

Abstract
The human islet amyloid polypeptide (hIAPP) is the primary component in the toxic islet amyloid deposits in type-2 diabetes. hIAPP self-assembles to aggregates that permeabilize membranes and constitutes amyloid plaques. Uncovering the mechanisms of amyloid self-assembly is the key to understanding amyloid toxicity and treatment. Although structurally similar, hIAPP's rat counterpart, the rat islet amyloid polypeptide (rIAPP), is non-toxic. It has been a puzzle why these peptides behave so differently. We combined multiscale modelling and theory to explain the drastically different dynamics of hIAPP and rIAPP: The differences stem from electrostatic dipolar interactions. hIAPP forms pentameric aggregates with the hydrophobic residues facing the membrane core and stabilizing water-conducting pores. We give predictions for pore sizes, the number of hIAPP peptides, and aggregate morphology. We show the importance of curvature-induced stress at the early stages of hIAPP assembly and the α-helical structures over β-sheets. This agrees with recent fluorescence spectroscopy experiments.
Amyloidoses are a class of diseases including Type II Diabetes Mellitus, Alzheimer's and Parkinson's. These diseases are characterized by the conversion of peptides or proteins from their soluble functional states into fibrillar aggregates1. The formation and buildup of these dense amyloid plaques has been widely accepted as the primary cause of disease (amyloid hypothesis)2.
The amyloid hypothesis is challenged by the observation that a large number of Alzheimer's patients3 with amyloid plaques in the brain show no symptoms4,5,6,7. Similarly, amyloid deposition in type II diabetes is evident in only about 90% of patients8,9 and the remaining 10% do not develop any significant amyloid deposition10. This evidence substantiates the hypothesis that soluble, small-sized oligomers rather than mature fibrils may be the root of the pathogenic state. Thus, understanding the early steps of their formation, oligomeric interactions, and inhibition are among the key issues and prerequisites to develop effective therapies11,12,13. In general, the mechanism(s) by which amyloid intermediates cause cytotoxicity and disease remains unresolved. Nevertheless, the inhibition of amyloid toxicity by a common antibody, independent of the location of the oligomers in extracellular or intracellular compartments, clearly supports a common mechanism in areas which are accessible via extra-and intracellular regions, such as cell membranes14,15,16,17,18.
In general, amyloid-forming peptides are amphipathic. They may cause membrane perturbation through changes in bilayer fluidity, generate protein-stabilized pores (poration), lay on one leaflet of the membrane (carpeting), or remove lipid components from the bilayer by a detergent-like mechanism19. A recent study has also suggested a two-step mechanism for amyloid β induced disruption and pore formation with properties such as ion selectivity20. There is, however, no consensus regarding which membrane perturbations are the most relevant to disease21. It has also been shown that the formation of amyloid fibers is a separate process from membrane disruption22 and that the composition of membrane and ions may have a significant influence membrane disruption23. Brender et al.24provide an excellent review of membrane disruption induced by IAPP.
Islet Amyloid Polypeptide (IAPP) is the primary component in the pancreatic islet amyloid deposits observed in type 2 diabetes25,26. Human IAPP (hIAPP) is a 37 amino acid peptide produced in the islet β-cells and co-secreted with insulin. Like other amyloid-forming peptides, hIAPP has been observed permeabilizing lipid membranes27,28,29,30,31. The toxic oligomeric state of IAPP and its interaction with membranes has recently gained attention4,32,33, but its transient nature makes experimental characterization highly elusive34. It is known that addition of IAPP results in unspecific pore formation in membrane conductance experiments35. Yet, some studies point to the critical role of hIAPP monomers in membrane interactions36,37. Whether monomers, dimers or some larger structure are the most important in triggering membrane damage remains to be established.
Molecular dynamics (MD) is the only available technique for obtaining protein data at atomistic spatial resolution and microsecond, or finer, temporal resolution. MD simulations provide an invaluable tool to study the early steps of membrane-bound IAPP oligomerization and its effect on membrane integrity. There are several in silico studies of IAPPs, but, thus far the most extensive ones have focused on the free state structure in solution38,39,40. All Atom (AA) and Coarse Grained (CG) MD studies of IAPP in membrane41,42 addressing the conformational equilibria of IAPP monomeric state have been also reported43. In these studies, the β-strand conformation was assumed as the initial state34. None of these studies, however, could single out the IAPP oligomers population and the variation of the membrane structure during self-assembly. To address this, we chose a different approach and performed a multiscale modeling study of the self-assembly and oligomerization of monomeric α-helical IAPP peptides in a model lipid bilayer (palmitoy-oleyl-phosphatidylcoline (POPC) in a microsecond time scale. We employed a combination of atomistic MD (to explore the conformation of the assembled proteins) and CG simulations (to unveil the dynamics of protein aggregation in membrane) and used a mapping procedure to switch between the different levels of representations. Based on the outcome of simulations, we developed a thermodynamic model for aggregate formation. By combining the computational and analytical approaches, we demonstrate and explain the drastically different dynamics of oligomerization of hIAPP and its non-amyloidogenic rat variant (rIAPP). Our results underscore the importance of electrostatic dipolar interactions for determining the shape of membrane-bound hIAPP aggregates as well as their impact on membrane physical properties.
Results
Self-assembly of hIAPP in a POPC lipid bilayer
We start by comparing the CG simulations of 36 hIAPP and as control 36 rIAPP peptides in α-helix conformations (26 μs at 300 K) in a POPC membrane at 1:44 peptide-lipid molar ratio (Fig. 1). This ratio and secondary structure were chosen in order to simulate typical experimental conditions44,36. All hIAPP peptides remained fully inserted in the membrane spanning from one leaflet to the other. They diffused laterally and self-assembled to form aggregates.
Figure 1. Self-assembly of membrane-embedded hIAPP or rIAAP give rise to differently structured peptide aggregates.

Top view from a simulation of peptide aggregation at 300 K for hIAPP (red) and rIAPP (green). Snapshots were taken at 1.0 μs (a,e), 7.0 μs (b,f), 13.0 μs (c,g), 20.0 μs (d,h) respectively. Lipids: blue. Yellow shows the lysine residues.
Short time behavior
Dimers, trimers and tetramers started to form within the first 10 ns. At ~6 μs, dimers and trimers had virtually disappeared and only clusters made of 4–6 peptides were present. Once formed, these clusters diffused as a single unit and peptides did not dissociate from them. Repeated simulations at different protein concentrations exhibit a size concentration dependence behavior.
Longer time behavior
Larger aggregates started to form at ~12 μs and after ~20 μs (protein/lipid ratio of 1:44), a large, crescent shaped assembly containing 26 hIAPP molecules and a smaller circular one containing 10 hIAPP had formed: at greater peptide concentration the overall size of the aggregates is larger.
Repeated simulations showed that hIAPP aggregates consisting of less than ~10 peptides preferred circular shapes and larger aggregates grew linearly to form crescent-like aggregates. We determined the basic stable structural unit to be a pentameric aggregate. Aggregate growth occurred through the formation of edge-fused pentameric clusters. This finding is in agreement with AFM experiments (both circular and pentameric aggregates have been observed) that have also indicated the possibility of a similar growth mechanism28,45. Growth of hIAPP and rIAPP aggregates is shown in Fig. 2. We will discuss the physical mechanism determining the stability and shape of hIAPP aggregates later on.
Figure 2. hIAPP aggregation gives rise to larger peptide assemblies than rIAPP within a μs timescale.

Peptide aggregation in a POPC membrane as a function of time for 36 molecules of rIAPP and hIAPP in 20 μs CG simulations.
Next, we compared the behavior of rIAPP (used here as a negative control) with hIAPP. As figure 1 show, hIAPP forms large aggregates (edge-fused pentameric assemblies). For rIAPP, the aggregate size appears to be limited and the preferred shape is more circular. Another important qualitative difference between hIAPP and rIAPP is that hIAPP peptides (monomers and assemblies) always stayed within the membrane, but rIAPP peptides moved out of the bilayer and emerged onto its surface. Upon moving toward the surface, the rIAPP peptides helped form membrane defects that allowed water to permeate into the bilayer. This behavior was always observed in repeated simulations.
The CG approach provides information about peptide and membrane dynamics, but not about the secondary structure of membrane-embedded hIAPP peptides. To study that, we performed extensive atomistic MD simulations of pentameric assemblies. First, a comparison of the hIAPP secondary structures at the beginning and at the end of the simulations showed that only residues 14 to 27 (sequence NFLVHSSNNFGAIL) remained α-helical. The rest partially unfolded to random coils. The secondary structure of the remaining residues in the assembly did not change during 0.3 μs of atomistic MD simulations. This confirms the stability of hIAPP pentamers: No dissociation of pentamers or even an indication of any of the hIAPP peptides moving out of the membrane was seen in repeated simulations. The helical wheel representation of hIAPP in a pentameric assembly (Fig. 3) provides a qualitative explanation of their stability: Hydrophobic residues become oriented toward the surrounding lipids.
Figure 3. Membrane-embedded hIAPP peptides associate into α-helical pentamers.

(A) Pentameric assembly of membrane-bound hIAPP peptides obtained at the end of a 0.3 μs MD simulation at 300 K. Green: polar, white: non-polar, cyan: polar ionizable. (B) Helical wheel representation of hIAPP side chains 14–27. (C) Pentameric aggregate. The vector in the xy-plane represents the hydrophobic moment89. The helix dipole moment (not shown) is along the z-axis. Color code (b and c): yellow: hydrophobic (large), light blue: polar ionizable, magenta: polar; gray: hydrophobic (small); pink: polar.
MD simulations of the pentameric assembly at 300 K did not reveal any tendency for the peptide to adopt β-sheet structures. These are traditionally considered the precursors of amyloid formation in solution, but not necessarily of oligomers interacting with membranes37,46,47. To assess if the transition from α-helical to β-sheet structures is hindered by energy barriers, we carried out an additional 80 ns MD simulation at 400 K. Only a 1.8% increase in β-sheet content was observed.
Figure 4 illustrates an ion-channel like water permeable pore formed by five hIAPP peptides (in red). The pore allowed for water molecules to diffuse across the bilayer. Of the total 12 water molecules inside the hIAPP aggregate, four persisted inside for the entire 0.3 μs. In the case of rIAPP (right panel), of the total 24, just one remained inside (hydrogen-bonded to the residues with a longer persistence). The main difference is the water molecules' distribution inside the pore: Water molecules inside the hIAPP pore appear to be confined in a restricted region because of the tight connections between peptides. For rIAPP, they are distributed randomly to fill the empty space generated by structural rearrangements of the replicas. We also performed 0.3 μs full atomistic MD simulation of rIAPP assemblies at 300 K. The lack of secondary structure stability is clearly evident, and α-helix/random coil/β-turn conformational transitions occur at a higher rate as compared to hIAPP. Snapshots are shown in Figs. 4 and 5. The structural changes and motions of rIAPP peptides occurred through movement of the N-terminus (residues 1–15) across lipid-water interface. The observed differences between hIAPP and rIAPP are in agreement with NMR studies that show that hIAPP and rIAPP have very different orientations in a membrane48.
Figure 4. hIAPP pentamers resemble barrel-shaped channels whereas the corresponding rIAPP assemblies look like loose funnels.

Snapshots from AA-MD simulations of hIAPP (red) and rIAPP (green) pentamers after 300 ns. Color code: white-surf-lipids, white-CPK-water.
Figure 5. Conformational preferences of monomeric hIAPP/rIAPP dictate the structural features of the pentameric assemblies.

Snapshots of hIAPP (red) and rIAPP (green) at monomeric state after 40 ns of MD simulation. POPC lipids are shown in white and water molecules in cyan.
The stability of the 10- and 26-meric hIAPP assemblies at the end of the CG simulation was addressed by setting up a 40 ns of all atom MD simulation at 300 K, using the configuration from the last frame of the 26 μs CG simulation as the starting point. We performed an additional 20 ns all atom MD simulation at 400 K to help the system to overcome energy barriers hindering further association-dissociation events. For the first 40 ns, the α-helix content remained unchanged at about 11%. Water molecules were able to permeate through the 26-meric aggregate, but with a distinctly higher efficiency in comparison with the pentameric aggregate. We infer this to be due to the larger size and mobility of hIAPP aggregates. We then increased the temperature to 400 K for additional 20 ns and observed an increase in the β-sheet content from 3% to 9%.
Thermodynamic model for hIAPP self-assembly within lipid bilayer
We developed a simple thermodynamic model to study the stability and morphological changes of hIAPP aggregates. The basic assumptions: 1) Aggregates may consist of i) isolated monomers, ii) pentagonal aggregates and iii) aggregates of edge-fused pentagons. Since fused pentagons can be formed by adding (one-by-one) three peptides to a pentagonal assembly, aggregates contain 5, 8,… 3n + 2,…. monomers. Smaller aggregates (n<5) exist in dilute systems and are of not of interest here. 2) Two regular pentagons sharing a common edge are rotated by π/5 with respect to each other. Although regular pentagonal tiling is not space filling in two dimensions, to keep the model simple we did not take into account the factors arising from incomplete packing. 3) Equilibrium requires the chemical potential of peptides in different aggregates to be the same, 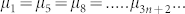 and
and 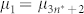 where
where 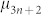 and
and 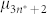 are the chemical potentials of linear and cyclic assemblies containing n and n* fused pentagons, respectively. The general form of
are the chemical potentials of linear and cyclic assemblies containing n and n* fused pentagons, respectively. The general form of  is
is
where 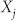 is the fraction of structures with aggregation number j, and
is the fraction of structures with aggregation number j, and 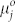 is the corresponding free energy per molecule. An analogous expression holds for
is the corresponding free energy per molecule. An analogous expression holds for 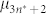 .
.
Each trimer forms 2 internal bonds with its own peptides and 4 bonds with the neighbouring trimers. The associated mean energy is -6nα, where -α is the strength of each intermolecular bond. Due to the two free ends in a linear aggregate, and since there are 4 missing bonds at the ends, we have  .
.
The above is strictly local. To include non-local forces, we introduce electrostatics. Our MD simulations showed that the dipole moment of the hIAPP peptides is perpendicular to the membrane surface and in the range 300–500 D. This makes dipolar effects the most important interactions. Two identical parallel dipoles inserted into a lipid bilayer interact repulsively: 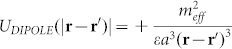 , where
, where 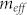 is the effective dipole moment of the protein repeat unit, ε is the bilayer dielectric permittivity and a is the mean distance between two nearby repeat units along the peptide. Summing over a line of n identical dipoles gives the dipolar energy (see Supplementary Material)
is the effective dipole moment of the protein repeat unit, ε is the bilayer dielectric permittivity and a is the mean distance between two nearby repeat units along the peptide. Summing over a line of n identical dipoles gives the dipolar energy (see Supplementary Material)
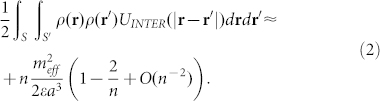 |
where 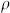 the density per unit length. Adding
the density per unit length. Adding 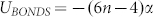 and Equation (2), and dividing by the number of peptides in a generic aggregate (3n + 2), we find an expression for the chemical potential:
and Equation (2), and dividing by the number of peptides in a generic aggregate (3n + 2), we find an expression for the chemical potential:
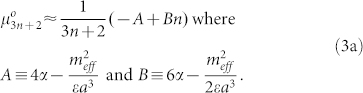 |
When the dipole is perpendicular to the membrane plane, self-assembly is hindered by competition with short-range adhesion forces. To trigger aggregation, we must have 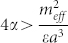 . An analogous calculation for a cyclic aggregate made of n* repeat units gives (see Supplementary Material)
. An analogous calculation for a cyclic aggregate made of n* repeat units gives (see Supplementary Material)
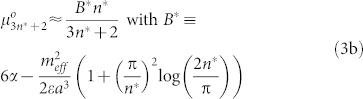 |
The numerical values of A, B and B* (in Equations. 3a and 3b) obey A<B* <B.
Figure 6 shows that the chemical potential of the circular aggregate is almost independent of the aggregation number n, while that of an open aggregate decreases monotonously. At small n, circular aggregates are more stable than linear ones, but beyond a critical value the situation is reversed. This is a consequence of the non-local electrostatic interactions and it provides mechanism to relieve the electrostatic stress.
Figure 6. Kinetics obtained from the thermodynamic model shows the transient nature of aggregates.

Variation of the chemical potentials of linear (full line) and cyclic (dashed line) vs the aggregation number n. At the intersection, linear and cyclic aggregate have the same energy. Inset: Relative concentration of cyclic aggregates vs. the total protein concentration C. Curves have been calculated for different values of the temperature scaled dipolar energy 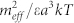 . From the top to the bottom:
. From the top to the bottom: 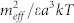 = 1, 2, 3.
= 1, 2, 3.
Combining the entropic and enthalpic contributions to the chemical potential (Equations 3a,b and 1) and equating the chemical potentials of the aggregate and free monomers, we calculate the concentrations of linear and cyclic aggregates 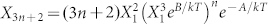 and
and 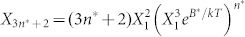 , respectively. A, B and B* are defined by Equations. (3a,b). Eliminating the still unknown monomer concentration
, respectively. A, B and B* are defined by Equations. (3a,b). Eliminating the still unknown monomer concentration 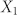 by using mass conservation for peptides, we numerically calculated the fraction of linear and cyclic aggregates as a function of the total peptide concentration C. The plot of
by using mass conservation for peptides, we numerically calculated the fraction of linear and cyclic aggregates as a function of the total peptide concentration C. The plot of 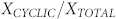 vs C is shown in the inset of Figure 6. At low concentrations, the fraction of cyclic aggregates is zero. At the critical protein concentration,
vs C is shown in the inset of Figure 6. At low concentrations, the fraction of cyclic aggregates is zero. At the critical protein concentration, 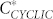 , cyclic structures begin to form. Their concentration grows rapidly reaching a maximum at
, cyclic structures begin to form. Their concentration grows rapidly reaching a maximum at 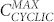 . The concentration of circular aggregates decreases slowly with protein concentration, long fibers being more favoured. The maximum becomes less pronounced and shifts toward higher concentrations upon increasing dipole moment.
. The concentration of circular aggregates decreases slowly with protein concentration, long fibers being more favoured. The maximum becomes less pronounced and shifts toward higher concentrations upon increasing dipole moment.
The existence of an optimum size for cyclic structures is further reinforced if we use the notion of spontaneous curvature. As discussed at the beginning of this section, the repeat units of hIAPP aggregates are fused pentamers. Each unit introduces a spontaneous bending of π/5. In order to form a closed circle, one must accommodate 10 fused pentamers (or 32 peptides). Such a structure has a zero internal strain, while larger or smaller cycles are less stable.
Kinetic control of the growing aggregate morphology
The simple model developed in the previous section relies on the assumption of a linear growth of the aggregate through continuous insertion of monomers at the growing free ends. MD simulations show a more complex mechanism involving a branching mechanism often observed in worm-like micellar systems49. Branching strongly increases with local supersaturation, as also modelled in a recent paper50. This parasitic process competes in a concentration-dependent way with the formation of closed regular toroids as discussed in the previous section. The formation of branched assemblies explains the slow and erratic kinetics of toroids birth and death in MD simulations we describe below.
In order to verify if the distribution of the different aggregates is consistent with the prediction of our thermodynamic model, we performed a series of CG simulations at 300 K in the microsecond time range. Calculations were performed at different lipid/protein ratios (from 1/13 to 1/44). The most interesting outcomes were:
The shape of the hIAPP aggregates is not linear. This feature is consistent with the notion of spontaneous curvature stemming from the pentameric geometry of the repeat units. Moreover, the growth of the aggregates exhibits frequent branching points, a clear indication of comparable lipid-peptide and peptide-peptide interactions (Fig. 7 b, c).
Formation of closed toroids is a rare event. This observation is consistent with the large activation energy required to bend an aggregate made of self-repelling dipoles into a circle. High peptide concentrations favor closure (Fig. 7).
Once the toroid has been closed, it is thermodynamically stable because the energy gain associated to the sealing of the two free ends. This process was investigated by leaving a closed toroid in contact with a dispersion of monomers (protein/lipid ratio 1/13) for 2 μs at 300 K (Fig. 7d).
Figure 7.

(a) snapshot at 10 μs (protein/lipid ratio 1/44); (b) snapshot at 8 μs (protein/lipid ratio 1/19); (c) snapshot at 6 μs (protein/lipid ratio 1/24); (d) snapshot taken after 1 μs from the toroid formation (protein/lipid ratio 1/13).
Membrane curvature generated by hIAPP assemblies
Insertion of a protein into a membrane perturbs lipid packing and results in local bending deformations. To minimize energetically unfavorable deformations, proteins tend to cluster51. These elastic forces have to be added to other contributions such as hydrophobicity and electrostatics to understand the origin of protein aggregation in membranes. The intricate coupling between shape and aggregation has been studied both analytically52 and computationally53. When the bilayer-spanning impurity is anisotropic (e.g. wedge shaped), geometric anisotropy induces anisotropic interactions among the impurities resulting in linear rather than globular aggregates54,55. Curvature stress has been suggested to account for the early stages of hIAPP-mediated membrane damage56,57,58.
hIAPP aggregates have a semi-toroid structure with cone-shaped cross-section (top side depth of about 43 Å and down side depth of about 22 Å), mainly due to the electrostatic repulsions between charges localized in the water-exposed region of the outer monolayer. Cone shapes have also been indicated in aggregates of amyloid β51. This geometry suggests a considerable aggregate-membrane bending coupling. The MD simulation of the 26-meric hIAPP assembly performed at 400 K showed significant curvature stress induced by the aggregate as shown in Fig. 8. This geometry can be explained by considering the electrostatic repulsions of the positively charged residues in the same side of the bilayer. We observed an increase in membrane fluctuations along the z-axis in the MD simulations of 26 hIAPP molecules.
Figure 8. Annular cone-shaped hIAPP assemblies cause membrane curvature.

Snapshots (cross-section) from the MD simulation of 26 hIAPP molecules in POPC bilayer at 400 K.
Although the experimental data reported in Fig. 9 involve larger time and space scales than those investigated by our MD simulations, results are fully consistent. Well-founded analytical theories for the shape of multi-component vesicles predict a sphere to ellipsoid transition due to the segregation of the component with the greatest intrinsic curvature in the more curved regions of the vesicle59. The effect is enhanced in the case of strong incompatibility among the two vesicles components (segregation). Both incompatibility and spontaneous bilayer curvature were observed in all atoms simulations in the case of hIAPP but not with rIAPP.
Figure 9. AFM in liquid of 1 μm2 containing POPC/POPS (95/5 molar ratio) LUVs.

Left panel: imagine of vesicles in PBS buffer. Right panel: imagine of vesicles after incubation with hIAPP 5 μM. rIAPP did not show induce any changes in the vesicles shape (not shown).
Discussion
Previous theoretical considerations of oligomer and fibril growth in aqueous solutions have revealed some of the general principles governing the transition from monomeric proteins to ordered fibrillar aggregates60,61,62. However, increasing evidence supports the hypothesis that hIAPP permeabilizes membranes either through the formation of pores or through detergent-like damage of the bilayer. These mechanisms are not mutually exclusive. A recent report suggested a leakage mechanism wherein a porous protein aggregate is formed by stochastic nucleation of hIAPP molecules63. Such states have been referred to as less structurally defined chaotic pores64,65, a view reconcilable with the nonspecific nature of channel-like aggregates.
Our atomistic simulations performed at the critical intermediate steps along the CG simulation allowed: i) the capture of the structural details about the relevant intermediate peptide assemblies, and ii) construction of a thermodynamic model to study the importance of dipole interactions in determining size and morphology of the aggregates. CG simulations provided a mechanistic description of the interactions of transmembrane α-helices with lipid bilayers66 as well as evidence that a water-permeable pentameric assembly of hIAPP α-helices is a critical intermediate towards reconciliation with the predictions of the proposed thermodynamic model. The number of helices constituting the pore as well as its effective diameter predicted from our simulations are in agreement with previous experimental studies28,38. The multiscale approach allowed us to study the detailed mechanisms and to develop a minimal thermodynamic model for analysing the transient nature of toroidal aggregates and aggregate shapes. The balance among concentration-related entropy, unfavorable edge effects of a self-assembled small aggregates and repulsive dipolar interactions among the dipoles make the circular aggregates a metastable state that can be observed only in a narrow window of protein/lipid ratios. At lower concentrations smaller oligomers (from monomers to pentamers) prevail, while at higher values fibrils dominate over the toroidal aggregates.
The predictions of the model when compared with CG simulations by increasing hIAPP concentration above the threshold protein concentration showed that hIAPP pentamers spontaneously evolve into a large cyclic assembly.
Importantly, this structure is able to render the membrane water-permeable and, concomitantly, to induce significant curvature into the membrane. It is tempting to speculate that this assembly may involve more than one peptide-based mechanisms for membrane-damage, but a conclusive demonstration would require a separate study. Parallel simulations performed on the non-toxic, non-amyloidogenic rat derivative rIAPP were significantly different from hIAPP: No compact ordered large aggregates were observed (Fig. 1).
There are also some important differences between our CG simulations and experimental observations previously reported. First, the time scale of the events: Experimental measurements require an initial diffusion to the membrane surface, and could be rate-determining (peptides are inserted into the membrane). The second difference is that the relatively small size of our system precludes the observation of large long-lived pores or other breakage mechanisms in the membrane. Thus, the increase in permeability of water and ions observed in our simulations may be a prelude to a multifaceted disruption of the membrane.
Most of the studies thus far have focussed on dimer formation, typically by investigating IAPPs in solutions31,32,33. Two recent computational studies using very different methods have, however, suggested the importance of trimers67,68. Our analytical model shows how trimers are the most important building blocks in the formation of the stable fused pentameric superstructures. Our CG simulations confirm this mechanism.
Finally, using the structural differences between hIAPP and rIAPP observed in MD simulations, we speculate that α-helices may be the dominant structure under certain conditions. Using a simple model, Dias et al.69 have shown that β-sheets, since they have the ability to lower hydrophobic energy, tend to be the favored ones in solution – this has also been seen in many simulations31,32,33,34,35. In a membrane environment, however, the free energy difference between α-helices and β-sheet may become much smaller, although the conformations are separated by free energy barrier that can be large; here, we saw only a small increase in β-sheet formation even at an elevated temperature. This is in excellent agreement with the fluorescence correlation spectroscopy experiments of Magzoub and Miranker70 who found that in membranes, IAPPs prefer α-helices over β-sheet, and that toxicity is related to the α-helical conformations. hIAPP is able to form aggregates that minimize the hydrophobic mismatch with the bilayer and peptide-peptide H-bonds that stabilize the pore and allow for a small number of waters to penetrate. Such stabilization mechanism does not exist for rIAPP and hence the peptides escape to the membrane surface. The stability of hIAPP assemblies also allows for diffusion of ions through the channel that may disrupt ion homeostasis and contribute to neuronal death as has been suggested in the case of amyloid-β71. Whether or not the hIAPP channel is selective remains a topic for future studies.
Methods
We performed a combination of coarse grained (CG) and atomistic molecular dynamics (MD) simulations at 300, 350 and 400 K. Reverse mapping72 was used to connect the different length scales. This protocol allowed for longer simulation times and detailed studies of the different intermediate states. The NMR structures of hIAPP bound to sodium dodecylsufate (SDS) micelles (pdb ID: 2KB8)73 and rIAPP in dodecylphosphocholine (DPC) micelles (pdb ID: 2KJ7)74 were first energy minimized using the method of steepest descents (the first structures in the PDB files were used). Pre-equilibration was done using atomistic MD in the NVT ensemble. This was followed by an equilibration simulation run of 40 ns. All simulations, CG and MD, were carried out using Gromacs 475 software and methodological issues76 related to neighborlist updates and charge groups were taken into account. GROMOS 53A677 force field was used in all atomistic MD. This force-field has been show to perform well in simulations of membrane-protein systems78,79. Coarse-grained models based on the MARTINI80,81,82 force field were constructed based on the final conformations.
For the lipid bilayer, a pre-equilibrated (100 ns) 128 lipid POPC membrane83 hydrated by 1500 water molecules was used as a starting point. This bilayer was replicated to obtain a membrane with 1596 lipids, energy minimized by the steepest descent algorithm (1000 steps), and equilibrated in the NpT ensemble for 100 ns. Finally, systems consisting of rIAPP or hIAPP and a bilayer were constructed. Peptides were inserted inside the membrane perpendicularly to the membrane surface using Gromacs tool genbox. The box containing a single protein was then replicated and the lateral size adjusted to achieve the desired concentration. The CG systems were hydrated by ~23,000 water molecules in the presence of 0.1 M NaCl. Cl− counterions were added to preserve charge neutrality. This system was again energy minimized by the steepest descent algorithm and equilibrated in the NpT ensemble for 100 ns. The starting configurations were obtained by extracting a portion of the bilayer containing the corresponding structure, maintaining the same protein/lipid ratio, from the CG simulation; a section of the bilayer within a specified radius from the center of mass of a chosen aggregate was cut and reverted to an atomistic representation. As for pentamers, we selected and reverted three different aggregates to test the reproducibility of the structural properties investigated. Peptide chains had same orientations (in agreement with NMR experiments showing the tendency of the single peptide to penetrate in membrane through the N-terminal).
In atomistic MD, the SPC (simple point charge) water model84 was used. The time step was set to 1 fs and the temperature was kept constant using the Nose-Hoover algorithm85,86 with a time constant of 0.1 ps. Periodic boundary conditions were applied. The Parrinello-Rahman algorithm87 was applied for semi-isotropic pressure coupling (1 bar). The particle-mesh Ewald (PME) algorithm88 was used for electrostatics. In the CG simulations, we used the Berendsen weak coupling thermostat and barostat algorithms89 with coupling constants of 0.3 ps and 3.0 ps, respectively. The following CG-systems were used in production runs: 36 hIAPP/rIAPP peptides for 26 μs at 300 K. A 13 μs simulation at 300 K with 104 peptides. Several concentration ratios protein/lipids have been also tested (1:13, 1:19, 1:24, 1:44).
MD production runs were as follows: Monomeric hIAPP or rIAPP embedded in a POPC membrane was simulated for 40 ns. Pentameric hIAPP/rIAPP assemblies obtained at the end of 6 μs CG simulation were simulated at 300 K for 0.3 μs. The temperature was increased to 400 K for 80 ns. hIAPP semi-toroidal 10-meric and the hIAPP semi-toroidal 26-meric aggregates were simulated for 40 ns at 300 K. The structure at the end of 26 μs CG simulation was simulated. The temperature was increased to 400 K for 20 ns (after 40 ns at 300 K).
Atomic Force Microscopy (AFM) measurements were performed in liquid at room temperature using a Digital Instruments nanoscope, model IIIa. Images were acquired in tapping mode with a fluid cell filled with phosphate buffer solution (pH 7.4, 100 mM NaCl). Where present, hIAPP or rIAPP concentration was 5 μM. Model membranes, containing a mixture of 1,2-palmitoil-oleil-sn-glycero-3-phosphocholine (POPC) and 1,2-palmitoil-oleil-sn-glycero-3-phospho-L-serine (POPS) (molar ratio 95/5) were prepared as described elsewhere57. The choice of this lipid composition is critical to prevent the formation of a Supported Lipid bilayer (planar bilayer) and to keep the LUVs intact although they are bound to the silica surface.
Author Contributions
M.K., C.L.R., A.R., D.M. and M.P. designed the experiments, performed the analysis and wrote the manuscript. M.P. performed the simulations.
Supplementary Material
Supplementary Information
Acknowledgments
We thank NSERC of Canada (MK) and the Ontario Early Researcher Award Program (MK) for financial support. SharcNet [www.sharcnet.ca] and CINECA (Bologna, Italy) are acknowledged for computational resources. We thank Prof. Luisa D'Urso for AFM measurements.
References
- Merlini G. & Bellotti V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 349, 583–596 (2003). [DOI] [PubMed] [Google Scholar]
- Hardy J. & Selkoe D. J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). [DOI] [PubMed] [Google Scholar]
- Dickson D. W. The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 56, 321–339 (1997). [DOI] [PubMed] [Google Scholar]
- Abedini A. & Raleigh D. P. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys. Biol. 6, 015005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Y., Ma B. & Nussinov R. Polymorphism in Alzheimer Abeta amyloid organization reflects conformational selection in a rugged energy landscape. Chem. Rev. 110, 4820–4838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis N. F., Wu C., Shea J. & Bowers M. T. Human islet amyloid polypeptide monomers form ordered beta-hairpins: a possible direct amyloidogenic precursor. J. Am. Chem. Soc. 131, 18283–18292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein S. L. et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nature Chem. 1, 326–331 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppener J. W., Ahren B. & Lips C. J. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 343, 411–19 (2000). [DOI] [PubMed] [Google Scholar]
- Kahn S. E., Andrikopoulos S. & Verchere C. B. Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 48, 241–253 (1999). [DOI] [PubMed] [Google Scholar]
- Haataja L., Gurlo T., Huang C. J. & Butler P. C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 29, 303–316 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy A. & Lim M. H. Structural Characterization and Inhibition of Toxic Amyloid-β Oligomeric Intermediates. Biophys. J. 105, 287–288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojevic V. & Melacini V. Stoichiometry and Affinity of the Human Serum Albumin-Alzheimer's Aβ Peptide Interactions. Biophys. J. 100, 183–192 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raditsis V., Milojevic V. & Melacini V. Aβ Association Inhibition by Transferrin. Biophys. J. 105, 473–480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe C. G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. of aging. 4, 570–575 (2006). [DOI] [PubMed] [Google Scholar]
- Kayed R. et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 (2003). [DOI] [PubMed] [Google Scholar]
- Demmester N. et al. Apoptosis induced in neuronalcells by C-terminal amyloid fragments is correlated with theiraggregation properties in phospholipid membranes. Mol. Membr. Biol. 17, 219–228 (2000). [DOI] [PubMed] [Google Scholar]
- Kawahara M., Kuroda Y., Arispe N. & Rojas E. Alzheimer's amyloid, human islet amylin, and prion peptide fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 275, 14077–14083 (2000). [DOI] [PubMed] [Google Scholar]
- Lin H., Bhatia R. & Lal R. Amyloid protein forms ion channels: implications for Alzheimer's disease pathology. FASEB. J. 15, 2433–2444 (2001). [DOI] [PubMed] [Google Scholar]
- Hebda J. A. & Miranker A. D. The interplay of Catalysis and Toxicity by Amyloid intermediates on Lipid Bilayers: Insights from Type II Diabetes. Ann. Rev. Biophys. 38, 125–152 (2009). [DOI] [PubMed] [Google Scholar]
- Sciacca M. F. et al. Two-Step Mechanism of Membrane Disruption by Ab through Membrane Fragmentation and Pore formation. Biophys. J. 103, 702–710 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield S. M. & Lashuel H. Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem. 49, 5628–5654 (2010). [DOI] [PubMed] [Google Scholar]
- Brender J. R. et al. Amyloid Fiber Formation and Membrane Disruption are Separate Processes Localized in Two Distinct Regions of IAPP, the Type-2-Diabetes-Related Peptide. J. Am. Chem. Soc. 130, 6424–6429 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacca M. F. M. et al. Cations as Switches of Amyloid-Mediated Membrane Disruption Mechanisms: Calcium and IAPP. Biophys. J. 104, 172–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brender J. R., Salamekh S. & Ramamoorthy A. Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem Res. 45, 454–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G. J. et al. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. USA 84, 8628–8632 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P., Andersson A. & Westermark G. T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826 (2011). [DOI] [PubMed] [Google Scholar]
- Harroun T. A., Bradshaw J. P. & Ashley R. H. Inhibitors can arrest the membrane activity of human islet amyloid polypeptide independently of amyloid formation. FEBS. Lett. 507, 200–204 (2001). [DOI] [PubMed] [Google Scholar]
- Hirakura Y., Yiu W. W., Yamamoto A. & Kagan B. L. Amyloid peptide channels: blockade by zinc and inhibition by Congo red (amyloid channel block). Amyloid 7, 194–199 (2000). [DOI] [PubMed] [Google Scholar]
- Mirzabekov T. A., Lin M. C. & Kagan B. L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 271, 1988–1992 (1996). [DOI] [PubMed] [Google Scholar]
- Anguiano M., Nowak R. J. & Lansbury P. T. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry 41, 11338–11343 (2002). [DOI] [PubMed] [Google Scholar]
- Kawahara M., Kuroda Y., Arispe N. & Rojas E. Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 275, 14077–14083 (2000). [DOI] [PubMed] [Google Scholar]
- Weise K., Radovan D., Gohlke A., Opitz N. & Winter R. Interaction of hIAPP with model raft membranes and pancreatic beta-cells: cytotoxicity of hIAPP oligomers. ChemBioChem. 11, 1280–1290 (2010). [DOI] [PubMed] [Google Scholar]
- Meier J. J. et al. Inhibition of human IAPP fibril formation does not prevent beta-cell death: evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Endocrinol. Metab. 291, E1317–24 (2006). [DOI] [PubMed] [Google Scholar]
- Nath A., Miranker A. D. & Rhoades E. A membrane-bound antiparallel dimer of rat islet amyloid polypeptide. Angew. Chem. 50, 10859–10862 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quist A. et al. Amyloids form membrane pores: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 102, 10427–10432 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel M. F. M. et al. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 356, 783–789 (2006). [DOI] [PubMed] [Google Scholar]
- Prakash R., Nanga R. P. R., Brender J. R., Vivekanandan S. & Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta 1808, 2337–2342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laghaei R., Mousseau N. & Wei G. Effect of the disulfide bond on the monomeric structure of human amylin studied by combined Hamiltonian and temperature replica exchange molecular dynamics simulations. J. Phys. Chem. B 114, 7071–7077 (2010). [DOI] [PubMed] [Google Scholar]
- Dupuis N. F., Wu C., Shea J. E. & Bowers M. T. The amyloid formation mechanism in human IAPP: dimers have β-strand monomer-monomer interfaces. J. Am. Chem. Soc. 133, 7240–7243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A. S. et al. Stable and metastable states of human amylin in solution. Biophys. J. 99, 2208–2216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews M. N. & Winter R. Comparing the structural properties of human and rat islet amyloid polypeptide by MD computer simulations. Biophys. Chem. 156, 43–50 (2011). [DOI] [PubMed] [Google Scholar]
- Rivera E., Straub J. & Thirumalai D. Sequence and crowding effects in the aggregation of a 10-residue fragment derived from islet amyloid polypeptide. Biophys. J. 96, 4552–4560 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Wei G., Su H., Nordenskiöld L. & Mu Y. Effects of cholesterol on pore formation in lipid bilayers induced by human islet amyloid polypeptide fragments: A coarse-grained molecular dynamics study. Phys. Rev. E 84, 1–8 (2011). [DOI] [PubMed] [Google Scholar]
- Lee C., Sun Y. & Huang H. W. How Type II Diabetes-Related Islet Amyloid Polypeptide Damages Lipid Bilayers. Biophys. J. 102, 1059–1068 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalisi S. et al. Self-Assembling Pathway of HiApp Fibrils within Lipid Bilayers. ChemBiochem 11, 1856–1859 (2010). [DOI] [PubMed] [Google Scholar]
- Sajith A., Jayasinghe S. A. & Langen R. Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta 1768, 2002–2009 (2007). [DOI] [PubMed] [Google Scholar]
- Soong R., Brender J. R., Macdonald P. M. & Ramamoorthy A. Association of Highly Compact Type II Diabetes Related Islet Amyloid Polypeptide Intermediate Species at Physiological Temperature Revealed by Diffusion NMR Spectroscopy. J. Am. Chem. Soc. 131, 7079–7085 (2009). [DOI] [PubMed] [Google Scholar]
- Nanga R. P. R., Brender J. R., Xu J., Veglia G. & Ramamoorthy A. Structures of Rat and Human Islet Amyloid Polypeptide IAPP1-19 in Micelles by NMR Spectroscopy. Biochemistry 47, 12689–12697 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelschlaeger C., Schopferer M., Scheffold F. & Willenbacher N. Linear-to-Branched Micelles Transition: A Rheometry and Diffusing Wave Spectroscopy (DWS) Study. Langmuir 25, 716–723 (2009). [DOI] [PubMed] [Google Scholar]
- Tang M. & Carter W. C. Branching Mechanisms in Surfactants Micellar Growth. J. Phys. Chem. B 117, 2898–2905 (2013). [DOI] [PubMed] [Google Scholar]
- Pannuzzo M., Milardi D., Raudino A., Karttunen M. & La Rosa C. Analytical model and multiscale simulations of Aβ peptide aggregation in lipid membranes: towards a unifying description of conformational transitions, oligomerization and membrane damage. Phys.Chem. Chem. Phys. 15, 8940–8951 (2013). [DOI] [PubMed] [Google Scholar]
- Zimmerberg J. & Kozlov M. M. How proteins produce cellular membrane curvature. Nature Rev. Mol. Cell. Biol. 7, 9–19 (2006). [DOI] [PubMed] [Google Scholar]
- Bahrami A. H. & Jalali M. A. Vesicle deformations by clusters of transmembrane proteins. J. Chem. Phys. 134, 085106 (2011). [DOI] [PubMed] [Google Scholar]
- Dommersnes P. G. & Fournier J. B. N-body study of anisotropic membrane inclusions: Membrane mediated interactions and ordered aggregation. Eur. Phys. J. B. 12, 9–12 (1999). [Google Scholar]
- Iglic A. et al. On the role of anisotropy of membrane constituents in formation of a membrane neck during budding of a multicomponent membrane. J. Biomech. 40, 579–585 (2007). [DOI] [PubMed] [Google Scholar]
- Ramamoorthy A., Thennarasu S., Lee D-K., Tan A. & Maloy L. Solid-State NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 91, 206–216 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacca M. F. M., Pappalardo M., Milardi D., Grasso D. M. & La Rosa C. Calcium-activated membrane interaction of the islet amyloid polypeptide: Implications in the pathogenesis of type II diabetes mellitus. Arch. Biochem. Biophys. 477, 291–298 (2008). [DOI] [PubMed] [Google Scholar]
- Sciacca M. F. M. et al. Are fibril growth and membrane damage linked processes? An experimental and computational study of IAPP(12–18) and IAPP(21–27) peptides. New J. Chem. 34, 200–207 (2010). [Google Scholar]
- Seifert U. & Lipowsky R. The morphology of vesicles. In Lipowsky, R. & Sackmann, E. editors ″Structure and Dynamics of Membranes.″ p.403–463. Elsevier, Amsterdam (1995).
- Straub J. E. & Thirumalai D. Toward a molecular theory of early and late events in monomer to amyloid fibril formation. Ann. Rev. Phys. Chem. 62, 437–463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappalardo M. et al. A molecular dynamics study on the conformational stability of prp 180–193 helix ii prion fragment. Chem. Phys. Lett. 390, 511–516 (2004). [Google Scholar]
- Milardi D., Pappalardo M., Pannuzzo M., La Rosa C. & Grasso D. M. The role of the Cys2-Cys7 disulfide bridge on the early steps of Islet Amyloid Polypeptide aggregation. Chem. Phys. Lett. 463, 396–399 (2008). [Google Scholar]
- Last N. B., Rhoades E. & Miranker A. D. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc. Natl. Acad. Sci. USA 108, 9460–9465 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory S. M., Cavenaugh A., Journigan V., Pokorny A. & Almeida P. F. F. A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys. J. 94, 1667–1680 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsen P. H. A chaotic pore model of polypeptide antibiotic action. Biophys. J. 94, 1549–1550 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond P. & Sansom M. S. P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 128, 2697–2704 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Yu X., Liang G. & Zheng J. Heterogeneous triangular structures of human islet amyloid polypeptide (amylin) with internal hydrophobic cavity and external wrapping morphology reveal the polymorphic nature of amyloid fibrils. Biomacromolecules 12, 1781–94 (2011). [DOI] [PubMed] [Google Scholar]
- Mo Y., Lu Y., Wei G. & Derreumaux P. Structural diversity of the soluble trimers of the human amylin(20–29) peptide revealed by molecular dynamics simulations. J. Chem. Phys. 130, 125101 (2009). [DOI] [PubMed] [Google Scholar]
- Dias C. L., Karttunen M. & Chan H. S. hydrophobic interactions in the formation of secondary structures in small peptides. Phys. Rev. E 84, 041931 (2011). [DOI] [PubMed] [Google Scholar]
- Magzoub J. & Miranker A. D. Concentration-dependent transitions govern the subcellular localization of islet amyloid polypeptide. FASEB J. 26, 1228 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S. P., Farhangrazi Z. S., Ying H. S., Yeh C.-H. & Choi D. W. Enhancement of Outward Potassium Current May Participate in β-Amyloid Peptide-Induced Cortical Neuronal Death. Neurobiol. Dis. 5, 81–88 (1998). [DOI] [PubMed] [Google Scholar]
- Rzepiela, et al. Reconstruction of atomistic details from coarse grained structures. J. Comp. Chem. 31, 1333–1343 (2010). [DOI] [PubMed] [Google Scholar]
- Patil S. M., Xu S., Sheftic S. R. & Alexandrescu A. T. Dynamic alpha-helix structure of micelle-bound human amylin. J. Biol. Chem. 284, 11982–11991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanga R. P. R. et al. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J. Am. Chem. Soc. 131, 8252–8261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Kutzner C., Van der Spoel D. & Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory. Comput. 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- Wong-ekkabut J., Miettinen M. S., Dias C. & Karttunen M. Static charges cannot drive a continuous flow of water molecules through a carbon nanotube. Nature Nanotech. 5, 555–557 (2010). [DOI] [PubMed] [Google Scholar]
- Oostenbrink C., Soares T. A., van der Vegt N. F. & van Gunsteren W. F. Validation of the 53A6 GROMOS force field. Biophys. J. 34, 273–284 (2005). [DOI] [PubMed] [Google Scholar]
- Wong-ekkabut J. & Karttunen M. Assessment of common simulation protocols for simulations of nanopores, membrane proteins & channels. J. Chem. Theory Comput. 8, 2905–2911 (2012). [DOI] [PubMed] [Google Scholar]
- Cino E. A., Choy W.-Y. & Karttunen M. Comparison of secondary structure formation using 10 different force fields in microsecond molecular dynamics simulations. J. Chem. Theory Comput. 8, 2725–2740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L. et al. The MARTINI coarse grained force field: extension to proteins. J. Chem. Theory Comp. 4, 819–834 (2008). [DOI] [PubMed] [Google Scholar]
- Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P. & de Vries A. H. The MARTINI forcefield: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007). [DOI] [PubMed] [Google Scholar]
- Marrink S. J., de Vries A. H. & Mark A. E. Coarse grained model for semi-quantitative lipid simulations. J. Phys. Chem. B 108, 750–760 (2004). [Google Scholar]
- de Jong D. H., Singh G., Bennett W. F. D., Arnarez C., Wassenaar T. A., Schäfer L. V., Periole X., Tieleman D. P. & Marrink S. J. Improved parameters for the Martini coarse-grained protein force field. J. Chem. Theory Comput. 9, 687–697 (2013). Parameters at http://md.chem.rug.nl/cgmartini/index.php/force-field-parameters/lipids. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F. & Hermans J. Interaction Models for Water in Relation to Protein Hydration, in Intermolecular Forces, edited by Pullman, B. 331–342 D. Reidel Publishing Company, Dordrecht (1981). [Google Scholar]
- Nosé S. A. Unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511 (1984). [Google Scholar]
- Hoover W. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985). [DOI] [PubMed] [Google Scholar]
- Parrinello M. & Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- Darden T., York D. & Pedersen L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089 (1993). [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A. & Haak J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information