

Abstract
The seasonal outbreaks of human rotavirus (RV) infection occur every winter. Most patients are diagnosed clinically by a rapid latex agglutination detection kit or polymerase chain reaction assays for RV from stool samples, but some problems have been reported on the specificity and sensitivity of such rapid detection assays. To ratify these issues, a sensitive, specific, simple, and rapid nucleic acid based diagnostic method is expected to be introduced and the reverse transcription loop-mediated isothermal amplification (RT-LAMP) was developed to detect the RV in human stool samples by incubation at 60 °C for 1 h and amplification was confirmed by electrophoretic laddering, restriction enzyme digestion, and hydroxynapthol blue discoloration. The assay established in this study was found to detect only the RVs and no cross-reaction with other viruses, demonstrating its high specificity. By using serial samples dilution as template, the detection limit of LAMP was 10 times more than that of PCR. The results showed the potential clinical feasibility of RT-LAMP as a useful diagnostic tool for the detection of RV with high sensitivity in comparison to conventional RT-PCR.
Keywords: Human, Rotavirus, Group A, NSP4 gene, RT-LAMP
Introduction
The human group A rotavirus (RVA) infection has become a major public health concern worldwide, with more than 80 % of the deaths occurring in the developing countries of South Asia and sub-Saharan Africa [18]. It is a major cause of acute dehydrating gastroenteritis in neonates. Of the 7 groups identified in rotaviruses (RVs), three groups [A, B, C] are known to affect human beings [15]. Of these, RVA is predominant in humans and its rapid accurate diagnosis is of paramount importance for the prevention of virus transmission, management of disease progression and neonatal fatality [13].
In the absence of suitable or convenient cell culture isolation system, human RVA diagnosis is based on serological tests like latex agglutination test, enzyme immunoassays, reverse transcription-polymerase chain reaction (RT-PCR) [11, 12, 27, 28]. Nucleic-acid-based detection techniques are currently the more widely accepted methods for detecting RVA. However, these assays, whether qualitative or quantitative, are relatively time-consuming, labor-intensive, and costly requiring specialized laboratory equipment. In resource-limited settings, the cost and technology requirement limit their universal application.
A new technique known as loop-mediated isothermal amplification (LAMP) introduced by Notomi et al. [17] provided a breakthrough into all these limitations and has been opted for detection of several important human and animal pathogens in the past [1, 3, 4, 8, 20, 26, 28]. It has been proven to be both cost and time effective, making it possible to have a reliable and confirmatory result within a short span of time. The LAMP reaction uses a set of four to six specific primers which recognizes distinct sequences on the target. It is estimated that target sequence got amplified up to 109 copies in less than an hour [17]. The non-structural protein-4 (NSP4) of RV, the first viral enterotoxin has been shown the potential as a suitable target for epidemiologic surveillance of RV infections in human beings [24]. Perusal of literature revealed that at present no human RVA detection assay using this methodology has been reported. We describe here development of a simple reverse transcription-loop-mediated isothermal amplification (RT-LAMP) method for rapid and sensitive detection of human RVA targeting consensus region of segment 10 encoding the NSP4 protein. The RT-LAMP assay was further evaluated on clinical samples collected from hospitalized children or Out Patient Department (OPD) patients visited Government Hospitals in the foothills of temperate western Himalayan region of India.
Materials and Methods
Clinical specimens
Sixty-two diarrhoeic faecal samples collected from patients who either admitted to Govt. Hospital, Haldwani (Uttarakhand) or visited OPD for treatment between the age group of 2 days to 45 years were used for evaluation in this study. Suspensions (10 % w/v) of faecal samples were made in 0.2 mM phosphate buffer saline (PBS) (pH 7.4; Sigma-Aldrich, St. Louis, USA) and centrifuged at 2,000 g for 20 min to remove coarse particulate matter and the upper aqueous layer was then filtered through 0.22 μm syringe filters and stored at −20 °C until required.
Nucleic acid extraction and cDNA synthesis of clinical samples
Total RNA was extracted from 500 μL of 10 % faecal suspension in PBS using an equal volume of TriReagent-LS (Sigma-Aldrich, St. Louis, USA). The RNA was eluted with nuclease free water (NFW) in a final volume of 25 μL. The isolated RNA was assessed qualitatively and quantitatively using Nanodrop Spectrophotometer (ND-1000, Thermo-Scientific, USA) and stored at −70 °C until further use. Reverse-transcription for cDNA synthesis from viral RNA was performed using 0.2 μg/μL random hexamer (Fermentas, Lithuania). Initially, 50–100 ng of viral RNA, 0.1 μg random hexamer, and 2 μL of dimethyl sulphoxide (DMSO) were added to PCR tube followed by incubation of the reaction mixture at 95 °C for 5 min for denaturing the RNA strands. The mixture was immediately snap chilled on ice followed by the addition of 4 μL of 5× RT buffer, 2 μL of 10 mM dNTPs (Fermentas, Lithuania), 40 U RNase Inhibitor (Ambion, USA) and 200 U MMLV-RT (Promega, USA) and kept at 37 °C for 90 min. The enzyme was denatured at 80 °C for 5 min at the end of the incubation step.
Designing of LAMP primers
To design an assay that can detect Human RVA, 64 individual sequences of the NSP4 gene of most common RVA strains from different geographical locations within India and the rest of the world were retrieved from the NCBI nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore). The sequences were assembled using the ClustalW program of Lasergene 6.0 software (DNASTAR Inc, USA) for the alignment of all the retrieved sequences. Through NSP4 nucleotide sequences alignment analysis, the conserved regions of NSP4 gene of Human RVA (GenBank accession number: HQ853486) were used for generating the set of primers. The two outer (F3 and B3) and two inner (FIP-forward inner primer and BIP-backward inner primer) primers were designed according to the guidelines provided by PrimerExplorerV4 (http://loopamp.eiken.co.jp/). In inner primers set, FIP consisted of F1c, complementary to the F1 sequence, and F2 sequence, and BIP consisted of the B1 sequence and B2c, complementary to the B2 sequence. The outer primers, F3 and B3 were located outside F2 and B2. The oligonucleotides sequences designed within the conserved regions of NSP4 gene are provided in Table 1. All primers were custom synthesized (Integrated DNA Technologies (IDT) Inc., India).
Table 1.
Group A human rotavirus NSP4 gene based oligonucleotides used in RT-LAMP and RT-PCR assays
| Assays | Primers | Sequences (5′–3′) |
|---|---|---|
| RT-LAMP | FIP | CTCTTACATTGCTGCAGTCACT-AGAATGGGAGAGTGGAAG |
| RT-LAMP | BIP | CGGCGGAGTTCTTCACAGTA-CTGACTGTGGCTTCTCAG |
| RT-LAMP/RT-PCR | F3 | AAAGAACGTGAAAACGCTAG |
| RT-LAMP/RT-PCR | B3 | CGGGATTAAGGCTTGAG |
Optimization of the LAMP assay
The plasmid constructs of partial Human RVA NSP4 gene (206 bp) was used as standard for the optimization of LAMP reaction conditions. In brief, the Human RV positive diarrhoeic faecal sample (UKD-5) retrieved from Indian Veterinary Research Institute, India was processed for viral ribonucleic acid (RNA) extraction using TriReagent-LS (Sigma, St. Louis, USA) followed by cDNA synthesis as described above. The partial length NSP4 gene of human RVA (GenBank accession number: HQ440219) was amplified by RT-PCR using forward and reverse primers: (5′-AAAGAACGTGAAAACGCTAG-3′ and 5′- CGGGATTAAGGCTTGAGC-3′), respectively. The specific RT-PCR amplicon (206 bp) was gel-purified by using QIAquick Gel Extraction kit (Qiagen, Germany) and cloned into pJET1.2/blunt cloning vector (Fermentas, Lithuania). The clones were screened for the presence of the insert by colony PCR and restriction digestion with 40 U BglII enzymes (Fermentas, Lithuania). The positive clones were used further as standard for evaluation of the sensitivity and specificity of the LAMP assay.
The optimized LAMP reaction mixture for a total of 25 μL volume contained 2 mM of hydroxynaphthol blue (HNB) dye, 40 pmol each of FIP and BIP, 5 pmol each of F3 and B3, 8 mM MgSO4, 1.4 mM deoxynucleoside triphosphates, 0.8 M betaine and 8 U of Bst DNA polymerase (New England Biolabs) in supplied buffer {20 mM Tris–HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4 and 0.1 % Tween-20}. The annealing temperature was checked from 55 to 65 °C with different incubation time durations for the best amplification of target sequence.
Analysis of the LAMP products
Visual detection
For naked-eye visualization, 2 mM of HNB dye (Sigma-Aldrich, St. Louis, USA) was added to the reaction mixture at the time of setting up of reaction. A change in the color of the reaction solution from violet to light sky blue indicate a positive reaction, while in case of negative reaction, there is no change in color.
Gel electrophoresis
The amplified LAMP reaction products were resolved in a 1.5 % agarose gel stained with ethidium bromide (0.5 μg/mL) for the confirmation of specific target sequence amplification. The LAMP reaction products were visualized and documented using Transilluminator-UV®300 (UVP Inc., Upland, USA).
Restriction enzyme digestion
The restriction enzyme, AluI (Fermatas, Lithuania) was used to digest LAMP reaction products to confirm amplified target sequence specificity. The amplified LAMP reaction products containing AluI restriction site at 89 bp, was incubated with AluI enzyme at 37 °C for 4 h. Digested products were analyzed in 1.5 % agarose gel electrophoresis.
Sensitivity and specificity of LAMP assay
For calculation of sensitivity, tenfold dilutions (10−1–10−7) of NSP4 gene plasmid construct were used in LAMP reaction as well as in the conventional PCR. The LAMP reactions were performed as mentioned earlier. The conventional PCR reaction mixtures consisted of 5 μL 10× PCR buffer, 2 mM MgCl2, 0.5 μL 10 mM dNTP, 0.2 μM of F3 and B3 primers, 1.25 U of KOD Hot Start DNA Polymerase (Novagen, Germany) with volume made up to 25 μL with NFW. The PCR reaction was performed with initial denaturation at 95 °C for 4 min to activate the KOD Hot Start DNA Polymerase followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 52 °C for 20 s, and extension at 70 °C for 30 s, followed by a final incubation at 70 °C for 10 min. The LAMP and PCR amplified products were resolved in 1.5 % agarose gel and visualized in Transilluminator-UV®300 (UVP Inc., Upland, USA).
The specificity of the LAMP primers was checked using RNA extracted from enteric viruses viz. picobirnavirus, group B and D RVs.
Evaluation of LAMP assay in field samples
To determine the assay’s performance on Human RVA isolates from the field, diarrhoeic faecal samples (n = 62) received from Govt. Medical College Hospital, Haldwani, Uttarakhand were tested using both the diagnostic NSP4 gene based conventional RT-PCR and RT-LAMP assays.
Results
Optimization of LAMP assay
The LAMP reactions were optimized using partial length NSP4 gene plasmid construct of human RVA as standard, where we got the best detectable amplification at annealing temperature of 62 °C within 60 min, followed by termination of the reaction at 85 °C for 5 min. Also, this assay successfully amplified the target sequence of the NSP4 gene in the human RV clinical samples as revealed by electrophoretic laddering in 1.5 % agarose gel electrophoresis (Fig. 1).
Fig. 1.
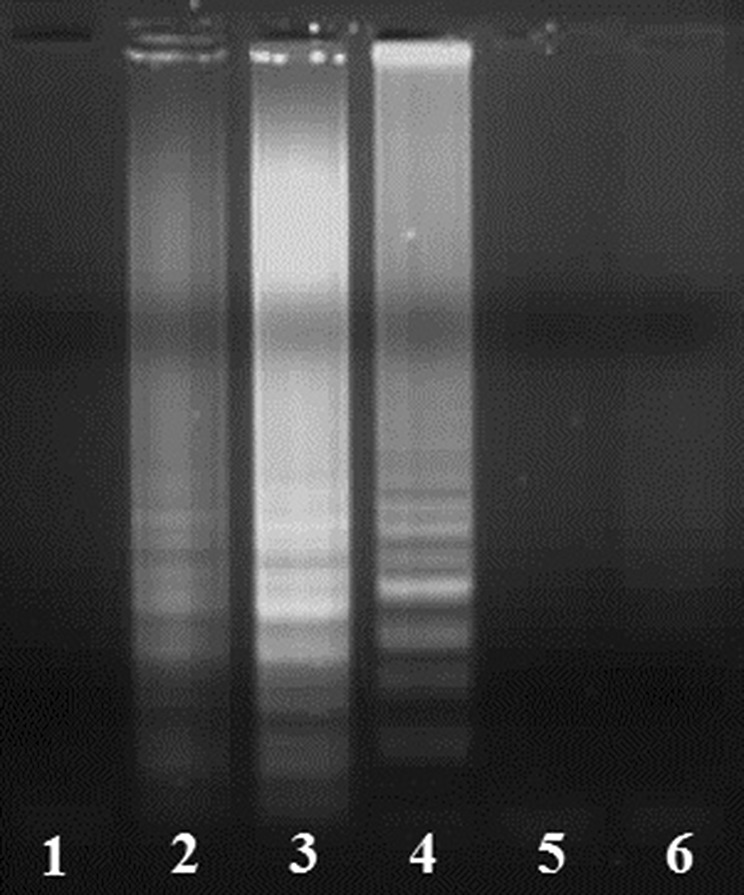
LAMP products resolved in 1.5 % agarose gel during optimization of LAMP reaction by varying the annealing temperatures. Lanes1–5 shows LAMP products obtained at annealing temperatures of 58, 60, 62, 63, 65 °C, respectively, and Lane 6 no template control (NTC)
Scrutiny of LAMP products
In the naked-eye visualization reaction, sky blue color obtained through intercalating dye HNB (Fig. 2a) corresponded exactly with the generation of products with the same ladder-like appearance. Further confirmation was done by restriction enzyme AluI digestion of LAMP reaction products where it produced predictable digested product sizes of 89 and 117 bp in 1.5 % agarose gel electrophoresis (Fig. 2b).
Fig. 2.

Analysis of LAMP products. a Naked-eye-visualization of HNB dye colored RT-LAMP amplified products of Human RVA clinical samples. Tubes1, 2, 4, 5 Human RVA positive samples (sky blue color); Tube3, 6, 7 Human RVA negative samples (violet color); Tube8 no template control (NTC). b Restriction enzyme analysis of LAMP products in 1.5 % agarose gel. Lane M 100-bp DNA ladder (Fermentas); Lane 1 LAMP products of Human RVA NSP4 gene without AluI digestion; Lane 2 LAMP products digested with AluI (arrowpoints 89 and 117 bp digests). (Color figure online)
Specificity of LAMP assay
The LAMP primers specificity was determined by checking the cross-reactivity with picobirnavirus, group B and D RVs. The primer sets demonstrated 100 % specificity for human RVA by amplifying members of all human RVA and no amplification in all other viruses tested (Fig. 3).
Fig. 3.

Inspections of specificities of LAMP primers with respect to other enteric viruses. Lanes1, 2, 5 Human RVA samples; Lane3 Picobirnavirus; Lane4 Group D rotavirus; Lane 6 Group B rotavirus. All the LAMP products were resolved in 1.5 % agarose after amplification at 62 °C for 1 h
Sensitivity comparison of conventional PCR and LAMP assays
The sensitivity for both conventional RT-PCR and LAMP assays was determined using log dilution of partial length NSP4 gene plasmid construct. LAMP assay detected 1.26 × 104 copy number of plasmid (Fig. 4a) whereas RT-PCR could detect only 1.26 × 105 copy number (Fig. 4b). Both these assays were also evaluated on diarrhoeic cases (n = 62) in human patients from Haldwani (Uttarakhand) and it was found that both these assays detected RV genome in 59.67 % (37/62) of the clinical cases.
Fig. 4.

Sensitivity comparison of conventional RT-PCR and RT-LAMP assays using human RVA partial length NSP4 gene plasmid construct. aLaneM 100-bp DNA ladder (Fermentas); Lanes2–8 LAMP products using plasmid log dilutions starting from 1 log to 7 log dilutions. bLaneM 100-bp DNA ladder (Fermentas); Lanes 2–8 RT-PCR products using plasmid log dilutions starting from 1 log to 7 log dilutions. The RT-PCR amplicon size (206 bp) is in good agreement with the expected product size. All products were electrophoresed on a 1.5 % agarose gel
Discussion
The RV infection is a leading cause of public health concern particularly in the developing countries. An early identification of RV is, therefore, critical for control of disease and to minimize losses to the human life. The RV clinical diagnosis mainly relies on antigen capture ELISA and nucleic acid based amplification techniques. Of these, RT-PCR has been proved to be a powerful, sensitive and robust tool for RV detection [9, 10, 14, 15, 21–23], but transferring RT-PCR to the field demands sophisticated and costly instrumentations. LAMP has been proved simple and efficient technique in detection of numbers of human and animal pathogens [1–5, 8, 16, 19, 20, 26, 28, 29]. Being simple, efficient and cost-effective technique, LAMP is considered as a more suitable field test for diagnosis of microbes. The naked eye visual detection of positive LAMP reactions is based on the turbidity produced due to the formation of white precipitate of magnesium pyrophosphate by-product. An increase in the turbidity of the LAMP reaction mixture due to the production of white precipitate correlates with the amount of DNA synthesized. Other DNA intercalating chemicals such as SYBR green [20], Picogreen [4], propidium iodide [7] and calcein [25] have also been used for clear visual differentiation between positive and negative reactions. More recently HNB, a metal indicator for easy detection of the positive LAMP reaction has come up with superior quality with regard to reducing contamination risks compared to other available dyes [6].
In this study, LAMP primers were designed targeting the conserved region of NSP4 gene of human RVA and reaction mixture conditions were optimized at annealing temperature of 62 °C with 60 min incubation. The LAMP products were detected and analyzed in three ways i.e. naked eye visual detection by change in color from violet to light sky blue in the reaction mixture due to intercalating HNB dye, resolving the LAMP products in 1.5 % agarose gel and further confirmation of products by restriction digestion by AluI. The reaction products observed in agarose gel as a ladder-like pattern were due to the formation of stem-loop DNAs of varying stem length and cauliflower-like structures with multiple loops formed by sequentially inverted repeats of the target sequence. Of these detection methods, the naked visualization way of detection is more suitable for detection of Human RVA in the field (Fig. 2a). The advantages of this method are due to its simple operation, rapid reaction, and potential for visual interpretation without instrumentation. This could make the technique far more suitable for field deployment.
The sensitivity of LAMP assay calculated using the partial length NSP4 gene plasmid construct of human RVA as standard was 1.26 × 104 copy numbers and it was found to be 100 % specific in amplification of only human RVA when checked with other enteric viruses. After going through optimization, sensitivity and specific barrier checks, this assay along with RT-PCR was evaluated on clinical samples (n = 62) obtained from patients either hospitalized or visited hospital for treatment in Haldwani (Uttarakhand). Comparative evaluation results of conventional RT-PCR and RT-LAMP assay showed 100 % specificity and correlation over the number of samples tested where both detected 59.67 % samples positive for RV genome. However, the sensitivity of RT-LAMP was found 10 times more than conventional RT-PCR assay.
The results from many of the past studies are based on the use of real time turbidimeter, thermal cycler and UV-transilluminator for the detection of pathogens, making this assay more expensive with dependence on expensive instruments, ultimately limiting the wide use of this procedure, especially in clinical laboratory of developing country. Therefore, the naked eye visualization method of LAMP product combined with more sensitive in comparison to RT-PCR makes this assay more suitable for field applications.
Overall, the results confirm that in comparison to conventional RT-PCR, RT-LAMP was consistently easy, rapid, cost-effective, and more sensitive. The development of LAMP assay enables detection of RVA in children within an hour and without requirement of sophisticated instruments and could therefore be invaluable in field diagnosis. As the assay is relatively economic and reliable, most probably LAMP will more widely integrate into routine molecular techniques. The LAMP assay provides a number of benefits for the diagnosis of RV. Since the assay is sensitive and rapid, and the isothermal amplification strategy used is not reliant upon expensive equipment, it is particularly suited for “front line” diagnosis of RV infection in modestly equipped laboratories, in field stations or in mobile diagnostic units.
Acknowledgments
The authors are thankful to the Director, Uttarakhand State Biotechnology Department for the financial support through project and Director, Indian Veterinary Research Institute (IVRI) for infrastructural support to carry out the research work. The authors are also thankful to Dean, Govt. Medical College Haldwani (Uttarakhand) for granting the permission for sampling.
References
- 1.Cai T, Lou G, Yang J, Xu D, Meng Z. Development and evaluation of real-time loop-mediated isothermal amplification for hepatitis B virus DNA quantification: a new tool for HBV management. J Clin Virol. 2008;41:270–276. doi: 10.1016/j.jcv.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 2.Chinsangaram J, Akita GY, Castro AE, Osburn BI. PCR detection of group A bovine rotaviruses in feces. J Vet Diag Invest. 1993;5:516–521. doi: 10.1177/104063879300500403. [DOI] [PubMed] [Google Scholar]
- 3.Curtis KA, Rudolph DL, Owen SM. Rapid detection of HIV-1 by reverse-transcription, loop-mediated isothermal amplification (RT-LAMP) J Virol Methods. 2008;151:264–270. doi: 10.1016/j.jviromet.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Dukes JP, King DP, Alexandersen S. Novel reverse transcription loop-mediated isothermal amplification for rapid detection of foot-and-mouth disease virus. Arch Virol. 2006;151:06–1093. doi: 10.1007/s00705-005-0708-5. [DOI] [PubMed] [Google Scholar]
- 5.Fukuta S, Iida T, Mizukami Y, Ishida A, Ueda J, Kanbe M, Ishimoto Y. Detection of Japanese yam mosaic virus by RT-LAMP. Arch Virol. 2003;148:1713–1720. doi: 10.1007/s00705-003-0134-5. [DOI] [PubMed] [Google Scholar]
- 6.Goto M, Honda E, Ogura A, Nomoto A, Hanaki K. Colorimetric detection of loop mediated isothermal amplification reaction by using hydroxynaphthol blue. Biotechniques. 2009;46:167–172. doi: 10.2144/000113072. [DOI] [PubMed] [Google Scholar]
- 7.Hill J, Beriwal S, Chandra I, Paul VK, Kapil A, Singh T, Wadowsky RM, Singh V, Goyal A, Jahnukainen T, Johnson JR, Tarr PI, Vats A. Loop-mediated isothermal amplification assay for rapid detection of common strains of Escherichia coli. J Clin Microbiol. 2008;46:2800–2804. doi: 10.1128/JCM.00152-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong TC, Mai QL, Cuong DV, Parida M, Minekawa H, Notomi T, Hasebe F, Morita K. Development and evaluation of a novel loop-mediated isothermal amplification method for rapid detection of severe acute respiratory syndrome coronavirus. J Clin Microbiol. 2004;42:1956–1961. doi: 10.1128/JCM.42.5.1956-1961.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hussein HA, Frost E, Talbot B, Shalaby M, Cornaglia E, el-Azhary Y. Comparison of polymerase chain reaction and monoclonal antibodies for G-typing of group A bovine rotavirus directly from fecal material. Vet Microbiol. 1996;51:1–7. doi: 10.1016/0378-1135(95)00202-2. [DOI] [PubMed] [Google Scholar]
- 10.Isegawa Y, Nakagomi O, Nakagomi T, Ishida S, Uesugi S, Ueda S. Determination of bovine rotavirus G and P serotypes by polymerase chain reaction. Mol Cell Probes. 1993;7:4–277. doi: 10.1006/mcpr.1993.1041. [DOI] [PubMed] [Google Scholar]
- 11.Kelkar SD, Bhide VS, Ranshing SS, Bedekar SS. Rapid ELISA for the diagnosis of rotavirus. Indian J Med Res. 2004;119:60–65. [PubMed] [Google Scholar]
- 12.Lauren J, Stockman LJ, Staat MA, Holloway M, Bernstein DI, Kerin T, Hull J, Yee E, Gentsch J, Parashar UD. Optimum diagnostic assay and clinical specimen for routine rotavirus surveillance. J Clin Microbiol. 2008;46:1842–1843. doi: 10.1128/JCM.02329-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Banyai K, Brister JR, Buesa J, Esona MD, Estes MK, Gentsh JR, Iturriza-Gomara M, Johne R, Kirkwood CD, Martella V, Mertens PPC, Nakagomi O, Parreno V, Rahman M, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Patton JT, Desselberger U, Van Ranst M. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG) Arch Virol. 2011;156:13–1397. doi: 10.1007/s00705-011-1006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagamine K, Watanabe K, Ohtsuka K, Hase T, Notomi T. Loop-mediated isothermal amplification reaction using a nondenatured template. Clin Chem. 2001;47:1742–1743. [PubMed] [Google Scholar]
- 15.Nagamine K, Hase T, Notomi T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes. 2002;16:223–229. doi: 10.1006/mcpr.2002.0415. [DOI] [PubMed] [Google Scholar]
- 16.Nemoto M, Ohta M, Tsujimura K, Bannai H, Yamanaka T, Kondo T, Matsumura T. Direct detection of equine herpesvirus type 1 DNA in nasal swabs by loop-mediated isothermal amplification (LAMP) J Vet Med Sci. 2011;73:1225–1227. doi: 10.1292/jvms.11-0065. [DOI] [PubMed] [Google Scholar]
- 17.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parashar UD, Gibson CJ, Bresse JS, Glass RI. Rotavirus and severe childhood diarrhea. Emerg Infect Dis. 2006;12:304–306. doi: 10.3201/eid1202.050006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parida M, Posadas G, Inoue S, Hasebe F, Morita K. Real-time reverse transcription loop-mediated isothermal amplification for rapid detection of West Nile virus. J Clin Microbiol. 2004;42:3–257. doi: 10.1128/JCM.42.1.257-263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parida M, Horioke K, Ishida H, Dash PK, Saxena P, Jana AM, Islam MA, Inoue S. Rapid detection and differentiation of dengue virus serotypes by a real-time reverse transcription loop-mediated isothermal amplification assay. J Clin Microbiol. 2005;43:03–2895. doi: 10.1128/JCM.43.6.2895-2903.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parwani AV, Rosen BI, Flores J, McCrae MA, Gorziglia M, Saif LJ. Detection and differentiation of bovine group rotavirus serotypes using polymerase chain reaction-generated probes to the VP7 gene. J Vet Diagn Invest. 1992;4:148–158. doi: 10.1177/104063879200400206. [DOI] [PubMed] [Google Scholar]
- 22.Reynolds DJ, Morgan JH, Chanter N, Jones PW, Bridger GC, Debney TG, Bunch KJ. Microbiology of calf diarrhea in southern Britain. Vet Rec. 1986;119:34–39. doi: 10.1136/vr.119.2.34. [DOI] [PubMed] [Google Scholar]
- 23.Taniguchi K, Wakasugi F, Pongsuwanna Y, Urasawa T, Ukae S, Chiba S, Urasawa S. Identification of human and bovine rotavirus serotypes by polymerase chain reaction. Epidemiol Infect. 1992;109:303–312. doi: 10.1017/S0950268800050263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.TdeM Tavares, Brito WM, Fiaccadori FS, Freitas ER, Parente JA, Costa PS, Giugliano LG, Andreasi MS, Soares CM, Cardoso D. Molecular characterization of the NSP4 gene of human group A rotavirus samples from the West Central region of Brazil. Mem Inst Oswaldo Cruz. 2008;103(3):4–288. doi: 10.1590/s0074-02762008000300011. [DOI] [PubMed] [Google Scholar]
- 25.Tomita N, Mori Y, Kanda H, Notomi T. Loop mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat Protoc. 2008;3:2–877. doi: 10.1038/nprot.2008.57. [DOI] [PubMed] [Google Scholar]
- 26.Toriniwa H, Komiya T. Rapid detection and quantification of Japanese encephalitis virus by real-time reverse transcription loop mediated isothermal amplification. Microbiol Immunol. 2006;50:7–379. doi: 10.1111/j.1348-0421.2006.tb03804.x. [DOI] [PubMed] [Google Scholar]
- 27.Tsunemitsu H, Jiang B, Saif LJ. Detection of group C rotavirus antigens and antibodies in animals and humans by enzyme-linked immunosorbent assays. J Clin Microbiol. 1992;30:2129–2134. doi: 10.1128/jcm.30.8.2129-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoneyama T, Kiyohara T, Shimasaki N, Kobayashi G, Ota Y, Notomi T, Totsuka A, Wakita T. Rapid and real-time detection of hepatitis A virus by reverse transcription loop-mediated isothermal amplification assay. J Virol Methods. 2007;145:162–168. doi: 10.1016/j.jviromet.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 29.Yousif AY, Anderson J, Chard-Bergstrom C, Bustamante A, Muenzenberger M, Austin K, Kapil S. Evaluation of a Latex Agglutination Kit (Virogen Rotatest) for detection of bovine rotavirus in fecal samples. Clin Diagn Lab Immunol. 2001;8:496–498. doi: 10.1128/CDLI.8.3.496-498.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]