

Abstract
West Nile virus (WNV) is an emerging neurotropic flavivirus that has recently spread to America and Southern Europe via an enzootic/epizootic bird-mosquito-bird transmission cycle. The virus can occasionally infect humans through mosquito bites, and man-to-man transmission has also been reported via infected blood or organ donation. In the human host, WNV causes asymptomatic infection in about 70%-80% of cases, while < 1% of clinical cases progress to severe neuroinvasive disease; long-term neurological sequelae are common in more than 50% of these severe cases. The pathogenesis of the neuroinvasive form of WNV infection remains incompletely understood, and risk factors for developing severe clinical illness are largely unknown. The innate immune response plays a major role in the control of WNV replication, which is supported by the fact that the virus has developed numerous mechanisms to escape the control of antiviral interferons. However, exaggerated inflammatory responses lead to pathology, mainly involving the central nervous system. This brief review presents the salient features of innate host responses, WNV immunoevasion strategies, and WNV-induced immunopathology.
Keywords: West Nile virus infection, Innate immunity, Antigen presenting cells, Inflammation, Interferon and cytokines, Central nervous system
INTRODUCTION
West Nile virus (WNV) is a lipid-enveloped virus that contains a single stranded, positive sense RNA genome. The virus is introduced into the host by an infected vector (mosquitoes generally belonging to the genus Culex) during its blood meal. WNV was originally found in Africa and in the Middle East but has recently reached America[1,2], where it has spread throughout the United States. In the last 15 years, WNV has also caused several human outbreaks in southern Europe[3-5].
Most individuals infected with WNV remain asymptomatic. In 20%-30% of cases, WNV causes a mild flu-like illness. In such cases, symptoms appear suddenly and may include malaise, eye pain, headache, myalgia, gastrointestinal discomfort and rash[6]. Less than 1% of infected individuals develop neurological symptoms, including aseptic meningitis, febrile convulsion in children, encephalitis or myelitis, the last of which causes acute flaccid paralysis[7-9]. Long-term neurological sequelae are common in more than 50% of neuroinvasive cases. The virus can infect neurons in areas as diverse as the cerebral cortex, basal ganglia and thalami, as well as the brainstem and cerebellum. Currently, risk factors for developing severe clinical illness are unknown. However, it is clear that WNV central nervous system (CNS) disease occurs with increased frequency in immunocompromised individuals and the elderly[10-12].
Innate immune responses are believed to be crucial for the control of WNV replication, as a potent and rapid type I interferon (IFN) response is essential for the successful control of WNV infection in mice[13]. As a first line of defence, the host cell senses the presence of the virus by pathogen recognition receptors (PRRs), such as toll-like receptors (TLRs) and retinoic acid-inducible gene (RIG)-I like receptors. Binding of viral components to these receptors activates adaptor proteins, which in turn activate transcription factors, and induces a release of soluble mediators, including type I IFNs[14,15]. Members of RIG-I like receptor family (RIG-I and melanoma differentiation-associated gene, MDA5) and TLR family (TLR3 and TLR7) are the major innate host sensors of WNV infection. RIG-I and MDA5 are cytosolic RNA helicases that recognize ssRNA and dsRNA. RIG-I and MDA5 transmit their signal through a common adaptor molecule, IFN-promoter stimulator (IPS)-1, thus activating transcription factors such as IFN regulatory factor (IRF) 3 and IRF7 to induce the transcription of type I IFN and antiviral genes. TLR3 and TLR7 are expressed primarily in endosomes and are activated by dsRNA and ssRNA, respectively. Engagement of TLR7 leads to the activation of a signalling pathway involving an intracellular adaptor protein, myeloid differentiation primary response gene 88 (MyD88), the activation of IRF7 and the induction of type I IFNs. TLR3 activates the adaptor molecule TIR-domain-containing adapter-inducing IFN-β and induces alternative pathways that lead to the activation of the transcription factors IRF3 and nuclear factor κB (NF-κB), which consequently induce type I IFNs and inflammatory cytokines, respectively.
Antigen presenting cells (APCs) are among the first cells that encounter the virus after infection; WNV is injected intradermally by a mosquito bite and most likely initially replicates in Langerhans dendritic cells (DCs). The infected Langerhans cells migrate to draining lymph nodes from which the virus enters the bloodstream[16]. Primary viremia disseminates the virus to the reticuloendothelial system (macrophagic cells), where replication further augments viremia (secondary viremia), followed by spread in various organs including the brain. Monocytes and polymorphonuclear leukocytes (PMNLs) are readily recruited and activated following infection in rodent models[17].
Thus, cells of the innate immune system and their receptors are the first to encounter WNV after infection in the host, and the interaction between the virus and factors of innate immunity likely determines the outcome of the infection. In addition, macrophages (Mφs) constitute an important fraction on the inflammatory infiltrate observed in the CNS of WNV infected patients[18], suggesting that cells of innate immunity can also contribute to immunopathology in the course of WNV infection.
INTERPLAY BETWEEN CELLS OF INNATE IMMUNITY AND WNV
Despite the potentially critical role of APCs during WNV infection, few studies have addressed the effect of WNV infection on APCs obtained from humans. Human myeloid DCs (mDCs) have been shown to be among the targets of WNV infection. Production of tumor necrosis factor (TNF)-α and IFN-α in infected mDCs requires viral replication[19,20] (Figure 1). Conversely, plasmacytoid DCs (pDCs) are resistant to infection but are clearly activated upon contact with the virus through stimulation of endosomal TLRs. Upon activation with WNV, pDCs release higher amount of IFN-α than mDCs[19]. It has been demonstrated that glycosylated strains of WNV use DC-SIGN (a C-type lectin that binds high-mannose N-linked glycans present on the surface of viral glycoproteins) as an attachment receptor to bind mDCs, leading to enhanced infection in cell cultures[20]. This finding suggests that glycosylated strains of WNV, mainly belonging to lineage I, exhibit an increased capability to infect mDCs and thus higher pathogenicity.
Figure 1.
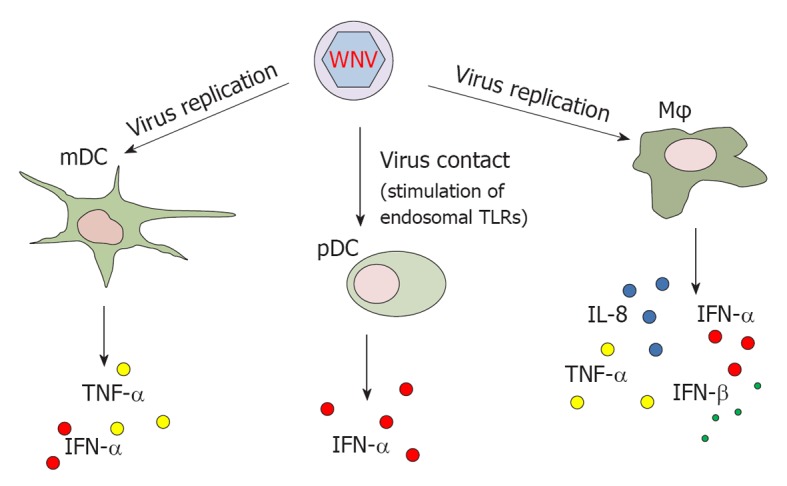
West Nile virus stimulates the production of interferons and pro-inflammatory cytokines in human antigen presenting cells. TLR: Toll-like receptors; WNV: West Nile virus; mDC: Myeloid dendritic cells; pDC: Plasmacytoid dendritic cells; Mφ: Macrophages; TNF: Tumor necrosis factor; IFN: Interferon; IL: Interleukin.
Human monocytes and monocyte-derived Mφs also undergo productive infection upon in vitro incubation with WNV[21]. Interestingly, these cells are infected without gross cytopathic effects, suggesting that they possess effective defence mechanisms against WNV[22]. The lack of cell deterioration upon WNV infection in monocytes/Mφs also suggests that these cells play a significant role as a reservoir in initial (or secondary) viral replication and dissemination. Upon WNV infection, monocyte-derived Mφs release interleukin (IL)-8, IFN-α, IFN-β and TNF-α[22,23]. However, in Mφ cultures activated by LPS and IFN-γ, WNV infection down-modulates the secretion of IL-1β and IFN-β and inhibits the JAK/STAT signalling pathway[23], as a potential strategy employed by the virus to evade the host response (see below).
Notably, monocyte-derived Mφs from elderly individuals show increased susceptibility to WNV infection and augmented expression levels of TLR3 upon infection, as compared to young subjects. Once stimulated with the virus, cells from the elderly also secrete higher levels of IFN-β and IL-6[24]. This in vitro model of WNV infection suggests that the age-associated impairment of the innate immune response to WNV may contribute to increased severity of this viral infection in older individuals.
IFNs AND WNV
Type I IFNs represent a major innate immune control and comprise various IFN-α and one IFN-β, which are secreted by leukocytes and parenchymal cells during viral infections[25]. These cytokines induce an antiviral state by up-regulating genes with direct and indirect antiviral functions. Type I IFNs also link innate and adaptive immunity by inducing DC maturation and by directly activating B and T cells[26].
As mentioned in the previous section, human mDCs, pDCs and monocyte-derived Mφs secrete type I IFNs upon contact with WNV[19-21]. The role of these antiviral mediators upon WNV infection and the role of the pathways involved in IFN secretion have been elucidated only in animal models. Studies in mice indicate that type I IFNs play a crucial role in the early control of WNV infection. Mice lacking IFN-α/β receptor are highly vulnerable to WNV, and uncontrolled viral replication occurs with rapid dissemination to the CNS and 100% mortality[13]. In addition, it has been observed that pre-treatment or treatment with type I IFNs in vitro inhibits WNV replication in Vero cells[27,28]. Additionally, treatment of primary murine neurons in vitro with IFN-β either before or after infection increased neuronal survival independent of its effect on WNV replication[13]. Altogether, these findings in animals and in vitro cultured cells support a crucial role for type I IFNs in the early phases of WNV infection by preventing viral replication and protecting infected neurons from death.
Cells recognize WNV and respond by producing type I IFNs through the endosomal receptors TLR3 and TLR7, thus activating the adaptor MyD88 and transcription factors IRF3 and IRF7 (Figure 2). This response has been demonstrated in rodent models of infection, as mice with genetic defects in any one of these receptors[29,30], adaptor[31] or transcription factors[32,33] have a higher mortality rate with experimental WNV infection (reviewed in[34]).
Figure 2.
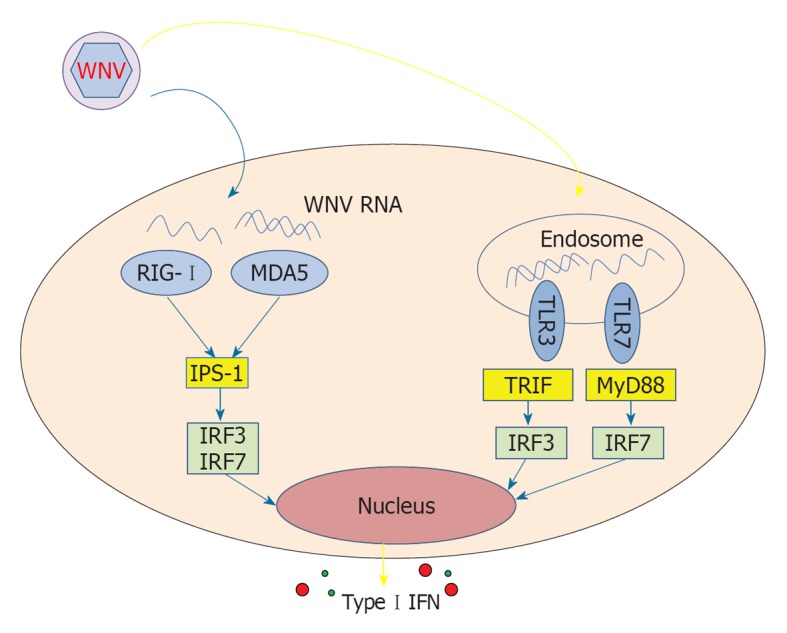
Receptors of innate immunity, adaptors and transcription factors involved in recognition of West Nile virus. WNV: West Nile virus; IFN: Interferon; IPS-1: IFN-β promoter stimulation-1; IRF: IFN regulatory factor; MDA5: Melanoma differentiation-associated protein 5; MyD88: Myeloid differentiation primary response gene 88; RIG; Retinoic acid-inducible gene; TLR: Toll-like receptor; TRIF: TIR-domain-containing adapter-inducing interferon-β.
WNV RNA can also induce the release of type I IFNs by triggering RIG-I, which appears to be involved in the early phases of response to the virus[35]. MDA5, belonging to the RIG-I receptor family, is also involved in sensing WNV RNA; abrogation of both RIG-I and MDA5 pathways blocks activation of the antiviral response to WNV, while such an effect is not as evident if only one of the two pathways is ablated[36]. In line with these findings, infected mice lacking IPS-1, the central adaptor for RIG-I and MDA5, display uncontrolled inflammation that is coupled with the failure to protect against WNV infection[37]. Thus, TLR3 and TLR7, as well as RIG-I and MDA5, are activated by WNV and appear to induce redundant IFN-mediated responses that trigger downstream effective adaptive responses.
The regulation of IFN responses could be more complex than indicated by the present understanding. Increasing evidence indicates a crucial antiviral role for the inflammasome, a cytoplasmic multi-protein complex that recruits inflammatory caspases and triggers their activation[38]. For example, recent evidence shows that caspase-12, an important component of the inflammasome signalling, plays an important role in WNV infection by influencing RIG-I activity and type I IFN release[39]. The role of other inflammasome complex proteins in influencing the release of type I IFNs during WNV infection has not been investigated.
IFN type II, i.e., IFN-γ, is mainly produced by CD8+ T cells, it is also secreted by γδ T cells and natural killer cells and may contribute to innate immune control of viral infections. In vivo, IFN-γ restricts early WNV dissemination to the CNS; mice deficient in either IFN-γ or the IFN-γ receptor show a higher peripheral viral load, augmented entry into the CNS and increased lethality[40,41]. Notably, no major deficits of adaptive immunity were found in these studies, suggesting that IFN-γ plays mainly an early innate role in the control of WNV infection.
In recent years, a third type of IFN has been described. Originally termed IL-28a/b and IL-29, these proteins have been re-classified as IFN-λs, based on the similar modes of induction and the antiviral activities that they share with the type I and type II IFNs[25]. In support to their antiviral role, IFN-λ3 has recently been identified as key cytokine in the control of a flavivirus infection, i.e., hepatitis C virus[42]. The role of these mediators during the course of other flaviviruses is relatively unknown; only one study has examined the role of IFN-λ in the control of WNV to date. Similar to type I IFN, IFN-λ prevents infection by WNV virus-like particles in susceptible cells but fails to inhibit viral replication in cells infected prior to the addition of this cytokine[43].
INHIBITION OF IFN-INDUCED RESPONSES BY WNV
WNV has successfully evolved countermeasures to overcome host innate immunity and productively infect host cells by using a combination of two strategies: (1) passive evasion of the interaction with cellular PRRs and/or (2) active inhibition of different steps of the intracellular pathways that lead to type I IFN production and signalling.
Passive evasion of PRR activation
WNV may regulate the time of induction of the host cell antiviral response by modulating the activation of IRF3 during early phases of infection. WNV does not actively inhibit the RIG-I pathway but rather delays IRF3 activation, possibly by preventing host cells from sensing viral replication shortly after infection[35,44], thus allowing the virus to replicate to high titers before the host cells can mount an effective antiviral response.
Active inhibition of type I IFN production and signalling
WNV attenuates type I IFN response by targeting multiple steps of the induction and signalling cascade, and a number of nonstructural viral proteins (NSs), such as NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5 have been implicated in this process[45,46]. WNV NS1, a protein secreted from infected cells, inhibits TLR3 signalling by preventing IRF3 and NF-κB nuclear translocation[47], and NS2A has been identified as an inhibitor of IFN-β gene transcription[48].
WNV also targets essential elements of the IFN signalling pathway and thus prevent the induction of antiviral genes. Type I IFN signalling initiates when IFNα/β bind to type I receptors (IFNRs) on the cell surface. This process results in the activation of JAK1 and Tyk2, phosphorylating STAT1 and STAT2, which, in association with IRF9, form a heterotrimeric complex known as IFN-stimulated gene (ISG) factor 3. This complex translocates to the nucleus where it induces hundreds of ISGs. The expression of WNV NSs prevents the accumulation of IFNR1 in multiple cells through a non-canonical protein degradation pathway, contributing to the inhibition of the IFN response[49].
The NS5 codified by the virulent lineage I strains of WNV can function as an efficient IFN antagonist by preventing the phosphorylation and nuclear translocation of STAT1[50], while NS4B inhibits JAK1 and TyK2 phosphorylation thus blocking the STAT1 and STAT2 signalling cascade and the subsequent ISG expression. WNV infection actively promotes a redistribution of cholesterol within the cells, which contributes to the down-regulation of the IFN-stimulated JAK-STAT antiviral response to infection and thus facilitates viral replication and survival[51]. Furthermore, characteristic membranous structures induced during WNV replication are connected to viral immune evasion mechanisms, providing partial protection from the IFN-induced antiviral protein MxA[52].
However, viral control of the IFN signalling cascade is not complete, as demonstrated by occurrence of IFNα/β induction and ISG expression during WNV infection. WNV may attenuate or modulate the innate antiviral response, and the ability of only pathogenic lineage I WNV isolates to inhibit the JAK/STAT signalling pathways indicates the importance of this fine modulation as a feature of WNV pathogenesis[53].
ROLE OF INNATE IMMUNITY IN THE PATHOGENESIS OF THE NEUROINVASIVE FORM OF WNV INFECTION
Despite its severity, the pathogenesis of the neuroinvasive form of WNV infection remains incompletely understood. Knowledge in this field relies almost completely on studies in murine models, while the roles of innate mechanisms in inducing protection or causing pathology in human WNV disease are still poorly known. The increased risk of severe WNV infections for immunosuppressed patients[12,54] and the successful infection outcome in a transplant recipient by the modulation of the immunosuppressive regimen[55] suggest that an intact immune system is essential for the control of WNV infection. On the other hand, it is generally recognized that a major hallmark of WNV pathogenesis is neuroinflammation[56,57], which is caused by exaggerated innate and acquired immune responses.
WNV is believed to first multiply in mDCs and monocytes/Mφs before spreading to the brain[58], and recent evidence indicates that early viral replication in myeloid APCs has a crucial pathogenetic role; silencing such replication in Mφs and mDCs effectively suppresses virus-induced encephalitis in mice[59]. Mechanisms underlying this clear-cut effect could rely on (1) an increased viral burden induced by infected APCs, which would be sufficient for the virus to cross the blood-brain barrier, or (2) WNV-infected Mφs acting as “Trojan horses” to carry the virus into the brain[60]. Accumulation of inflammatory monocytes into the brain and their differentiation to Mφs and microglia can also worsen neuroinflammation and CNS injury, as demonstrated in a murine model of non-lethal WNV infection[61]. As an additional pathogenetic role of infected APCs, recognition of WNV nucleic acid in monocytes/microglia by TLR3 leads to the production of TNF-α, which results in a loss of tight junctions, allowing the entry of WNV and immune cells into the perivascular space of the brain in mice[56]. Further, increased levels of macrophage migratory inhibitory factor (MIF) (a potent pro-inflammatory mediator and chemotactic factor that is produced by activated Mφs) have been found in the serum and CSF of WNV-infected patients, and abrogation of MIF in WNV-infected mice mitigates clinical disease by inducing a remarkably reduced number of infiltrating WNV-infected leukocytes in the CNS[62]. Thus, activation of cells of the monocyte/Mφ system by WNV appears to result in important neuropathological consequences, and exaggerated innate responses may cause inflammation, altering the blood brain barrier permeability and allowing the virus to enter the CNS.
On the other hand, early monocytosis induced by WNV in a murine model of infection appears to be protective against lethal disease[63]. Further murine studies on WNV infection indicate a protective role for Mφs[64] and for TLR3, the latter being essential for restricting WNV replication in neurons and protecting the host from lethal encephalitis[29]. Finally, CCR5, a chemokine receptor expressed on Mφs and T cells, is a critical antiviral agent and survival determinant in WNV infection in mice that acts by regulating the trafficking of leukocytes to the infected brain[65]. These controversial studies suggest that monocyte/Mφ involvement and TLR simulation may contribute to inducing protection or causing immunopathology during WNV neuroinvasive disease in mice.
In addition to monocytes/Mφs, other cells belonging to the innate immune system may contribute to the pathogenesis of neuroinvasive WNV infection. For example, PMNLs predominate in the CSF of patients with WNV meningitis and encephalitis in 40% of cases[8] and are recruited shortly after infection into the CNS in an experimental model of WNV infection[17]. In infected mice, the expression of PMNL-recruiting chemokines was dramatically elevated in early phases after infection and PMNLs were quickly recruited to sites of WNV infection. Depletion of PMNLs prior to WNV challenge paradoxically lowered viremia and enhanced survival[66], suggesting that these cells have a pathogenic role in the early phases of WNV infection. Mechanisms that underlie the contribution of PMNLs to the pathogenesis of WNV infection may include the efficient replication of WNV in PMNLs; these cells may act as a virus reservoir, as PMNLs are the predominant cell type recruited to the site of infection and carry the highest amount of virus[66].
As part of the innate response, two important cell types within the CNS respond to infection, i.e., microglia and astrocytes. These cells have been found to be infected in tissue sections from patients with WNV meningoencephalitis[67]. WNV-infected human astrocytes are capable of releasing matrix metalloproteinase 1, 3 and 9, which contribute by disrupting the blood brain barrier and degrading tight junction proteins[68].
In addition to glial cells, which are classically considered to be the predominant source of pro-inflammatory mediators in the CNS during WNV infection, WNV-infected neurons release pro-inflammatory mediators, contributing to neuronal cell death and glial cell activation[69]. Additionally, pro-inflammatory chemokines, such as IFN-γ inducible protein 10, monocyte chemoattractant protein-5 and monokine induced by IFN-γ, are important triggers of inflammation in the brain, and their early up-regulation in the CNS is followed by the up-regulation of TNF-α at the same site in a rodent model of WNV infection[57]. Further, treatment of infected neuronal cells with antibodies blocking TNF-α and other pro-inflammatory mediators results in a significant reduction of WNV-mediated neuronal death[69], suggesting that such mediators play a major role in the pathogenesis of WNV infection in the CNS.
However, pro-inflammatory factors also possess a crucial role in defence against WNV, and leukocyte trafficking into the brain induced by TNF-α protects mice against lethal infection[70]. Altogether, contradictory findings regarding the role of innate responses to WNV infection in mice have been reported; early responses appear to be beneficial or harmful depending on the model. Different experimental settings, including the virus passage history, virus inoculation route and dose, time between the infection and the experiments and potential diverse inflammatory response to WNV in different murine strains, may account for these contradictory findings. Early control of WNV by innate responses would likely effectively restrict WNV dissemination, while continuous triggering and/or excessive reactivity of innate receptors to the virus may contribute to enhanced inflammation, which is known to be a main contributor to WNV neuropathology as a result of CNS invasion.
CONCLUSION
The innate immune response is considered to be a major controller of WNV replication, a notion that is also supported by the fact that the virus has developed numerous mechanisms to escape the control of antiviral IFNs. However, exaggerated innate immune responses appear to be detrimental and lead to neuropathology. Importantly, the role of aging in enhancing the WNV-induced innate immune response has recently been clarified in an in vitro model of infection[24]. Nevertheless, the mechanisms triggering protection or pathology during natural WNV infection are largely unclear.
The interplay between WNV and innate responses has been mainly studied in animal models, while studies on the effect of WNV on human cells of innate immunity are restricted to in vitro cultured cells. All of the abovementioned models have a common limitation, i.e., the transmission of the virus does not occur by a typical route. This limitation leads to two major biases: (1) a lack of transmission of saliva and potential symbionts with the mosquito bite, and (2) a lack of “natural” stimulation of Langerhans DCs and/or antimicrobial peptides at the inoculation site. Thus, further immunological studies in individuals undergoing natural infection are required to better understand the immunopathogenesis of WNV disease, as elucidating the immunopathological mechanisms is essential to inform novel approaches to combat this infection.
Footnotes
Supported by RFO of University of Bologna, the grant “Fondi Finalizzati Lab P3” from Regione Emilia-Romagna; the grant “Ricerca Finalizzata RF-2009-1539631” from the Italian Ministry of Health
P- Reviewer Zhang LW S- Editor Jiang L L- Editor A E- Editor Zheng XM
References
- 1.Centers for Disease Control and Prevention (CDC) West nile virus disease and other arboviral diseases - United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:510–514. [PubMed] [Google Scholar]
- 2.Petersen LR, Fischer M. Unpredictable and difficult to control--the adolescence of West Nile virus. N Engl J Med. 2012;367:1281–1284. doi: 10.1056/NEJMp1210537. [DOI] [PubMed] [Google Scholar]
- 3.Reiter P. West Nile virus in Europe: understanding the present to gauge the future. Euro Surveill. 2010;15:19508. [PubMed] [Google Scholar]
- 4.Magurano F, Remoli ME, Baggieri M, Fortuna C, Marchi A, Fiorentini C, Bucci P, Benedetti E, Ciufolini MG, Rizzo C, et al. Circulation of West Nile virus lineage 1 and 2 during an outbreak in Italy. Clin Microbiol Infect. 2012;18:E545–E547. doi: 10.1111/1469-0691.12018. [DOI] [PubMed] [Google Scholar]
- 5.Papa A. West Nile virus infections in Greece: an update. Expert Rev Anti Infect Ther. 2012;10:743–750. doi: 10.1586/eri.12.59. [DOI] [PubMed] [Google Scholar]
- 6.Lim SM, Koraka P, Osterhaus AD, Martina BE. West Nile virus: immunity and pathogenesis. Viruses. 2011;3:811–828. doi: 10.3390/v3060811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turtle L, Griffiths MJ, Solomon T. Encephalitis caused by flaviviruses. QJM. 2012;105:219–223. doi: 10.1093/qjmed/hcs013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis LE, DeBiasi R, Goade DE, Haaland KY, Harrington JA, Harnar JB, Pergam SA, King MK, DeMasters BK, Tyler KL. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- 9.Kramer LD, Li J, Shi PY. West Nile virus. Lancet Neurol. 2007;6:171–181. doi: 10.1016/S1474-4422(07)70030-3. [DOI] [PubMed] [Google Scholar]
- 10.Murray K, Baraniuk S, Resnick M, Arafat R, Kilborn C, Cain K, Shallenberger R, York TL, Martinez D, Hellums JS, et al. Risk factors for encephalitis and death from West Nile virus infection. Epidemiol Infect. 2006;134:1325–1332. doi: 10.1017/S0950268806006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray KO, Koers E, Baraniuk S, Herrington E, Carter H, Sierra M, Kilborn C, Arafat R. Risk factors for encephalitis from West Nile Virus: a matched case-control study using hospitalized controls. Zoonoses Public Health. 2009;56:370–375. doi: 10.1111/j.1863-2378.2008.01197.x. [DOI] [PubMed] [Google Scholar]
- 12.Iwamoto M, Jernigan DB, Guasch A, Trepka MJ, Blackmore CG, Hellinger WC, Pham SM, Zaki S, Lanciotti RS, Lance-Parker SE, et al. Transmission of West Nile virus from an organ donor to four transplant recipients. N Engl J Med. 2003;348:2196–2203. doi: 10.1056/NEJMoa022987. [DOI] [PubMed] [Google Scholar]
- 13.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barton GM, Medzhitov R. Linking Toll-like receptors to IFN-alpha/beta expression. Nat Immunol. 2003;4:432–433. doi: 10.1038/ni0503-432. [DOI] [PubMed] [Google Scholar]
- 15.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 16.Johnston LJ, Halliday GM, King NJ. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J Invest Dermatol. 2000;114:560–568. doi: 10.1046/j.1523-1747.2000.00904.x. [DOI] [PubMed] [Google Scholar]
- 17.Bréhin AC, Mouriès J, Frenkiel MP, Dadaglio G, Desprès P, Lafon M, Couderc T. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+B220-BST-2+ leukocyte population. J Immunol. 2008;180:6760–6767. doi: 10.4049/jimmunol.180.10.6760. [DOI] [PubMed] [Google Scholar]
- 18.Kelley TW, Prayson RA, Ruiz AI, Isada CM, Gordon SM. The neuropathology of West Nile virus meningoencephalitis. A report of two cases and review of the literature. Am J Clin Pathol. 2003;119:749–753. doi: 10.1309/PU4R-76JJ-MG1F-81RP. [DOI] [PubMed] [Google Scholar]
- 19.Silva MC, Guerrero-Plata A, Gilfoy FD, Garofalo RP, Mason PW. Differential activation of human monocyte-derived and plasmacytoid dendritic cells by West Nile virus generated in different host cells. J Virol. 2007;81:13640–13648. doi: 10.1128/JVI.00857-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martina BE, Koraka P, van den Doel P, Rimmelzwaan GF, Haagmans BL, Osterhaus AD. DC-SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN-alpha and TNF-alpha. Virus Res. 2008;135:64–71. doi: 10.1016/j.virusres.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Rios M, Zhang MJ, Grinev A, Srinivasan K, Daniel S, Wood O, Hewlett IK, Dayton AI. Monocytes-macrophages are a potential target in human infection with West Nile virus through blood transfusion. Transfusion. 2006;46:659–667. doi: 10.1111/j.1537-2995.2006.00769.x. [DOI] [PubMed] [Google Scholar]
- 22.Yeung AW, Wu W, Freewan M, Stocker R, King NJ, Thomas SR. Flavivirus infection induces indoleamine 2,3-dioxygenase in human monocyte-derived macrophages via tumor necrosis factor and NF-κB. J Leukoc Biol. 2012;91:657–666. doi: 10.1189/jlb.1011532. [DOI] [PubMed] [Google Scholar]
- 23.Kong KF, Wang X, Anderson JF, Fikrig E, Montgomery RR. West nile virus attenuates activation of primary human macrophages. Viral Immunol. 2008;21:78–82. doi: 10.1089/vim.2007.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong KF, Delroux K, Wang X, Qian F, Arjona A, Malawista SE, Fikrig E, Montgomery RR. Dysregulation of TLR3 impairs the innate immune response to West Nile virus in the elderly. J Virol. 2008;82:7613–7623. doi: 10.1128/JVI.00618-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Weerd NA, Nguyen T. The interferons and their receptors--distribution and regulation. Immunol Cell Biol. 2012;90:483–491. doi: 10.1038/icb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo YJ, Hahm B. Type I interferon modulates the battle of host immune system against viruses. Adv Appl Microbiol. 2010;73:83–101. doi: 10.1016/S0065-2164(10)73004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson JF, Rahal JJ. Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg Infect Dis. 2002;8:107–108. doi: 10.3201/eid0801.010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crance JM, Scaramozzino N, Jouan A, Garin D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: antiviral compounds active against pathogenic flaviviruses. Antiviral Res. 2003;58:73–79. doi: 10.1016/s0166-3542(02)00185-7. [DOI] [PubMed] [Google Scholar]
- 29.Daffis S, Samuel MA, Suthar MS, Gale M, Diamond MS. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol. 2008;82:10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, Anderson JF, Flavell RA, Fikrig E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity. 2009;30:242–253. doi: 10.1016/j.immuni.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M, Diamond MS. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J Virol. 2010;84:12125–12138. doi: 10.1128/JVI.01026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Diamond MS. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol. 2008;82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daffis S, Samuel MA, Keller BC, Gale M, Diamond MS. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007;3:e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daffis S, Suthar MS, Gale M, Diamond MS. Measure and countermeasure: type I IFN (IFN-alpha/beta) antiviral response against West Nile virus. J Innate Immun. 2009;1:435–445. doi: 10.1159/000226248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fredericksen BL, Gale M. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J Virol. 2006;80:2913–2923. doi: 10.1128/JVI.80.6.2913-2923.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol. 2008;82:609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang P, Arjona A, Zhang Y, Sultana H, Dai J, Yang L, LeBlanc PM, Doiron K, Saleh M, Fikrig E. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat Immunol. 2010;11:912–919. doi: 10.1038/ni.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shrestha B, Wang T, Samuel MA, Whitby K, Craft J, Fikrig E, Diamond MS. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J Virol. 2006;80:5338–5348. doi: 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang T, Scully E, Yin Z, Kim JH, Wang S, Yan J, Mamula M, Anderson JF, Craft J, Fikrig E. IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J Immunol. 2003;171:2524–2531. doi: 10.4049/jimmunol.171.5.2524. [DOI] [PubMed] [Google Scholar]
- 42.Kelly C, Klenerman P, Barnes E. Interferon lambdas: the next cytokine storm. Gut. 2011;60:1284–1293. doi: 10.1136/gut.2010.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma D, Jiang D, Qing M, Weidner JM, Qu X, Guo H, Chang J, Gu B, Shi PY, Block TM, et al. Antiviral effect of interferon lambda against West Nile virus. Antiviral Res. 2009;83:53–60. doi: 10.1016/j.antiviral.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fredericksen BL, Smith M, Katze MG, Shi PY, Gale M. The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu WJ, Wang XJ, Mokhonov VV, Shi PY, Randall R, Khromykh AA. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J Virol. 2005;79:1934–1942. doi: 10.1128/JVI.79.3.1934-1942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo JT, Hayashi J, Seeger C. West Nile virus inhibits the signal transduction pathway of alpha interferon. J Virol. 2005;79:1343–1350. doi: 10.1128/JVI.79.3.1343-1350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson JR, de Sessions PF, Leon MA, Scholle F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J Virol. 2008;82:8262–8271. doi: 10.1128/JVI.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu WJ, Chen HB, Wang XJ, Huang H, Khromykh AA. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J Virol. 2004;78:12225–12235. doi: 10.1128/JVI.78.22.12225-12235.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evans JD, Crown RA, Sohn JA, Seeger C. West Nile virus infection induces depletion of IFNAR1 protein levels. Viral Immunol. 2011;24:253–263. doi: 10.1089/vim.2010.0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laurent-Rolle M, Boer EF, Lubick KJ, Wolfinbarger JB, Carmody AB, Rockx B, Liu W, Ashour J, Shupert WL, Holbrook MR, et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol. 2010;84:3503–3515. doi: 10.1128/JVI.01161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackenzie JM, Khromykh AA, Parton RG. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe. 2007;2:229–239. doi: 10.1016/j.chom.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Hoenen A, Liu W, Kochs G, Khromykh AA, Mackenzie JM. West Nile virus-induced cytoplasmic membrane structures provide partial protection against the interferon-induced antiviral MxA protein. J Gen Virol. 2007;88:3013–3017. doi: 10.1099/vir.0.83125-0. [DOI] [PubMed] [Google Scholar]
- 53.Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J Virol. 2006;80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inojosa WO, Scotton PG, Fuser R, Giobbia M, Paolin A, Maresca MC, Brunello A, Nascimben E, Sorbara C, Rigoli R, et al. West Nile virus transmission through organ transplantation in north-eastern Italy: a case report and implications for pre-procurement screening. Infection. 2012;40:557–562. doi: 10.1007/s15010-012-0263-4. [DOI] [PubMed] [Google Scholar]
- 55.Morelli MC, Sambri V, Grazi GL, Gaibani P, Pierro A, Cescon M, Ercolani G, Cavrini F, Rossini G, Capobianchi MR, et al. Absence of neuroinvasive disease in a liver transplant recipient who acquired West Nile virus (WNV) infection from the organ donor and who received WNV antibodies prophylactically. Clin Infect Dis. 2010;51:e34–e37. doi: 10.1086/655146. [DOI] [PubMed] [Google Scholar]
- 56.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 57.Garcia-Tapia D, Hassett DE, Mitchell WJ, Johnson GC, Kleiboeker SB. West Nile virus encephalitis: sequential histopathological and immunological events in a murine model of infection. J Neurovirol. 2007;13:130–138. doi: 10.1080/13550280601187185. [DOI] [PubMed] [Google Scholar]
- 58.Diamond MS, Shrestha B, Mehlhop E, Sitati E, Engle M. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 2003;16:259–278. doi: 10.1089/088282403322396082. [DOI] [PubMed] [Google Scholar]
- 59.Ye C, Abraham S, Wu H, Shankar P, Manjunath N. Silencing early viral replication in macrophages and dendritic cells effectively suppresses flavivirus encephalitis. PLoS One. 2011;6:e17889. doi: 10.1371/journal.pone.0017889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samuel MA, Diamond MS. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol. 2006;80:9349–9360. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arjona A, Foellmer HG, Town T, Leng L, McDonald C, Wang T, Wong SJ, Montgomery RR, Fikrig E, Bucala R. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. 2007;117:3059–3066. doi: 10.1172/JCI32218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, Murphy PM. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol. 2011;186:471–478. doi: 10.4049/jimmunol.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ben-Nathan D, Huitinga I, Lustig S, van Rooijen N, Kobiler D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch Virol. 1996;141:459–469. doi: 10.1007/BF01718310. [DOI] [PubMed] [Google Scholar]
- 65.Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bai F, Kong KF, Dai J, Qian F, Zhang L, Brown CR, Fikrig E, Montgomery RR. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J Infect Dis. 2010;202:1804–1812. doi: 10.1086/657416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, et al. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J Virol. 2007;81:10933–10949. doi: 10.1128/JVI.02422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verma S, Kumar M, Gurjav U, Lum S, Nerurkar VR. Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology. 2010;397:130–138. doi: 10.1016/j.virol.2009.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar M, Verma S, Nerurkar VR. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J Neuroinflammation. 2010;7:73. doi: 10.1186/1742-2094-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J Virol. 2008;82:8956–8964. doi: 10.1128/JVI.01118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]