

Abstract
AUY922 is a potent synthetic Hsp90 antagonist that is moving steadily through clinical trials against a small range of cancers. To identify protein markers that might measure the drug’s effects, and to gain understanding of mechanisms by which AUY922 might inhibit the proliferation of leukemia cells, we characterized AUY922’s impacts on the proteomes of cultured Jurkat cells. We describe a robust and readily assayed proteomics fingerprint that AUY922 shares with the flagship Hsp90 inhibitors 17-DMAG and radicicol. We also extend our proteomics findings, demonstrating that an unrelated antagonist of protein folding potentiates the anti-proliferative effects of AUY922. Results provide a set of candidate biomarkers for responses to AUY922 in leukemia cells, and suggest new modalities for enhancing AUY922’s anti-cancer activities.
Keywords: Hsp90 inhibitors, AUY922, 17-DMAG, radicicol, cancer, proteomics, spectrum counting, label free quantitation, biomarkers
Introduction
Due to their widespread roles in diverse physiological processes, the 90-kDa heat shock proteins (Hsp90) continue to generate great interest as pharmacological targets. Small molecules that inhibit Hsp90 hold promise in treating a wide range of cancers (1, 2), neurodegenerative diseases(3, 4), in suppressing immune function(5–8), and as experimental tools for studying Hsp90-dependent cellular processes. At this writing, Hsp90 inhibitors are featured in more than 80 ongoing or completed clinical trials (ClinicalTrials.gov).
The selective tumoricidal activities of Hsp90 inhibitors are widely thought to reflect Hsp90’s roles in chaperoning signal transduction(9). Related models postulate that cancer cells experience both exaggerated pro-growth signals from activated oncogenes, and exaggerated apoptotic signals from DNA damage and other neoplastic abnormalities(10). Hsp90 inhibitors are thought to disrupt this delicate state of balance by indirectly attacking multiple Hsp90-dependent pro-growth pathways. Additionally, mutant kinases and hyperactive oncoproteins require exaggerated Hsp90 support(11–15), thus making cancer cells even more vulnerable to the “oncogenic shock” caused by Hsp90 inhibitors.
AUY922 is a synthetic resorcinylic isoxazole amide that shows all of the hallmarks associated with Hsp90 inhibitors. Crystal structures show that AUY922 binds to the ATP-binding site within Hsp90’s N-terminal domain(16). Competitive fluorescence polarization assays, assays of inhibitory kinetics, and calorimetry assays demonstrate that AUY922 has low-nanomolar affinities for Hsp90’s cytosolic isoforms(16). In contrast, AUY922 binds less avidly to Hsp90’s GRP94 and TRAP-1 isoforms, and shows negligible in vitro affinities for all other proteins tested to date(16). AUY922’s assignment as a highly specific Hsp90 inhibitor is supported by its effects in inducing the expression of Hsp70(16), in depleting various Hsp90-dependent “client” proteins(16–18), and in disrupting Hsp90’s association with its p23 subunit(19, 20). Due to its high affinity and selectivity for Hsp90 and its potent activities against cancer cells, AUY922 is an especially promising candidate for Hsp90-directed therapies. Thus, more than 20 ongoing clinical trials are characterizing AUY922 in individual and combinatorial treatments against various cancers.
In pursuit of the models noted above, AUY922’s impacts on cellular signal transduction machineries have been assessed in a wide range of cancer cell lines. The most frequently reported responses include the induction of isoforms of Hsp70 and Hsp90, and the depletion of the canonical signaling kinases Akt1/2, ErbB-2/HER2/EGRF2/Neu, and Erk1/2. Less frequently, some 50 other pro-growth signaling proteins (primarily other kinases and downstream transcription factors) have been utilized as reporters of AUY922 mechanisms. Results seem to vary somewhat among cell lines and individual studies, with some AUY922-induced changes in protein expression being highly conserved, while others seem to be cell-line-specific. Missing from this focus on signal transduction, however, is a wider appreciation of AUY922’s impact on cellular physiology, independent of a priori Western blotting targets.
In this work, we characterized AUY922’s impacts on cultured Jurkat leukemia cells, and compare this impact to those of two flagship Hsp90 inhibitors, 17-DMAG and radicicol. We describe a robust and readily assayed proteomics fingerprint that is highly conserved among the three drugs. Based upon AUY922’s effects on this cell line’s proteome, we also characterized AUY922’s effects in the presence of an unrelated antagonist of protein folding, observing significant potentiation of AUY922’s anti-proliferative activity. Results provide a set of candidate markers for AUY922-elicited responses in leukemia cells, and suggest new modalities for enhancing AUY922’s anti-cancer activities.
Materials and Methods
Reagents
The Jurkat leukemia cell line E6.1 was obtained from ATCC. Stocks of 17-DMAG (LC Laboratories D-3440), radicicol (Cayman Chemicals 13089), and AUY922 (Selleck Chemicals S1069) were dissolved in DMSO (Sigma D2650) and stored at −80 degrees C. Stocks of L-azetedine-2-carboxylic acid (AZC; Sigma A0760) were formulated in water. Cellular proliferation was assayed using Cell Titer Aqueous One Solution Cell Proliferation Assay (MTS; Promega G3581), following the manufacturer’s protocols. Antibodies and their respective sources included: mouse monoclonal anti-human β-actin (Sigma A5441); rabbit polyclonal anti-human poly-ADP ribose polymerase (Cell Signaling Technology 9542); rabbit monoclonal anti-human Cdk6 (Epitomics 3524-1); rabbit monoclonal anti-human Cdk1(Epitomics 3787-1), rabbit monoclonal anti-human Dnmt1 (Epitomics 2788-1); rabbit monoclonal anti-human DDX5 (Epitomics 5567-1); polyclonal goat anti-human UNR (Santa Cruz Biotechnology SC-79293); polyclonal rabbit anti-human MCM7 (Santa Cruz Biotechnology SC-22782); monoclonal mouse anti-human UHRF1 (Santa Cruz Biotechnology SC-166898); and polyclonal rabbit anti-human eIF4A1 (Abcam AB-31217).
Cell culture, treatment, assay, and harvest
Jurkat cells were cultured at 37 degrees C in a humidified CO2 incubator (5% CO2) in RPMI media (Pierce 89984) supplemented with fetal bovine serum (Pierce 89986), 200 mM L-glutamine, 500 μg/ml streptomycin, and 100 U/ml penicillin. Cells were seeded in fresh media at densities of 100,000 cells/ml for proliferation assays or 250,000 cells/ml for protein assays, and cultured for 24 h prior to treatments with Hsp90 inhibitors. Drug concentrations and durations of treatment were as described for individual assays, with three or more biological replicates performed for each experiment. Cellular proliferation was measured using MTS assays following the manufacturer’s recommended protocols. Cellular proteins were harvested by washing cells washed three times in PBS, and followed by lysis in RIPA. Lysates were quantified by Bradford assays and characterized by SDS-PAGE and Western blotting.
For analysis by mass spectrometry, cell lysates were precipitated with acetone/TCA, redissolved in 8 M urea, 100 mM TrisHCl pH = 8.5 at 21 degrees C, reduced at 21 degrees C for 20 min using 5 mM tris(2-carboxyethyl)phosphine, and alkylated with 10 mM iodoacetamide for 15 min in the dark at room temperature. Samples were then diluted with three volumes of 100 mM TrisHCl, and digested with 4 μg/ml trypsin for overnight at 37 degrees C. Digested samples were acidified to 1% trifluoroacetic acid, desalted using C18 affinity tips (OMIX), and analyzed by liquid chromatography and tandem mass spectrometry (LC-MS/MS).
For LC-MS/MS, samples were analyzed on a hybrid LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to a New Objective PV-550 nanoelectrospray ion source and an Eksigent NanoLC-2D chromatography system. Peptides were analyzed by trapping on a 2.5 cm pre-column in a vented column configuration (5 μm Magic C18 AQ), followed by analytical separation on a 75 μm ID 15-cm fused silica column (5 μm Magic C18 AQ ) terminated with an integral fused silica emitter. Peptides were eluted using a 2.5–28% ACN/0.1% formic acid gradient performed over 116 min at a flow rate of 300 nL/min. During elution, samples were ionized by nanospray, and analyzed using a “top-6” methodology, consisting of one full-range FT-MS scan (nominal resolution of 60,000 FWHM, 360 to 1400 m/z, using lock mass 445.1200), and up to six data-dependent MS/MS scans performed in the linear ion trap. MS/MS settings included: a trigger threshold of 8,000 counts, monoisotopic precursor selection (MIPS), isolation width of 2 m/z, normalized collision energy of 35%, activation Q of 0.25 for 30 msec, and rejection of parent ions that had unassigned charge states, that were previously identified as contaminants on blank gradient runs, or that had been previously selected for MS/MS (dynamic exclusion at 150% of the observed chromatographic peak width.
Centroided ion masses were extracted using the extract_msn.exe utility from Bioworks 3.3.1 and were used for database searching with Mascot v2.2.04 (Matrix Science) and X! Tandem v2007.01.01.1 (www.thegpm.org). Searches against the IPI.Human database (v3.87, 91,464 sequences) used the following search parameters: 5 ppm parent ion mass tolerance, 0.6 Da fragment ion tolerance, one missed tryptic cleavage, pyroglutamate cyclization of N-terminal Gln, oxidation of Met, formylation or acetylation of the protein N-terminus, and iodoacetamide alkylation of Cys. Peptide and protein identifications were validated using Scaffold v2.2.00 (Proteome Software) and the PeptideProphet algorithm (21), using Scaffold’s high-mass accuracy scoring option. Protein acceptance thresholds were equal to or greater than 99% protein probability, based upon at least 2 peptides, each identified with at least 50% certainty. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Searches included validations against a reversed-sequence database, generating estimates of false positive rates of 0.0% among 931 proteins identified in the AUY022 dataset, 0.5% among 921 proteins identified in the 17-DMAG dataset, and 0.4% among the 900 proteins identified in the radicicol dataset. Differences in spectral counts were assessed using Scaffold (unnormalized total spectrum counts, with the minimum spectrum count value set to zero) and Student’s t-testing (two tails, equal variance), and were subjected to additional validation criteria as described in Results.
Differences in protein expression were also estimated on the basis of peptide peak intensities, by using the LFQ algorithm of MaxQuant v1.2.2.5(22). Proteins were identified by using Andromeda to search the IPI.HUMAN database (v3.87, as above) and Contaminants databases, wherein settings included 2 missed trypsin cleavages, parent ion tolerances of 20 ppm and 1 ppm (first and second searches, respectively), fragment ion tolerance of 0.5 Da, and peptide and protein FDR of 0.01. Protein quantifications were based upon 2 or more unique unmodified or carbamoidomethylated-Cys peptides, using 2-min thresholds to match peaks between four replicative LC-MS/MS analyses performed for each of three biological replicates. Differences in protein levels were assessed using t-testing algorithms embedded in Microsoft Excel, using 2 tails and unequal variance as per the recommendations of Ruxton.(23) Differences in protein expression were also subjected to additional validation criteria as described in Results. Scatter plots and Venn analyses were performed using Perseus 1.2.0.16 (Max Planck Institute of Biochemistry). GO analyses were performed using David (http://david.abcc.ncifcrf.gov/) and REVIGO (http://revigo.irb.hr/). Dose curves and IC50 values were analyzed using GraphPad Prism software (GraphPad, Inc., San Diego, CA) using non-linear regression analysis (least squares fit, no weighting, no constraints).
Results
To determine the inhibitory potential of AUY922 in cultured Jurkat leukemia cells, cellular proliferation was measured in the presence of 1.0 nM to 1000 nM AUY922 (Fig. 1A). Results indicated EC50 values of 10 nM, 7.3 nM, and 8.6 nM when cellular proliferation was measured at 24, 48, and 72 hr post-treatment, respectively. These EC50 values for AUY922 were consistent with those previously reported among a wide range of cultured cancer cell lines(16–18). The AUY922 dose-response curve was very steep, with cells proliferating at near-control levels at 3.5 nM AUY922, but declining to only 32% of control values at 12 nM AUY922. This dose-response curve was consistent with AUY922’s potency, and was notable in comparison to the shallower dose-curve responses of the less-potent flagship compounds 17-DMAG and radicicol (cf. Fig 1. versus Fig. 2 below). Notably, dosages between 35 and 900 nM entirely prevented Jurkat cell proliferation, but without reducing cellular metabolic assay below the initial culture seed density. Thus, these dosages of AUY922 seemed to be more cytostatic than cytotoxic for this cell line.
Figure 1. Effects of AUY922 on cultured Jurkat leukemia cells.

Panel 1A, Cell proliferation after 24, 48, or 72 hr incubation in the presence of the indicated concentrations of AUY922 (n=3). Panel 1B, Cdk6 levels after 0–8 hr of treatment with 150 nM 17-DMAG. Panel 1C, Cdk6 levels after 24 hr of treatment with the indicated concentrations of AUY922. Panel 1D, Densitometry of Cdk6 levels remaining after 24 hr of treatment with the indicated concentrations of AUY922 (n=3). Panel 1E, PARP cleavage after 24 treatment with the indicated concentrations of AUY922.
Figure 2.
Structures of AUY922, 17-DMAG, and radicicol.
To further investigate the impacts of AUY922, we first used the well studied Hsp90 inhibitor 17-DMAG to validate the kinase Cdk6 (24, 25) as a reporter of Hsp90 inhibition in the Jurkat cells. In cultures treated with 150 nM 17-DMAG, Cdk6 expression declined to undetectable levels within 8 hrs after Hsp90 inhibition (see Supporting Information). This demonstrated that Cdk6 was a sensitive reporter of Hsp90 inhibition in Jurkat cells.
Using Cdk6 to report Hsp90 inhibition, we assessed the inhibitory potential of AUY922 in Jurkat cells. Dose curve analyses (Fig. 1B and 1C) showed that the EC50 for Cdk6 depletion was comparable to that observed versus cell proliferation (11 nM and 10 nM, respectively), with 15 nM AUY922 depleting 50% of Cdk6 levels within 24 hr. As was observed for inhibition of proliferation, the dose curve for Cdk6 depletion was notably steep. We also assessed apoptosis in AUY922-treated cells, using cleavage of poly (ADP-ribose) polymerase (PARP) as a reporter of caspase 3/7 activation(26). Results indicated only moderate levels of PARP cleavage after 24-hr treatment with 1.0 nM to 128 nM AUY922 (Fig. 1D), suggesting that lower dosages were largely sub-apoptotic. This interpretation was consistent with our observations above that AUY922 appeared to be cytostatic rather than cytotoxic in this cell line.
We used similar assays of cellular proliferation, depletion of Cdk6, apoptosis, and cellular cytotoxicity to select matching dosages for two flagship Hsp90 inhibitors, namely 17-DMAG and radicicol (Fig. 2). From these assays, 11 nM AUY922, 30 nM 17-DMAG, and 60 nM radicicol represented each drug’s EC50 for Cdk6 depletion in Jurkat cultures (Fig. 3 & 4).
Figure 3. Effects of 17-DMAG on cultured Jurkat leukemia cells.

Panel 3A, Cell proliferation after 24, 48, or 72 hr incubation in the presence of the indicated concentrations of 17-DMAG (n=3). Panel 3B, Cdk6 levels after 24 hr of treatment with the indicated concentrations of 17-DMAG. Panel 3C, Densitometry of Cdk6 levels remaining after 24 hr of treatment with the indicated concentrations of 17-DMAG (n=3). Panel 3D, PARP cleavage after 24 treatment with the indicated concentrations of 17-DMAG.
Figure 4. Effects of radicicol on cultured Jurkat leukemia cells.

Panel 4A, Cell proliferation after 24, 48, or 72 hr incubation in the presence of the indicated concentrations of radicicol (n=3). Panel 4B, Cdk6 levels after 24 hr of treatment with the indicated concentrations of radicicol. Panel 4C, Densitometry of Cdk6 levels remaining after 24 hr of treatment with the indicated concentrations of radicicol (n=3). Panel 4D, PARP cleavage after 24 treatment with the indicated concentrations of radicicol.
We next assessed the proteomics fingerprint induced by each drug. For these assays, Jurkat cell cultures were treated for 24 hr with 5-7X each drugs’ EC50 for Cdk6 depletion. Trypan blue uptake assays confirmed that more than 80% of the cells were viable at this dosage. Subsequently, two label-free proteomics methods were used to characterize changes in protein expression. In the first method, we utilized label-free spectrum counting assays(27), comparing the number of MS/MS spectra assigned to individual proteins from treated vs. untreated cultures. In the second method, the same datasets were analyzed using the intensities of individual peptide ions to derive each protein’s normalized “LFQ” intensity(28).
Comparison of the effects of AUY922, 17-DMAG, and radicicol showed that this family of drugs had highly conserved impacts on the proteomes of Jurkat cells. When 17-DMAG was compared to radicicol, there was a strong correlation in the proteins that were up regulated or down regulated by each drug (Fig. 5). When the changes in protein expression induced by 17-DMAG were compared to those induced by AUY922, there was an equally strong correlation in the proteins up-regulated or down-regulated by these two drugs (Fig. 5). Extending this analysis, we found that the effects of AUY922 were strongly correlated with those of radicicol as well (Fig. 5). Based upon the highly conserved proteomics fingerprints of AUY922, 17-DMAG, and radicicol, and the well documented mechanism described for the latter, we concluded that the primary mechanism of by which AUY922 inhibited the proliferation of cultured leukemia cells was inhibition of Hsp90.
Figure 5. Comparison of proteomes from cells treated with AUY922 vs. 17-DMAG or radiciol.

Cells were treated overnight each drug as indicated, and their proteomes were analyzed by spectrum counting (upper panels) and LFQ peak intensities (lower panels). For each protein, the average log2 of the expression ratio in treated vs. control cultures was calculated (three biological replicates for each drug) and compared. Significant changes in protein expression are indicated in red, with non-significant changes shown in grey. R-squared values represent fits to just those protein ratios showing significant changes.
A conserved Hsp90-inhibition proteomics fingerprint was also apparent upon set analysis of proteins that demonstrated statistically significant drug-induced changes in protein expression (Fig. 6). Spectrum counting assays identified 30 changes in protein expression that were common to all three drugs. The LFQ peak intensity method identified 64 changes in protein expression that were common to all three drugs. Among these proteins, 20 were common to both assay techniques. This shared proteomics fingerprint reinforced our conclusion that AUY922 acted primarily as an Hsp90 inhibitor in suppressing the proliferation of Jurkat leukemia cells, and provided a short list of protein responses that could most easily and most reproducibly be detected (Supporting Information).
Figure 6. Reproducibility of inhibitor-induced changes in protein expression.
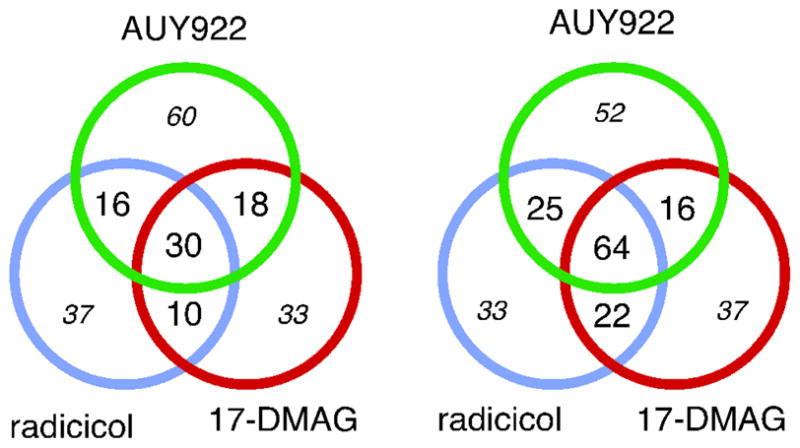
Cells were treated overnight each drug as indicated, their proteomes were analyzed by spectrum counting (left) and LFQ peak intensities (right), as indicated. Numbers indicate the number of proteins showing significant changes in expression. Bold font indicates conserved responses. Italics font indicates non-conserved responses that might be caused by experimental variance or statistical weakness.
Numerous changes in protein expression were common to two of the three drugs, but escaped detection in cells treated with the third drug. Upon manual review, it was apparent that many of these escaped proteins had been previously described as being Hsp90-regulated (Supporting Information). Moreover, volcano plots showed that while many individual proteins seemed to show large differences in their spectrum count values between treated vs. untreated cultures, many of these differences did not survive statistical testing (Supporting Information). In contrast, the LFQ peak intensity values appeared to be more robust statistically, but they appeared to suffer limitations in discovering changes in weakly expressed proteins. This was consistent with our appreciation of the strengths and limitations of both techniques(27). We concluded that many of the protein responses common between two of three Hsp90 inhibitors were likely valid, but were not robust enough to be detected without fail in all 9 out of the 9 experiments assayed.
In contrast to these conserved responses, our spectrum counting assays and LFQ peak intensity assays identified 130 responses and 122 responses, respectively, that were associated with only one of the three individual inhibitors (enumerated by italics font in Fig. 6). In the absence of confirmatory effects from another drug, the potential for statistical weaknesses or for experiment-dependent effects meant that these non-conserved changes could not be regarded as real. Because additional studies would be required to justify conclusions regarding the validity of these effects, these proteins were not considered further, and they were not included in the analyses described below.
To study the more conserved responses, we compiled and assessed a dataset of 150 proteins whose expression was altered by two out of our three Hsp90 inhibitors, drawing from both the spectrum counting and LFQ datasets (Supplementary Information). Only two of these proteins showed discrepancies in the effects reported among the Hsp90 inhibitors, that is, “up-regulated” by one inhibitor, but “down-regulated” by another (e.g., Fig. 5); these two proteins were eliminated from our list, and were not subjected to further analysis.
As was observed in the drug-vs-drug comparisons above, concordant results were obtained when results from spectrum counting assays were compared to results from LFQ intensity assays of peptide ion peak areas: proteins that were identified as significantly up-regulated using spectrum counting assays were also found to be up-regulated using LFQ peak area assays (Fig. 7A). Similarly, down-regulated proteins were identified as such by both assay techniques. This correlation held true across the whole set of 148 proteins, even for those proteins whose altered expression was statistically validated by only one of the two methods (Fig. 7B). This strengthened our conclusion that these 148 proteins were differentially regulated by Hsp90 inhibition.
Figure 7. Comparison of results from spectrum counting assays vs. peak intensity assays.

Panel 7A, protein expression was measured in treated vs. control cells using spectrum counting and LFQ peak intensities, and the average ratios (n=3) obtained from each assay method were compared. Individual points represent proteins showing significant changes in response to two or more Hsp90 inhibitors, via either assay. Panel 7B, Venn analysis of proteins showing significant changes in response to two or more Hsp90 inhibitors, via either assay.
We randomly selected 7 of our differentially expressed proteins for further analysis by Western blotting (Fig. 8). Results demonstrated that: (i) all seven of the validation proteins were depleted by each of the three drugs; (ii) this depletion was strongly dose-dependent; and (iii) each protein’s dose-dependence reflected our estimates of the relative efficacy of the individual drugs. These results provided a validation of our larger set of 148 inhibitor-induced changes in protein expression, and confirmed our estimates of the relative efficacies among the three drugs.
Figure 8. Validation of 7 inhibitor-induced protein responses.

Jurkat cultures were treated for 24 hr with the indicated concentrations of each inhibitor and assayed by Western blotting.
Analysis of Jurkat responses to Hsp90 inhibition revealed enrichment in several GO categories (Supporting Information). Enriched processes included DNA-dependent metabolic processes such as DNA-dependent DNA replication, largely due to the down-regulation of a whole cassette of MCM DNA replication licensing proteins, as well as several proteins involved in the recognition and repair of DNA damage. GO analyses also revealed enrichment in proteins involved in programmed cell death pathways, reflecting both the up-regulation and down-regulation of several proteins involved in these processes. Additionally, our set of 148 inhibitor-responsive proteins was enriched in proteins with roles in carboxylic acid biosynthetic process that included cellular amino acid biosynthetic processes (glutamine), reflecting the down regulation of several ATP-binding biosynthetic enzymes and the up-regulation of two biosynthetic enzymes that do not bind ATP.
Though not identified via automated GO analyses, the set of inhibitor responsive proteins also included three enzymes involved in tRNA synthesis, 5 ATP-dependent RNA helicases, and 6 kinases known to be Hsp90 dependent. Additional inhibitor-sensitive proteins included several proteins involved in regulating the cytoskeleton and in regulating vesicle trafficking, as well as both the heavy and light chains of the neutral amino acids transporter heteromer.
GO analyses of the inhibitor-induced changes in protein expression also showed a significant enrichment in molecular chaperones, including Hsp90 machineries, Hsp70 and its subunits, multiple subunits from the trimeric ring complex cytosolic chaperonins (a.k.a. CCT or TRIC), two mitochondrial chaperonins, and a small number of other chaperone-related proteins (Supporting Information). From this set of 29 chaperone-related proteins, 27 were up-regulated by Hsp90 inhibition, while only two (cyclophilin A and importin alpha 1) were down-regulated. Because induction of chaperones by Hsp90 inhibitors is widely regarded to reflect dysregulation of the Hsp90-Hsf1 complex, we compared (Supporting Information) our induced chaperone proteome to that regulated by Hsf1 (29). From the set of 27 up-regulated chaperones, 22 had been previously shown to be repressed by silencing expression of the Hsf1 transcription factor. This suggested that their up-regulation in response to Hsp90 inhibition might have reflected the previously described “short-circuiting” of Hsp90-mediated repression of Hsf1 activity(30, 31). We also noted inhibitor-induced up-regulation of 28 Hsf1-regulated proteins that were not chaperone-related, consistent with global inhibitor-induced dysregulation of HSF1-regulated gene products.
In contrast to the cytosolic and mitochondrial chaperones noted above, we did not consistently detect reproducible up-regulation of the ER-resident chaperones endoplasmin (GRP94) or BiP (GRP78) within our set of 148 signature proteins. While this was consistent with previous work showing that genes for these chaperones are not compromised by Hsf1 silencing, some induction of GRP94 was apparent in the AUY922-treated cells (p=0.042). This result seemed inconsistent with previous work demonstrating that AUY922 has 25–fold to 67-fold more affinity for cytosolic Hsp90 that for GRP94(16), particularly in light of the relatively low dosages used here (75 nM), suggesting explanations other than direct inhibition of GRP94 by AUY922.
While chaperone up-regulation in our drug-treated Jurkat cells might have resulted from short-circuiting of Hsp90’s regulation of Hsf1, the global up-regulation of chaperones that we observed and the magnitude of that response also suggested to us that Hsp90 inhibition was inducing bona fide protein folding stress in these cells. Thus, we characterized the impacts of AUY922 in conjunction with another protein folding antagonist, L-azetidine-2-carboxylic acid (AZC). AZC is a four-carbon-ring analogue of L-proline (Fig. 9) that is readily taken up by cells and misincorporated into protein structures during translation(32, 33). Misincorporation of AZC into the polypeptide backbone alters protein structure and stability(34–36), and induces chaperones residing in both the cytoplasm and the ER(37–42). Because the only mechanism described for AZC’s toxicity to date is its antagonism of protein folding, we used it to exacerbate protein folding in Jurkat cells in the presence versus absence of AUY922.
Figure 9.

Structure of Proline or L-azetidine-2-carboxylic acid (AZC), incorporated into peptide trimers
For these characterizations, Jurkat cells were pretreated with AZC to slightly destabilize their proteomes. Subsequently, AUY922 was applied and cellular proliferation assayed. In the absence of AZC, 7.9 nanomolar AUY922 slightly stimulated Jurkat proliferation (Fig. 10), an effect that was consistent throughout our studies. Like AUY922, moderate doses of AZC (0.2 mM) slightly stimulated cell proliferation in the absence of AUY922. In contrast, sequential applications of AZC followed by AUY922 dramatically reduced cellular proliferation (Fig. 10). This demonstrated that AUY922 did not induce a chaperone response capable of restoring AZC-induced protein folding burdens. Instead, AZC significantly potentiated AUY922’s anti-proliferative effect. This potentiation was highly reproducible, and was apparent throughout a range of drug concentrations (Fig. 10). Because AZC concentrations below 0.7 mM AZC were not inhibitory to Jurkat proliferation, the observed potentiation of AUY922 was not simply an additive effect at the low dosages used; i.e., the two drugs appeared to have synergistic effects at low dosages. We concluded that AZC increased cellular requirements for Hsp90 function, and that this requirement for extra Hsp90 function enhanced the anti-proliferative activity of AUY922.
Figure 10. AZC-induced protein folding burdens potentiate AUY922’s inhibition of cellular proliferation.

Jurkat cultures were pre-treated for 10 hr with indicated concentrations of AZC, followed by treatments with indicated concentrations of AUY922. After 38 hr additional incubation (48 hr total), metabolic activity was measured as described in Methods. Results show the averages and SEM from three independent biological replicates. Asterisks denote statistically significant differences in proliferation ( p < 0.05, *; p < 0.01, **; p < 0.001, ***).
Discussion
One goal of this study was to determine the extent to which the proteomics fingerprint of the novel anti-cancer compound AUY922 overlaps those of flagship Hsp90 inhibitors. We find that AUY922 shares a highly conserved proteomics fingerprint with 17-DMAG and radicicol. This conserved fingerprint indicates that AUY922 inhibits the proliferation of Jurkat cells via the same mechanism as 17-DMAG and radicicol, namely the inhibition of Hsp90 and cellular process that depend upon Hsp90 function. Vis-à-vis, this conserved fingerprint strongly argues against an off-target mechanism for AUY922’s inhibition of Jurkat proliferation. This finding reinforces our current understanding of AUY922’s anti-Hsp90 activities in vivo and in vitro (see Introduction and references therein).
A second goal was to identify proteins that might serve as biomarkers of Hsp90 inhibition in leukemia cells. Using three different Hsp90 inhibitors and two label-free assay techniques, we identify a of set reproducible and readily assayed protein responses to low levels of Hsp90 inhibition in cultured Jurkat T cells. The validity of these responses is supported by several relationships within this study and from the literature. (i) Sixty four of the AUY922 responses we describe are also shared with either 17-DMAG or radicicol. (ii) A random subset of seven proteins that our mass spectrometry assays identified as inhibitor-sensitive readily demonstrate dose-dependent, inhibitor-induced depletion in confirmatory Western blot assays (Fig. 8). (iii) Many of the protein responses we observed are consistent with our understanding of Hsp90 and Hsp90 inhibition(43, 44). (iv) Many of the inhibitor-induced alterations in protein expression that we observe in cultured leukemia cells are also seen among other experimental cell lines treated with Hsp90 inhibitors [see Supplementary Material, references (45, 46), and discussion below].
Our studies add to the developing atlas of cellular responses to Hsp90 inhibition. To date, the largest segments of the Hsp90 inhibitor-responsive proteome have been elucidated from studies in yeast(47, 48), and from two recent landmark studies that used SILAC techniques to interrogate five different human cell lines treated with Hsp90 inhibitors(45, 46). Comparisons among those five cell lines suggest hundreds of highly conserved cellular responses to Hsp90 inhibition(46). This comparison also reveals that other cellular responses are less well conserved: for instance 28% of the protein responses seen in CAL27 are not seen in the COLON205 cell lines(46). Comparing our results to these previous studies, we see a comparable overlap between responses in Jurkat cells vs. cell lines used in previous SILAC assays; 86 out of our 148 protein responses (58%) were also seen in one or more of the cell lines from those studies. We also present 62 novel responses specific to Hsp90 inhibition in leukemic T cells. Thus, our studies support an emerging picture of Hsp90-inhibitor responses that are conserved, and other responses that are specific to cell lineage or status (or whose quantification reflects technical subtleties).
As AUY922 moves steadily through clinical trials, there is a need for robust proteome and peptide markers to monitor its effects on cancer cells. From this perspective, it is important to appreciate that our studies are the first LC-MS/MS characterizations to be performed using low dosages of Hsp90 inhibitors (i.e., 75 nM AUY922, 150 nM 17-DMAG, and 300 nM radiciciol). These dosages are markedly lower than those recently used to deeply probe other Hsp90-regulated proteomes (i.e., 50 micromolar 17-DMAG (46) and 5–10 micromolar geldanamycin (45)). By design, our lower drug dosages provided multiple benefits. (i) Low dosages elicited only the most sensitive responses to Hsp90 inhibition. (ii) Low dosages minimized off-target effects, cellular mortality effects, and apoptosis effects (e.g., Fig. 1D). (iii) Since Hsp90 inhibitors have proven to be frustratingly toxic in clinical trials, the low dosages used in the current study may best mirror clinical applications. It is also noteworthy that Hsp90 inhibitors are widely anticipated to hold their greatest promise when utilized in combinatorial treatments with other anti-cancer drugs, and these treatments will likely utilize Hsp90-inhibitor dosages lower than those previously used to maximize cellular responses in vitro. (iv) These low dosages also compare favorably with our appreciation of the pharmokinetic profiles for 17-DMAG and AUY922(16, 20).
Previous studies have characterized some 56 Western blot responses to AUY922. In work presented here, we add to these responses some 64 proteins whose responses to low dosages of AUY922 can be readily detected by mass spectrometry. In addition to the mainstays Hsp90alpha and Hsp90beta, we note the strong induction of the chaperones SerpinH1 (NP_001226), DnaJB1 (NP_006136), FKBP52 (NP_002005), and mitochondrial chaperonin 10 (NP_002148). In our hands, these chaperone responses were more reproducibly detected by LC-MS/MS than the Hsp70 isoforms most commonly used to report Hsp90 inhibition. Additionally, the Hsp90-dependent kinases Cdk6 and Cdk1 also demonstrated exceptionally robust performance in LC-MS/MS assays, making them strong candidates for further development as high-throughput quantitative biomarkers. Another twelve proteins also gave robust responses to AUY922 treatments, and to other Hsp90 inhibitors in all six out of our six assays (Supplementary Information). Among these proteins, we also note four very robust leukemia-cell responses that have not been described previously as proteomics reporters of Hsp90 inhibition. Among these, two spectrin isoforms (NP_003119 and NP_001123910) were induced two-fold to four-fold in six out of six assays, consistent with previous work suggesting chaperone-like roles for spectrin(49). Our most-robust leukemia-specific responses also included the ~50% down-regulation of DNA methyltransferase (NP_001124295), consistent with previous work identifying DNMT1 as a probable Hsp90 client protein(50). We also observed the profound depletion of the RIAM Rap1-GTP-interacting adaptor molecule (a.k.a., amyloid precursor protein-binding family B member 1-interacting protein, NP_061916), an effect that appears to be unique to the current study. While the T-cell specific Lck and ZAP70 kinases were frequently detected, their down-regulation was not as statistically robust as the responses described above. In addition to the proteins highlighted above, the other 136 conserved protein responses we report (Supporting Information) may also represent candidates for LC-MS/MS assays to monitor Hsp90 inhibition in vivo. It should be noted, however, that the in vivo responses among diverse leukemias may differ significantly from those of cultured Jurkat cells, highlighting the need to transition proteomics assays of Hsp90 inhibitors into whole-organism models of cancer.
All three Hsp90 inhibitors used in our study induced the expression of a diverse cassette of molecular chaperones. This well-described effect is often ascribed to Hsp90’s role in repressing Hsf1 (30, 31), where inhibition of Hsp90 de-represses Hsf1 to induce the global up-regulation of Hsf1-regulated chaperones. Our results support this model, describing both the up-regulation of Hsf1-regulated chaperones, and of other Hsf1-regulated proteins.
Lost within this explanation, however, is the potential that Hsp90 inhibitors might create bona fide protein folding burdens in treated cells, despite a concomitant induction of chaperone expression. Testing this, we demonstrate that while low doses of AUY922 up-regulate numerous chaperones in Jurkat cells, this does not alleviate mild protein folding burdens induced by the proline analog AZC. Instead, AZC significantly sensitizes Jurkat cells to Hsp90 inhibition (Fig. 10). This finding invites speculation that some of AUY922’s cytostatic activity in these cells may result from general proteotoxicity. This observation joins an increasing body of evidence and opinion suggesting that Hsp90 inhibitors may target the neoplastic protein folding burdens created by the hundreds of non-oncogenic mutant “passenger” proteins present in the typical cancer cell(51–64). However, this does not diminish the potential importance of Hsp90-depenent oncoproteins, since the two mechanisms are not mutually exclusive. In either case, our observation that the AZC protein folding antagonist sensitizes leukemia cells to Hsp90 inhibition invites consideration of additional combinatorial modalities for AUY922 therapies.
Supplementary Material
Acknowledgments
This work was supported in part by the Oklahoma Center for the Advancement of Science and Technology (HR10-072 to SDH), the Oklahoma Agricultural Experiment Station (Project 1975 to RLM), the NIH (R01 CA125392 to RLM), the Sarkeys Foundation (RLM) and the Oklahoma University Cancer Institute (RLM). The authors thank Daniel Smith and Maurie Balch for critical reading of the manuscript. Mass spectrometry analyses were performed in the DNA/Protein Resource Facility at Oklahoma State University, using resources supported by the NSF MRI and EPSCoR programs (award #0722494 to SDH).
Abbreviations
- 17-DMAG
17-Dimethylaminoethylamino-17-demethoxygeldanamycin
- AZC
L-azetedine-2-carboxylic acid
- DMSO
dimethyl sulfoxide
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- PBS
phosphate buffered saline
- RIPA
radioimmunoprecipitation cell lysis buffer
- TCA
trichloroacetic acid
- LC-MS/MS
liquid chromatography and tandem mass spectrometry
- FDR
false discovery rate
- EC50
effective concentration 50%
- LFQ
label-free quantitation via the MaxQuant algorithm
Footnotes
Supporting Information Available: A supplemental Excel file is available free of charge via the Internet at http://pubs.acs.org, and consists of individual spreadsheets containing: (i) ratios and p values for proteins showing significant differential expression; (ii) bioinformatic GO analyses and comparisons to the Hsf1-regulated proteome; (iii) comparisons between the differentially expressed proteomes observed in this study versus previous work; (iv) raw spectrum counting values; (v) raw LFQ values; (vi) an image of the Cdk6 depletion time course; (vii) volcano plots of p values vs. expression values for each drug using each assay technique.
References
- 1.Barrott JJ, Haystead TA. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013;280(6):1381–96. doi: 10.1111/febs.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–55. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo W, Sun W, Taldone T, Rodina A, Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener. 2010;5:24. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao H, Michaelis ML, Blagg BS. Hsp90 modulation for the treatment of Alzheimer’s disease. Adv Pharmacol. 2012;64:1–25. doi: 10.1016/B978-0-12-394816-8.00001-5. [DOI] [PubMed] [Google Scholar]
- 5.Stuehler C, Mielke S, Chatterjee M, Duell J, Lurati S, Rueckert F, Einsele H, Bargou RC, Topp MS. Selective depletion of alloreactive T cells by targeted therapy of heat shock protein 90: a novel strategy for control of graft-versus-host disease. Blood. 2009;114(13):2829–36. doi: 10.1182/blood-2009-06-224600. [DOI] [PubMed] [Google Scholar]
- 6.Sugita T, Tanaka S, Murakami T, Miyoshi H, Ohnuki T. Immunosuppressive effects of the heat shock protein 90-binding antibiotic geldanamycin. Biochem Mol Biol Int. 1999;47(4):587–95. doi: 10.1080/15216549900201633. [DOI] [PubMed] [Google Scholar]
- 7.Yorgin PD, Hartson SD, Fellah AM, Scroggins BT, Huang W, Katsanis E, Couchman JM, Matts RL, Whitesell L. Effects of geldanamycin, a heat-shock protein 90-binding agent, on T cell function and T cell nonreceptor protein tyrosine kinases. J Immunol. 2000;164(6):2915–23. doi: 10.4049/jimmunol.164.6.2915. [DOI] [PubMed] [Google Scholar]
- 8.Yun TJ, Harning EK, Giza K, Rabah D, Li P, Arndt JW, Luchetti D, Biamonte MA, Shi J, Lundgren K, Manning A, Kehry MR. EC144, a synthetic inhibitor of heat shock protein 90, blocks innate and adaptive immune responses in models of inflammation and autoimmunity. J Immunol. 2011;186(1):563–75. doi: 10.4049/jimmunol.1000222. [DOI] [PubMed] [Google Scholar]
- 9.Theodoraki MA, Caplan AJ. Quality control and fate determination of Hsp90 client proteins. Biochim Biophys Acta. 2012;1823(3):683–8. doi: 10.1016/j.bbamcr.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma SV, Settleman J. Exploiting the balance between life and death: targeted cancer therapy and “oncogenic shock”. Biochem Pharmacol. 2010;80(5):666–73. doi: 10.1016/j.bcp.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Bijlmakers MJ, Marsh M. Hsp90 is essential for the synthesis and subsequent membrane association, but not the maintenance, of the Src-kinase p56(lck) Mol Biol Cell. 2000;11(5):1585–95. doi: 10.1091/mbc.11.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartson SD, Ottinger EA, Huang W, Barany G, Burn P, Matts RL. Modular folding and evidence for phosphorylation-induced stabilization of an hsp90-dependent kinase. J Biol Chem. 1998;273(14):8475–82. doi: 10.1074/jbc.273.14.8475. [DOI] [PubMed] [Google Scholar]
- 13.Scholz G, Hartson SD, Cartledge K, Hall N, Shao J, Dunn AR, Matts RL. p50(Cdc37) can buffer the temperature-sensitive properties of a mutant of Hck. Mol Cell Biol. 2000;20(18):6984–95. doi: 10.1128/mcb.20.18.6984-6995.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholz GM, Hartson SD, Cartledge K, Volk L, Matts RL, Dunn AR. The molecular chaperone Hsp90 is required for signal transduction by wild-type Hck and maintenance of its constitutively active counterpart. Cell Growth Differ. 2001;12(8):409–17. [PubMed] [Google Scholar]
- 15.Hartson SD, Barrett DJ, Burn P, Matts RL. Hsp90-mediated folding of the lymphoid cell kinase p56lck. Biochemistry. 1996;35(41):13451–9. doi: 10.1021/bi961332c. [DOI] [PubMed] [Google Scholar]
- 16.Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68(8):2850–60. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 17.Gaspar N, Sharp SY, Eccles SA, Gowan S, Popov S, Jones C, Pearson A, Vassal G, Workman P. Mechanistic evaluation of the novel HSP90 inhibitor NVP-AUY922 in adult and pediatric glioblastoma. Mol Cancer Ther. 2010;9(5):1219–33. doi: 10.1158/1535-7163.MCT-09-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KH, Lee JH, Han SW, Im SA, Kim TY, Oh DY, Bang YJ. Antitumor activity of NVP-AUY922, a novel heat shock protein 90 inhibitor, in human gastric cancer cells is mediated through proteasomal degradation of client proteins. Cancer Sci. 2011;102(7):1388–95. doi: 10.1111/j.1349-7006.2011.01944.x. [DOI] [PubMed] [Google Scholar]
- 19.Baldo B, Weiss A, Parker CN, Bibel M, Paganetti P, Kaupmann K. A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. J Biol Chem. 2012;287(2):1406–14. doi: 10.1074/jbc.M111.294801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, Quadt C, Buckler A, Cozens R, Drysdale MJ, Garcia-Echeverria C, Chene P. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10(2):R33. doi: 10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 22.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 23.Ruxton GD. The unequal variance t-test is an underused alternative to Student’s t-test and the Mann-Whitney U test. Behavioral Ecology. 2006;17(4):688–90. [Google Scholar]
- 24.Srethapakdi M, Liu F, Tavorath R, Rosen N. Inhibition of Hsp90 function by ansamycins causes retinoblastoma gene product-dependent G1 arrest. Cancer Res. 2000;60(14):3940–6. [PubMed] [Google Scholar]
- 25.Georgakis GV, Li Y, Rassidakis GZ, Medeiros LJ, Younes A. The HSP90 inhibitor 17-AAG synergizes with doxorubicin and U0126 in anaplastic large cell lymphoma irrespective of ALK expression. Exp Hematol. 2006;34(12):1670–9. doi: 10.1016/j.exphem.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Duriez PJ, Shah GM. Cleavage of poly(ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem Cell Biol. 1997;75(4):337–49. [PubMed] [Google Scholar]
- 27.Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev Proteomics. 2010;7(1):39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 28.Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, Akira S, Wiegand M, Hochrein H, O’Keeffe M, Mann M. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32(2):279–89. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 29.Page TJ, Sikder D, Yang L, Pluta L, Wolfinger RD, Kodadek T, Thomas RS. Genome-wide analysis of human HSF1 signaling reveals a transcriptional program linked to cellular adaptation and survival. Mol Biosyst. 2006;2(12):627–39. doi: 10.1039/b606129j. [DOI] [PubMed] [Google Scholar]
- 30.Kim HR, Kang HS, Kim HD. Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life. 1999;48(4):429–33. doi: 10.1080/713803536. [DOI] [PubMed] [Google Scholar]
- 31.Ali A, Bharadwaj S, O’Carroll R, Ovsenek N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol. 1998;18(9):4949–60. doi: 10.1128/mcb.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fowden L, Richmond MH. Replacement of proline by azetidine-2-carboxylic acid during biosynthesis of protein. Biochimica et Biophysicia Acta. 1963;71:459–61. [Google Scholar]
- 33.Trasko CS, Franzblau C, Troxler RF. Incorporation of L-azetidine-2-carboxylic acid into hemoglobin S in sickle erythrocytes in vitro. Biochim Biophys Acta. 1976;447(4):425–35. doi: 10.1016/0005-2787(76)90080-0. [DOI] [PubMed] [Google Scholar]
- 34.Cameron PH, Chevet E, Pluquet O, Thomas DY, Bergeron JJ. Calnexin phosphorylation attenuates the release of partially misfolded alpha1-antitrypsin to the secretory pathway. J Biol Chem. 2009;284(50):34570–9. doi: 10.1074/jbc.M109.053165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz TW. Effect of amino acid analogs on the processing of the pancreatic polypeptide precursor in primary cell cultures. J Biol Chem. 1988;263(23):11504–10. [PubMed] [Google Scholar]
- 36.Tan EM, Ryhanen L, Uitto J. Proline analogues inhibit human skin fibroblast growth and collagen production in culture. J Invest Dermatol. 1983;80(4):261–7. doi: 10.1111/1523-1747.ep12534593. [DOI] [PubMed] [Google Scholar]
- 37.Guan JC, Yeh CH, Lin YP, Ke YT, Chen MT, You JW, Liu YH, Lu CA, Wu SJ, Lin CY. A 9 bp cis-element in the promoters of class I small heat shock protein genes on chromosome 3 in rice mediates L-azetidine-2-carboxylic acid and heat shock responses. J Exp Bot. 2010;61(15):4249–61. doi: 10.1093/jxb/erq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee Y, Nagao RT, Lin CY, Key JL. Induction and Regulation of Heat-Shock Gene Expression by an Amino Acid Analog in Soybean Seedlings. Plant Physiol. 1996;110(1):241–8. doi: 10.1104/pp.110.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trotter EW, Kao CM, Berenfeld L, Botstein D, Petsko GA, Gray JV. Misfolded proteins are competent to mediate a subset of the responses to heat shock in Saccharomyces cerevisiae. J Biol Chem. 2002;277(47):44817–25. doi: 10.1074/jbc.M204686200. [DOI] [PubMed] [Google Scholar]
- 40.Van Rijn J, Wiegant FA, Van den Berg J, Van Wijk R. Heat shock response by cells treated with azetidine-2-carboxylic acid. Int J Hyperthermia. 2000;16(4):305–18. doi: 10.1080/02656730050074078. [DOI] [PubMed] [Google Scholar]
- 41.Li WW, Hsiung Y, Zhou Y, Roy B, Lee AS. Induction of the mammalian GRP78/BiP gene by Ca2+ depletion and formation of aberrant proteins: activation of the conserved stress-inducible grp core promoter element by the human nuclear factor YY1. Mol Cell Biol. 1997;17(1):54–60. doi: 10.1128/mcb.17.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watowich SS, Morimoto RI. Complex regulation of heat shock- and glucose-responsive genes in human cells. Mol Cell Biol. 1988;8(1):393–405. doi: 10.1128/mcb.8.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartson SD, Matts RL. Approaches for defining the Hsp90-dependent proteome. Biochim Biophys Acta. 2012;1823(3):656–67. doi: 10.1016/j.bbamcr.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samant RS, Clarke PA, Workman P. The expanding proteome of the molecular chaperone HSP90. Cell Cycle. 2012;11(7):1301–8. doi: 10.4161/cc.19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma K, Vabulas RM, Macek B, Pinkert S, Cox J, Mann M, Hartl FU. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol Cell Proteomics. 2012;11(3):M111 014654. doi: 10.1074/mcp.M111.014654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Z, Moghaddas Gholami A, Kuster B. Systematic identification of the HSP90 candidate regulated proteome. Mol Cell Proteomics. 2012;11(6):M111 016675. doi: 10.1074/mcp.M111.016675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131(1):121–35. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 48.Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, Krogan N, Cagney G, Mai D, Greenblatt J, Boone C, Emili A, Houry WA. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120(5):715–27. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 49.Bhattacharyya M, Ray S, Bhattacharya S, Chakrabarti A. Chaperone activity and prodan binding at the self-associating domain of erythroid spectrin. J Biol Chem. 2004;279(53):55080–8. doi: 10.1074/jbc.M406418200. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res. 2008;6(5):873–83. doi: 10.1158/1541-7786.MCR-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA. Impact of deleterious passenger mutations on cancer progression. Proc Natl Acad Sci U S A. 2013;110(8):2910–5. doi: 10.1073/pnas.1213968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130(6):986–8. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 54.Travers J, Sharp S, Workman P. HSP90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today. 2012;17(5–6):242–52. doi: 10.1016/j.drudis.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 55.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130(6):1005–18. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, Davies FE. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110(7):2641–9. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
- 57.Davenport EL, Zeisig A, Aronson LI, Moore HE, Hockley S, Gonzalez D, Smith EM, Powers MV, Sharp SY, Workman P, Morgan GJ, Davies FE. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia. 2010;24(10):1804–7. doi: 10.1038/leu.2010.168. [DOI] [PubMed] [Google Scholar]
- 58.Heimberger T, Andrulis M, Riedel S, Stuhmer T, Schraud H, Beilhack A, Bumm T, Bogen B, Einsele H, Bargou RC, Chatterjee M. The heat shock transcription factor 1 as a potential new therapeutic target in multiple myeloma. Br J Haematol. 2012 doi: 10.1111/bjh.12164. [DOI] [PubMed] [Google Scholar]
- 59.Lamoureux F, Thomas C, Yin MJ, Kuruma H, Beraldi E, Fazli L, Zoubeidi A, Gleave ME. Clusterin inhibition using OGX-011 synergistically enhances Hsp90 inhibitor activity by suppressing the heat shock response in castrate-resistant prostate cancer. Cancer Res. 2011;71(17):5838–49. doi: 10.1158/0008-5472.CAN-11-0994. [DOI] [PubMed] [Google Scholar]
- 60.Lawson B, Brewer JW, Hendershot LM. Geldanamycin, an hsp90/GRP94-binding drug, induces increased transcription of endoplasmic reticulum (ER) chaperones via the ER stress pathway. J Cell Physiol. 1998;174(2):170–8. doi: 10.1002/(SICI)1097-4652(199802)174:2<170::AID-JCP4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 61.Neznanov N, Gorbachev AV, Neznanova L, Komarov AP, Gurova KV, Gasparian AV, Banerjee AK, Almasan A, Fairchild RL, Gudkov AV. Anti-malaria drug blocks proteotoxic stress response: anti-cancer implications. Cell Cycle. 2009;8(23):3960–70. doi: 10.4161/cc.8.23.10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget. 2011;2(3):209–21. doi: 10.18632/oncotarget.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S, Ince TA. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A. 2011;108(45):18378–83. doi: 10.1073/pnas.1115031108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zaarur N, Gabai VL, Porco JA, Jr, Calderwood S, Sherman MY. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006;66(3):1783–91. doi: 10.1158/0008-5472.CAN-05-3692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.