridges (3). However, the nature of these bridges is not entirely clear.
The stacked architecture of the Golgi is stable in animal cells but becomes dynamic during cell division (4). When cells enter mitosis, the Golgi stacks are disassembled into many small vesicles, allowing partitioning of the Golgi components into the two daughter cells. After successful chromosome segregation, stacked cisternae are rebuilt to restore proper Golgi functions. In an effort to identify components required for post-mitotic reassembly of Golgi cisternae, a peripheral Golgi protein termed GRASP65 (Golgi reassembly stacking protein of 65 kDa) was identified (5). GRASP65 localizes to the cis-Golgi through its N-terminal myristoylation and interaction with GM130, a cis-Golgi marker (6, 7). Modification of GRASP65 on mitotic Golgi fragments by N-ethylmaleimide in vitro and the microinjection of antibodies against GRASP65 into mitotic cells inhibit the stacking of reformed Golgi cisternae (5, 8). Depletion of GRASP65 results in a reduced number of cisternae in each Golgi stack (9, 10), suggesting a direct role of GRASP65 in Golgi stacking. GRASP65 is localized mainly on the cis-side of the Golgi (5), whereas GRASP55, a closely related homolog of GRASP65, was found to stack the medial- and trans-Golgi (11). Similar to GRASP65, GRASP55 is anchored to the Golgi membranes via myristoylation, but GRASP65 interacts with GM130, and GRASP55 binds golgin-45, a medial/trans-Golgi resident protein (12). The molecular behavior of GRASP55 in stacking Golgi membranes resembles that of GRASP65 (10, 13).
Both GRASP65 and GRASP55 consist of an N-terminal GRASP domain composed of two tandem PDZ domains with high sequence homology and a C-terminal serine/proline-rich (SPR)5 domain (14). Biochemical experiments have indicated that GRASP proteins form oligomers and that the GRASP domain is sufficient for homotypic interactions (8, 10, 14). The PDZ domain is known to be a peptide-binding module with a hydrophobic cleft formed between an α-helix and a β-strand as a binding pocket (15). Mutagenesis studies have suggested that the PDZ domains of GRASP proteins may bind to an internal ligand (16, 17). A crystal structure of the GRASP domain of GRASP55 was reported recently (17). The structure reveals canonical folding of the PDZ domains, but no relevant intermolecular interactions were identified.
The oligomerization of GRASP proteins is affected by phosphorylation, which in turn influences Golgi stacking, consistent with a role of GRASP proteins in cell cycle-dependent regulation of Golgi stacking. GRASP65 is a substrate for Cdc2 and Plk (Polo-like kinase) (18), whereas GRASP55 can be phosphorylated by ERK2 (19). Even though multiple phosphorylation sites have been identified in both proteins (14, 19–21), mostly in the SPR domain, the molecular basis for phosphorylation-regulated disassembly of GRASP oligomers is not clear.
In addition to Golgi stacking, GRASP proteins have been implicated in lateral fusion of cisternae to form ribbon-like Golgi structures in mammalian cells (22, 23) and in unconventional secretion pathways (24–26). The membrane-tethering activity appears to be critical for the functions of GRASP proteins. To understand the mechanism of GRASP-mediated membrane tethering, we determined the crystal structures of the GRASP domain of GRASP65 and GRASP55. In both structures, the GRASP domain forms a dimer through homotypic interactions between the two PDZ2 domains; the tail of the GRASP domain (i.e. the immediate extension of the GRASP domain into the C-terminal region) also associates with the PDZ1 domain from the neighboring molecule. In vitro experiments confirmed that these two interfaces play an important role in mediating the oligomerization of GRASP proteins and membrane tethering of Golgi.
MATERIALS AND METHODS
Molecular Cloning, Mutagenesis, and Antibodies
For bacterial expression of GRASP proteins, fragments of rat GRASP65 (residues 1–228, 1–210, 1–206, 12–111, or 111–204) and rat GRASP55 (residues 1–215) were amplified and cloned into the pET30-TEV/LIC vector (Novagen), which contains an N-terminal His6 tag. For expression in mammalian cells, full-length rat GRASP65 or residues 1–210 were PCR-amplified with a C-terminal Myc tag and ligated into the pcDNA4/TO vector (Invitrogen) using HindIII and XhoI. Rat GRASP55-Myc was cloned similarly using KpnI and XhoI. All point mutations were generated using the QuikChange site-directed mutagenesis kit (Stratagene). All constructs were confirmed by DNA sequencing.
Expression, Purification, and Crystallization of GRASP Proteins
All bacterial expression constructs were transformed into the Escherichia coli BL21(DE3) strain (Novagen). Cells were grown at 37 °C to an A600 of 0.6 and induced with 0.3 mm isopropyl β-d-thiogalactopyranoside for 16 h at 16 °C. Harvested cells were lysed by sonication in binding buffer (50 mm Tris (pH 8.0), 500 mm NaCl, and 4 mm imidazole). The lysates were clarified by centrifugation, and the protein was isolated using nickel-nitrilotriacetic acid resin (Qiagen). The His6 tag was cleaved by tobacco etch virus protease at 20 °C for 12 h, and the protein was further purified by ion-exchange chromatography (HiTrap Q HP, GE Healthcare), followed by gel filtration (Superdex 200, GE Healthcare) in buffer containing 50 mm Tris (pH 8.0) and 200 mm NaCl. Fractions containing GRASP proteins were pooled and concentrated to 10 mg/ml. The purified GRASP proteins were crystallized by the vapor diffusion method. Crystals of GRASP65 (residues 1–228 or 1–210) grew in reservoir buffer containing 0.1 m BisTris (pH 5.5) and 25% PEG 3350 at room temperature. Crystals of GRASP55(1–215) were obtained in buffer containing 0.1 m Tris (pH 8.5), 25% PEG 3350, and 0.2 m (NH4)2Ac at room temperature.
Data Collection and Structure Determination
Diffraction data for GRASP65 and GRASP55 crystals were collected at 100 K on beamline BL17U1 at the Shanghai Synchrotron Radiation Facility and beamline 3W1A at the Beijing Synchrotron Radiation Facility, respectively. For cryoprotection, crystals of GRASP65 were soaked in buffer containing mother liquor supplemented with 20% glycerol. No cryoprotectant was used for crystals of GRASP55 prior to data collection due to the fragility of the crystals. All datasets were processed using the HKL2000 package (31). Both structures were determined by molecular replacement using the GRASP55 structure (Protein Data Bank code 3RLE (17)) as a search model. The preliminary models were refined using the Phenix program (32), and manual adjustments of residue positions were performed in Coot (33). The data collection and refinement statistics are summarized in Table 1. All figures for the structures were prepared using PyMOL.
TABLE 1.
X-ray crystallographic data and refinement statistics for the crystals of GRASP65 and GRASP55
| Crystals |
||
|---|---|---|
| GRASP65 | GRASP55 | |
| Data collection | ||
| Space group | P21212 | P43212 |
| Wavelength (Å) | 0.9795 | 0.9793 |
| Unit cell dimensions | ||
| a (Å) | 44.99 | 83.08 |
| b (Å) | 104.29 | 83.08 |
| c (Å) | 37.93 | 145.15 |
| α, β, γ | 90°, 90°, 90° | 90°, 90°, 90° |
| Molecules/ASUa | 1 | 2 |
| Resolution (Å)b | 2.2 (2.28-2.20) | 2.7 (2.82-2.70) |
| Completeness (%)b | 99.3 (96.0) | 98.2 (93.2) |
| Redundancyb | 6.4 (5.6) | 10.9 (10.1) |
| No. of total reflections | 60,785 | 156976 |
| No. of unique reflections | 9540 | 14,443 |
| I/σb | 17.5 (2.4) | 10.7 (1.6) |
| Rsymb,c | 6.3 (27.4) | 8.3 (56.5) |
| Refinement statistics | ||
| Resolution (Å) | 2.2 | 2.7 |
| No. of reflections | 9498 | 14,320 |
| Rwork/Rfree (%)d,e | 20.33/26.46 | 20.24/28.99 |
| No. of atoms | ||
| Protein | 1549 | 3231 |
| Ligand/ion | 2 | 0 |
| Water | 139 | 91 |
| B-factors (Å2) | ||
| Protein | 32.37 | 68.34 |
| Water | 34.27 | 46.17 |
| r.m.s.d. | ||
| Bond length (Å) | 0.008 | 0.008 |
| Bond angle | 1.169° | 1.112° |
| Ramachandran analysis | ||
| Most favored (%) | 79.6 | 80.2 |
| Additional allowed (%) | 17.4 | 16.1 |
| Generously allowed (%) | 3.0 | 3.7 |
| Disallowed (%) | 0 | 0 |
a ASU, asymmetric unit; r.m.s.d., root mean square deviation.
b Values in parentheses are for the highest resolution shell.
c Rsym = Σ|I − 〈I〉|/Σ〈I〉, where I is the observed intensity, and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections.
d R = Σhkl|Fobs| − |Fcalc|/Σhkl|Fobs|.
e Rfree was calculated from 5% of the reflections excluded from refinement.
Analytical Ultracentrifugation
Sedimentation velocity experiments were performed in a ProteomeLab XL-1 protein characterization system (Beckman Coulter, Inc.). All absorbance data were collected at a speed of 142,249 × g in an An-60 Ti rotor at 4 °C. A set of 999 scans was collected at 30-s intervals. Purified proteins were prepared in 500 mm NaCl and 50 mm Tris (pH 8.0) at a concentration of 50 μm. Data were analyzed using the programs SEDFIT and SEDPHAT (34, 35).
Mammalian Cell Culture, Transfection, and Brefeldin A (BFA) Treatment
HeLa or COS-7 cells were maintained at 37 °C with 5% CO2 in DMEM containing 10% fetal bovine serum and passaged every 2–3 days. For BFA sensitivity experiments, 60–70% confluent cells were split onto coverslips and transfected with various GRASP constructs using X-tremeGENE HP (Roche Applied Science). After 24 h, varying concentrations of BFA (Sigma) were added to the medium for 30 min. After washing out BFA, the cells were recovered for 2 h in fresh medium without BFA. Cells on the coverslips were used in subsequent indirect immunofluorescence microscopy experiments, and the remaining cells were harvested to monitor the expression levels of various GRASP constructs.
Cross-linking Experiments
HeLa cells were transfected with various GRASP constructs using Lipofectamine 2000 (Invitrogen). After 24 h, cells were washed with PBS and incubated with 400 μm ethylene glycol bis(succinimidyl succinate) (EGS) in PBS (pH 8.2) for 30 min at 37 °C. The reactions were quenched for 15 min with 1 m Tris (pH 7.5). Cells were then harvested and lysed with SDS-PAGE loading buffer. The total lysates were analyzed by SDS-PAGE and immunoblotting.
Immunofluorescence Microscopy
For immunofluorescence microscopy studies, transfected cells were fixed for 10 min at room temperature with 4% formaldehyde (Bio Basic Inc.) and permeabilized with 0.1% Triton X-100 (Bio Basic Inc.). The cells were washed twice with PBS and probed with primary antibodies, including mouse anti-Myc antibody (Sigma) diluted 1:400 or rabbit anti-GM130 antibody (BD Biosciences) diluted 1:400, for 45 min in PBS containing 3% calf serum, followed by incubation with various fluorophore-conjugated secondary antibodies for an additional 45 min (Alexa Fluor 488-conjugated anti-rabbit or Alexa Fluor 594-conjugated anti-mouse (1:500 dilution); Invitrogen). In some cases, the cells were then stained with DAPI for 5 min. To monitor the organelle morphologies in transfected cells, anti-calreticulin antibody (Abcam) or anti-cytochrome c antibody (BD Biosciences) was used. All images were captured on a Leica TCS SP5 confocal microscope with a 63×/1.40 numerical aperture Plan Apochromat oil immersion objective lens using Leica LAS AF version 1.3.1 build 525 software. Changes were made linearly across the entire image using Adobe Photoshop. To categorize Golgi morphology, serial images were captured using a Carl Zeiss Axio Imager Z1 upright fluorescence microscope with a 40×/1.40 numerical aperture objective lens.
RESULTS
Crystal Structure of the GRASP Domain of GRASP65
We first engineered an E. coli expression construct that encodes rat GRASP65(1–228) and the N-terminal His6 tag. This region of GRASP65 includes the entire GRASP domain and a small portion of the SPR domain in which several phosphorylation sites have been identified. Crystals of GRASP65 were obtained ∼1 month after the crystallization setup. A 2.2-Å dataset was collected, and the structure was determined by molecular replacement (Table 1), but only residues 12–210 were visible in the electron density. SDS-PAGE analysis of the crystals revealed that the protein was slightly degraded (supplemental Fig. S1A). We modified the construct to exclude residues 211–228. The same crystals of GRASP65 were obtained within a few days, indicating that the original crystals contained a fragment covering residues 1–210.
In the structure of the GRASP domain of GRASP65, each of the two PDZ domains adopts a canonical PDZ fold (Fig. 1B) with a β-sandwich of five β-strands and two α-helices (supplemental Fig. S1B). The PDZ1 and PDZ2 domains are virtually superimposable (Fig. 2A), consistent with their high sequence homology. Only one GRASP65 molecule is present in the asymmetric unit, but a GRASP65 dimer of interest is generated by a crystallographic 2-fold axis (Fig. 1B). In the dimer, the PDZ2 domains interact in a way that positions the peptide-binding pockets facing each other. In addition, the dimers are linked through interactions between the two C-terminal tails (CTs) of one dimer and two peptide-binding pockets of the PDZ1 domains in the next dimer (Fig. 1C). These results suggest that GRASP65 utilizes two distinct interfaces in the GRASP domain, both involving the peptide-binding pocket of the PDZ domain, to achieve oligomerization.
FIGURE 1.

Crystal structures of the GRASP domains of GRASP65 and GRASP55. A, domain structure of GRASP65 and GRASP55. The asterisk indicates the region enriched with phosphorylation sites. B, structure of the GRASP domain of rat GRASP65(1–210). The PDZ1 domains in the dimer are shown in blue and orange, and the PDZ2 domains are shown in cyan and yellow. A molecular surface is superimposed on the ribbon representation of the dimer. The CTs following the PDZ2 domains are highlighted. Zinc and chloride ions are shown as green and gray spheres, respectively. A different view of the dimer is shown below. C, crystal packing of the GRASP domain of GRASP65. The ribbon representations of six symmetry mates are colored as in B and labeled A–F. D, same as described for B, but with the GRASP domain of rat GRASP55(1–215). The PDZ1 domains are shown in dark green and purple, and the PDZ2 domains are shown in light green and violet. E, crystal packing of the GRASP domain of GRASP55. The ribbon representations of six symmetry mates are colored as in D and labeled A–F. See also supplemental Fig. S1.
FIGURE 2.
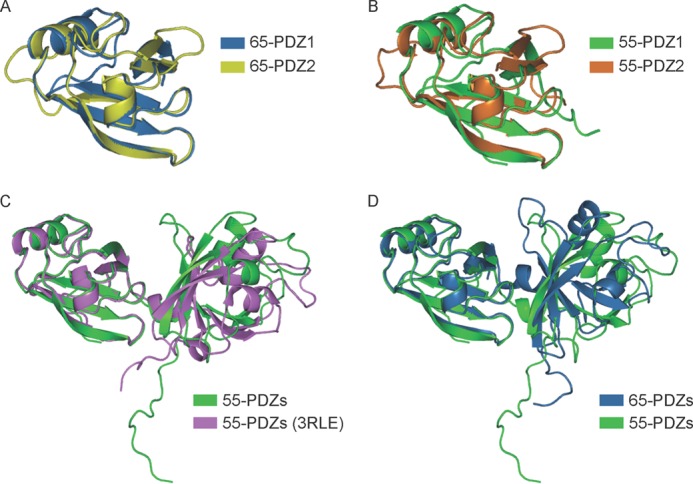
Structural comparisons of the GRASP proteins. Shown is the superposition of the PDZ domains of GRASP65 (A), GRASP55 (B), GRASP55 determined here and previously (C) (17), and GRASP65 and GRASP55 (D) in ribbon representation. The PDZ domains are colored as indicated.
Crystal Structure of the GRASP Domain of GRASP55
We determined the crystal structure of the GRASP domain (residues 1–215) of rat GRASP55 at 2.7-Å resolution (Table 1). This construct of GRASP55 is very similar to GRASP65(1–210) and seven residues longer than the one used in previous studies, in which the structure of the tandem PDZ domains of GRASP55 was reported (17). The conformation of the PDZ domains in our structure of GRASP55 is nearly identical to that in the previous structure (Fig. 2C). The structure is also very similar to that of GRASP65 (Fig. 2D), suggesting that GRASP55 may have the same types of homotypic interactions. In fact, a non-crystallographic dimer is seen in the structure (Fig. 1D), in which the two PDZ2 domains interact as in the structure of GRASP65. The symmetry operation reveals that, similar to GRASP65, the GRASP55 dimer inserts the two CTs into the PDZ1 domain pockets in the neighboring dimers (Fig. 1E); only in this case, one dimer engages two other dimers. These results suggest that GRASP65 and GRASP55 utilize a similar mode of oligomerization.
PDZ2-PDZ2 Interface
The dimerization of the GRASP domain mediated by PDZ2-PDZ2 interactions is symmetric and highly conserved between GRASP65 and GRASP55 (Fig. 3, A and B). The interaction buries ∼450 Å2 of total surface area, which is relatively modest compared with the typical interfaces of a stable interaction (>1000 Å2). β-Strand 5 (residues 189–200 for GRASP65 and residues 190–201 for GRASP55) of one of the PDZ2 domains forms extensive antiparallel interactions with the same β-strand of the PDZ2 domain in the other protomer (Fig. 3, A and B). α-Helix 2 in the two protomers meets on top of β5, contributing a Phe (Phe-149 in GRASP65 and Phe-150 in GRASP55) from each side to pack against a Tyr (Tyr-195 in GRASP65 and Tyr-196 in GRASP55) in β5 (Fig. 3, A and B). However, an Asp (Asp-147 in GRASP65 and Asp-148 in GRASP55) in one of the PDZ2 domains is positioned in close proximity to its equivalent residue in the other protomer, creating electrostatic repulsion that possibly weakens the dimer formation. Notably, the typical peptide-binding pocket of the PDZ domain is localized in a cleft between β5 and α2; the formation of a GRASP dimer prevents peptide recognition by the PDZ2 domain.
FIGURE 3.

Homotypic interactions of the PDZ2 domain. A, stereoview of the PDZ2-PDZ2 interface in the GRASP65 dimer. The two protomers are shown in cyan and yellow, respectively, and relevant amino acids are indicated. Interacting elements from the yellow protomer are labeled with asterisks. B, same as described for A, but with the GRASP55 dimer. The two protomers are shown in green and purple, respectively. C, the sizes of wild-type (wt) GRASP65(1–210) (theoretical molecular mass of 25.76 kDa) and the indicated mutants (all at 50 μm) were determined by analytical ultracentrifugation. D, wild-type or mutant GRASP65 (residues 1–210) was subjected to gel filtration on a Superdex 200 column. mAU, milli-absorbance units. E, HeLa cells were transfected with wild-type or mutant GRASP65-Myc and treated in the absence or presence of 400 μm EGS. The lysates were analyzed by SDS-PAGE and immunoblotting (IB) with anti-Myc antibodies. Asterisks indicate the number of GRASP65 molecules in the cross-linked product. Black arrowheads indicate degraded GRASP65, and the white arrowhead indicates cross-linked product containing degraded protein. A short exposure of the immunoblot with anti-Myc antibodies shows the levels of GRASP65. The level of β-actin was monitored as a loading control. Molecular mass markers are shown in kilodaltons. See also supplemental Fig. S2.
To test the dimerization properties of the GRASP domain, we performed analytical ultracentrifugation with wild-type or mutant GRASP65 (residues 1–210). Wild-type GRASP65 behaved as a monomer (Fig. 3C), which is consistent with the relatively small interface and the potential charge repulsion. However, some dimers were formed when Asp-147 was mutated to Asn (Fig. 3C), presumably due to the removal of repulsion and generation of reciprocal hydrogen bonds by the new side chains. The tendency of dimer formation can be disrupted by additional substitution of Phe-149 with Ala (Fig. 3C). Similar changes in the molecular weights were observed by gel filtration chromatography (Fig. 3D). When an additional 18 residues were included at the C terminus, an increase in dimerization/oligomerization was observed, even without the D147N mutation (supplemental Fig. S2, A and B). We tested GRASP55(1–215) using the same methods with equivalent mutations, but both the wild-type and mutant proteins exhibited very little dimerization in solution (supplemental Fig. S2, C and D), suggesting an even weaker dimer interaction than that of GRASP65.
To gain further insight into the PDZ2-PDZ2 interactions in a cellular context, we performed chemical cross-linking experiments in mammalian cells. When HeLa cells expressing Myc-tagged full-length GRASP65 were treated with EGS, an amine-reactive bifunctional cross-linker, GRASP65 dimers were observed (Fig. 3E). Similar to our in vitro results, the D147N mutation resulted in an increased level of cross-linking, whereas the F149A mutation abolished cross-linking (Fig. 3E). We also found that, unlike the full-length protein, a fragment of GRASP65 containing residues 1–210 yielded nearly no cross-linked dimer (supplemental Fig. S3A). Only when the D147N mutation was introduced were some dimers observed upon EGS treatment, but these interactions were disrupted when Phe-149 was mutated (supplemental Fig. S3A). To test the possibility that GRASP65 cross-links to a nonspecific protein, HeLa cells were cotransfected with Myc- and HA-tagged GRASP65 and treated with EGS. Both tagged proteins were detected in the cross-linked product (supplemental Fig. S3B), the molecular weight of which matches a dimer, suggesting that the cross-linking less likely involves other proteins. Collectively, in light of the crystal structures, these results suggest that the GRASP domain forms a weak dimer between the PDZ2 domains and that residues following the GRASP domain may contribute to the homotypic interactions of the GRASP proteins.
PDZ1-CT Interface
The CTs of the GRASP domains of GRASP65 and GRASP55 associate with the PDZ1 domain of a neighboring molecule. The interactions are similar in that the hydrophobic pocket in the PDZ1 domain is formed by β5 and α2, and part of the CT forms an antiparallel β-sheet with β5 (a conventional feature of PDZ domain-mediated peptide binding), but different residues are involved. In GRASP65, Tyr-209 in the CT is surrounded by Leu-54 of α2 and Val-99 of β5 (Fig. 4A). Phe-16 in β1 also facilitates binding (Fig. 4A). In contrast, Ile-213 and Leu-215 in the CT of GRASP55 interact with Leu-55 and Leu-59 in α2 and Ile-100 in β5 (Fig. 4B). In addition, Lys-54 in α2 reaches Glu-208 in the CT to form a salt bridge (Fig. 4B).
FIGURE 4.

PDZ1-CT interactions. A, stereoview of the PDZ1-CT interface in the GRASP65 structure. A ribbon representation of the PDZ1 domain is shown in blue, and the stick representation of the CT is colored in cyan. Relevant amino acids are indicated. The residue from the CT is labeled with an asterisk. B, same as described for A, but with the GRASP55 structure. A ribbon representation of the PDZ1 domain is shown in dark green, and the stick representation of the CT is colored in light green. C, HeLa cells were transfected with wild-type (wt) or mutant GRASP65-Myc and treated in the absence or presence of 400 μm EGS. The lysates were analyzed by SDS-PAGE and immunoblotting with anti-Myc antibodies. Asterisks indicate the number of GRASP65 molecules in the cross-linked product. The black arrowhead indicates degraded GRASP65, and the white arrowhead indicates cross-linked product containing degraded protein. The levels of β-actin were monitored as a loading control. D, same as described for C, but with wild-type or mutant GRASP55-Myc.
The interactions between the PDZ1 domain and CT appear to be weak, as purified GRASP domains exist as monomers in solution. As expected, the oligomerization states of GRASP65 were not altered when the CT was truncated (supplemental Fig. S2, A and B). To test the PDZ1-CT contacts in cells, we transfected HeLa cells with Myc-tagged wild-type GRASP65 or mutant Y209A and treated cells with EGS. Unlike wild-type GRASP65, fewer cross-linked dimers were seen with Y209A (Fig. 4C). Similarly, the I213A/L215A mutations in GRASP55 reduced the level of cross-linking (Fig. 4D). These results suggest that the PDZ1 domains and CT form weak, but important, interactions to facilitate oligomerization of the GRASP proteins.
Membrane Tethering by GRASP Proteins in Cells
To gain mechanistic insights into Golgi membrane stacking in cells, we treated cells with BFA to temporarily disrupt Golgi assembly and assessed the GRASP-mediated tethering of Golgi membranes with wild-type or mutant GRASP65 and GRASP55. BFA inhibits the activation of Arf1 GTPases, which are required for vesicular trafficking at the Golgi, causing subsequent absorption of the Golgi into the endoplasmic reticulum (27, 28). To carefully evaluate Golgi morphology, cells were categorized into three groups: intact, mildly fragmented, or scattered Golgi (Fig. 5A). When control cells were treated with BFA, a majority of the cells contained scattered Golgi (Fig. 5B). These defects in Golgi morphology were dose-dependent (Fig. 5C) and reversible when BFA was washed out (Fig. 5D). As expected, when the cells were transfected with wild-type full-length GRASP65 or GRASP55, the Golgi fragmentation induced by BFA was alleviated (Fig. 5B), but the morphologies of other organelles in these cells, such as the endoplasmic reticulum and mitochondria, were unaltered (supplemental Fig. S4). Thus, membrane tethering of GRASP proteins can be tested by judging the states of Golgi organization in response to BFA. Notably, expression of GRASP65 conferred sufficient resistance to BFA, even when BFA was added at 15 μg/ml, but with equivalent amounts of GRASP55, the same protection of the Golgi was seen only at a BFA concentration of 0.2 μg/ml (Fig. 5C). These results imply that GRASP65 has a stronger tendency to oligomerize, consistent with our in vitro analysis.
FIGURE 5.

BFA sensitivity assay in mammalian cells. A, the Golgi morphology in COS-7 cells was categorized using anti-GM130 antibodies with indirect immunofluorescence and confocal microscopy. Scale bar = 7.5 μm. B, COS-7 cells were transfected with GRASP65-Myc or GRASP55-Myc and stained with anti-Myc and anti-GM130 antibodies. The Golgi morphology of Myc-positive cells was analyzed as described for A in the absence or presence of 5 μg/ml BFA. At least 300 cells were categorized for each sample. All graphs are representative of three repetitions. Untransfected cells were used as a control. The levels of the indicated GRASP proteins analyzed by immunoblotting (IB) with anti-Myc antibodies are show below. The level of β-actin was monitored as a loading control. C, cells expressing GRASP65-Myc (left) or GRASP55-Myc (right) were treated with a gradient of BFA and analyzed as described for B. D, cells were treated with 5 μg/ml BFA, washed, and analyzed as described for B. See also supplemental Fig. S3.
Using the BFA sensitivity assay, we tested mutants that affect the PDZ2-PDZ2 interface. Cells expressing the F149A mutant of GRASP65 or the F150A mutant of GRASP55 had less intact Golgi (Fig. 6, A and B; see supplemental Fig. S5 for expression levels and supplemental Fig. S6 for localization), suggesting a defective ability of these mutant GRASP proteins to tether Golgi membranes. In contrast, the D148N mutant of GRASP55 led to a significant improvement in Golgi morphology compared with the wild-type protein (Fig. 6C), even though the same mutation caused very little dimerization of GRASP55(1–215) in vitro (supplemental Fig. S2, C and D). The F150A mutation in the GRASP55 D148N mutant abolished the enhanced tethering (Fig. 6D).
FIGURE 6.

Golgi membrane stacking mediated by the GRASP proteins. Wild-type (wt) or mutant GRASP proteins were subjected to the BFA sensitivity assay. BFA concentrations of 5 and 0.5 μg/ml were used for GRASP65 and GRASP55, respectively. A–D, mutations related to the PDZ2-PDZ2 interface. E–G, mutations related to the peptide-binding pocket of the PDZ1 domain. H and I, mutations related to the CT. For each sample, at least 300 cells were categorized as described in the legend to Fig. 5A. All graphs are representative of three repetitions. See also supplemental Fig. S4.
We transfected cells with mutants related to the PDZ1-CT interface and monitored the Golgi morphology in the absence or presence of BFA. Leu-54 in GRASP65 and Leu-59 in GRASP55 are located in the peptide-binding pocket of the PDZ1 domain. We found that substitution with Ala reduced Golgi tethering compared with the wild-type protein (Fig. 6, E and G). Similarly, C-terminal mutants Y209A in GRASP65 and I213A/L215A in GRASP55 failed to efficiently support Golgi tethering (Fig. 6, H and I). However, when Leu-57 in α2 of GRASP65, which is near the pocket but facing the opposite direction, was replaced with Ala, Golgi tethering was not affected (Fig. 6F). Collectively, the results of the BFA sensitivity assay suggest that the structural information derived from the GRASP domains is relevant to the Golgi tethering observed with the full-length proteins and functionally significant in vivo.
Zinc Finger in GRASP65
The BFA sensitivity assay suggested that GRASP65 may form stronger oligomers than GRASP55. When the structures of these two GRASP proteins are compared, the relative position between the two PDZ domains is different (Fig. 2D). The PDZ domains are closer to each other in GRASP65, rendering a more compact configuration than in GRASP55. Unexpectedly, a zinc finger-like structure is observed in the linker region between the PDZ domains of GRASP65 (Fig. 1B). Atomic absorbance spectrum analysis of the purified GRASP65 protein confirmed the presence of the zinc atom. The configuration of the zinc finger resembles the classical Cys2-His2, where His-17 and His-19 in β1 and Cys-102 in β5 participate in coordinating the zinc ion. The fourth site, which is normally occupied by a cysteine, is substituted with a chloride ion (Fig. 7A). The zinc finger may stabilize the linker between the two PDZ domains, which is supported by the formation of a short α-helix in the linker of GRASP65 but not GRASP55 (Fig. 7A). A sequence comparison of GRASP65 and GRASP55 reveals that GRASP55 has a Leu at the position of the second His residue (His-19 in GRASP65) (Fig. 7B), explaining the absence of the zinc finger in GRASP55. However, when the H19L mutant of GRASP65 was subjected to EGS cross-linking, no appreciable defects were observed (Fig. 7C), suggesting that the zinc finger of GRASP65 plays a minor role in oligomerization.
FIGURE 7.
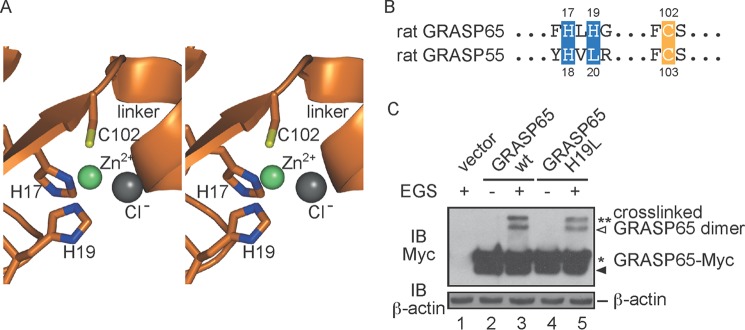
Zinc finger in GRASP65. A, stereoview of the zinc finger. Residues engaging the zinc ion are shown in stick representation. The zinc and chloride ions are shown as green and gray spheres, respectively. B, sequence alignment of the residues forming the zinc finger. C, HeLa cells were transfected with wild-type (wt) or mutant GRASP65-Myc and treated in the absence or presence of 400 μm EGS. The lysates were analyzed by SDS-PAGE and immunoblotting (IB) with anti-Myc antibodies. Asterisks indicate the number of GRASP65 molecules in the cross-linked product. The black arrowhead indicates degraded GRASP65, and the white arrowhead indicates cross-linked product containing degraded protein. The level of β-actin was monitored as a loading control.
DISCUSSION
The dynamic organization of Golgi cisternae plays an important role in Golgi biogenesis and function. Our results provide important insights into the mechanism of GRASP-mediated Golgi membrane stacking. We showed that the tandem PDZ domains can form a dimer via interactions between the two PDZ2 domains and that residues following the PDZ2 domain in one GRASP molecule can insert into the peptide-binding pocket of the PDZ1 domain of another GRASP. However, biochemical analysis suggested that these two sets of interactions are relatively weak, i.e. nearly not detectable in vitro using only purified GRASP domains. It is conceivable that the effective concentrations of the GRASP proteins on Golgi membranes are much higher than under the conditions we tested in vitro (29). In addition, the anchorage of the GRASP proteins onto the membranes may properly orient the molecules to yield efficient oligomerization. Nevertheless, the observation of similar ways of binding for two different proteins in two different crystal forms suggests that these interactions are likely of physiological relevance. In addition, mutagenesis studies using full-length proteins confirmed that disruption of either interface would drastically undermine the ability of GRASP proteins to stack Golgi membranes.
PDZ domains are well known molecular modules that mediate protein-protein interactions. The GRASP domain of GRASP65, which contains two tandem PDZ domains, is responsible for oligomerization and Golgi membrane stacking (14). A recent report proposed that the PDZ1 domain of GRASP65 is sufficient for homotypic interactions (30). Using gel filtration chromatography, we were able to confirm that the purified PDZ1 domain of GRASP65, but not the PDZ2 domain, is dimeric in solution (supplemental Fig. S2E). As the purified tandem PDZ domains appear to be monomeric under the same conditions, the absence of the PDZ2 domain could expose some hydrophobic surfaces in the PDZ1 domain, causing nonspecific dimerization of the PDZ1 domain. Our data support the notion that the GRASP domain, including the extended CT, is the minimal fragment for efficient oligomerization of the GRASP proteins and that the two PDZ domains cooperate to achieve dimerization and oligomerization.
The peptide-binding pockets of both PDZ domains in the GRASP proteins are formed by α2 and β5. A typical ligand peptide is predicted to form antiparallel β-strand interactions with β5 and inserts hydrophobic side chains between α2 and β5. Our crystal structures show that the CT of the GRASP proteins interacts with the PDZ1 domain in just such a manner; even the unconventional PDZ2-PDZ2 association generates an antiparallel β-sheet using the two β5s and utilizes hydrophobic packing via a Phe from α2. In the previous structural studies of GRASP55, the PDZ1 domain was speculated to bind to an internal ligand, residues 196–199 of β5 in the PDZ2 domain, whereas the PDZ2 domain engaged the C terminus of GM130 (17). Consistent with this proposal, hydrophobic residues from α2 and β5 of the PDZ1 domain (Leu-59 and Ile-100 in GRASP55) and part of β5 of the PDZ2 domain (residues 196–199) were shown to be critical for the tethering ability of GRASP55 (17). In addition, the residues involved in the GRASP65-GM130 association were mapped to β5 in the PDZ2 domain (residues 189–201) (6). Our new mutagenesis data (GRASP65 L54A and GRASP55 L59A) verified the importance of the PDZ1 pocket. In contrast, the additional mutations, including GRASP65 Y209A and GRASP55 I213A/L215A, suggest that the CT of the GRASP domain is an internal ligand. Furthermore, the combination of the D147N and F149A mutations in GRASP65 (Asp-148 and Phe-150 in GRASP55) strongly supported the homotypic dimerization of the PDZ2 domains. Consistently, it has been pointed out previously that Phe-150 in GRASP55 occludes the PDZ2 pocket (17). Although some of the PDZ2 domains may bind to the pairing golgins during the initial anchoring of the GRASP proteins, the PDZ2 domains may mainly self-associate to mediate dimerization of the GRASP proteins.
Based on the results presented here, the GRASP proteins may mediate the tethering of Golgi membranes in two different ways. One possibility is that GRASP molecules from the same membrane are connected into oligomers through PDZ1-CT interactions and zipper up two apposing membranes via PDZ2-PDZ2 association (Fig. 8A). In this model, the organization of the GRASP molecules represents the packing of the GRASP65 crystal structure. Alternatively, the GRASP molecules may form PDZ2-mediated dimer units on the same membrane and bridge the apposing membranes by reaching the PDZ1 domains on the other side with their CTs (Fig. 8B). This configuration of the GRASP proteins correlates partly with the GRASP55 crystal structure. Because of the difficulties in observing physiological oligomers in our assay, it remains unclear which model the GRASP proteins may utilize. In cells, efficient Golgi stacking may even involve a combination of both mechanisms. Our analytical ultracentrifugation, gel filtration, and in vivo cross-linking experiments also suggest that part of the SPR region, at least the residues immediately after the GRASP domain, provides additional interactions for the oligomerization of GRASP proteins.
FIGURE 8.
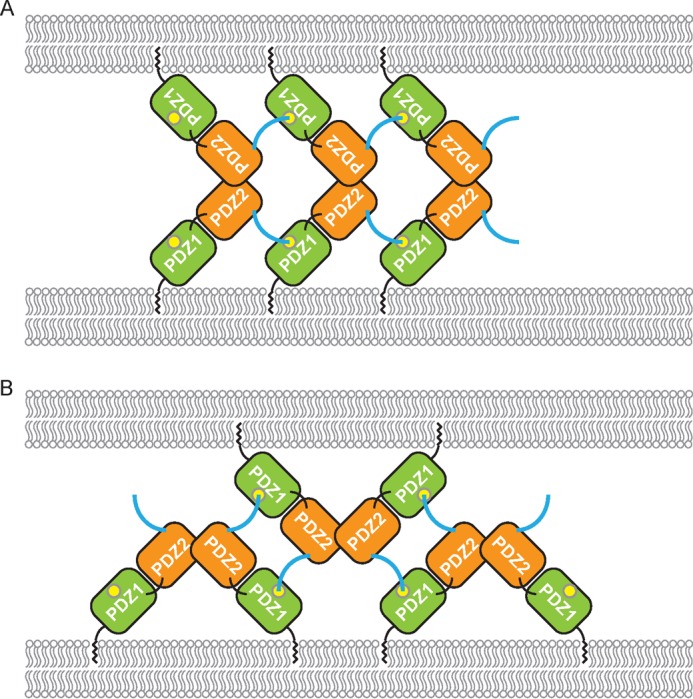
Models for GRASP-mediated membrane tethering. A, PDZ1-CT interactions cluster GRASP molecules on one membrane, and PDZ2-PDZ2 interactions bring apposing membranes together. The PDZ1 and PDZ2 domains are colored in green and orange, respectively. The CT of the GRASP domain is shown in cyan, and the peptide-binding pocket is shown as a yellow circle. B, same as described for A, but the PDZ2-PDZ2 interactions dimerize GRASP molecules on one membrane, and PDZ1-CT interactions bridge apposing membranes. See also supplemental Fig. S5.
Mitotic phosphorylation of the GRASP proteins is known to dissemble the oligomers. The region encompassing the phosphorylation sites may interact with a negatively charged surface and release upon the addition of negatively charged phosphates. Because most of these phosphorylation sites are clustered in the region close to the CT of the GRASP domain, detachment of the phosphorylated region may simultaneously displace the CT from the PDZ1 domain, weakening oligomerization. Our crystal structures of the GRASP proteins implicate a few potential binding surfaces for the phosphorylation sites (supplemental Fig. S7).
In general, the mechanism for GRASP oligomerization and subsequent Golgi membrane stacking could represent a common feature of membrane-tethering events in which weak interactions join forces in a regular pattern. The mechanism resembles that of the cell adhesion molecule cadherin, in which homotypic contacts in trans promote lateral clustering in cis through an otherwise weak interface (29), and could apply to other membrane-tethering systems.
Acknowledgments
We thank S. Hubbard for critical reading of the manuscript and Y. Wang for materials and helpful suggestions and discussions.
This work was supported by National Basic Research Program of China 973 Program Grant 2010CB911800 and National S&T Major Project on Major Infectious Diseases Grants 2012ZX10001006-002 and 2012ZX10001-008 from the Ministry of Science and Technology of China (to X. L.) and by National Basic Research Program of China 973 Program Grant 2010CB833702, National Science Foundation of China Grant 31225006, and an International Early Career Scientist grant from the Howard Hughes Medical Institute (to J. H.).

This article contains supplemental Figs. S1–S7.
The atomic coordinates and structure factors (codes 4KFW and 4KFV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- SPR
- serine/proline-rich
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BFA
- brefeldin A
- EGS
- ethylene glycol bis(succinimidyl succinate)
- CT
- C-terminal tail.
REFERENCES
- 1. Rambourg A., Clermont Y. (1990) Three-dimensional electron microscopy: structure of the Golgi apparatus. Eur. J. Cell Biol. 51, 189–200 [PubMed] [Google Scholar]
- 2. Farquhar M. G. (1985) Progress in unraveling pathways of Golgi traffic. Annu. Rev. Cell Biol. 1, 447–488 [DOI] [PubMed] [Google Scholar]
- 3. Cluett E. B., Brown W. J. (1992) Adhesion of Golgi cisternae by proteinaceous interactions: intercisternal bridges as putative adhesive structures. J. Cell Sci. 103, 773–784 [DOI] [PubMed] [Google Scholar]
- 4. Warren G. (1993) Membrane partitioning during cell division. Annu. Rev. Biochem. 62, 323–348 [DOI] [PubMed] [Google Scholar]
- 5. Barr F. A., Puype M., Vandekerckhove J., Warren G. (1997) GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 91, 253–262 [DOI] [PubMed] [Google Scholar]
- 6. Barr F. A., Nakamura N., Warren G. (1998) Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J. 17, 3258–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bachert C., Linstedt A. D. (2010) Dual anchoring of the GRASP membrane tether promotes trans pairing. J. Biol. Chem. 285, 16294–16301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y., Seemann J., Pypaert M., Shorter J., Warren G. (2003) A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. EMBO J. 22, 3279–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sütterlin C., Polishchuk R., Pecot M., Malhotra V. (2005) The Golgi-associated protein GRASP65 regulates spindle dynamics and is essential for cell division. Mol. Biol. Cell 16, 3211–3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xiang Y., Wang Y. (2010) GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J. Cell Biol. 188, 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shorter J., Watson R., Giannakou M. E., Clarke M., Warren G., Barr F. A. (1999) GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 18, 4949–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Short B., Preisinger C., Körner R., Kopajtich R., Byron O., Barr F. A. (2001) A GRASP55-Rab2 effector complex linking Golgi structure to membrane traffic. J. Cell Biol. 155, 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pfeffer S. R. (2001) Constructing a Golgi complex. J. Cell Biol. 155, 873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y., Satoh A., Warren G. (2005) Mapping the functional domains of the Golgi stacking factor GRASP65. J. Biol. Chem. 280, 4921–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doyle D. A., Lee A., Lewis J., Kim E., Sheng M., MacKinnon R. (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85, 1067–1076 [DOI] [PubMed] [Google Scholar]
- 16. Sengupta D., Truschel S., Bachert C., Linstedt A. D. (2009) Organelle tethering by a homotypic PDZ interaction underlies formation of the Golgi membrane network. J. Cell Biol. 186, 41–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Truschel S. T., Sengupta D., Foote A., Heroux A., Macbeth M. R., Linstedt A. D. (2011) Structure of the membrane-tethering GRASP domain reveals a unique PDZ ligand interaction that mediates Golgi biogenesis. J. Biol. Chem. 286, 20125–20129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin C. Y., Madsen M. L., Yarm F. R., Jang Y. J., Liu X., Erikson R. L. (2000) Peripheral Golgi protein GRASP65 is a target of mitotic Polo-like kinase (Plk) and Cdc2. Proc. Natl. Acad. Sci. U.S.A. 97, 12589–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jesch S. A., Lewis T. S., Ahn N. G., Linstedt A. D. (2001) Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol. Biol. Cell 12, 1811–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Preisinger C., Körner R., Wind M., Lehmann W. D., Kopajtich R., Barr F. A. (2005) Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J. 24, 753–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sengupta D., Linstedt A. D. (2010) Mitotic inhibition of GRASP65 organelle tethering involves Polo-like kinase 1 (PLK1) phosphorylation proximate to an internal PDZ ligand. J. Biol. Chem. 285, 39994–40003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Puthenveedu M. A., Bachert C., Puri S., Lanni F., Linstedt A. D. (2006) GM130- and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 8, 238–248 [DOI] [PubMed] [Google Scholar]
- 23. Feinstein T. N., Linstedt A. D. (2008) GRASP55 regulates Golgi ribbon formation. Mol. Biol. Cell 19, 2696–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Manjithaya R., Anjard C., Loomis W. F., Subramani S. (2010) Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 188, 537–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duran J. M., Anjard C., Stefan C., Loomis W. F., Malhotra V. (2010) Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 188, 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dupont N., Jiang S., Pilli M., Ornatowski W., Bhattacharya D., Deretic V. (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lippincott-Schwartz J., Donaldson J. G., Schweizer A., Berger E. G., Hauri H. P., Yuan L. C., Klausner R. D. (1990) Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell 60, 821–836 [DOI] [PubMed] [Google Scholar]
- 28. Klausner R. D., Donaldson J. G., Lippincott-Schwartz J. (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu Y., Vendome J., Shapiro L., Ben-Shaul A., Honig B. (2011) Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature 475, 510–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang D., Yuan H., Wang Y. (2010) The role of GRASP65 in Golgi cisternal stacking and cell cycle progression. Traffic 11, 827–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bernstein A. (2008) An HIV/AIDS vaccine: where do we go from here? Trends Microbiol. 16, 553–554 [DOI] [PubMed] [Google Scholar]
- 32. Craig R. B., Summa C. M., Corti M., Pincus S. H. (2012) Anti-HIV double variable domain immunoglobulins binding both gp41 and gp120 for targeted delivery of immunoconjugates. PLoS ONE 7, e46778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang M. Y., Yuan T., Li J., Rosa Borges A., Watkins J. D., Guenaga J., Yang Z., Wang Y., Wilson R., Li Y., Polonis V. R., Pincus S. H., Ruprecht R. M., Dimitrov D. S. (2012) Identification and characterization of a broadly cross-reactive HIV-1 human monoclonal antibody that binds to both gp120 and gp41. PLoS ONE 7, e44241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]