

ENDOCRINOLOGY'S ENIGMA
Not all subjects with vitamin D deficiency (VDD), as defined by presently accepted criteria,[1,2] have clinical, biochemical or radiological features suggestive of rickets/osteomalacia. This paradox has remained unresolved, though a genetic basis has been suggested for this clinical riddle.[3] Genetic polymorphism alone can explain just 5-10% variation in serum 25-hydroxy vitamin D (25OHD) levels. However, in clinical practice and population-based studies, one often encounters serum 25OHD levels <5 ng/ml suggesting severe VDD, without raised parathormone (PTH), bone turnover markers or characteristic bone scan.[4] At the same time, one can see evidence of florid rickets/osteomalacia at the serum 25OHD levels of 10-13 ng/ml.[5] These large differences cannot be explained satisfactorily by our current understanding of vitamin D-PTH-calcium-bone axis.
PTH MEDIATED ADAPTATION
Current literature and text book teaching suggest that vitamin D deficiency is linked with a parallel decrease in calcium absorption. This leads to systemic adaptation, mediated by an increase in PTH, which, by increasing conversion of serum 25OHD to active vitamin D metabolites, increases calcium absorption. Simultaneously, PTH itself releases calcium from bone by causing bone resorption [Figure 1]. This restores the calcium homeostasis, which is essential for many physiological functions. This theory, however, does not explain the marked variation seen in most population-based studies, where about 50% of subjects with serum 25OHD levels below 10 ng/ml do not show an adaptive rise in serum PTH.[6,7] Concern about this confusing, yet significant, controversy has been expressed by experts.[1] Most studies try to explain this by a variation in calcium intake,[5] or by genetic polymorphism.[3,4,5,6,7,8] However, this phenomenon is seen equally in populations with adequate[7] and low calcium intake.[6] Can there be a better explanation?
Figure 1.
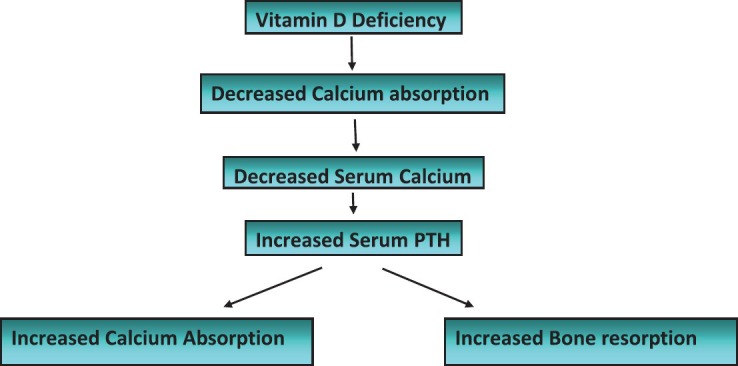
Systemic adaptation to vitamin D deficiency (Currently believed mechanism)
THE FALLACY OF SYSTEMIC ADAPTATION FIRST
There are certain fallacies of the theory of systemic adaptation to vitamin D deficiency, especially if we assume that systemic adaptation is the first response to this hormonal deficiency. This theory of adaptation is based upon an increase in PTH, which leads to increased calcium absorption, indirectly through generation of active vitamin D metabolites. Literature search reveals that there is paucity of studies assessing effect of PTH on calcium absorption directly.[9,10] In subjects with primary hyperparathyroidism, hypercalcemia is maintained by increased renal calcium re-absorption and bone resorption.[11] Similarly, in patients with hypoparathyroidism, hypocalcemia is caused by renal calcium wasting, rather than a decrease in calcium absorption.[12] However, PTH levels may affect calcium absorption indirectly, through enhanced generation of active vitamin D metabolites, which affect transcription of the calcium sensing receptor (CaSR).[13] In a study carried out in vitamin D-treated hypoparathyroid patients, calcium absorption was normal and comparable with normal controls.[14] This fallacy is further highlighted by studies in the CaSR and PTH knockout mouse. In this murine model, PTH is absent: As per the systemic adaptation theory, calcium absorption should not rise in the presence of hypocalcemia. On the contrary, these animals show increase in calcium absorption with hypercalcemic in response to oral calcium diet.[15] How can this be explained?
THE INTESTINAL CALCISTAT
Garg and Mahalle[16] explain this paradigm by invoking a local adaptive mechanism hypothetically named “intestinal calcistat.” “Intestinal calcistat” envisages the interaction of calcium sensing receptor (CaSR) with local intestinal vitamin-D and vitamin D receptors. The authors postulate that CaSR, present on the intestinal brush border, helps to sense intraluminal calcium present in the intestine, and modifies calcium absorption, by interacting with vitamin D system in intestinal cells. This is done according to the body's requirement and maintains calcium homeostasis. Calcium absorption occurs by active transcellular and passive paracellular process. This hypothesis further suggests that activation of CaSR in intestinal cells by dietary calcium decreases vitamin D-dependent active transcellular absorption and increases less effective vitamin D-independent passive paracellular calcium absorption. In support of this hypothesis, a recent study reported that CaSR present in renal tubules increase paracellular calcium re-absorption independent of serum PTH.[17] Moreover, CaSR and PTH knockout mouse show exaggerated hypercalcemic response to activated vitamin D metabolites. This is explained by an increase in TRPV-6 expression, which is responsible for increase in calcium absorption, even in the absence of PTH.[18] This may be due to removal of suppressive effect of CaSR on vitamin D system in intestinal cells.[18] This further supports the existence of a local intestinal mechanism (“intestinal calcistat”) for regulation of calcium homeostasis in absence of PTH.
Till this local adaptation is maintained, there will be sufficient calcium absorption to maintain calcium homeostasis, irrespective of serum 25OHD levels, there will be no systemic manifestations of VDD. On the other hand, if this local adaptation fails, systemic adaptation will come into play. Interaction of CaSR with different genetic polymorphisms of vitamin D system, which decides the generation or responsiveness of active vitamin D metabolites (“intestinal calcistat”) will decide the level of serum 25OHD, at which this local adaptation fails in a particular individual. This possibly explains the observed differences in PTH response to same levels of serum 25OHD [Figure 2].
Figure 2.
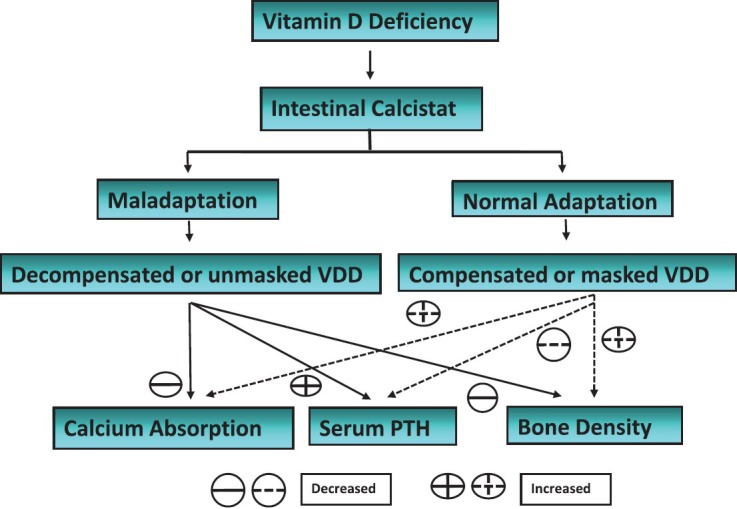
Local and systemic adaptation to vitamin D deficiency (Proposed mechanism)
HYPERCALCEMIA EXPLAINED
The systemically generated active vitamin D metabolites, secreted in response to PTH, largely act on bones to increase release of calcium, rather than increasing calcium absorption. This also explains the observed hypercalcemia with vitamin D intoxication, where hypercalcemia is mediated by bone resorption rather than increased calcium absorption.[19] However, active metabolites may modify calcium absorption indirectly through their effect on CaSR gene expression.[13] Vitamin D response elements (VDRE) have been identified in the promoter region of CaSR, which increase expression of CaSR. This physiologic process leads to increased renal calcium absorption and passive paracellular calcium absorption. With this background, hypercalcemia of vitamin D intoxication will respond best with drugs that inhibit bone resorption[20] or inhibit renal calcium absorption through action on CaSR (calcimimetics).[21]
This hypothesis also raises questions about traditional wisdom of explaining hypercalcemia associated with granulomatous disease. Increased calcium absorption has been suggested as a probable reason, secondary to enhanced generation of active vitamin D metabolites in granulomatous tissue.[22] There are no clinical studies to support this. Further studies are required to confirm or refute this explanation. According to the current hypothesis, hypercalcemia encountered in these conditions should be due to the effect of active vitamin D metabolites on bone, and should respond to bisphosphonates.
PEDIATRIC POINTS TO PONDER
Among patients with vitamin-resistant rickets type-II (due to mutation in vitamin D receptor), eucalcemic levels can be maintained by high calcium intake, which according to this hypothesis facilitates a vitamin D-independent increase in passive paracellular calcium absorption. However, this does not explain the dependency of calcium absorption on vitamin D from infancy until the end of puberty in these patients.[23] It may be possible that maturation of “intestinal calcistat” takes place during puberty. It is well-known that placental lactogen and prolactin increases calcium absorption through a vitamin D-independent mechanism.[24] Similarly, vitamin D-independent calcium absorption is also upregulated by estrogen.[25] These hormones may help in maturation of “intestinal calcistat” during puberty. If this is true, it will help in our understanding of increased risk of rickets in infants, children, and adolescents before puberty, where vitamin D is required for adequate calcium absorption. However, all these projections require further studies to help in better understanding of vitamin D-PTH-calcium-bone axis.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 Report on Dietary Reference Intakes for Calcium and Vitamin D from the Institute of Medicine: What Clinicians Need to Know. J Clin Endocrinol Metab. 2011;96:53–8. doi: 10.1210/jc.2010-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hollick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, Treatment, and Prevention of Vitamin D Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:1911–30. doi: 10.1210/jc.2011-0385. [DOI] [PubMed] [Google Scholar]
- 3.Garg MK, Kalra S. Rickets: Turns and twist in Gordian Knot. Indian J Endocrinol Metab. 2013;17:1–4. doi: 10.4103/2230-8210.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: Consequences for bone loss and fractures and therapeutic implications. Endocrinology. 2001;22:477–501. doi: 10.1210/edrv.22.4.0437. [DOI] [PubMed] [Google Scholar]
- 5.Aggarwal V, Seth A, Aneja S, Sharma B, Sonkar P, Singh S, et al. Role of calcium deficiency in development of nutritional rickets in Indian children: A case control study. J Clin Endocrinol Metab. 2012;97:3461–6. doi: 10.1210/jc.2011-3120. [DOI] [PubMed] [Google Scholar]
- 6.Marwaha RK, Tandon N, Garg MK, Kanwar R, Narang A, Sastry A, et al. Vitamin D status in healthy Indians aged 50 years and above. J Assoc Physicians India. 2011;59:706–9. [PubMed] [Google Scholar]
- 7.Valcour A, Blocki F, Hawkins DM, Rao SD. Effects of age and serum 25-OH-vitamin D on serum parathyroid hormone levels. J Clin Endocrinol Metab. 2012;97:3989–95. doi: 10.1210/jc.2012-2276. [DOI] [PubMed] [Google Scholar]
- 8.McGrath JJ, Saha S, Burne TH, Eyles DW. A systematic review of the association between common single nucleotide polymorphisms and 25-hydroxyvitamin D concentrations. J Steroid Biochem Mol Biol. 2010;121:471–7. doi: 10.1016/j.jsbmb.2010.03.073. [DOI] [PubMed] [Google Scholar]
- 9.Reeve J, Hesp R, Vealle N. Effects of therapy on rate of absorption of calcium from gut in disorders of calcium homoeostasis. BMJ. 1974;3:310–3. doi: 10.1136/bmj.3.5926.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torizumi K, Yamada R, Ota K, Ebisuno S. A study of the intestinal 47Ca absorption test employing a scintillation camera. Radioisotopes. 1987;36:227–31. doi: 10.3769/radioisotopes.36.5_227. [DOI] [PubMed] [Google Scholar]
- 11.Maruani G, Hertig A, Paillard M, Houillier P. Normocalcemic primary hyperparathyroidism: Evidence for a generalized target-tissue resistance to parathyroid hormone. J Clin Endocrinol Metab. 2003;88:4641–8. doi: 10.1210/jc.2002-021404. [DOI] [PubMed] [Google Scholar]
- 12.De Sanctis V, Soliman A, Fiscina B. Hypoparathyroidism: From diagnosis to treatment. Curr Opin Endocrinol Diabetes Obes. 2012;19:435–42. doi: 10.1097/MED.0b013e3283591502. [DOI] [PubMed] [Google Scholar]
- 13.Canaff L, Hendy GN. Human calcium-sensing receptor gene. vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J Biol Chem. 2002;277:30337–50. doi: 10.1074/jbc.M201804200. [DOI] [PubMed] [Google Scholar]
- 14.Mortensen L, Hyldstrup L, Charles P. Effect of vitamin D treatment in hypoparathyroid patients: A study on calcium, phosphate and magnesium homeostasis. Eur J Endocrinol. 1997;136:52–60. doi: 10.1530/eje.0.1360052. [DOI] [PubMed] [Google Scholar]
- 15.Kantham L, Quinn SJ, Egbuna OI, Baxi K, Butters R, Pang JL, et al. The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab. 2009;297:915–23. doi: 10.1152/ajpendo.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garg MK, Mahalle N. Calcium homeostasis, and clinical or subclinical vitamin D deficiency – Can a hypothesis of “Intestinal Calcistat” explain it all? Med Hypotheses. 2013 doi: 10.1016/j.mehy.2013.04.035. In press. [DOI] [PubMed] [Google Scholar]
- 17.Loupy A, Ramakrishnan SK, Wootla B, Chambrey R, de la Faille R, Bourgeois S, et al. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest. 2012;122:3355–67. doi: 10.1172/JCI57407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egbuna O, Quinn S, Kantham L, Butters R, Pang J, Pollak M, et al. The full-length calcium-sensing receptor dampens the calcemic response to 1alpha, 25(OH)₂ vitamin D3 in vivo independently of parathyroid hormone. Am J Physiol Renal Physiol. 2009;297:720–8. doi: 10.1152/ajprenal.00164.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nordin BE. Evolution of the calcium paradigm: The relation between vitamin D, serum calcium and calcium absorption. Nutrients. 2010;2:997–1004. doi: 10.3390/nu2090997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orbak Z, Doneray H, Keskin F, Turgut A, Alp H, Karakelleoglu C, et al. Vitamin D intoxication and therapy with alendronate (case report and review of literature) Eur J Pediatr. 2006;165:583–4. doi: 10.1007/s00431-005-0069-9. [DOI] [PubMed] [Google Scholar]
- 21.Brown EM. Clinical utility of calcimimetics targeting the extracellular calcium-sensing receptor (CaSR) Biochem Pharmacol. 2010;80:297–307. doi: 10.1016/j.bcp.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Fuss M, Pepersack T, Gillet C, Karmali R, Corvilain J. Calcium and vitamin D metabolism in granulomatous diseases. Clin Rheumatol. 1992;11:28–36. doi: 10.1007/BF02207080. [DOI] [PubMed] [Google Scholar]
- 23.Tiosano D, Hadad S, Chen Z, Nemirovsky A, Gepstein V, Militianu D, et al. Calcium absorption, kinetics, bone density, and bone structure in patients with hereditary vitamin D-resistant rickets. J Clin Endocrinol Metab. 2011;96:3701–9. doi: 10.1210/jc.2011-1432. [DOI] [PubMed] [Google Scholar]
- 24.Fudge NJ, Kovacs CS. Pregnancy up-regulates intestinal cal-cium absorption and skeletal mineralization independently of the vitamin D receptor. Endocrinology. 2010;151:886–95. doi: 10.1210/en.2009-1010. [DOI] [PubMed] [Google Scholar]
- 25.Van Cromphaut SJ, Rummens K, Stockmans I, VanHerck E, Dijcks FA, Ederveen AG, et al. Intestinal calcium transporter genes are upregulated by estrogens and the reproductive cycle through vitamin D receptor-in-dependent mechanisms. J Bone Miner Res. 2003;18:1725–36. doi: 10.1359/jbmr.2003.18.10.1725. [DOI] [PubMed] [Google Scholar]