

Abstract
Paragangliomas are rare neuroendocrine neoplasms arising in extra-adrenal chromaffin cells of autonomic nervous system and histologically akin to chemodectomas. They are rare, affecting about 1 in 2,000,000 population. It is a generic term applied to tumors of paraganglia regardless of the location. In rare instances, paragangliomas present around and involve the pancreas, thereby mimicking any one of the more common primary pancreatic lesions. Pancreatic paraganglioma is an extremely rare tumor. It grows slowly, so radical resection is recommended to achieve curability with good prognosis. These neoplasms present considerable diagnostic difficulty not only for the clinician and radiologist but also for the pathologist. Here, we report a case of a 55-year-old woman who presented with a left-sided abdominal swelling for 3 months duration, initially having clinical suspicion of an ovarian tumor. The radiological imaging revealed a lesion in the tail of pancreas with a differential diagnosis of pancreatic carcinoma and metastatic tumor. Only after exploratory laparotomy, the diagnosis was made as a rare case of pancreatic paraganglioma on the basis of histological examination and immunohistochemistry.
Keywords: Chromogranin A, immunohistochemistry, pancreas, paraganglioma
INTRODUCTION
Paragangliomas are tumors of paraganglia that are widely distributed from skull to bottom of the pelvis. They are commonly found in abdomen, pelvis, and in head and neck region. In abdominal region, most paragangliomas are located in adrenal glands or along the aorta. Pancreatic paragangliomas are rarely reported with clear-cut female preponderance and all described cases being adults of more than 40 years of age.
CASE REPORT
A 55-year-old lady presented to our hospital with a complaint of epigastric discomfort and swelling in left side of the abdomen for preceding 3 months along with slight dragging sensation. Her family history and past history was unremarkable. The physical examination of abdomen revealed a palpable, non-tender lump over left hypochondrium extending up to midline and mild tenderness in epigastrium. No palpable lymphadenopathy of supraclavicular, cervical, and inguinal region were detected. She was normotensive. Her total blood count was within normal limit, except low hemoglobin level. Biochemical investigations for urea, creatinine, and RBS showed normal physiological values. Urine examination for vanil mandelic acid (VMA) was within normal limits. Liver function tests and Chest X-ray were normal. The Ultrasonography revealed a lesion in the tail of pancreas with differential diagnosis of pancreatic malignancy and metastatic tumor. Further imaging by CECT revealed the lesion as multicystic, size of 17 cm × 19 cm [Figure 1] with an impression of a malignant pancreatic tumor. In color Doppler study, the tumor appeared to be highly vascular. The patient underwent exploratory laparotomy. On gross examination, the tumor was well-circumscribed, round, like a football with glistening surface reddish-brown in color and measured 17 cm × 19 cm. On cut section, the tumor revealed a thickened capsule of size 1.2 cm. The entire tumor mass underneath the capsule showed extensive necrosis, cystic, and hemorrhagic areas [Figure 2]. Microscopically, the tumor composed of oval to round cells arranged in an organoid and trabecular pattern, separated by a delicate vascular network, giving an appearance of classic Zellballen pattern [Figure 3]. The tumor cells have abundant acidophilic cytoplasm containing granules with moderate variation in size and shape and insignificant mitosis. Areas of variable necrosis and hemorrhage were noted [Figure 4]. Immunohistochemistry for chromogranin A showed characteristic punctate pattern of staining by cytoplasmic granules of tumor cells [Figure 5] and S-100 nuclear immunoreactivity shown by thin lining of sustentacular cells encircling the nests. The cytokeratin was non-reactive and Ki67 labeling was <2%. Therefore, morphology, immunohistochemical features were consistent with extra adrenal paraganglioma. The patient's post-operative course was unremarkable. There was no evidence of disease recurrence at 10 months of follow up with ultrasonography post-operatively.
Figure 1.
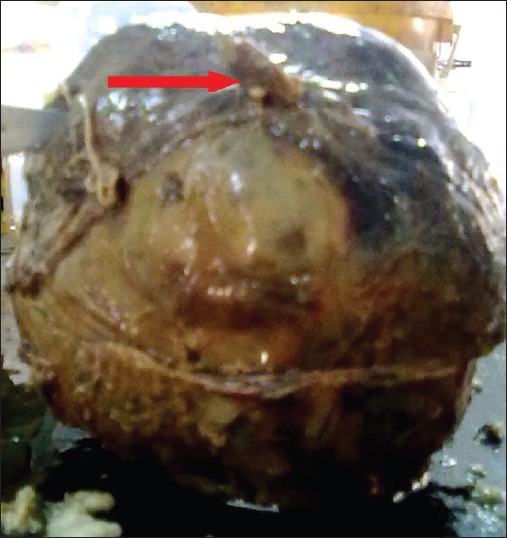
On gross examination, tumor measures 17× 19 cm,[2] well circumscribed, round, like a football, having variable nodularity externally with glistening surface, focal change of color from brown to chocolate. A tube-like structure on the top [indicated by big red arrow] was the stalk, attaching the tumor mass from pancreatic tissue
Figure 2.
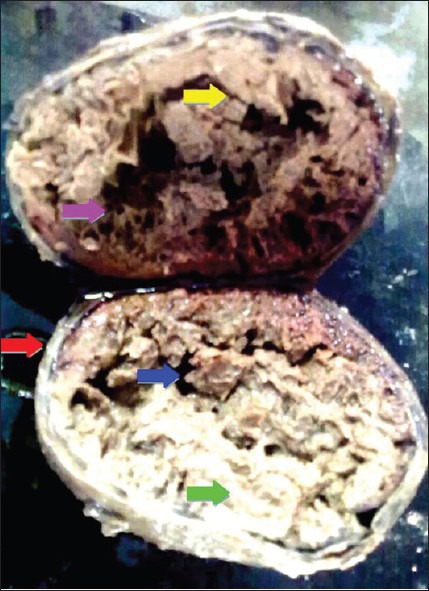
On cut section, the tumor revealed a thickened capsule of size 1.2 cm [red arrow]. The entire tumor mass underneath the capsule showed extensive necrosis, cystic degeneration [green arrow] and hemorrhagic areas forming dry layers, like those of thin nets [pink] and septae [yellow], containing empty spaces [blue arrow] within them
Figure 3.
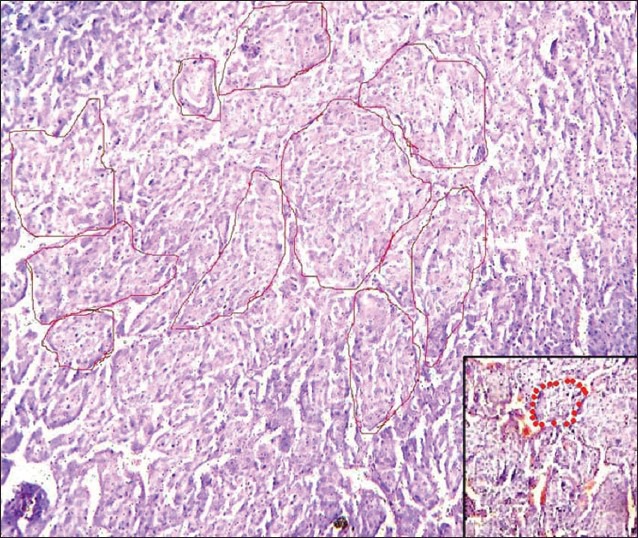
H and E section under light microscopy (×10) shows large, oval cells arranged in organoid and trabecular pattern separated by a delicate vascular network, which appear as red dots in a magnified view (see inset). The whole appearance, which is classically described as Zellballen pattern, meaning ‘well-defined nests,’ is prominent with encircling red lines
Figure 4.
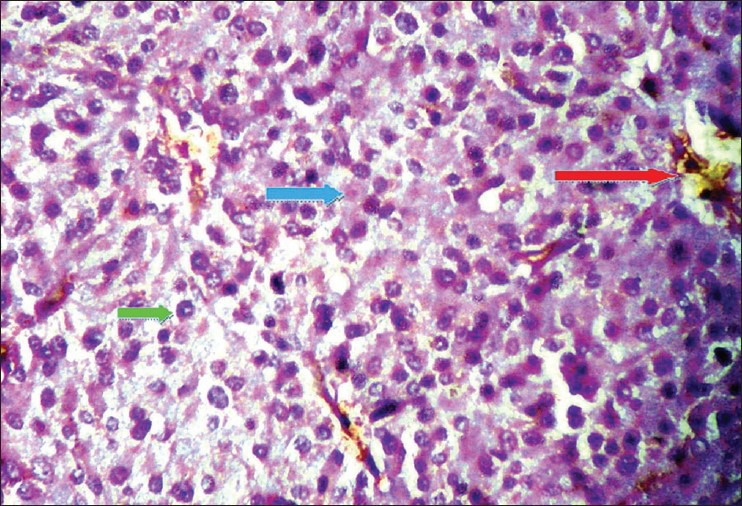
H and E section under light microscopy (×40) shows tumor cells have abundant acidophilic cytoplasm containing granules [blue arrow] with moderate variation in nuclear size and shape, some showing inclusion like structures [green arrow] and insignificant mitosis. Areas of necrosis, hemorrhage [red arrow] and cystic degeneration
Figure 5.
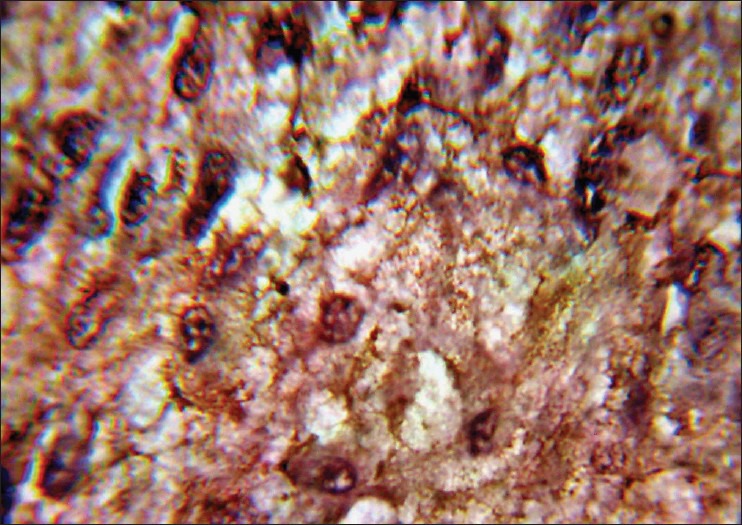
Immunohistochemistry for chromogranin A displayed positive immunoreactivity, a characteristic punctate pattern of staining by cytoplasmic large vesicle granules of tumor cells
DISCUSSION
Paragangliomas are rare neoplasms originating from paraganglia within the ganglia of the sympathetic trunk and of the coeliac, renal, suprarenal, aortic, and hypogastric plexuses. The paraganglia are clusters of neuroendocrine cells associated with the sympathetic and parasympathetic nervous system. Tumors arising from the chromaffin cells of the adrenal medulla are called pheochromocytomas. Extra-adrenal paraganglioma arising within the pancreas is an extremely rare entity with only 12 cases reported in the literature.[1] Paraganglioma is found more commonly in retroperitoneal location. It is equivocal whether paraganglioma of the pancreas represents true visceral origin with derivation from ectopic paraganglia or extension from a retroperitoneal tumor. In our case, the neoplasm was involving the pancreatic tail. Paragangliomas are reported at multiple locations throughout the pancreas, including the head and body,[2,3] but no case of paraganglioma in pancreatic tail has been reported in literature so far. Pancreatic paragangliomas are generally non-functional,[4] in contrast to tumors arising from other sites, where functional activity is more common. In tumors associated with symptomatic hypertension, headache, palpitations, and sweating, catecholamine secretion is reported in 30-60% of cases. Non-secretory tumors may present with abdominal pain or palpable mass or may be incidentally discovered during radiological imaging of the abdomen. Radiologically, paragangliomas show a homogeneous or heterogeneous hyper-enhancing soft tissue mass with cystic areas on CT and hypervascularity in color Doppler ultrasonography.[4] However, these features are not specific for paragangliomas, and a pancreatic neoplasm is a feasible differential diagnosis. Careful assessment of routine histology may raise the suspicion of paraganglioma and prompt immunohistochemical evaluation leads to confirmation.[5] In our case, pancreatic neoplasms were excluded by histology and immunohistochemistry. Malignancy is uncommon, and there is no definite histological criterion to differentiate between benign and malignant counterparts.[5] The use of an immunohistochemical panel, in addition to routine histology, can confirm the diagnosis of a paraganglioma and can give an indication of the likely prognosis for a patient.[6] Malignancy can only be definitively diagnosed with the development of metastases at sites where paraganglionic tissue is not normally found.[7] Histological features suggestive, but not diagnostic of malignant behavior, include mitotic activity, vascular invasion, and necrosis. Up to 50% malignancies are reported in retroperitoneal paragangliomas.[3]
Surgery for removal of the lesion remains the primary modality of treatment of paragangliomas. When unresectable, parangangliomas are treated with octreotide, which may ameliorate symptoms of catecholamine excess and stabilizes tumor size. Nevertheless, the pancreatic paragangliomas presented in the literature to date show a generally favorable long term outcome.
CONCLUSION
Pancreatic paraganglioma is a very rare entity with limited cases reported. When tumors are non-secretory and the intra-pancreatic location is variable, the diagnosis of paraganglioma pre-operatively is speculative, and malignant tumor is a feasible differential diagnosis. Surgical resection is necessary for histological assessment, and ancillary study like immunohistochemistry is crucial. Pancreatic paragangliomas are generally benign; however, the risk of malignant transformation justifies aggressive management and any resultant post-operative morbidity.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.He J, Zhao F, Li H, Zhou K, Zhu B. Pancreatic paraganglioma: A case report of CT manifestations and literature review. Quant Imaging Med Surg. 2011 doi: 10.3978/j.issn.2223-4292.2011.08.02. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mills Stacey E, Carter Darryl, Greenson Joel K, Oberman Harold A, Reuter Victor E, Stoler Mark H. 4th ed. Philadelphia: Lippincott Williams and Wilkins; 2004. Paraganglioma: Sternberg's Diagnostic Surgical Pathology; p. 67880. [Google Scholar]
- 3.Lightfoot N, Santos P, Nikfarjam M. Paraganglioma mimicking a pancreatic neoplasm: A case report. JOP. J Pancreas (Online) 2011;12:259–61. [PubMed] [Google Scholar]
- 4.Kim SY, Byun JH, Choi G, Yu E, Choi EK, Park SH, et al. A Case of primary paraganglioma that arose in the pancreas: The color doppler ultrasonography and dynamic CT features. Korean J Radiol. 2008;9:S18–21. doi: 10.3348/kjr.2008.9.s.s18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Awasthi NP, Kumari N, Krishnani N, Goel A. ′Functional′ paraganglioma of ureter: An unusual case. Indian J Pathol Microbiol. 2011;54:405–6. doi: 10.4103/0377-4929.81631. [DOI] [PubMed] [Google Scholar]
- 6.Kliewer KE, Wen DR, Cancilla PA, Cochran AJ. Paragangliomas: Assessment of prognosis by histologic, immunohistochemical, and ultrastructural techniques. Hum Pathol. 1989;20:29–39. doi: 10.1016/0046-8177(89)90199-8. [DOI] [PubMed] [Google Scholar]
- 7.Ardon H, Plets C, Sciot R, Calenbergh FV. Paraganglioma of the cauda equina region: A report of three cases. Surg Neurol Int. 2011;2:96. doi: 10.4103/2152-7806.82989. [DOI] [PMC free article] [PubMed] [Google Scholar]