

Abstract
Over the past several decades we have learned a great deal about microglia and innate brain immunity. While microglia are the principle innate immune cells, other cell types also play a role, including invading macrophages, astrocytes, neurons, and endothelial cells. The fastest reacting cell is the microglia and despite its name, resting microglia (also called ramified microglia) are in fact quite active. Motion photomicrographs demonstrate a constant movement of ramified microglial foot processes, which appear to be testing the microenvironment for dangerous alteration in extracellular fluid content. These foot processes, in particular, interact with synapses and play a role in synaptic function. In event of excitatory overactivity, these foot processes can strip selected synapses, thus reducing activation states as a neuroprotective mechanism. They can also clear extracellular glutamate so as to reduce the risk of excitotoxicity. Microglia also appear to have a number of activation phenotypes, such as: (1) phagocytic, (2) neuroprotective and growth promoting, or (3) primarily neurodestructive. These innate immune cells can migrate a great distance under pathological conditions and appear to have anatomic specificity, meaning they can accumulate in specifically selected areas of the brain. There is some evidence that there are several types of microglia. Macrophage infiltration into the embryonic brain is the source of resident microglia and in adulthood macrophages can infiltrate the brain and are for the most part pathologically indistinguishable from resident microglia, but may react differently. Activation itself does not imply a destructive phenotype and can be mostly neuroprotective via phagocytosis of debris, neuron parts and dying cells and by the release of neurotrophins such as nerve growth factor (NGF) and brain derived neurotrophic factor (BDNF). Evidence is accumulating that microglia undergo dynamic fluctuations in phenotype as the neuropathology evolves. For example, in the early stages of neurotrauma and stroke, microglia play a mostly neuroprotective role and only later switch to a neurodestructive mode. A great number of biological systems alter microglia function, including neurohormones, cannabinoids, other neurotransmitters, adenosine triphosphate (ATP), adenosine, and corticosteroids. One can appreciate that with aging many of these systems are altered by the aging process itself or by disease thus changing the sensitivity of the innate immune system.
Keywords: Immune surface receptors, immunoexcitotoxicity, innate immunity, microglia, microglial priming
SUMMARY OF PART I: IMMUNITY PRIMER
In part one, we explored the anatomy and functional activation of the various components of the systemic immune system. Basically, systemic immunity is divided into an innate system and an adaptive system. These two systems do not act independently, but rather exhibit intimate interactions so as to initiate a coordinated attack. The innate system reacts rapidly, acting as the first line of defense when the interior of the body is invaded, whereas the adaptive system reacts slower, but with far greater specificity – that is, it can identify individual molecular differences in invading organisms and other antigens.
Next to the central nervous system (CNS), the immune system is one of the most complex systems in the body. This complexity extends to its individual components, especially the various cells involved in immunity. The innate immune system contains a number of specialized immune cells located in tissues lining the respiratory air passages, the gastrointestinal (GI) tract's mucosa and the mucosal lining of the urogenital tract. It is here that most invasions take place. Combined with the physical barriers of the skin and mucosal cell layers, these innate immune cells provide a formable protective defense.
The adaptive immune system utilizes a system of presentation of invading antigens that involves specialized molecules such as complement, to aid in molecular identification for future reference. Macrophages within tissues play a major role in transporting the antigen to the regional lymph nodes so that they can be presented to lymphocytes where tagging for future reference occurs. Certain lymphocytes store memories of the antigen for later identification, should the same antigen reinvade the body. This then allows for very rapid responses, since the complex identification and recognition process can be avoided.
Of special importance to this discussion is the communication between the systemic immune system and the brain's own special innate immune system. Previous beliefs about the brain being an immunologically privileged site have now been modified once it was realized that this immune barrier was incomplete.
The brain does not contain its own adaptive immune system, but rather can transport antigens to regional cervical lymph nodes that can process the antigen and then direct activated lymphocytes and other immune cells back to the brain substance. Immune chemokines from microglia and astrocytes can direct systemic immune cells to the brain as well. In addition, the vagus nerve acts as a rapid response system to turn on brain innate immune cells when inflammation occurs within the GI tract or abdominal cavity. This two-way communication between the brain and the systemic immune system is critical. One can easily see that when the body is invaded, the brain must be rapidly informed of the invasion so that it can gear up its innate immune system in preparation of a possible invasion.
This gut – brain and systemic immune axis each play a major role in a number of neurological disorders, especially in the case of neurodegenerative diseases. We now know that when an infection or inflammatory condition occurs systemically, microglia are activated within specific brain areas within minutes. In fact, even minor surgical procedures have been shown to trigger microglial activation within the brain. This is especially important in treating patients with multiple injuries, since microglial activation worsens the neurological picture if prolonged or too intense.
Along this line, it should be appreciated that the immune system, both systemic and within the CNS, contains a great number of fail-safe systems and feedback controls to prevent over-activation of the immune process. In addition, complex cell signaling mechanisms are utilized to switch the brain's microglia so as to tailor the immune cell to the particular function that is needed. For example, with brain trauma microglia migrate to the site of the injury and are switched to a phagocytic phenotype so as to remove cellular debris. They can then be switched to a reparative phenotype to aid in repairing the damage. It is now apparent that dysfunction of this switching mechanism can play a major role in a number of neurological disorders should the microglia become stuck in a neurodestructive mode.
It is also important to appreciate that systemic immune cells, particularly macrophages, frequently enter the brain from the blood stream and once inside the brain take on the appearance and function of resident microglia. Under certain conditions, such as with brain trauma and CNS infections, large numbers of neutrophils, lymphocytes, mast cells, and macrophages enter the CNS. The functional state of these immune cells can make the difference between progressive brain destruction or a reversal of immunoexcitotoxicity. For example, macrophages can assume a primarily cytotoxic phenotype called M1 or an antiinflammatory phenotype called M2. Likewise, lymphocytes can be either cytotoxic or suppress brain cell inflammation and injury. The antiinflammatory lymphocytes, called regulatory T-cells or Tregs, are now recognized to play a critical role in protecting the brain. In fact, we now know that early assumptions concerning the pathological role of invading lymphocytes in experimental allergic encephalomyelitis (EAE), for example, were mistaken and that these were mostly Tregs attempting to combat the out of control inflammatory response.
Finally, several hormones and neurotransmitters also play a critical role in controlling immune reactions. One of the critical control systems is the immune–hypothalamo–pituitary axis, with corticosteroids playing a major role. Links between systemic immune activation and microglia within the hypothalamus trigger a release of corticosteroids, which under most conditions, dampens the immune reaction, both systemically and within the brain – but not always. Under certain conditions, these hormones can worsen inflammation.
Frequently forgotten is the fact that norepinephrine, epinephrine, and acetylcholine play a major role in dampening inflammation, especially within the CNS. A loss of acetylcholine levels in the Alzheimer's brain, for example, would significantly enhance any inflammatory reaction that is ongoing and may explain in part why acetylcholine agonists improve symptoms. With this brief review, now let us examine more closely the innate immune reactions within the CNS.
PART II: MICROGLIA
In 1932 Pio del Rio-Hortega introduced the concept of the microglia with incredible accuracy in the publication Cytology and Cellular Pathology of the Nervous system, citing many characteristics that would not be agreed upon until over half a century later. For instance, he accurately postulated that microglia entered the brain during early neurodevelopment and that these cells were of mesodermal origin. Further, that the guiding structures for the embryonic precursors included blood vessels and white matter tracts, which allowed these cells to migrate to all areas of the brain.
He was also the first to suggest that in the undisturbed brain microglia assumed a ramified (resting) morphology and upon disturbance or encountering specific brain pathology, they were morphed into an amoeboid phenotype. Del Rio-Hortega suggested that these cells could not only migrate within the brain toward areas of injury, but also could proliferate and carry out phagocytosis. The only error he made was to assume that microglia were evenly distributed throughout the brain. We now know that there is a heterogeneous distribution, with the highest concentrations in the gray matter and particularly within the limbic areas (hippocampus, amygdala, and entorhinal cortex) and substantia nigra, which has the highest microglial density of any area of the brain.[89,92]
Largely ignored until quite recently, microglia have now become the focus of a great deal of research. Newer studies suggest that microglia form a part of the tripartite synapse along with astrocyte foot processes and the synapse itself. Time lapse two-cell microphotography, extending up to 10 hours of observation, demonstrate that ramified microglia, the so-called resting phenotype, actually are quite active, with continuous extension and retraction of foot processes around the synaptic neuropile.[111] Active phagocytosis of cellular debris and chemicals was observed, suggesting a state of continuous surveillance of the microenvironment. This allows the microglia to maintain homeostasis and react rapidly when significant microenviromental threats exist. Microglia, along with astrocytes, also play a major role in maintaining low levels of extracellular glutamate, so as to prevent excitotoxicity and disturbed neural signals.
These newer studies also demonstrate that microglia are not uniform in their activity but exist as a number of subtypes, which offers an extensive program of activation states in response to a wide number of dynamic pathological conditions. In addition, microglia, as mentioned, are heterogenous in their distribution. Microglia found along brain–blood vessels are often found in an activated state and form a particular immunological barrier for the brain in conjunction with the blood–brain barrier (BBB).[88] Microglia are also concentrated at sites of incomplete BBB function, such as the circumventricular organs (CVOs), such as the organum vasculosum of the lamina terminalis, subcommissural organ, subfornical organ, area postrema, posterior pituitary, median eminence, pineal, and choroid plexus, as these are sites of entry of blood-borne invaders and even large molecules.[20]
Under pathological conditions, resident microglia may not only proliferate, but also can migrate great distances and accumulate near the sites of pathologic damage. Activation stages of microglia are not all or none, rather they are capable of assuming gradations of activation, which also includes priming phenotypes [Figure 1]. Likewise, we see phenotypes that are in various states of activity, including primarily phagocytic and neuroprotective; combined cytotoxic and neuroprotective or predominantly cytotoxic and destructive.[98,125] It is not clear whether these phenotypes are based purely on individual subtypes of microglia, or various activation states of a uniform type of microglia in response to the microenvironmental cues. Also likely are dynamic fluctuations in these activation phenotypes during the evolving pathological process. A number of stimulatory molecules within the microenvironment seem to determine the activation state, and one can see that as the microenvironment changes, the activation state of the microglia changes as well.
Figure 1.
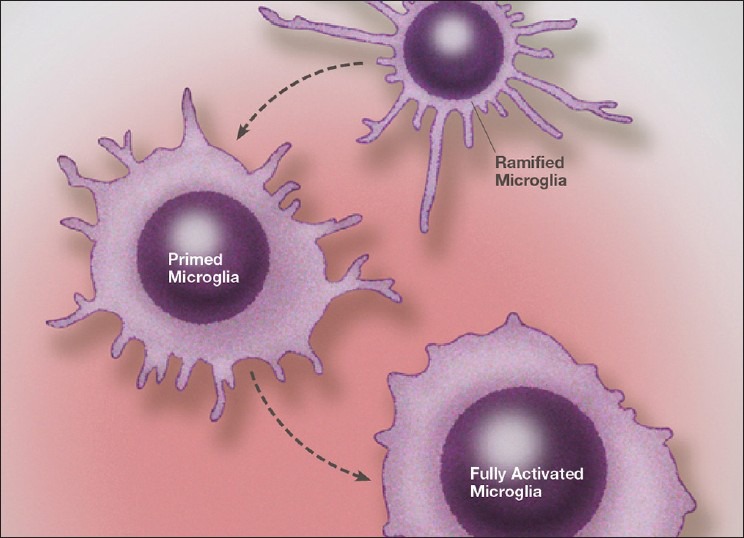
Illustration of the stages of microglial activation from a resting (ramified) state, to a primed state and finally to a fully activated phenotype. The exact phenotype and physiology of each stage of activation is determined by a number of extraneuronal molecules and environmental conditions
We are learning that much like the peripheral immune system, the brain's innate immune system is enormously complex. Further complexity is added when we consider the interaction between the innate immune system and the adaptive immune system composed of invading immune cells and immune factors from the periphery.
Developmental anatomy of microglia and of the innate CNS immune system
It is now accepted that microglia are derived from mesodermal/mesenchymal tissues, primarily myeloid cells from the bone marrow. These cells migrate to the CNS during the first trimester and throughout the early part of the second trimester in the human. There is evidence that during these stages microglia consist of two populations, which include (a) myeloid/mesenchymal cells and (b) a developmental and transitory forms of fetal macrophages.[35] These cells enter the fetal brain, traveling along blood vessels and nerve tracts and once within the parenchyma of the brain transform into resting microglial phenotypes. A population of amoeboid microglia is observed during later embryogenesis that appears to play an important role in synaptic pruning. For example, a recent study by Paolicelli et al. demonstrated a central role for microglia in synaptic pruning and circuit development in the developing embryonic brain.[116] Previous evidence suggested that resting microglia constantly monitor synaptic function and play a role in neurotransmission, but also were capable of synaptic stripping, even in the adult brain.
Clinical Correlation: Fractalkine, a cytokine modulator of microglial proinflammatory activation, and its receptor, located on microglial cell membranes, in general suppress inflammatory signaling. In the Paolicelli et al. study they found that microglia via stimulation by fractalkines, modulated synaptic pruning during neural development and that deficiencies in fractalkine or its receptor led to excessive synaptic connectivity and abnormal brain development. This condition also produced excessive sensitivity to excitatory neurotransmission and excitotoxicity-related disorders, such as seizures. While other factors are playing a role in embryonic brain pruning, such as the compliment cascade components C1q and C3, during the early stages of neurodevelopment, fractalkine is the primary factor.
While macrophages enter the developing brain in significant numbers; in the adult brain such an invasion is quite rare in the undisturbed brain. Under pathological conditions invasion of leukocytes is dramatically increased.
Molecular pathophysiology of microglia
Microglia membrane receptors, second messengers, and cell signaling
There many cell signaling pathways from receptors on the cell surface connecting to the genes in the nucleus. These signaling pathways are called second messengers, a collective term referring to a number of molecules in the signaling pathway. When the cell surface receptor is engaged by its specific stimulating extracellular activator, the intracellular signaling pathway is activated leading directly to the nuclear genes. These genes or gene, when activated, lead to the production of proteins that are made in the cytoplasm directed by mRNA, which copies the sequence of the gene in the nucleus and then travels to the cytoplasm to participate in the manufacture of the gene-specific protein on the ribosomes. These proteins then direct certain activities of the cell. These signaling pathways exist in all cells but there are some that are very important in microglia, which are discussed as follows.
Signaling Pathways and MyD88, a major coordinating signaling protein: Immune cell receptors, like most cell membrane receptors, are linked to intracellular signaling molecules that carry the messages to effector systems and nuclear gene activators. This process is called cell signaling and is central to the activation and suppression of molecular pathways in all cells. Cells contain thousands of cell signaling molecules, which are linked into functional pathways and when stimulated cause the cell to perform specific physiologic roles. Central to immune activation responses is the cell signaling adaptor molecule, myeloid differentiation primary-response protein 88 (MyD88), which acts like a coordinating molecule (called an adaptor molecule) linking surface receptors to immune cell signaling pathways within the cytoplasm. Mice lacking MyD88 cannot produce tumor necrosis factor-alpha (TNF-α) or Interleukin 12 (IL-12) when exposed to bacterial antigens.[138] Individuals with a genetically determined deficiency in MyD88 are subject to recurrent life-threatening infections of viral, bacterial and fungal types. Not all immune receptors are linked to MyD88.
Signaling Pathways containing Janus kinases: Another cell signaling pathway in immune cells and microglia involve the Janus kinases. Critical to immune function are the Janus family of kinases, Jak1, Jak 2, Jak3, and Tyk2, which are involved in cell growth, survival, development, and differentiation of immune cells.[66] These are special enzymes that transduce cytokine-mediated signals from the immune cell membrane to internal cell signaling pathways, such as JAK-STAT pathway, which in turn impart immune competence to the cell. The critical nature of these cell signaling molecules is seen with defects in Jak3, which leads to combined immunodeficiency syndrome.
Signaling Pathway From Cell Membranes to a gene through NFkB: One essential cell signaling link from the cell membrane is directed to Nuclear Factor kappa B (NFkB), which activates genes controlling the production and secretion of proinflammatory cytokines such as TNF-α. Activation of toll-like receptors (TLRs) and other immune receptors on the surface of microglia, macrophages, and other immune cells are linked to NFkB activation. This cell signaling molecule represents a major controlling mechanism of inflammation.
-
Signaling Pathways that reduce the inflammatory response of microglia and other immune cells:
- Other cell signaling mechanisms play an important role in reducing inflammation and potentially dangerous states of immune activation. For instance, MAPK phosphatase 1 (MKP1), is a major negative modulator of TLR-induced inflammation. You will recall the TLRs are a family of receptors on the cell membrane of immune cells that interact with antigens and other immune chemicals to initiate immune activation. By modulating these receptors, MAPK phosphatase 1 can fine-tune the immune reaction.
- Another immune response regulator is called a suppressor of cytokine signaling 1 (SOCS1), which is an encoded gene. When the SOCS1 gene is activated by extracellular produced cytokines, such as IL-2, IL-3, interferon-gamma (INF-γ), or GM-CSF, the microglial SOCS1 gene reduces the activation of proinflammatory cytokine cell signaling. That is, it also dampens the immune response. Another such immune inhibitory molecule TREM2, is found on the surface of myeloid cells (primarily monocytes and macrophages) from the bone marrow. This protein interacts with another protein produced by the TYROBP gene, which in turn reduces the proinflammatory signaling of microglia, macrophages and dendritic cells. Mutations of this gene, which have been found to be common in Alzheimer's cases, impairs this immune dampening effect, thus leading to chronic brain inflammation, as seen in many neurodegenerative diseases.[72]
There are a growing number of second messenger cell signaling molecules essential for modulating immune reactions. For a detailed discussion of immune cells signaling, see reference.[2]
Molecular pathophysiology of microglia
Immune receptors and microglia reactions
-
Immune Receptors leading to molecular activation: A great number of surface membrane receptors are seen on microglia, which not only act as “on/off” signals, but also modulate their responses to the microenvironment. For example, proinflammatory cytokines and glutamate can activate microglia toward a cytotoxic role and lead to destruction of synapses, dendrites, and whole neurons if not interrupted [Figure 2]. Other signals, acting through immune suppressing molecules, such as fractalkines, can switch the microglia to a resting and reparative state [Figure 3]. Pattern recognition receptors (PRRs) respond to pathogen associated molecular pattern (PAMPs) and damage associated molecular patterns (DAMPs), the last two of which are produced by pathogenic organisms and products of neuronal damage, respectively. What this means is that the particular molecular pattern of either the microorganism (PAMPs) or molecular signature of the debris of damaged cells (DAMPs) can react with specific activating receptors (PRRs) on the cell surface of microglia and lead to activation of the microglia's immune function. PAMPs activate various receptors and include TLRs, which trigger the release of cell surface activation molecules such as major histocompatibility complex class II (MHC class II), B7-1, B7-2, and CD40[5] [Figure 4]. These released molecules in turn stimulate the release of innate proinflammatory cytokines such as TNF-α, IL-1ß, IL-6, IL-12, IL-17, IL-23 as well as antiinflammatory cytokines IL-4, IL-10, and TGF-ß from microglia and invading macrophages.
One can easily see that either chronic activation of the brain's innate immune cells or even a condition where the cells are unable to switch off their immune activation, can lead to progressive destruction of synapses, dendrites or even the entire neurons or complex networks of neurons, as we see in neurodegenerative diseases. Examples of chronic microglial activation would include long-term systemic immune activation, as with autoimmune diseases and chronic infections (periodontal infections, chronic viral infections, Lyme disease, etc.). Neurotoxic metal accumulation within microglia or astrocytes, as seen with aluminum, lead, triethyl tin, and mercury exposure, also leads to continuous activation of microglia/astrocytes and macrophages.
Immune receptors leading to microglia phagocytosis: Stimulation of PRRs by PAMPs and DAMPs can also activate microglia phagocytosis.[88] When microglia are in the phagocytic phenotype they can either do so in a mode that releases neurodestructive cytokines or other such molecules or one that is not inflammatory. During phagocytic activity one sees greatly increased upregulation of cell-surface costimulatory molecules, which aid in engulfing the invaders or debris. The engulfed bacteria or viruses are then destroyed by a “respiratory burst” through upregulation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a producer of high levels of superoxide.[152] The respiratory burst of highly concentrated free radicals within the immune cells (macrophages, microglia, and polymorphonuclear white blood cells [WBCs]) kills the microorganisms and then allows the proteosomal enzymes to eliminate the debris.
Upregulation of Fc receptors by pathogens: Exposure to pathogenic organisms also upregulates Fc receptors on microglia and macrophages, which aid phagocytosis by opsonizing the antigen. Opsionization is a process whereby antigens are attached to larger molecules (opsonins) that make phagocytosis easier by giving the phagocytic microglia a larger target to grasp. Microglial responses to cellular debris from dying cells either mounts a phagocytic response without a release of proinflammatory cytokines or combines phagocytosis with proinflammatory cytokine release.[70,71,132]
-
Toll-Like Receptors as Antigen-Specific Receptors: Human microglia recognize bacteria, fungi, viruses, parasites, and the host itself, by utilizing specific toll-like receptors 1-9 (TLRs 1-9) located on the microglia cell membrane. Like peripheral macrophages, brain microglia demonstrate specific ligand reactions to TLR subtypes. For example, lipopolysaccharides (LPS), which make up the outer wall of Gram-negative bacteria, react specifically with TLR4, petidoglycan (PGN) (from cell walls of Gram-positive bacteria) reacts with TLR2, unmethylated CpG-DNA (viruses) reacts with TLR9, polyI: C (synthetic double-strand RNA) responds with TLR3 and West Nile virus RNA signals via TLR3[113,169] [Figure 5].
In experimental studies, vaccinations and in human infections, TLR4 is of vital importance. In conjunction with CD14, TLR4 makes up the primary receptor for LPS the cell wall component of Gram-negative organisms [Figure 6]. LPS is also found in many human vaccines. This receptor is upregulated when the brain is inflamed, as in the case of endotoxemia.[94] CD14 is a helper protein that aids the immune TLR in reacting to the particular antigen. Once the TLR activates the microglia, the exact reaction phenotype (phagocytic, reparative or cytotoxin/inflammatory) depends on the signaling message traveling through the cell signaling pathways. That is, the particular molecular pattern of the antigen (via PRRs, such as TLRs) determines the reaction state of the immune cell.
Cellular debris has a different molecular pattern and operates through Damage Associated Recognition Receptors (DAMPs). What activates the DAMPs is that the debris contains either internal cellular components that normally would not be in contact with the immune cell surface receptor or a mutated molecule is formed from the damaging process that is then recognized as a foreign molecule. Again, enzymes within the proteasome and lysomes digest the foreign protein and debris and either remove it from the body or utilize the proteins for cell repair.
In some instances, as with persistent viruses and neurotoxic metal accumulation, the offending antigen cannot be removed and thus acts as a constant source of immune stimulation. We see this with aluminum accumulation, where the highest concentration is within microglia. This would keep the microglia in an activated proinflammatory state, which could lead to a chronic state of immunoexcitotoxicity and subsequent neurodegeneration.
Clinical Correlation: There is growing evidence that excessive or overreactive responses to certain vaccines or groups of vaccines are responsible for many of the complications associated with vaccinations, including encephalomyelitis, seizures, autism spectrum disorders, and the recently described macrophagic myofascitiis.[7,14,16,65] The mechanism for these untoward reactions could be based both on the intense, prolonged peripheral stimulation by the aluminum-containing adjuvants (even lasting several years) or localized microglial priming/activation by immune adjuvant molecules accumulating within astrocytes and microglia within affected brain areas. Studies have shown that aluminum and mercury both accumulate within the brain following vaccination.[61,126]
Both metals can cause prolonged microglial activation and mercury has been shown to kill astrocytes, its primary site of accumulation.[13,36,105] A loss of astrocytes can result in not only a massive release of stored glutamate upon astrocyte cell death, but also a long-term loss of one of the brain's major regulators of extracellular glutamate. The use of aluminum adjuvants in vaccines should be discouraged and in my opinion, such vaccines should be used only in cases where no alternative is available and in the prevention of very high-risk, communicable diseases of childhood. A discussion of the vaccine issue is beyond the scope of this paper, but growing evidence suggest we should reevaluate the number of and manner in which these vaccines are given.
Neuropathic Pain: Clinical Correlation: TLR4 has also been implicated with neuropathic pain, for example, as that associated with transection of the L5 nerve root, thus suggesting microglial reactions to DAMPs. A number of studies have linked the production of chronic pain to a triggering of immune-excitotoxic pathways, which are initiated by activated microglia.[103,134,151] Microglial activation in spinal ganglion and central pain pathways trigger the release of proinflammatory cytokines and this increases glutamate receptor sensitivity with prolonged activation of these receptors. This immunoexcitotoxic reaction increases nociceptive signaling, resulting in chronic, unrelenting pain syndromes.[83]
-
Microglia and TLR Molecular specificity: Free gangliosides and sialic acid-containing glycosphingolipids found in neural membranes, are known to activate microglia via TLR4, offering another way cellular debris can activate microglia.[85] This explains the frequent finding of reactive antibodies to these neuronal components with head injuries and strokes. It is possible that this produces a state of microglial priming, which would mean that subsequent systemic immune activation from any cause, trauma, surgery, autoimmune disease, etc., would increase immunoexcitotoxic reactions in the damaged areas.
TLR7 is vital for microglial reactions to viruses such as the herpes simplex virus[60] [Figure 7]. Besides being integral players in the release of proinflammatory cytokines, chemokines and interferons, TLR2, TLR4 and TLR9 on the microglial surface induce microglial production of IL-6 and IL-10, IL-6 and TNF-α and TNF-α and nitric oxide (NO), respectively.[88] The proinflammatory cytokines increase the production of a number of free radicals, especially the super oxide radical, which in itself is a rather mild radical. Yet, when it combines with NO, it forms the very powerful peroxynitrite radical, which is found in high concentration is a number of neurodegenerative diseases. So, the increase in NO seen with activation of these immune receptors becomes very important.
Other Microglial actions: Microglia also contain a number of scavenger receptors, which can identify modified lipoproteins and various polyanionic ligands. These include scavenger receptors of class A1 (SR-A1), SR-B1, and CD36 that are differentially regulated by microglia in response to various pathologies.[88] These receptors play a major role in defense against bacteria and can be upregulated in response to injury and cytokines as well. That is, they respond to a wide assortment of pathological insults.
-
· Scavenger receptors.
-
Scavenger receptors are upregulated in Alzheimer's disease (AD) in reaction to amyloid beta (Aß).[3] In this way, as the amyloid progressively accumulates and is deposited in the extracellular space it acts as a source of continued microglial stimulation, with resulting release of a number of toxic molecules. Activated microglia are known to accumulate within amyloid plague in the Alzheimer brain.Another important scavenger receptor in neurodegenerative diseases, receptor for advanced glycation end-products (RAGE), recognizes advanced glycation end products produced as a reaction to elevated glucose levels in the brain. The RAGE receptors on microglia are upregulated in the presence of Aß, which enhances plaque phagocytosis.[174]
-
-
MAC1 Receptor (The Integrin or Compliment Receptor)
- The MAC1 receptor (macrophage antigen complex 1), also called integrin CD11b/CD18 or complement receptor 3 (CR3), functions both as an adhesion molecule and a PRR capable of recognizing diverse ligands.[117] It also plays a critical role in phagocytosis activation in response to a number of compounds. Activation of this microglial receptor can trigger a respiratory burst and activation of neutrophils and macrophages.
Figure 2.
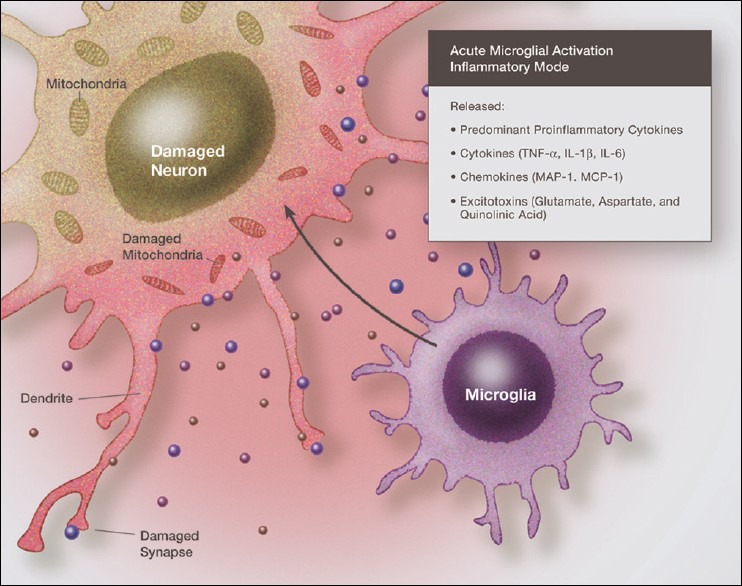
Illustration of fully activated microglia in a neurodestructive phenotype, with the release of proinflammatory cytokines, chemokines and excitatory amino acids, all acting in concert to damage the surrounding neurons
Figure 3.
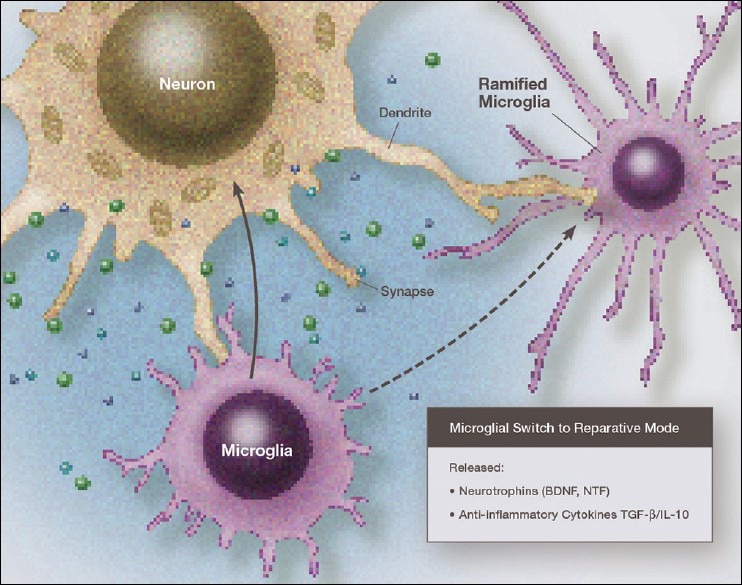
Illustration of a microglia in a primary reparative phenotype, which can be in either the resting mode or an activated-reparative mode, both of which release neurotrophic factors and antiinflammatory cytokines. This repairs the damage done during neurodestructive microglial activation
Figure 4.
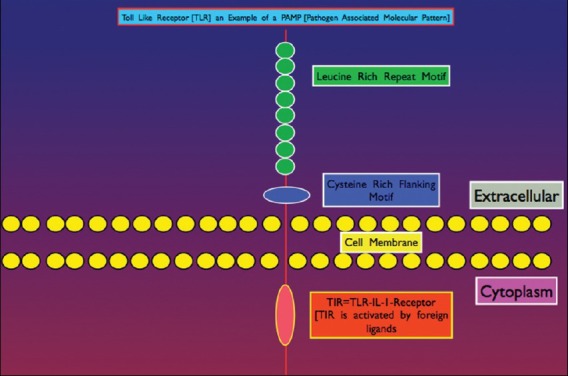
Illustration of general cellular anatomy of a TLR type Pattern Recognition Receptor (PRR). The external receptor interacts with specific Pathogen Associated Molecular Patterns (PAMPs) that translates signals to the intracellular signaling pathways for gene activation of defense mechanisms
Figure 5.
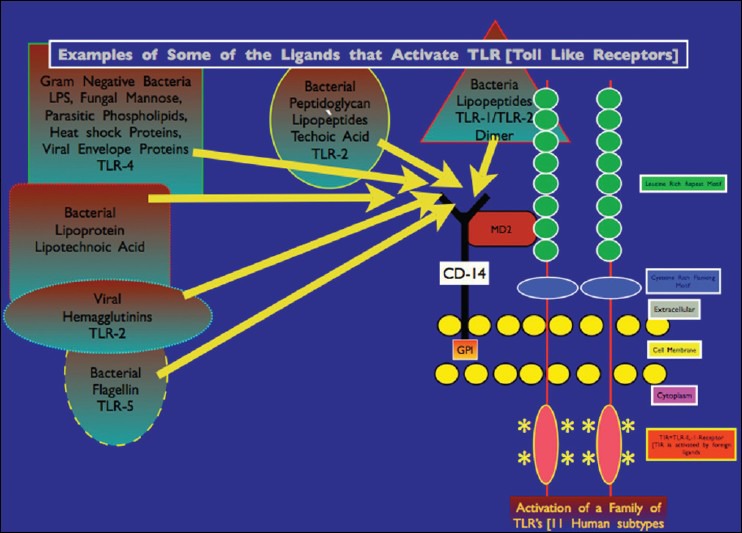
Illustration of various microorganism-related molecular ligands that can interact with specific TLRs leading to intracellular activation of defensive cell signaling mechanisms
Figure 6.
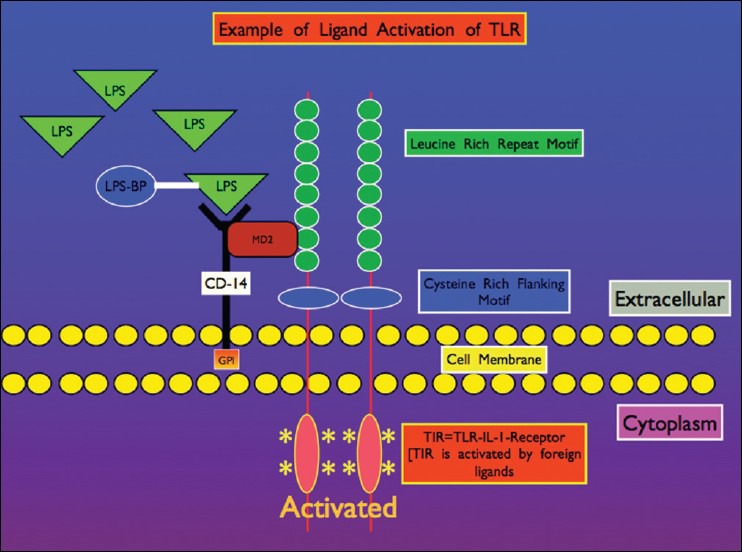
Illustration of lipopolysaccharide (LPS) Gram-negative cell wall molecular component interacting with TLR on microglial membrane surface, which along with co-stimulatory molecule CD14, activates defensive cell signaling. The IL-1 type pro-inflammatory cytokine receptor TIR, plays a major role in microglial activation
Figure 7.
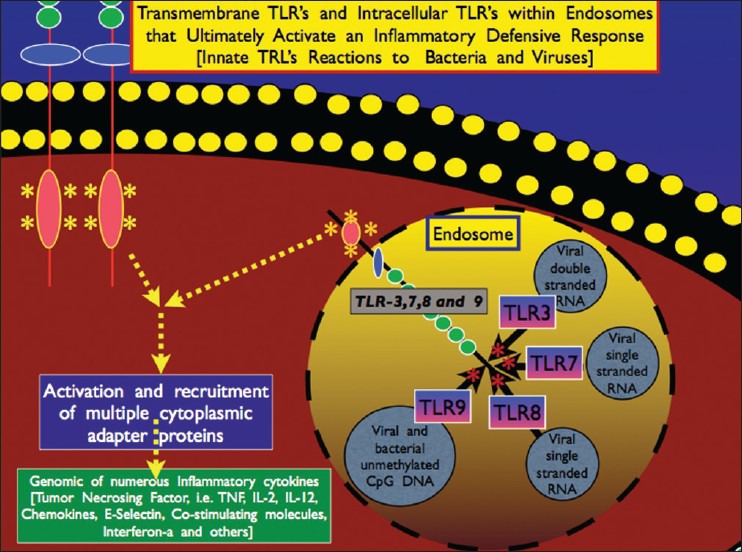
Illustration of intracellular TLRs found on the membrane of endosomes, which play a major role in neutralizing viruses I within the cell itself
How microglia are activated: Molecular mechanisms behind “microglial priming”
Through receptors
Cell signaling pathways can set in motion a series of molecular reactions that activate microglia. When an antigen is recognized by a receptor on the surface of the microglia cell, the receptor, through a series of conformational changes which requires energy, sets off a chain of molecular reactions that signal the genes in the microglia to produce proteins that activate the microglial cells to respond by producing proteins that react with the antigen and or initiate phagocytosis. For example, when PRRs in the microglial membrane recognize an antigen, NADPH oxidase is activated in response to antigen stimulation of the immune receptor. NADPH oxidase generates high levels of superoxide, which as discussed previously, combines with free NO to produce the powerful radical peroxynitrite. Superoxide can also break down into other powerful radicals, such as the hydroxyl radical.
Several PRRs can activate NADPH oxidase, the major source of microglial reactive oxygen species (ROS), especially the superoxide radical. NADPH oxidase is composed of several subunits that must be assembled for activity. These subunits are distributed between the cytosol and the membrane of intracellular vesicles. Once activated by complexing, they are translocated to the cell membrane. MAC1 appears to be crucial for assembly and activation of NADPH oxidase.[186] Intense activation of NADPH oxidase is seen in both Alzheimer's dementia and Parkinson's disease brains.[140]
Through elevated levels of ROS
The morphology and proliferation of microglia are to a large degree regulated by H202 generated by NADPH oxidase and, as a result, higher levels of ROS, in most instances, amplifies inflammatory responses.[9,101] By altering the level of intracellular ROS, one can prime microglia to respond in an exaggerated way to subsequent stimuli. In fact, NADPH oxidase is essential for microglial priming.[40] NADPH oxidase activation alone has been shown to be relatively benign, since the superoxide radical itself is rather weak. Cell killing requires the simultaneous activation of inducible nitric oxide synthase (iNOS).[100] This is because NADPH oxidase primarily produces oxygen radicals and iNOS generates nitrogen radicals, which when combined forms the very powerful reactive nitrogen species (RNS) peroxynitrite. High levels of peroxynitrite are found in most of the neurodegenerative diseases as well as strokes and neurotrauma. Several antibiotics, peptides, and small molecules known to inhibit NADPH oxidase activation demonstrate neuroprotective effects.[41]
Through chemokines and cytokines
Chemokines are a family of molecules released from microglia that attract other microglia or immune cells, even at great distances. Microglia contain a variety of receptors for chemokines and cytokines.[88] Chemokines released by neurons can signal microglia of impending danger, causing them to migrate toward the zone of injury. Damage to the CNS, including neurotrauma, ischemia/hypoxia, and infectious inflammation, trigger the release of the various chemokines not only from microglia and astrocytes, but also neurons.[12,50,51] One of the most potent triggers for chemokine release is a high level of glutamate. Chemokines, along with adenosine triphosphate (ATP) and glutamate, act as gradients for macrophage and microglia migration. Alteration of chemokine receptors is common with a number of neurodegenerative disorders such as AD and multiple sclerosis.[45,173] Chemokines, such as CXCL10 and CXCL12, by reacting with their respective microglial and astrocytic receptors, are linked through cell signaling pathways to excessive glutamate release and excitotoxicity under a number of pathological conditions.[39]
Clinical Correlation: Elevated levels of chemokines within the CNS are seen with a number of neurological conditions, including AD, amyotrophic lateral sclerosis (ALS), human immunodeficiency virus (HIV) dementia, ischemia, viral encephalitis, multiple sclerosis, and Parkinson's disease (PD).[31,82,115,121,178] Sometimes, chemokines can be neuroprotective by reducing inflammatory cytokine expression. For example, Donohue et al. found that patients with severe strokes had lower CX3CL1(fractalkine) levels at day one as compared with those with less severe strokes.[53] CX3CL1 levels were also lower at 180 days in patients with a poor stroke outcome. Interestingly, the association of fractalkine levels to outcome was independent of the severity of the stroke. High fractalkine levels were also associated with lower inflammatory markers, such as high-sensitivity C-reactive protein (CRP). Fractalkines suppress microglial activation.
Microglia contain receptors for a number of cytokines, both proinflammatory and antiinflammatory. One of the critical types of cytokine receptors for microglia activation is the IL-1ß receptors, which includes the subtypes IL1RI, IL-1RII, and IL-RIII.[93] Systemic macrophage surface receptor reaction to LPS, a substance found within the cell wall of Gram-negative bacteria, triggers the release of IL-1ß and this cytokine in turn activates brain microglia. While under resting conditions, with resting microglial, IL-1RIII has the strongest expression and upon LPS stimulation, one sees a significant increase in expression of IL-1RI and IL-1RII in activated microglia. Human microglia have been shown to have IL-1RI, IL-1RII, IL-5R, IL-6R, IL-8R, IL-9R, IL-10R, IL-12R, IL-13R, and IL-15R type receptors.[93] Systemic activation of IL-1Rs by any pathological process, rapidly activates brain microglia, and while being a major activator, it is not the only one. IL-1ß activation occurs systemically with LPS exposure, infections, trauma, and with autoimmune disorders.
How glutamate is produced by activated microglia
Another proinflammatory cytokine, TNF-α not only plays a critical role in inflammatory brain pathology, but is integral to immunoexcitotoxicity itself as it links the immune response to glutamate excitotoxicity[15,18] [Figure 8]. A number of immune cells produce TNF-α, including macrophages, microglia and to a lesser extent, astrocytes and neurons. TNF-α acts through two main receptors, TNFR1 and TNFR2. Like IL-1ß, TNF-α plays a significant role in microglial activation.[179] In general activation of the TNF-α subtype receptor TNFR1, triggers cell-signaling pathways that are predominantly neurodestructive.[102] These receptors are found mostly on neurons. TNFR2 subtype receptor is neuroprotective when stimulated and is found mostly on glia cells, which makes sense as high levels of TNF-α released during innate immune activation would otherwise damage the brain's principle immune cell, the microglia.
Figure 8.
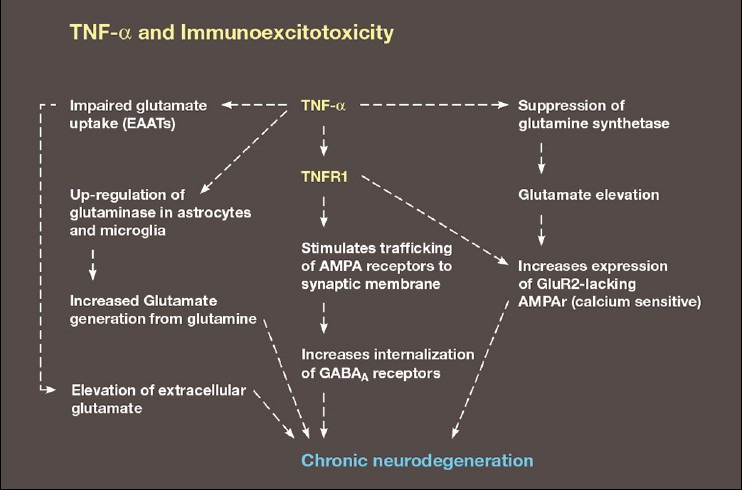
Schematic demonstrating the link between TNF -α and an enhancement of excitotoxicity via its interaction with glutamate transport proteins, upregulation of glutaminase, suppression of glutamine synthetase, increased trafficking of AMPA calcium-permeable receptors to the synaptic membrane and endocytosis of GABA receptors
Clinical Correlation: The concept of immunoexcitotoxicity is based on the interaction between inflammatory cytokines and glutamate excitotoxicity. In a previous paper, it was demonstrated that TNF-α had a number of effects on glutamate neurotransmission and excitotoxicity, including upregulation of astrocyte glutaminase (which converts glutamine into glutamate), increased trafficking of calcium-sensitive AMPA receptors to the synaptic membrane, inhibition of glutamate transport proteins (excitatory amino acid transporters [EAATs]), suppression of glutamate removal by Kreb's cycle enzymes and conversion of glutamate to glutamine (glutamate dehydrogenase and glutamine synthase) and increased trafficking of NMDA receptors.[18]
Based on this central effect of TNF-α, Tobinick et al. demonstrated dramatic, rapid improvement among 629 consecutive stroke patients following injection of the TNF-α-blocking drug etenercept given perispinally (Batson's plexus).[158] Statistically significant improvements were seen in a wide range of neurological deficits and symptoms, including motor impairment, spasticity, sensory impairment, cognition, behavioral function, aphasia, and pain. Incredibly, they noted a strong effect even in patients having strokes as long as 10 years previously. They found similar improvements among 12 patients with traumatic brain injury.
In a second group of patients, Tobinick and Gross, utilizing the same technique found that blocking of CNS TNF-α in a single case of severe AD produced rapid and sustained improvements in speech, agitation, and tests of spatial function.[157] Less impressive improvements were seen in memory function, but being a late stage case, one would expect severe loss of hippocampal neurons. Others have had similar results.[162]
The final effect of TNF-α depends on which TNF receptor is activated. Microglia contain predominantly TNFR2 type receptors that when stimulated by high levels of TNF-α, are protective, thus protecting the microglial cell from its own released cytokine. Neurons, in contrast, contain mostly TNFR1 subtype, which when stimulated by high levels of TNF-α triggers a series of cell signaling pathways that are neurodestructive. Very low, constitutive concentrations of TNF-α are considered neurotrophic.
Neurotransmitter and neuroregulatory receptors on microglia
Microglia contain a number of receptors for neurotransmitters and other neuroregulatory molecules, including those for glutamate, acetylcholine, dopamine, gamma-aminobutyric acid (GABA), adrenergic compounds, cannabinoids, opioids, substance P, vasoactive intestinal peptide, histamine, glucorticoid, somatostatin, angiotensin II, platelet activating factor (PAF), and neurotrophin.[88]
Ionotropic glutamate receptors
Glutamate is the most abundant neurotransmitter in the brain, contributing 90% of cortical neurotransmission and 50% of all brain neurotransmission. These receptors include three subtypes named NMDA receptors, AMPA receptors, and kainite receptors [Figure 9]. Each has its own particular physiological response to glutamate stimulation, providing the brain with a wide range of reaction options to a single neurotransmitter, glutamate. Interestingly, glutamate receptors reside on most neurons, even those utilizing other neurotransmitters, such as dopamine, serotonin, norepinephrine, and acetylcholine. There exists an interaction between these receptors so that each modulate the others functions. Being the most abundant receptor type, glutamate receptors play a major role in controlling the other neurotransmitter receptors.
Figure 9.
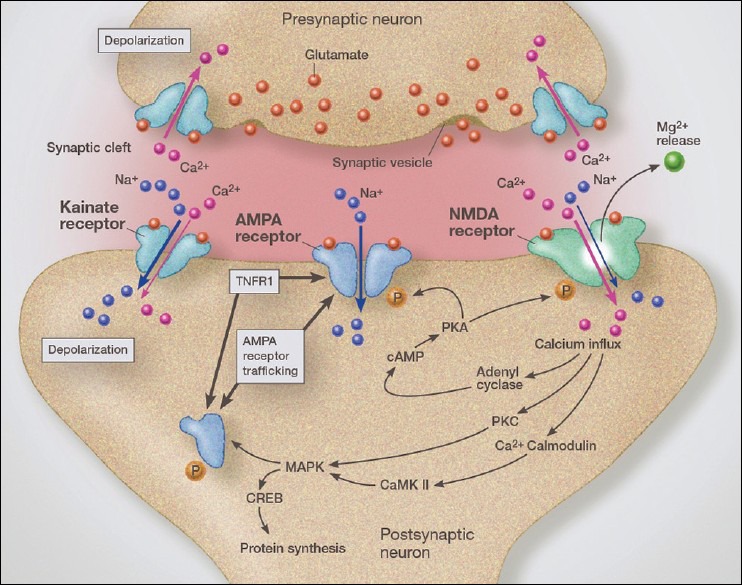
Illustration demonstrating the trafficking of AMPA calcium-permeable receptors from the endoplasmic reticulum to the synaptic lipid raft, thus increasing glutamate sensitivity and increasing the internal flow of calcium. We see a condition of crosstalk between TNFR1 receptors and AMPA receptor trafficking mechanisms
Glutamate receptors on neuronal dendrites and synapses play a major role controlling the strength of the synaptic impulse and does so by a variety of mechanisms. Of special importance is the role glutamate neurotransmission plays in learning and memory and behavioral control.
Interestingly, glutamate receptors are not limited to neuronal dendrites and synapses. Over the past decade, researchers have demonstrated a significant role for glutamate receptors in regulating microglial behavior. One role for microglia as a member of the tripartite synapse is to modulate extraneuronal concentrations of glutamate, especially in the vicinity of the synapse. This helps to sharpen the signal and prevent interfering “noise” caused by excess diffused glutamate from surrounding synapses. Cell surface receptors for glutamate are divided into inotropic receptors and metabotropic receptors. The ionotropic receptors regulate opening of various ionic channels, principally those for sodium and calcium and the metabotropic receptors act through G-protein-coupled receptors to modulate the inotropic receptors. Microglial inotropic glutamate receptors play a role in microglial migration as well as microglial activation. When sites of injury or pathology result in the release of high levels of glutamate, the diffused extracellular gradient attracts surrounding microglia to the site of injury.
Of the ionotropic glutamate receptors, AMPA receptors appear to be the most abundant and critically important for microglial reactivity. There is some evidence that NMDA receptors exist on microglia, most of which is indirect evidence. For example, blocking NMDA receptors in experimental models has been found to reduce microglial activation associated with ischemic insult or exposure to HIV protein.[148] Blocking inotropic glutamate receptors has been shown to reduce the size of the infarct and improve neurological outcomes in animal stroke models but has had little success in human studies, mainly because of side effects of the blocking drugs.[80] Recent studies may have further demonstrated the reason for failure of these drugs. Chen et al. found that the subtype of NMDA receptor was critical, with the NR2B subtype NMDA receptor triggering neuronal death and NR2A subtype being neuroprotective when activated.[37] One can see that blocking both receptors could be detrimental, as it would not only block the neurodestructive NR2B-containing NMDA receptor subtype, but also the NR2A-containing NMDA receptor neuroprotective subtype.
Stimulation of microglial AMPA receptors induces the release of TNF-α, which in a paracrine manner stimulates further microglial activation and migration. Glutamate receptors, in particular AMPA receptors, play a major role in microglial chemotaxis. By following gradients of extraneuronal glutamate, microglial cells migrate toward sites of pathological injury or damage.[95] With brain injury, massive amounts of glutamate are released from astrocyte stores. Also of interest is the finding that stimulation of AMPA receptors triggered rapid and extensive remodeling of the cytoskeleton (actin filaments) within microglia suggesting a role in regulation of microglial motility and phagocytosis when activated.[42]
Also of interest is the finding that under certain conditions, AMPA receptor stimulation can be neuroprotective and promote brain repair after a stroke.[43] Recovery after a stroke was improved by AMPAR stimulation, but only if done after 5-days from the stroke. At this stage, AMPAR stimulation mediated the release of brain derived neurotrophic factor (BDNF). Blocking AMPA receptors after the onset of this later period worsened the pathological damage. Prolonged activation of AMPA receptors, that is, after several weeks to months, again switches the microglia to a neurodestructive phenotype. Likewise, stimulating AMPA receptors during the first 5 days worsened the outcome and blocking these receptors during this time period improved brain injury, indicating that time of intervention is critical.
Metabotropic glutamate receptors
Metabotropic glutamate receptors (mGLuRs) are classed as noniontropic receptors and rather than utilizing ionic pores, such as the calcium pore seen with ionotropic glutamate receptors, this group of receptors is coupled to membrane G-proteins that act through cytoplasmic second messengers. Cloning studies have grouped them into three basic types. Group I mGLuRs, composed of metabotropic glutamate receptor subtypes 1 and 5 (mGluR1 and mGluR5), operate through activation of phospholipase C. In general, these enhance the activity of the iontropic glutamate receptors (NMDAR, AMPAR, and kainite receptors).
Group II metabotropic receptors include mGluR2 and mGluR3 and through their G-protein receptors inhibit adenylate cyclase. Most often they are inhibitory of the ionotropic receptors, but not always. Group III contains mGluR4, mGluR6, mGluR7, and mGluR8. They also inhibit adenyl cyclase and are considered to be inhibitory of ionotropic glutamate receptors. They are linked to the ionotropic receptors though cell-signaling molecules.
From a potential therapeutic viewpoint, mGLuRs exhibit a number of interesting properties. Group I mGluRs regulate LPS-induced microglial activation in primary cultures.[59] This links microglial activation by Gram-negative organisms as well as vaccines containing LPS as an adjuvant. Activation of group II mGLuRs triggers microglial activation along with cytotoxic release of TNF-α.[156] A more recent study clearly indicates that mGluR II and III stimulation suppresses the release of glutamate from microglia and prevents neurotoxicity associated with neuroinflammation.[106] In this instance microglia are moderating their own release of glutamate via these mGLuRs. Blocking these two receptors, Group II and Group III, under conditions of LPS immune stimulation greatly magnify associated glutamate excitotoxicity (immunoexcitotoxicity). In this study, group I mGLuRs played a very minor role in regulating glutamate release from microglia.
In essence, the mGLuRs are acting as modulators of inotropic receptors either enhancing or dampening their response. This adds an important layer in controlling the excitatory response to glutamate, so as to prevent excessive brain excitation, which can lead to neurodegeneration or seizures. In addition, they also aid in sharpening the signals during glutamate neurotransmission. As with the ionotropic glutamate receptors, the metabotropic receptors play an important role in controlling other neurotransmitter function and levels of activity.
Cholinergic receptors
Frequently overlooked in discussion on neurodegeneration is the critical role played by cholinergic systems in controlling inflammation, especially the α7-nicotinic receptors (α7nAchRs). Cholinergic pathways are impaired in both AD and PD, most likely as a result of the immunoexcitotoxic reactions in Ach-controlling areas of the brain, such as the nucleus basalis of Meynart.[109] This small nucleus contains a great number of glutamate receptors and when overstimulated can lead to destruction of the nucleus and a loss of brain cholinergic input. The basal nucleus of Meynart is the central source for the brain's cholinergic fiber system.
Clinical Correlation: Any condition that reduces brain acetylcholine, such as brain trauma, dietary restriction of choline, advanced aging or chronic neurodegenerative diseases that lowers brain acetylcholine, could worsen brain inflammation. One can see that a combination of poor diet and advanced aging in particular can worsen Alzheimer's dementia by removing or suppressing this antiinflammatory system. Likewise, improvements in some AD patients taking acetylcholine-enhancing medications, such as donepezil (Aricept) may be as a result of Ach's antiinflammatory effect. Chronic activation of these receptors, as occurs with smoking or nicotine patches, could impair microglial function in cases of cerebral infections, strokes, and other inflammatory pathologies. It would also explain how smoking reduces one's risk of PD and why nicotine patches may reduce risk.[27,87,124] Microglia containing α7nAcRs (acetylcholine nicotinic receptors), when activated generally suppress immune responses as part of the “cholinergic anti-inflammatory pathway.”[141]
Adrenergic receptors
Norepinephrine is known to suppress the release of NO, TNF-α, and IL-6 following immune stimulation with LPS and plays a critical role in protecting the brain from neuroinflammation.[58] Stimulation of ß2 adrenergic receptors (ARs) (ß2 ARs) suppresses microglial proliferation. Norepinephrine acting through ß1/2 receptors inhibits cyclic adenosine monophosphate (cAMP)-induced release of TNF-α.[108] ARs are instrumental in regulation of microglial migration and phagocytosis. In essence, like acetylcholine, norepinephrine and epinephrine play an important role in dampening immune activation and associated brain inflammation.
Adrenergic receptors and the locus ceruleus
This small collection of neurons, located in the posterior area of the rostral pons along the floor of the fourth ventricle, sends adrenergic (norepinephrine and epinephrine) projections widely throughout the CNS, thus making it the central controlling adrenergic nucleus (referred to as the locus ceruleus-noradrenergic [LC-NA] system). Stimulation of these neurons is generally excitatory for the brain, producing an arousal effect. Degeneration of the LC is an early event with AD and PD.[171,187] Recent studies have shown that norepinephrine suppresses microglial activation and migration, explaining how a loss of LC-NA system plays a significant role in these diseases.
Dopamine receptors
Functional dopamine receptors on microglial membranes have been identified in both human brain tissue and mouse brain. Chronic stimulation of microglial dopamine receptors enhanced migratory activity, and like ARs, attenuated LPS-induced NO release.[58] Cultures from elderly human microglia demonstrate dopamine receptor-induced chemotaxis. Interestingly, the substantia nigra has the highest concentration of microglia in the brain, which puts it at risk when they are activated.[92] We see this activation of substantia nigra microglia with exposure to certain agricultural chemicals such as rotenone and maneb, common chemical exposures linked to PD.[63,153] Activation of substantia nigra microglia either by rotenone or other agents used to produce experimental models of PD, increase the secretion of proinflammatory cytokines and excitotoxins (immunoexcitotoxicity) and this results in a selective degeneration of these pigmented neurons, as well as degeneration in other striatal neurons. Of interest has been the finding that inhibition of D4 dopamine receptors suppresses microglial activation in the spinal cord of transgenic models of ALS, slowing down disease progression.[154]
Clinical Correlation: ALS: A number of new studies considerably strengthen the case for an interaction between inflammation and excitotoxicity as the central mechanism in ALS. It has been determined that in experimental models of the disease microglial-triggered inflammation occurs in the presymptomatic stage of the disease, and microglial number increase as the disease progresses.[75] Interestingly, one also sees increased systemic inflammation with elevations in CD16+ monocytes in the peripheral blood as well as elevated levels of LPS (endotoxin).[183,184] This is important, as several studies have shown that stimulation of systemic immunity in ALS cases can worsen symptoms and cause the disease to advance more rapidly.[68]
Scanning studies of ALS patients demonstrate widespread microglial activation in the motor cortex, pons, dorsolateral prefrontal cortex, and thalamus.[161] The extent of the microglial activation was strongly correlated with the severity of the ALS. In addition, one sees high levels of ROS/RNS products released from the activated microglia. In ALS, oxidative and nitrogen stress begins very early in the course of the disease. The most toxic of the radicals is peroxynitrite, which powerfully inhibits mitochondrial energy production, something commonly seen in ALS.
The reason for the prolonged microglial activation in the affected areas of the brain and spinal cord in ALS is not specifically known, but we know that the most commonly linked initiating factors, such as pesticide/herbicide exposure, aluminum and/or mercury accumulation, repeated minor injuries and certain persistent viruses all are known to cause prolonged microglial activation in a neurodestructive mode.
The relationship of superoxide dismutase-1 to ALS
In the past, it was thought that there were two distinct ALS disease forms, one sporadic with normal levels of the antioxidant enzyme SOD1 and a familial form with mutated SOD1. Newer studies have offered some important surprises. First, the loss of motor neurons was not related to a loss of functional SOD1, an antioxidant molecule that neutralizes superoxide.[26] Instead, these studies indicated that the mutated SOD was acting as a toxic molecule itself. Mutated SOD1-containing microglia were found to be more intensely activated when stimulated than were wild-type microglia. These activated microglia released higher concentrations of NO and superoxide.[6] The mutated SOD, when released from astrocytes was shown to react with CD14+ receptors on microglia and via TLR2 and TLR4 coreceptors, thus triggering CNS inflammation by releasing cytokines. In essence, the mutated SOD1 was acting as a powerful microglial activator, much like a foreign antigen. A previous study found that SOD1 was frequently oxidized in sporadic cases of ALS and that this altered enzyme, like the mutated SOD1 in familial cases, also activated CD14+ receptors, thus linking all forms of ALS, sporadic and familial, to toxic, immune activating forms of SOD1.[74]
One might conclude incorrectly that motor neuronal death in ALS was caused by the immune activation alone, yet mouse models of ALS in which IL-1ß and TNF-α have been deleted still demonstrated continued neurodegeneration, indicating that another pathological process was in operation and linked to the immune effect.[128] The strongest link to motor neuron loss has been excitotoxicity primarily caused by impaired glutamate transport by EAATs, which allows dramatic elevations in extracellular glutamate. These glutamate transporters play a critical role in keeping extracellular levels of glutamate very low by shunting the glutamate into microglia and astrocytes. Should they fail, or be impaired in their function, glutamate can rise to neurotoxic levels. Because these transporters (EAATs) are redox sensitive, the free radicals and lipid peroxidation products produced by the inflammation impair the glutamate transporter's function and this causes a rise in extraneuronal glutamate levels. Rothstein has shown that a large percentage of ALS patients have defective glutamate transport.[130] All ALS animal models show impaired glutamate transport. This transport mechanism is present on both microglia and astrocytes, but astrocytes are the major storage reservoir for glutamate. Abnormal, mutated, or oxidized SOD1-containing astrocytes are seen prior to the onset of symptoms in animal models.[25]
Excitotoxicity and its relation to neurological diseases and ALS
The high level of ROS/RNS and lipid peroxidation resulting from the widespread inflammation impairs the glutamate transporters, in particular GLT-1 (EAAT2), and impairs other essential metabolic enzymes and enzymes used to clear excess glutamate, such as glutamine synthetase. A number of studies have demonstrated elevated CSF glutamate levels in ALS patients as compared with controls.[129] A follow-up of this earlier study found elevated CSF glutamate in 40% of 400 patients with sporadic ALS and that elevation of CSF glutamate correlated with disease severity.[145] It is important to appreciate that excitotoxicity is not always dependent on elevation of glutamate levels, since a number of conditions, such as mitochondrial dysfunction, hypoglycemia, heavy metal poisoning, and inflammation, can dramatically enhance neuronal sensitivity to glutamate – so much so as to allow even normal levels of extracellular glutamate to trigger excitotoxicity.
Impairment of EAAT2 expression in the ventral horn of the spinal cord of experimental models occurs in the presymptomatic period. Interestingly, EAAT2 levels were almost completely abolished at the end-stage of the disease.[79] This loss of the glutamate removal transporter action means that excitotoxicity is a major mechanism of motor neuron destruction. This destructive process is enhanced by inflammation via a number of mechanisms. Impairment of EAATs causes a slow neurodegeneration because of the gradual accumulation of glutamate to excess. This initiates the excitotoxic cascade in which excess calcium enters the neuron, triggering a number of neurodestructive processes, including production of free radicals and lipid peroxidation products, impaired mitochondrial energy production, and activation of inflammatory prostaglandins.
Overexpression of EAATs, thus increasing the efficiency of glutamate removal, in experimental animal models of ALS delays the onset of motor loss and prolongs survival. An analysis of motor cortex and spinal cord extracts from ALS patients demonstrated nearly complete loss of EAAT2 in 25% of patients and some impairment in up to 80%.[130] The resulting elevations in extraneuronal glutamate leads to the generation of high levels of ROS/RNS and lipid peroxidation products, which further impair glutamate transporters and suppresses mitochondrial energy production. Impaired energy generation markedly enhances excitotoxic sensitivity. One can easily envision a progressive neurodegenerative process from these interacting mechanisms that ends in widespread motor neuron loss.
Cannabinoid receptors
The cannabinoid system plays a critical role in modulating excitatory neurotransmission. Rodent microglial cells contain CB1 and CB2 receptors on activated microglia but are very low in ramified (resting) microglia.[10,11,150] The appearance of these receptors is quite rapid upon microglial activation. Activation of cannabinoid receptors on microglia reduce NO production and inhibit cytokine release resulting in a suppression of the inflammatory response.[28]
Experimental allergic encephalomyelitis
EAE is an animal model of multiple sclerosis and related inflammatory demyelinating disorders. A number of antigens have been used to produce the immune demyelination, including spinal cord homogenates, various myelin proteins (MBP, PLP, and MOG) or peptides of these proteins. The pathology produced varies to some degree based on the antigen used.
In the case of EAE, a model for human multiple sclerosis in animals, one sees upregulation of CB2 receptors induced by the synergistic action of IFN-γ and GM-CSF.[104] CB2 is also upregulated in microglia isolated from patients with AD, ALS, and AIDS-associated dementia.[180] In most cases, activation of cannabinoid receptors reduces microglial neurotoxicity while increasing microglial proliferation. This activation would switch the microglia from a proinflammatory cytokine-releasing phenotype into a phagocytic, neuroprotective phenotype.[55] This cannabinoid receptor activation is also demonstrated in the case of animal models of Huntington's disease in which stimulation of CB2 receptors reduced brain edema, neuroinflammation and death of striatal neurons.[114]
From this abbreviated review of microglial receptors, it becomes obvious that brain immune reactions are under a great number of regulatory controls and it is the interaction of these various receptors that determines the dynamic changes in microglial phenotypes and thus their function.
Microglial migration and motility
Microglia are not fixed immune cells, but are characterized by a high degree of motility in the ramified (resting) state and are capable of extensive migration upon activation. As demonstrated previously, motility of resting microglial foot processes allows the microglia to conduct ongoing surveillance of the synaptic microenvironment and make adjustments of the concentrations of excitatory neurotransmitters such as glutamate and aspartate. This surveillance is critical not only for protection against excitotoxicity, but also for signal sharpness, as previously mentioned.
At the interface of cerebral blood vessels, microglial motility allows rapid identification of invasion by microorganisms. This rapid identification process is especially important near zones where the BBB is deficient, as with the CVOs, which includes the area postrema, subfornical organ, organum vasculorum of the laminia terminalis, pineal region, subcommissural organ, median eminence, and neurohypophysis. These areas contain abundant microglia, which when exposed to invading organisms or systemic proinflammatory cytokines, are rapidly activated and migrate over great distances in an effort to protect the rest of the brain.[91] They also represent areas of easy entry for activated macrophages from the periphery.
Motility appears to be mostly controlled through purinergic receptors on the microglial surface membrane. Purinergic receptors respond to free ATP and adenosine and on neurons can initiate an excitatory response. A number of subtypes of purinergic receptors have been identified. ATP and adenosine, both stimulate motility in ramified (resting microglia) microglia and act through P2Y12 subtype purinergic receptors.[49] This receptor stimulation attracts the foot process toward the site of damage. Upon microglial activation, P2Y12 expression is reduced.
In the face of pathological injury, activated microglia migrate to the site of damage mostly by following a chemical gradient. Like motility, microglial migration is controlled by a set of receptors. Studies suggest that a combination of P2X4 and P2Y12 receptors, subtypes of purinergic receptors, are involved in microglial chemotaxis.[112] CD39 and CD73, cellular ectonucleotidase enzymes, are essential for degrading ATP for stimulation of chemotaxis. That is, these enzymes break down the ATP, releasing adenosine, which then activates particular purinergic-subtype receptors on the microglial surface. It appears that costimulation of purinergic and adenosine receptors are required for microglial migration. This costimulation becomes important when considering microglia migration to areas of significant neural injury and neuron death, as occurs with strokes, brain trauma, and neurodegenerative disorders. Under such conditions, high concentrations of ATP are released from the damaged neurons and glial cells and can act as activators of microglial migration and motility.
Other neurotransmitters are also involved in microglial migration, such as glutamate, dopamine and epinephrine via AMPA receptors, mGLuRs, dopamine receptors, and ARs, respectively.[58] Along with ATP, these neurotransmitters are also released with neural tissue damage. The high concentration of the ATP and neurotransmitters released from the dying cells diffuse into the surrounding tissue and establish a chemical gradient that attracts the microglia.
Chemokines are also potent chemoattractants and are released from neurons and microglia. Damaged neurons express the chemokine CCL21, which attracts microglia. Monocyte chemoattractant protein-1 (MCP-1) regulates migration of microglia, monocytes, and lymphocytes to the sites of inflammation in the CNS.[144] Cannabinoids, acting through CB2 receptors, can also act as stimulants for microglial migration.[166] They play a special role in increased microglial motility following ischemia.
A number of other factors also regulate microglial migration, but of special interest is nerve growth factor (NGF) and transforming growth factor-ß (TGF-ß) as both are involved in brain repair and reduction of neuroinflammation as well.[52] As the microglial transform to a reparative phenotype, release of these reparative NGF and TGF-B compounds can play a vital role in moving microglia to the sites of damage so as to enhance repair of the damage done.
Phagocytosis
Microglial cells are the main phagocytic cell type of the brain and can not only phagocytize dead cells, but also parts of neurons, for example, dendrites, and debris such as myelin and amyloid deposits. Microbes are recognized by TLRs and other PRRs, whereas apoptotic neurons utilize other receptors such as vitronectin and phosphotidylserine-mediated receptors.[176] Chloride channels play an important role in microglial phagocytosis and ciliary neurotropic factor increases phagocytosis via a Ca2+ pathway.
A major control mechanism for microglial phagocytosis is by way of the P2Y6 receptor, which is upregulated with neuronal damage.[81] This enhancement of a particular purinergic receptor indicates that purinergic receptors are important in several functions of microglia, which would be activated upon the release of ATP, which is associated with cellular damage.
Synaptic stripping and synaptic building
States of inflammation are often associated with removal of synapses and the process is driven by activated microglia. Neuronal electrical activity normally suppresses MHC class II activation on both astrocytes and microglia.[110] Yet, with immunoexcitotoxicity, where excitatory neurotransmission sensitivity is greatly increased, microglia can initiate synaptic stripping to reduce excitatory synaptic activity as a neuroprotective response.[160] This process principally removes excitatory glutamatergic synapses and spares inhibitory input.
When in a reparative phenotype, microglia can also promote synaptogenesis by secreting thrombospondins (TSPs), an extracellular matrix protein critical for synaptic formation.[34] As a result, microglia, when in a neuroprotective phenotype, are critical for restoration of neuronal connectivity following injury and after inflammatory reactions have terminated. The adaptive immune system, utilizing regulatory T-lymphocytes (CD4+, CD25+ Tregs), interacts with microglia to promote brain repair, again demonstrating the interaction between the innate immune system and the adaptive immune system. Tregs play a critical role in controlling brain inflammation and studies have shown that in diseases such as multiple sclerosis one sees a switch from infiltrating immune inhibitory Tregs to cytotoxic CD8+ T-lymphocytes during disease progression.[163] This switching process has also been shown in several neurodegenerative diseases, such as ALS and Alzheimer's dementia.[77] Microglial cells have a trophic function by upregulating the secretion of neurotrophins, such as BDNF. This functional switching allows microglia, while in this reparative phenotype, to repair the damage done during an immune attack. As shown in part I of this immune primer, CD200, fractalkines, IL-10, Il-4, IL-13, and TGF-ß also play critical roles in microglia switching to a reparative phenotype and in reducing brain inflammation.
Systemic immunity interactions with innate CNS immunity (Sickness behavior)
In the past it was assumed that the neurological and behavioral effects of viremia and sepsis were secondary to the infectious agent acting within the brain itself. It is now accepted that peripheral inflammation and immune activation secondarily effect brain function during the infectious process.[48] This secondary immune process has been named sickness behavior and is characterized by anorexia, hypersomnia, lethargy, reduced social interaction, reduced cognitive function, and weakness.
There are four major links between peripheral immunity and CNS immunity, one a rapid system and the other three of slower onset. The slower-onset systems involve a passive diffusion of proinflammatory cytokines into the brain via the CVOs – organum vasculosum of the lamina terminalis, subfornical organ, neurohypophysis, pineal gland, median eminence, and dorsal vagal complex.[2] Diffusion of proinflammatory cytokines into the CVO areas brings them into contact with ramified (resting) microglia, which are then activated and become mobile.
The BBB has an energy-dependent, saturable, carrier-mediated transport system for cytokines, primarily IL-1, IL-6, and TNF-α.[8,73] Finally, when endothelial cells making up the BBB come into contact with these peripheral cytokines, they secrete various immune molecules into the brain parenchyma, including NO, prostaglandin E2, IL-1, and IL-6, all proinflammatory cytokines known to affect neurological function.[57] There is some question as to the sufficiency of these secreted cytokines to actually affect behavior directly.
The rapid system operates through the vagus. Several lines of evidence implicate the vagus nerve in this fast process. For example, subdiaphragmatic sectioning of the vagus mitigates sickness behavior induced by peripheral immune activation.[22] IL-1 receptors are found on vagal paraganglion and with LPS injection i.p., expression of both IL-1 and IL-1 receptors is enhanced on the vagal paraganglion with an increase in vagal afferent activity. Hansen et al. found that intraperitoneal injection of IL-1ß increased IL-1ß mRNA levels in the hypothalamus, hippocampus, and brainstem and sectioning of the vagus blocked induction of IL-1ß mRNA in the hippocampus and brainstem, but only partially blocked responses in the hypothalamus, suggesting the existence of other pathways in this area.[76]
Activation of brain microglia following peripheral immune stimulation is most intense in the face of preexisting brain pathology and some feel that without this preexisting pathology peripheral immune stimulation will not lead to neurodegeneration. For example, using a ME7 model of prion disease, researchers found that injecting this protein into the brain prior to peripheral immune activation, produced an exaggerated proinflammatory response with intense microglial activation, extreme sickness behavior, and acceleration of neurodegeneration.[44,47]
Clinical Correlation: This may be the case in such conditions a chronic traumatic encephalopathy (CTE) and would explain why the phenomenon does not occur in every person suffering repeated minor concussions.[18] Silent neurological conditions occur in a significant number of individuals and may act to prime microglia upon the first exposure to a systemic inflammatory event.[175] For example, cases of silent multiple sclerosis lesions are thought to be present in as many as 25% of examined brains.[56] Other chronic conditions, such as latent infections, glial scarring, neurodevelopmental defects, illegal drugs use, exposure to pesticides/herbicides, and toxic metal accumulations (aluminum, mercury, cadmium, and lead) could also act as priming agents. Aging itself also results in microglial priming. Subsequent injuries, ischemia/hypoxia or infectious processes affecting the systemic immune system would then trigger an enhanced microglial activation state in the CNS, leading to progressive neurodegeneration. Microglial activation is quite rapid following systemic immune activation, usually within minutes and results in immunoexcitotoxicity.
MICROGLIAL PRIMING
As we see in peripheral immune cells, in particular macrophages, microglia can switch from a resting phenotype to a primed state by an initial immune stimulus that is not excessively intense. For example, a mild head injury or episode of hypoxia can switch microglia from its resting state to a functional condition in which the enzymes and genetic activation is upregulated, but the active immune molecules, primarily proinflammatory cytokines and chemokines, are not released. We see priming of microglia also with aging of the brain.
With a second immune stimulus, these primed microglia began to release proinflammatory cytokines and chemokines in concentrations much higher than would microglia that have not been primed. Systemic immune stimulation can prime brain microglia, which means that either subsequent brain disturbances or systemic immune activation would trigger a magnified immune response within the brain. Is essence, it strongly indicates that systemic immune activation can worsen and speed up CNS degenerative disorders, such as ALS, AD, and PD.
We see similar enhancement of neurodegeneration by peripheral infections in rodent models. The destructive process develops secondary to local microglial priming by the preexisting pathological process, such as a stroke, closed head injury, multiple sclerosis, AD, PD, prior brain surgery, or penetrating injury. The peripheral immune stimulation by way of the interacting process discussed earlier, fully activate the primed microglia in an exaggerated manner.[119]
Because humans are exposed to a number of immune events throughout life, such as multiple infections, persistent viruses, exposure to neurotoxic metals, exposure to pesticides/herbicides and fungicides, head injury, microinfarctions, chronic stress and aging, one can see that each of these episodes is associated with microglia priming and activation, leading to a progressive loss of neurons in the most vulnerable parts of the CNS, such as the hypothalamus, temporal lobes (hippocampus, striatal area, amygdala, and entorhinal cortex) and prefrontal cortex.
Those with the least efficient protective systems, such as glutamate transport systems, antioxidant network, cellular glutathione, low CD200 and fractakline and tissue iron binding would be at the greatest risk for one of the neurodegenerative diseases. Under such conditions, the microglia would remain activated in the affected zones and immunoexcitotoxicity would be triggered over a long period leading to neurodegeneration.
Aging and immunoexcitotoxicity
Since aging itself is associated with priming of microglia and elevations in inflammatory cytokines, one would expect peripheral immune stimulation under such conditions to worsen any coexisting clinical picture significantly.[118] Aged mice exposed to i.p. LPS demonstrate more severe anorexia, depression-like behavior, learning deficits, and memory impairment than do younger cohorts.[69] In addition, it takes aged individuals longer to get over the symptoms of sickness behavior than it does younger persons.
Immunoexcitotoxicity and behavioral disorders
Immunoexcitotoxicity is thought to play a significant, if not central role in many behavioral disorders in humans as well[46] [Figure 10]. It is known that individuals developing two or more infections, of any type (mild or serious bacterial or viral infections), over a 4-year period increase their risk of developing AD by 2-fold.[54] It also explains why systemic infections can worsen multiple sclerosis and accelerate the progression of neurodegenerative diseases.[78,142] This magnification process has been confirmed in animal models of neurological diseases.[69] The link between systemic inflammation and brain neurodegeneration also explains the finding that those with general ill health, especially in the later years of life, have a higher risk of cognitive decline. This link between systemic inflammation and neurodegeneration occurs because frail individuals suffer not only from repeated infections, but also from prolonged infections. This interaction with systemic immunity serves to prime the microglia, which means that subsequent immune events will result in a greatly magnified release of proinflammatory cytokines and release of excitotoxins. The resulting brain inflammation significantly magnifies glutamate receptor sensitivity by a number of mechanisms, such as AMPA and NMDA receptor trafficking, mitochondrial energy suppression, suppression of glutamate removal mechanisms (EAATs), stimulation of glutaminase (converts glutamine to glutamate) and impairment of enzymes metabolizing glutamate (glutamate dehydrogenase and glutamine synthase).[18,146]
Figure 10.
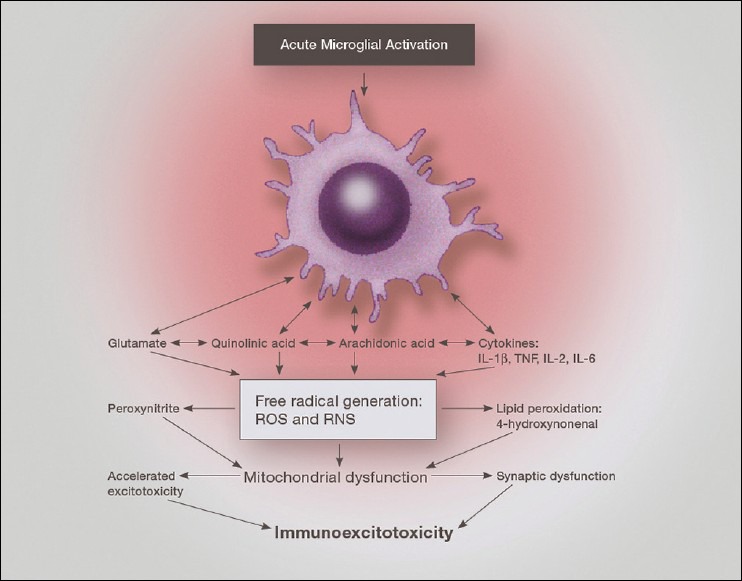
Illustration of the combined release of pro-inflammatory cytokines and chemokines along with excitatory amino acids during neurodestructive microglial activation. This demonstrates the interaction between immune factors, reactive oxygen and nitrogen species, lipid peroxidation species and excitotoxicity
Immunoexcitotoxicity and other diseases
A growing number of neurodegenerative disorders, including strokes, schizophrenia, autism, CTE, and neuropsychiatric disorders are associated with primed or activated microglia.[14,17,18,33,60,67,119] Studies have also shown that inflammatory bowel disorders can prime/activate brain microglia and cause or worsen neurologic disorders.[131,164]
Immunoexcitotoxicity is enhanced by vaccines
Also of concern is the present vaccine schedule for children, which entails a repetitive injection of concentrated powerful adjuvants during a period of intense brain pathway formation. The present US vaccine schedule recommended by the Center for Disease Control and Prevention (CDC), now mandated for most states, consist of giving small children up to age 6 years as many as 5-8 vaccines per office visit every 2 months for as total of 28 vaccines during the first year of life.[32] When given as multiple doses, for example, 6 doses to a small child during a single office visit, significant systemic immune activation occurs, with signs suggesting reactive brain inflammation–such as high pitched crying, fever, restlessness, and failure to eat.[14]
It is not the antigens that are of most concern, but rather the adjuvants, which initiate the most intense and prolonged immune stimulation. These adjuvants are designed to magnify the immune response, as the antigens (viruses and bacteria) themselves are too weak to provide protection. In addition, adjuvant additives, such as aluminum and mercury, are known powerful microglial activators and are neurotoxic, even in very small concentrations.[13,86,159] These neurotoxic metals have been shown to accumulate in the brain.[36,167] Because of the ability of immune adjuvants to prime microglia and trigger intense microglial activation with subsequent vaccinations, we see neurological side effects even in vaccines without aluminum or mercury.[13,24,36] Several studies have also shown that the aluminum adjuvant in human vaccines is distributed into the brain upon injection.[61,126]
Priming of the microglia
In the infant or small child, the priming event may come from a number of sources, such as vaccination of the mother during pregnancy or with intrauterine or early postbirth infections.[16,19] In other instances, the priming event may occur with the first vaccine inoculation, usually at birth (hepatitis B). Once primed, subsequent vaccinations, especially within months of the previous inoculation, will trigger full microglial activation and in the developing brain can result in abnormal pathways development.[1,14,15,133]
While natural infections can also produce this neurodestructive response, vaccinations produce higher levels of immune activation and the immune response can persist longer than natural infections – sometimes lasting years. In the case of the elderly, already having primed microglia, repeated, closely spaced, sequential vaccination can also present a potential hazard, since an exaggerated microglial response could worsen any preexisting neurological disorder (even silent ones) or initiate such a disorder.[17,48,119,120]
Control of microglia activation and deactivation
A great number of factors can activate microglia, which makes sense when you consider that it is the main defensive arm of the innate immune system and must react rapidly to a number of stimuli. Microglial activation occurs rapidly upon disturbance of the brain's homeostasis, usually within minutes. The major activator of microglia is IL-1ß, which can be released locally with pathological damage within the CNS or can be transported from the systemic circulation.[23] Activation of a PRR signals the intracellular inflammasomes and this cleaves pro-IL-1ß, leading to microglial release of additional IL-1ß, thus further activating surrounding microglia. Other important cytokines in microglial activation include INF-γ and TNF-α. Thrombin can also activate microglia by way of a protease activated receptor 1 (PAR-1) receptor.[29] This would be important in cases of brain hemorrhaging and a leaky BBB. Activation in most cases takes place, as discussed previously, by the interaction of PAMPs and DAMPs with PRRs, which can include a number of the types of surface receptors mentioned above.
Recent studies have shown that microglial activation can be anatomically selective in terms of both the brain anatomy affected and the microglial phenotype being expressed.[38] This would explain the ability of systemic immune activation to produce differing patterns of behavioral and neurological effects, such as obsessive–compulsive disorder, depression or schizophrenia.[33,67,147,168] The final clinical presentation would depend on the activating principle and other associated factors, such as the patterns of neural net injury. Microglia are not only heterogeneously distributed, but also appear to have specialized subgroups, which would give them differing patterns of activation.
Newer studies have shown that many of the traditionally used antipsychotic and antidepressive medications can suppress microglial activation and reduce inflammation and excitotoxicity.[107] In fact, both lithium and St John's Wort reduce excitotoxicity and the latter has been shown to be antagonistic to the NMDA receptor, which has been shown to play a major role in depression.[90,181]
Of equal importance in brain inflammation are mechanisms utilized to downregulate or terminate microglial activation. Of principle importance are the cytokines, IL-10, IL-4, fractalkine (CX3CL1), and CD200 (cluster of differentiation 200) and its receptor CD200R. Suppression of the last two modulators is necessary for microglial activation. The immune suppressant cytokine IL-4 modulates CD200 so as to reduce microglial activation.[99] Mice lacking CD200 receptors demonstrate an exaggerated proinflammatory release following stimulation with LPS or other CNS or systemic immune activators and deficiencies in CD200 are linked to Alzheimer's dementia and Parkinson's syndrome.[165,170,182] Aged mice were found to have less CD200R on the surface of their microglia as compared with younger animals. This loss of CD200 would make aged individual more susceptible to intense, prolonged microglial activation following immune activation from any cause.
IL-10 reduces microglial activation and lowers secreted levels of TNF-α, NO, ROS, and superoxide following LPS exposure (immune activation).[122] Another antiinflammatory cytokine is TGF-ß a soluble factor produced by astrocytes in a constitutive and inducible manner. TGF-ß inhibits microglial activation.[123] TGF-ß has also been shown to selectively induce apoptosis in microglia in a mixed glial culture of microglia, astrocytes, and oligodendrocytes.[177] A reduction in microglia reduces brain inflammation.
Recently, TREM2, a DAP12-associated receptor and modulator of TLRs, has attracted a lot of attention as a regulator of microglial activation.[62] While TREM2 is found in a number of cell types, in the brain, the highest expression is in microglia. Increased expression of TREM2 switches microglia to a phagocytic phenotype, which is a phenotype known to secrete none of the proinflammatory cytokines. That is, it is mostly neuroprotective. Reductions in TREM2 are associated with a high risk of AD.[84]
Should we be taking microglial inhibitors in our diets?
Streit et al. put forth the hypothesis that with aging, microglia become dystrophic and therefore lose much of their ability to protect the brain utilizing both their immune and neurotrophic functions.[149] This dystrophic microglia concept would argue against the neurotoxic effect of primed microglia with brain aging. Sierra et al. examined this hypothesis utilizing a transgenic mouse line and found that with aging the microglia actually increased their production of proinflammatory cytokines (IL-1ß, IL-6, IL-12, and TNFα) as well as antiinflammatory cytokines as compared with younger mice, which would be consistent with destructive effect of priming of microglia with aging.[143] The proinflammatory cytokine levels were higher than the antiinflammatory cytokines, resulting in an inflammatory condition. This demonstrates that aged microglia have a fully functioning neurotoxic capability and because they are primed, exhibit an exaggerated proinflammatory response.
With the finding of abnormal functioning of CD200-CD200R regulatory control in neurodegenerative diseases, one can appreciate the effect of age-related priming on accelerating neurodegenerative pathology since the normal switching off of the microglia would be impaired [Figure 11]. Abnormalities of T regulatory lymphocyte (Tregs) function, a major antiinflammatory mechanism discussed later, have been linked to several neurological conditions.[127,155] Abnormalities in these and other antiinflammatory molecules would mean that even low concentrations of proinflammatory molecules would have a greater impact on neuronal pathology because of the imbalance in the antiinflammatory and inflammatory systems.
Figure 11.
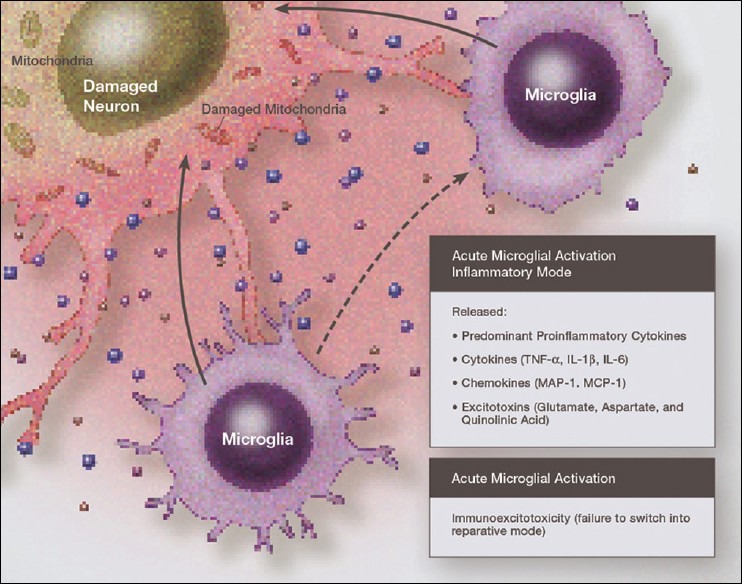
Illustration demonstrating chronic neurodegeneration resulting from a failure of activated microglia to switch to a resting state. Under such conditions immunoexcitotoxic cascades persist.
While microglial activation would appear to be a useful target in treating a number of neurological diseases, recent studies indicate that general suppression of microglia may not be a good idea, as certain phenotypes of microglia are neuroprotective. The timing is important when considering suppressing microglial activation. For example, it is thought that in the early course of ALS, microglia and macrophages are in the M2 protective mode of activation and as the disease progresses, they switch to an M1 cytotoxic mode.[21] The same pattern may also appear in AD. Tregs are thought to control microglia/macrophage switching from M1 (neurodestructive) to an M2 (neuroprotective) phenotype.[185]
Safer targets would be downstream mechanisms, such as inhibiting glutaminase, enhancing neuronal glutathione, antioxidant protection of critical enzymes, such as glutamine synthase and glutamate dehydrogenase; suppression of NADPH oxidase or iNOS, stimulation of group III mGLuRs (inhibitory), enhancing EAATs and suppression of prostaglandin E2 production. A number of studies have been done using these various approaches with evidence of considerable success in animal models and some limited human studies. A growing number of natural products, most extracted from plants, have shown an ability to alter immunoexcitotoxicity.
Immune cell adaptive responses in the CNS
Over 90 years ago it was shown that an implanted tumor in the brain of a rat was resistant to systemic immunity, thus establishing the idea that the brain was an “immunologically privileged site” and therefore isolated from systemic immunity. Over the past several decades growing evidence has supported the idea that this immune isolation is not absolute. It is true that among lymphocytes only activated lymphocytes can enter the CNS, but do so with reduced efficiency.[5,172]
Under normal conditions, once within the CNS parenchyma, the activated CD8+ cytotoxic T-cells are treated as being hostile and most undergo apoptosis or conversion to regulatory T-cells (Tregs). Neurons protected themselves from activated lymphocytes by upregulating and secreting TGF-ß, an inflammation-suppressing cytokine that converts CD8+ cytotoxic T-cells to immune suppressant regulatory T-cells (Tregs). Newer studies have shown a preponderance of Tregs in the face of CNS inflammation.[96,97] This indicates that neurons themselves are critical in immune modulation.
Lymphocyte migration into the CNS is regulated by chemokines and their receptors. In the normal brain, few lymphocytes are found in the brain parenchyma. Dramatic increased infiltration of T-cells occurs with strokes, brain trauma, and CNS infections. Lymphocytic infiltration into the CNS is increased in neurodegenerative disorders but not to the scale of brain trauma or infection. Gemechu and Bentivoglio found increased CNS lymphocyte infiltration with brain aging.[64]
Aging is associated with structural and functional alteration in the BBB, including alteration in the carrier-mediated mechanisms.[139] A leaky BBB allows normally excluded immune cells, as well as antibodies, to enter the brain parenchyma. Entry of T-cells into the brain parenchyma is also facilitated by chemokines secreted by reactive astrocytes and microglia such as monocyte chemotactic protein-1 (MCP-1) and macrophage inflammatory protein-1 (MIP-1).[4]
The fate of these entering T-cells depends on the state of brain inflammation and particular responses of lymphocyte interaction with glia cells. It appears that in most cases of prolonged brain inflammation, even with aging, T-cells are in a neuroprotective phenotype (Tregs).[30,135,137] This new concept has changed thinking regarding lymphocyte infiltration of the CNS in such diseases as multiple sclerosis, ALS, brain and spinal cord injury, and other neurodegenerative diseases.[136] It appears that in most cases of CNS inflammation, the preponderant T-cell phenotype consist of antiinflammatory Tregs, yet with disease progression, they may switch macrophages to a preponderant M1 cytotoxic phenotype.
SUMMARY
Over the past several decades we have learned a great deal about microglia and innate brain immunity. While microglia are the principle innate immune cells, other cell types also play a role, including invading macrophages, astrocytes, neurons, and endothelial cells. The fastest reacting cell is the microglia and despite its name, resting microglia are in fact quite active. Motion photomicrographs demonstrate a constant movement of ramified (resting) microglial foot processes, which appear to be testing the microenvironment for dangerous alteration in extracellular fluid content. These foot processes, in particular, interact with synapses and play a role in synaptic function. In event of excitatory overactivity, these foot processes can strip selected synapses, thus reducing activation states as a neuroprotective mechanism. They can also clear extracellular glutamate so as to reduce the risk of excitotoxicity.
Microglia also appear to have number of activation phenotypes, such as: principally phagocytic, principally neuroprotective and growth promoting or primarily neurodestructive. These innate immune cells can migrate a great distance under pathological conditions and appear to have anatomic specificity. There is some evidence that there are several types of microglia. Macrophage infiltration into the embryonic brain is the source of resident microglia and in adulthood macrophages can infiltrate the brain and are for the most part indistinguishable from resident microglia, but may react differently pathologically.
Activation itself does not imply a destructive phenotype and can be mostly neuroprotective via phagocytosis of debris, neuron parts and dying cells and by the release of neurotrophins such as NGF and brain BDNF. Evidence is accumulating that microglia undergo dynamic fluctuations in phenotype as the neuropathology evolves. For example, in the early stages of neurotrauma and stroke, microglia play a mostly neuroprotective role and only later switches to a neurodestructive mode.
A great number of biological systems alter microglia function, including neurohormones, cannabinoids, other neurotransmitters, ATP, adenosine, and corticosteroids. One can appreciate that with aging, many of these systems are altered by the aging process itself or diseases, thus changing the sensitivity of the innate immune system.
The realization that CNS microglia can be activated by stimulation of the systemic immune system is of great concern both in terms of natural disease and attempts to protect against disease by vaccination. It appears that not only the intensity of the immune stimulation is important, but also how closely spaced apart it occurs. Closely spaced, repetitive immune stimulation maximizes CNS innate immune activation and immunoexcitotoxicity.
ACKNOWLEDGMENT
The author would like to thank Dr. James Ausman for his invaluable suggestions concerning readability and continuity of the discussion. The author also thanks Dr. Dennis Malkasian for four of the illustrations, which have added significantly to the paper.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2013/4/1/118/118349
Disclaimer: The authors of this article has no conflict of interest to disclose, and has adhered to SNI's policies regarding human/animal rights, and informed consent. Advertisers in SNI did not ask for, nor did they receive access to this article prior to publication
REFERENCES
- 1.Aarum J, Sandberg K, Budd Haeberlein SL, Persson MA. Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci USA. 2003;100:15983–8. doi: 10.1073/pnas.2237050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Alarcon R, Fuenzalida C, Santibanez M, von Bernhardt R. Expression of scavenger receptors in glial cells. J Biol Chem. 2005;280:30406–15. doi: 10.1074/jbc.M414686200. [DOI] [PubMed] [Google Scholar]
- 4.Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cell: Different roles for microglia and astrocytes. Immunol Today. 2000;21:141–7. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- 5.Aloisi F, Ria F, Penna G, Adorini L. Microglia are more efficient than astrocytes in antigen processing and in the Th1 but not Th2 activation. J Immunol. 1998;160:4671–80. [PubMed] [Google Scholar]
- 6.Appel SH, Zhao W, Beers DR, Henkel JS. The microglial motor neuron dialogue in ALS. Acta Myol. 2011;30:4–8. [PMC free article] [PubMed] [Google Scholar]
- 7.Authier FJ, Cherin P, Creange A, Bonnotte B, Ferrer X, Abdelmoumni A, et al. Central nervous system disease in patients with macrophagic myofascitis. Brain. 2001;124(Pt 5):974–83. doi: 10.1093/brain/124.5.974. [DOI] [PubMed] [Google Scholar]
- 8.Banks WA, Kastin AJ. Blood to brain transport of interleukin links the immune and central nervous system. Life Sci. 1991;48:PL117–21. doi: 10.1016/0024-3205(91)90385-o. [DOI] [PubMed] [Google Scholar]
- 9.Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate released from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–13. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benito C, Kim WK, Chavarria I, Hillard CJ, Mackie K, Tolon RM, et al. A glial endogenous cannabinoid system is upregulated in the brains of macaques with simian immunoedeficiency virus-induced encephalitis. J Neurosci. 2005;25:2530–6. doi: 10.1523/JNEUROSCI.3923-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, et al. Cannabinoid CB 2 receptors and fatty acid amide hydrolase are selectively overexpressed in neurite plaque-associated glia in Alzheimer's disease brains. J Neurosci. 2003;23:11136–41. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HW. Ischemia-induced neuronal expression of microglial attracting chemokine Secondary Lymphoid-tissue Chemokine (SLC) Glia. 2001;34:121–33. doi: 10.1002/glia.1047. [DOI] [PubMed] [Google Scholar]
- 13.Blaylock RL. Aluminum induced immunoexcitotoxicity in neurodevelopmental and neurodegenerative disorders. Curr Inorg Chem. 2012;2:46–53. [Google Scholar]
- 14.Blaylock RL. A possible central mechanism in autism spectrum disorders, Part I. Altern Ther Health Med. 2008;14:46–53. [PubMed] [Google Scholar]
- 15.Blaylock RL. A possible central mechanism in autism spectrum disorders, Part 2: Immunoexcitotoxicity. Altern Ther Health Med. 2009;15:60–7. [PubMed] [Google Scholar]
- 16.Blaylock RL. Immunoexcitotoxicity as a central mechanism of autism spectrum disorders. In: Strunecka A, editor. Cellular and Molecular Biology of Autism Spectrum Disorders. Bentham Science Publishers, ebook; 2012. pp. 47–72. [Google Scholar]
- 17.Blaylock RL. Vaccines, depression, and neurodegeneration after age 50 years: Another reason to avoid the recommended vaccines. Med Veritas. 2008;5:1742–7. [Google Scholar]
- 18.Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy: A unifying hypothesis. Surg Neurol Int. 2011;2:107. doi: 10.4103/2152-7806.83391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blaylock RL, Strunecka A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr Med Chem. 2009;16:157–70. doi: 10.2174/092986709787002745. [DOI] [PubMed] [Google Scholar]
- 20.Bechmann I, Galea I, Perry VP. What is the blood-brain barrier (not)? Trend Immunol. 2007;28:5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A, et al. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun. 2011;25:1025–35. doi: 10.1016/j.bbi.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bluthe RM, Walter V, Parnet P, Laye S, Lestage J, Verrier D, et al. Lipopolysacchride induces sickness behavior in rats by a vagal mediated mechanism. C R Acad Sci III. 1994;317:499–503. [PubMed] [Google Scholar]
- 23.Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta inischemic brain damage. J Neurosci. 2001;21:5528–34. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks N. Specificity and reversibility of the inhibition by HgCl2 of glutamate transport in astrocytic cultures. J Neurochem. 1988;50:1117–22. doi: 10.1111/j.1471-4159.1988.tb10581.x. [DOI] [PubMed] [Google Scholar]
- 25.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–38. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 26.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, et al. Aggregation and motor neuron toxicity of an ALS linked SOD1 mutant independent from wild type SOD1. Science. 1998;281:1851–4. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 27.Buckingham SD, Jones AK, Brown LA, Sattelle DB. Nicotinic acetylcholine receptor signaling: Roles in Alzheimer's disease and amyloid neuroprotection. Pharmacol Rev. 2009;61:39–61. doi: 10.1124/pr.108.000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabral GA, Marciano-Cabral F. Cannabinoid receptors in microglia of the central nervous system: Immune functional relevance. J Leukoc Biol. 2005;78:1192–7. doi: 10.1189/jlb.0405216. [DOI] [PubMed] [Google Scholar]
- 29.Carreno-Muller E, Herrera AJ, de Pablos RM, Tomas-Carmardiel M, Venero JL, Cano JL, et al. Thrombin induces in vivo degeneration of nigral dopaminergic neurons along with the activation of microglia. J Neurochem. 2003;84:1201–14. doi: 10.1046/j.1471-4159.2003.01634.x. [DOI] [PubMed] [Google Scholar]
- 30.Carson MJ, Thrash JC, Walter B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in the neuronal death and survival. Clin Neurosci Res. 2006;6:237–45. doi: 10.1016/j.cnr.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cartier L, Hartley Q, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: Role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 32.CDC. [Last cited in 2013]. Available from: http://www.cdc.gov/vaccines/schedules/hcp/child.adolescent.html .
- 33.Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S. Glutamatergic dysfunction in OCD. Neuropsychopharmacology. 2005;30:1735–40. doi: 10.1038/sj.npp.1300733. [DOI] [PubMed] [Google Scholar]
- 34.Chamak B, Dobbertin A, Maliat M. Immunohistochemical detection of thrombospondin in microglia in the developing rat brain. Neuroscience. 1995;69:177–87. doi: 10.1016/0306-4522(95)00236-c. [DOI] [PubMed] [Google Scholar]
- 35.Chan WY, Kohsaka S, Rezaie P. The origin and cell linage of microglia: New concepts. Brain Res. 2007;53:344–54. doi: 10.1016/j.brainresrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Charleston JS, Body RI, Bolender RP, Mottet NK, Vhater ME, Brubacher TM. Changes in the number of astrocytes and microglia in the thalamus of the monkey Macaca fascicularis following long-term subclinical methylmercury exposure. Neurotoxicology. 1996;17:127–38. [PubMed] [Google Scholar]
- 37.Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, et al. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke. 2008;39:3042–8. doi: 10.1161/STROKEAHA.108.521898. [DOI] [PubMed] [Google Scholar]
- 38.Chen SK, Tvdik P, Peden E, Cho S, Wu S, Spangrude G, et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141:775–85. doi: 10.1016/j.cell.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho J, Nelson TE, Bajova H, Gruol DL. Chronic CXCL10 alters neuronal properties in rat hippocampal culture. J Neuroimmunol. 2009;207:92–100. doi: 10.1016/j.jneuroim.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. 2012;120:292–301. doi: 10.1111/j.1471-4159.2011.07572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi SH, Lee DY, Chung ES, Hong YB, Kim SU, Jin BK. Inhibition of thrombin-induced microglial activation and NADPH oxidase by minocycline protects dopaminergic neurons in the substantia nigra in vivo. J Neurochem. 2005;95:1755–65. doi: 10.1111/j.1471-4159.2005.03503.x. [DOI] [PubMed] [Google Scholar]
- 42.Christensen RN, Ha BK, Sun F, Bresnahan JC, Beattie MS. Kainate induces rapid redistribution of the actin cytoskeleton in amoeboid microglia. J Neurosci Res. 2006;84:170–81. doi: 10.1002/jnr.20865. [DOI] [PubMed] [Google Scholar]
- 43.Clarkson AN, Overman JJ, Zhong S, Mueller R, Lynch G, Carmichael ST. AMPA receptor-induced local brain derived neurotrophic factor signaling mediates motor recovery after a stroke. J Neurosci. 2011;31:3766–75. doi: 10.1523/JNEUROSCI.5780-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Combrinck MI, Perry VH, Cunningham C. Peripheral infection evokes exaggerated sickness behavior in pre-clinical murine prion disease. Neuroscience. 2002;112:7–11. doi: 10.1016/s0306-4522(02)00030-1. [DOI] [PubMed] [Google Scholar]
- 45.Correa JD, Starling D, Teixeira AL, Caramelli P, Silva TA. Chemokines in CSF of Alzheimer's disease patients. Arq Neuropsiqiatr. 2011;69:455–9. doi: 10.1590/s0004-282x2011000400009. [DOI] [PubMed] [Google Scholar]
- 46.Cortese BM, Phan KL. The role of glutamate in anxiety and related disorders. CNS Spectr. 2005;10:820–30. doi: 10.1017/s1092852900010427. [DOI] [PubMed] [Google Scholar]
- 47.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–84. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 50.De Haas AH, van Weering HR, de Jong EK, Boddeke HW, Biber KP. Neuronal chemokines: Versatile messengers in central nervous system cell interaction. Mol Neurobiol. 2007;36:137–51. doi: 10.1007/s12035-007-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Haas AH, Boddeke HW, Biber K. Region-specific expression of immune regulating proteins on microglia in healthy CNS. Glia. 2008;56:888–94. doi: 10.1002/glia.20663. [DOI] [PubMed] [Google Scholar]
- 52.De Simone R, Ambrosini E, Carnevale D, Ajmone-Cat MA, Minghetti L. NGF promotes microglial migration through the activation of its high affinity receptor: Modulation by TGF-ß. J Neuroinflamm. 2007;190:53–60. doi: 10.1016/j.jneuroim.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 53.Donohoe MM, Cain K, Zierath D, Shibata D, Tanzi PM, Becker KJ. Higher plasma fractalkine is associated with better 6-month outcome from ischemic stroke. Stroke. 2012;43:2300–6. doi: 10.1161/STROKEAHA.112.657411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dunn N, Mullee M, Perry VH, Holmes C. Association between dementia and infectious disease: Evidence from a case-control study. Alzheimer Dis Assoc Disord. 2005;19:91–4. doi: 10.1097/01.wad.0000165511.52746.1f. [DOI] [PubMed] [Google Scholar]
- 55.Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, et al. Stimulation of cannabinoid receptor 2(CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engell T. A clinical patho-anatomical study of clinically silent multiple sclerosis. Acta Neurol Scand. 1989;79:428–30. doi: 10.1111/j.1600-0404.1989.tb03811.x. [DOI] [PubMed] [Google Scholar]
- 57.Fabry Z, Fitzsimmons KM, Herlein JA, Moniger TO, Dobbs MB, Hart MN. Production of the cytokines interleukin 1 and 6 by murine brain microvessel endothelium and smooth muscle pericytes. J Neuroimmunol. 1993;47:23–34. doi: 10.1016/0165-5728(93)90281-3. [DOI] [PubMed] [Google Scholar]
- 58.Farber K, Pannasch U, Kettenmann H. Dopamine and noradrenalin control distinct functions in rodent microglial cells. Mol Cell Neurosci. 2005;29:128–38. doi: 10.1016/j.mcn.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 59.Farso MC, O’shea RD, Beart PM. Evidence group I mGLuR drugs modulate the activation profile of lipopolysaccharide-exposed microglia in culture. Neurochem Res. 2009;34:1721–8. doi: 10.1007/s11064-009-9999-3. [DOI] [PubMed] [Google Scholar]
- 60.Finberg RW, Knope DM, Kurt-Jones EA. Herpes simplex and toll-like receptors. Viral Immunol. 2005;18:457–65. doi: 10.1089/vim.2005.18.457. [DOI] [PubMed] [Google Scholar]
- 61.Flarend RE, Hem SL, White JL, Elmore D, Suckow MA, Rudy AC, et al. In vivo absorption of aluminum-containng vaccine adjuvants using 26Al. Vaccine. 1997;15:1314–8. doi: 10.1016/s0264-410x(97)00041-8. [DOI] [PubMed] [Google Scholar]
- 62.Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46. doi: 10.1016/j.coi.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003;23:6181–7. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gemechu JM, Bentivoglio M. T cell recruitment in the brain during normal aging. Front Cell Neurosci. 2012;6:38. doi: 10.3389/fncel.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gherardi RK, Coquet M, Cherin P, Belec L, Moretto P, Dreyfus PA, et al. Macrophagic myofascitis lesions assess long-term persistence of vaccine-derived aluminum hydroxide in muscle. Brain. 2001;124(Pt 9):1821–31. doi: 10.1093/brain/124.9.1821. [DOI] [PubMed] [Google Scholar]
- 66.Ghoreschi K, Larence A, O’shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gilmore JH, Frederik Jarskog I, Vadlamudi S, Lauder M. Prenatal infection and risk for schizophrenia: Il-1 beta, IL-6 and TNFalpha inhibit cortical neuron dendrite development. Neuropsychopharmacology. 2004;29:1221–9. doi: 10.1038/sj.npp.1300446. [DOI] [PubMed] [Google Scholar]
- 68.Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–31. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- 69.Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O’Connor J, et al. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology. 2007;33:2341–51. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gregory CD, Devitt A. The macrophage and the apoptotic cell: An innate immune interaction viewed simplistically? Immunology. 2004;113:1–14. doi: 10.1111/j.1365-2567.2004.01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grommes C, Lee CY, Wilkenson BL, Jiang Q, Koenigskech-Talboo JL, Varnum B, et al. Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J Neuroimmune Pharm. 2008;3:130–40. doi: 10.1007/s11481-007-9090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guerreiro R, Wojtas A, Bras J, Carrasquilo M, Rogaeya E, Majornie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–27. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol. 1993;47:169–76. doi: 10.1016/0165-5728(93)90027-v. [DOI] [PubMed] [Google Scholar]
- 74.Guzman A, Wood WL, Alpert E, Prasad MD, Miller RG, Rothestein JD, et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2007;104:12524–9. doi: 10.1073/pnas.0705044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocyte activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–56. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 76.Hansen MK, Nguyen KT, Goehler LE, Gaykema RP, Fleshner M, Maier SF, et al. Effects of vagotomy on liposacchride-induced brain interkeukin-1 beta protein in rats. Auton Neurosci. 2000;85:119–26. doi: 10.1016/s1566-0702(00)00230-7. [DOI] [PubMed] [Google Scholar]
- 77.He F, Balling R. The role of regulatory T cells in neurodegenerative diseases. Wiley Interdiscip Rev Syst Biol Med. 2013;5:153–80. doi: 10.1002/wsbm.1187. [DOI] [PubMed] [Google Scholar]
- 78.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:788–9. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci USA. 2002;99:1604–9. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoyte L, Barber PA, Buchan AM, Hill MD. The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med. 2004;4:131–6. doi: 10.2174/1566524043479248. [DOI] [PubMed] [Google Scholar]
- 81.Inoue K, Koizumi S, Kataoka A, Tozaki-Saitoh H, Tsuda M. P2Y 6 -evoked microglial phagocytosis. Int Rev Neurobiol. 2009;163:159–63. doi: 10.1016/S0074-7742(09)85012-5. [DOI] [PubMed] [Google Scholar]
- 82.Jaerve A, Muller HW. Chemokines in CNS injury and repair. Cell Tissue Res. 2012;349:229–48. doi: 10.1007/s00441-012-1427-3. [DOI] [PubMed] [Google Scholar]
- 83.Ji RR, Suter MR. p38MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jonsson T, Stefansson H, Steiunberg S, Josdottir I, Jonsson PV, Snaedal J, et al. Varient of TREM2 associated with the risk of Alzheimer's disease. N Eng J Med. 2013;368:107–16. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jou I, Lee JH, Park SY, Yoon HJ, Joe EH, Park EJ. Ganglosides trigger inflammatory responses via TLR4 in brain glia. Am J Pathol. 2006;168:1619–30. doi: 10.2353/ajpath.2006.050924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Juarez BI, Martinez MI, Montante M, Dufour L, Garcia E, Jimenez-Capdeville ME. Methylmercury increases glutamate extracellular levels in the frontal cortex of awake rats. Neurotoxicol Teratol. 2002;24:767–71. doi: 10.1016/s0892-0362(02)00270-2. [DOI] [PubMed] [Google Scholar]
- 87.Kawamata J, Suzuki S, Shimohama S. α7 nicotinic acetylcholine receptor mediated neuroprotection in Parkinson's disease. Curr Drug Targets. 2012;13:623–30. doi: 10.2174/138945012800399026. [DOI] [PubMed] [Google Scholar]
- 88.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 89.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J Neurosci. 2000;20:6309–16. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kumar V, Mdzinarishvili A, Kiewert C, Abbruscato T, Bickel U, van der Schyf CJ, et al. NMDA receptor-antagonistic properties of hyperforin, a constituent of St. John's Wort. J Pharmacol Sci. 2006;102:47–54. doi: 10.1254/jphs.fp0060378. [DOI] [PubMed] [Google Scholar]
- 91.Lacroix S, Feinstein D, Rivest S. The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol. 1998;8:625–40. doi: 10.1111/j.1750-3639.1998.tb00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–70. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 93.Lee YB, Nagal A, Kim SU. Cytokiness, chemokines, and cytokine receptors in human microglia. J Neurosci Res. 2002;69:94–103. doi: 10.1002/jnr.10253. [DOI] [PubMed] [Google Scholar]
- 94.Leong J, Zhou M, Jacob A, Wang P. Aging-related hyperinflammation in endotoxemia is mediated by the α2A-adrenoceptor and CD14/TKR4 pathways. Life Sci. 2010;86:740–6. doi: 10.1016/j.lfs.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu GJ, Nagarajah R, Banati RB, Bennett MR. Glutamate induces directed chemotaxis of microglia. Eur J Neurosci. 2009;29:1108–18. doi: 10.1111/j.1460-9568.2009.06659.x. [DOI] [PubMed] [Google Scholar]
- 96.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalogenic T cells suppresses EAE. Nat Med. 2006;12:518–25. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 97.Liu Y, Amarnath S, Chen W. Requirement of CD28 signaling in homeostasis of TGF-beta converted CD4+CD25+Tregs from thymic CD4+CD25-single positive T cells. Transplantation. 2006;82:953–64. doi: 10.1097/01.tp.0000232330.46688.37. [DOI] [PubMed] [Google Scholar]
- 98.Lynch MA. The Multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–56. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 99.Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand-receptor interaction modulates microglial activation in vivo and in vitro: A role for IL-4. J Neurosci. 2007;27:8309–13. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mander P, Brown GC. Activation of microglia NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J Neuroinflamm. 2005;2:20. doi: 10.1186/1742-2094-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mander PK, Jekabsone A, Brown GC. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol. 2006;176:1046–52. doi: 10.4049/jimmunol.176.2.1046. [DOI] [PubMed] [Google Scholar]
- 102.Mao XJ, Zhang XM, Zhang HL, Quezada HC, Mix E, Yang X, et al. TNF-alpha receptor 1 deficiency reduces antigen-presenting capacity of Schwann cells and ameliorates experimental autoimmune neurtitis in mice. Neurosci Lett. 2010;470:19–23. doi: 10.1016/j.neulet.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 103.Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–32. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- 104.Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–45. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- 105.Matyja E. Aluminum enhances glutamate-mediated neurotoxicity in organotypic cultures of rat hippocampus. Folia Neuropathol. 2000;38:47–53. [PubMed] [Google Scholar]
- 106.McMullan SM, Phanavanh B, Li GG, Barger SW. Metabotropic glutamate receptors inhibit microglial glutamate release. ASN Neuro. 2012 doi: 10.1042/AN20120044. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Michael-Titus AT, Bains S, Jeetle J, Whepton R. Imipramine and phenelzine decrease glutamate overflow in the prefrontal cortex: A possible mechanism of neuroprotection in major depression. Neuroscience. 2000;100:681–4. doi: 10.1016/s0306-4522(00)00390-0. [DOI] [PubMed] [Google Scholar]
- 108.Morioka N, Tanabe H, Inoue A, Dohi T, Nakata Y. Noradrenalin reduces the ATP-stimulated phosphorylation of p38 MAP kinase via beta-adrenergic receptors-cAMP protein kinaseA-dependent mechanism in rat spinal microglia. Neurochem Int. 2009;55:226–34. doi: 10.1016/j.neuint.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 109.Murry CL, Fibiger HC. Learning and memory deficits after lesions of the nucleus basalis magnocellularis: reversal by physostigmine. Neuroscience. 1985;14:1025–32. doi: 10.1016/0306-4522(85)90273-8. [DOI] [PubMed] [Google Scholar]
- 110.Neuman H, Boucraut J, Hahnel C, Misgeld C, Wekerle H. Neural control of MHC class II inductability in rat astrocytes and microglia. Eur J Neurosci. 1996;8:2582–90. doi: 10.1111/j.1460-9568.1996.tb01552.x. [DOI] [PubMed] [Google Scholar]
- 111.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglia cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 112.Obsawa K, Irino Y, Nakamura Y, Akazawa C, Inuone K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55:604–16. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- 113.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–24. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 114.Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, et al. Microglial CB 2 cannabinoid receptors are neuroprotective in Hunington's disease excitotoxicity. Brain. 2009;132:3152–64. doi: 10.1093/brain/awp239. [DOI] [PubMed] [Google Scholar]
- 115.Panaro MA, Cianciulli A. Current opinions and perspectives on the role of immune system in the pathogenesis of Parkinson's disease. Curr Pharm Des. 2012;18:200–8. doi: 10.2174/138161212799040574. [DOI] [PubMed] [Google Scholar]
- 116.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–8. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 117.Pei Z, Pang H, Qian L, Yang S, Wang T, Zhang W, et al. MAC-1 mediates LPS-induced production of superoxide by microglia: The role of pattern recognition receptors in dopaminergic neurotoxicity. Glia. 2007;55:1362–73. doi: 10.1002/glia.20545. [DOI] [PubMed] [Google Scholar]
- 118.Perry VH. The influence of systemic inflammation on inflammation in the brain: Implications for chronic neurodegenerative disease. Brain Behav Immunol. 2004;18:407–13. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 119.Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–7. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 120.Perry VH, Newman TA, Cunningham C. The impact of infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–12. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 121.Phani S, Re DB, Przedborski S. The role of the innate immune system in ALS. Front Pharmacol. 2012;3:150. doi: 10.3389/fphar.2012.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qian L, Hong JS, Flood PM. Role of microglia in inflammation-mediated degeneration of dopaminergic neurons: Neuroprotective effect of interleukin-10. J Neural Transm Suppl. 2006;70:367–71. doi: 10.1007/978-3-211-45295-0_56. [DOI] [PubMed] [Google Scholar]
- 123.Qian L, Wei SJ, Zhang D, Hu X, Xu Z, Wilson B, et al. Potent anti-inflammatory and neuroprotective effects of TGF-beta1 are mediated through the inhibition of ERK and p47phox-Ser345 phosphorylation and translocation in microglia. J Immunol. 2008;181:660–8. doi: 10.4049/jimmunol.181.1.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Quik M, Perez XA, Bordia T. Nicotine as a potential neuroprotective agent for Parkinson's disease. Mov Disord. 2012;27:947–57. doi: 10.1002/mds.25028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ransohoff RM, Perry VH. Microglial physiology: Unique stimuli, specific responses. Annu Rev Immunol. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 126.Redhead K, Quinlan GJ, Das RG, Guttteridge JM. Aluminum-adjuvanted vaccines transiently increase aluminum levels in murine brain tissues. Pharmacol Toxicol. 1992;70:278–80. doi: 10.1111/j.1600-0773.1992.tb00471.x. [DOI] [PubMed] [Google Scholar]
- 127.Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+regulatory T cells in an animal of Parkinson's disease. J Leukoc Biol. 2007;82:1083–94. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- 128.Rothstein JD. Current hypothesis for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(suppl):S3–9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- 129.Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornbiath DR, Drachman DB. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 130.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kunci RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 131.Scheid R, Teich N. Neurological manifestations of ulcerative colitis. Eur J Neurol. 2007;14:483–93. doi: 10.1111/j.1468-1331.2007.01718.x. [DOI] [PubMed] [Google Scholar]
- 132.Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2005;196:290–7. doi: 10.1016/j.expneurol.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 133.Schlett K. Glutamate as a modulator of embryonic and adult neurogenesis. Curr Top Med Chem. 2006;6:949–60. doi: 10.2174/156802606777323665. [DOI] [PubMed] [Google Scholar]
- 134.Scholz J, Woolf CJ. The neuropathic pain triad: Neurons, immune cells, and glia. Nature Neurosci. 2007;10:1361–8. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 135.Schwartz M. Beneficial autoimmune T cells and posttraumatic neuroprotection. Ann N Y Acad Sci. 2000;917:341–7. doi: 10.1111/j.1749-6632.2000.tb05400.x. [DOI] [PubMed] [Google Scholar]
- 136.Schwartz M, Kipnis J. Multiple sclerosis as a by-product of the failure to sustain protective autoimmunity: A paradigm shift. Neuroscientist. 2002;8:405–13. doi: 10.1177/107385802236966. [DOI] [PubMed] [Google Scholar]
- 137.Schwartz M, Shecter R. Protective autoimmunity functions by intracranial immunosurveilance to support the mind: The missing link between health and disease. Mol Psychiatry. 2010;15:342–54. doi: 10.1038/mp.2010.31. [DOI] [PubMed] [Google Scholar]
- 138.Seki E, Tsutsui H, Tsuji NM, Hayashi N, Adachi K, Nakano H, et al. Critcial roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J Immunol. 2002;169:3863–8. doi: 10.4049/jimmunol.169.7.3863. [DOI] [PubMed] [Google Scholar]
- 139.Shan GN, Mooradian AD. Age-related changes in the blood-brain barrier. Exp Gerontol. 1997;32:501–19. doi: 10.1016/s0531-5565(96)00158-1. [DOI] [PubMed] [Google Scholar]
- 140.Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, et al. Activation of NADPH oxidase in Alzheimer's disease brains. Biochem Biophys Res Commun. 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 141.Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zheng J, et al. Cholinergic modulation of microglial activation by α7 nicotinic receptors. J Neurochem. 2004;89:337–43. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 142.Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313–5. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55:412–24. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- 144.Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other ß-chemokines by resident glia and inflammatory cells in multiple sclerosis leisons. J Neuroimmunol. 1998;84:238–49. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- 145.Spreux-Varoquaux O, Bensimon G, Lacomblez L, Salachas F, Pradat PF, Le Forestier N, et al. Glutamate level in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci. 2002;193:73–8. doi: 10.1016/s0022-510x(01)00661-x. [DOI] [PubMed] [Google Scholar]
- 146.Strandberg TE, Pitkala KH, Linnavuori K, Tilvis RS. Cognitive impairment and infectious burden in the elderly. Arch Gerontol Geriatr Suppl. 2004;9:419–23. doi: 10.1016/j.archger.2004.04.053. [DOI] [PubMed] [Google Scholar]
- 147.Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflamm. 2011;8:94. doi: 10.1186/1742-2094-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Streit WJ, Morioka T, Kalehua AN. MK-801 prevents microglial reaction in rat hippocampus after forebrain ischemia. Neuroreport. 1999;57:146–8. doi: 10.1097/00001756-199202000-00006. [DOI] [PubMed] [Google Scholar]
- 149.Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45:208–12. doi: 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- 150.Stella N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes and astrocytomas. Glia. 2010;58:1017–30. doi: 10.1002/glia.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: The potential of NOX2 inhibitors. Cell Mol Life Sci. 2012;69:2409–27. doi: 10.1007/s00018-012-1015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Taetzsch T, Block ML. Pesticides, microglial NOX2, and Parkinson's disease. J Biochem Mol Toxicol. 2013;27:137–49. doi: 10.1002/jbt.21464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tanaka K, Okada Y, Kanno T, Otomo A, Yanagisawa Y, Shonguchi-Miyata J, et al. A dopamine receptor anagonist L-745,870 suppresses microglia activation in spinal cord and mitigates the progression in ALS model mice. Exp Neurol. 2008;211:378–86. doi: 10.1016/j.expneurol.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 155.Tani T, Tanaka K, Idezuka J, Nishizawa M. Regulatory T cells in paraneoplastic neurological syndromes. J Neuroimmunology. 2008;196:166–9. doi: 10.1016/j.jneuroim.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 156.Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglial metabotropic glutamate receptor mGLu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived Fa ligand. J Neurosci. 2005;25:2952–64. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tobinick EL, Gross H. Rapid improvement in verbal fluency and aphasia following perispinal etanercept in Alzheimer's disease. BMC Neurol. 2008;8:27. doi: 10.1186/1471-2377-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: An observational study involving 629 consecutive patients with perispinal etanercept. CNS Drugs. 2012;26:1051–70. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- 159.Tomljenovic L, Shaw CA. Aluminum vaccine adjuvants: Are they safe? Curr Med Chem. 2011;18:2630–7. doi: 10.2174/092986711795933740. [DOI] [PubMed] [Google Scholar]
- 160.Trapp BD, Wujek JR, Criste GA, Jalabi W, Yin X, Kidd GJ, et al. Evidence for synaptic stripping by cortical microglia. Glia. 2007;55:360–8. doi: 10.1002/glia.20462. [DOI] [PubMed] [Google Scholar]
- 161.Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–9. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 162.Tweedie D, Sambamuti K, Greig NH. TNF-alpha inhibition as a treatment strategy for neurodegenerative disorders: New drug candidates and targets. Curr Alzheimer Res. 2007;4:378–85. doi: 10.2174/156720507781788873. [DOI] [PubMed] [Google Scholar]
- 163.Venken K, Hellings N, Libau R, Stinissen P. Disturbed regulatory T cell homeostasis in multiple sclerosis. Trends Mol Med. 2010;16:58–68. doi: 10.1016/j.molmed.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 164.Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Argilelles S, Delgado-Cartes MJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: Potential risk factor in Parkinson's disease. J Neurochem. 2010;114:1687–700. doi: 10.1111/j.1471-4159.2010.06879.x. [DOI] [PubMed] [Google Scholar]
- 165.Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200R in Alzheimer's disease: A potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23:1398–405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Walton JR. Aluminum in hippocampal neurons from humans with Alzheimer's disease. Neurotoxicology. 2006;27:385–94. doi: 10.1016/j.neuro.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 168.Wang JQ, Mao L, Parelkar NK, Tang Q, Liu Z, Sarwar S, et al. Glutamate-regulated behavior, transmitter release, gene expression and addictive plasticity in the stratium: Roles of metabotropic glutamate receptors. Curr Neuropharmacol. 2003;1:1–20. [Google Scholar]
- 169.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–73. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 170.Wang XJ, Ye M, Zhang YH, Chen SD. CD200-CD200R regulation of microglia activation in the pathogenesis of Parkinson's disease. J Neuroimmune Pharmacol. 2007;2:259–64. doi: 10.1007/s11481-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 171.Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Curr Alzheimer Res. 2008;5:342–5. doi: 10.2174/156720508784533286. [DOI] [PubMed] [Google Scholar]
- 172.Wekerle H, Linington C, Lasserman H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci. 1986;9:271–7. [Google Scholar]
- 173.Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer's disease. PLoS One. 2012;7:e30525. doi: 10.1371/journal.pone.0030525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int J Alzheimers Dis 2012. 2012 doi: 10.1155/2012/489456. 489456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Windham BG, Griswold ME, Shibata D, Penman A, Catellier DJ, Mosley TH., Jr Covert neurological symptoms associated with silent infarcts from midlife to older age: The Atherosclerosis risk in communities study. Stroke. 2012;43:18–23. doi: 10.1161/STROKEAHA.111.643379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Witting A, Muller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by microglia/brain macrophages in vitro: Involvement of lectin-, integrin-, phosphotidylserine-mediated recognition. J Neurochem. 2000;75:1060–70. doi: 10.1046/j.1471-4159.2000.0751060.x. [DOI] [PubMed] [Google Scholar]
- 177.Xaio BG, Bai XF, Zhang GX, Link H. Transforming growth factor-beta1 induces apoptosis of rat microglia without relation to bcl-2 oncoprotein expression. Neurosci Lett. 1997;226:71–4. doi: 10.1016/s0304-3940(97)00234-6. [DOI] [PubMed] [Google Scholar]
- 178.Xia MQ, Hyman BT. Chemokines/chemokine receptors in the central nervous system and Alzheimer's disease. J Neurovirol. 1999;5:32–41. doi: 10.3109/13550289909029743. [DOI] [PubMed] [Google Scholar]
- 179.Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival though different transduction pathways. J Neurosci. 2002;22:3025–32. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, et al. COX2, CB2 and P2X-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yu Z, Ono C, Sora I, Tomita H. Effect of chronic lithium treatment on gene expression profile in mouse microglia and brain dendritic cells. Nihon Shinkei Seishin Yakurigaku Zasshi. 2011;31:101–2. [PubMed] [Google Scholar]
- 182.Zhan S, Wang XJ, Tian LP, Lu GQ, Zhang YJ, Ding JQ, et al. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson's disease. J Neuroinflamm. 2011;8:154. doi: 10.1186/1742-2094-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang R, Gascon R, Miller RG, Gelinas DF, Mass J, Lancero M, et al. MCP-1 chemokine receptor CCR2 is decreased on circulating monocytes in sporadic amyotrophic lateral sclerosis (sALS) J Neuroimmunol. 2006;179:87–93. doi: 10.1016/j.jneuroim.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 184.Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, et al. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis. J Neuroimmunol. 2009;206:121–4. doi: 10.1016/j.jneuroim.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhao W, Beers DR, Liao B, Henkel JS, Appel SH. Regulatory T lymphocytes from ALS mice suppresses microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol Dis. 2012;48:418–28. doi: 10.1016/j.nbd.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhou H, Liao J, Aloor J, Nie H, Wilson BC, Fessler MB, et al. CD11b/CD18 (MAC-1) is a novel surface receptor for extracellular double-stranded RNA to mediate cellular inflammatory responses. J Immunol. 2013;190:115–25. doi: 10.4049/jimmunol.1202136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zweig RM, Ross CA, Hedreen JC, Steele C, Cardillo JE, Whitehouse PJ, et al. The neuropathology of aminergic nuclei in Alzheimer's disease. Ann Neurol. 1988;24:233–42. doi: 10.1002/ana.410240210. [DOI] [PubMed] [Google Scholar]