

Key Points
PARP-1 controls TGF-β receptors on T cells.
PARP-1 regulates Treg generation.
Abstract
Transforming growth factor-β (TGF-β) receptors (TβRs) are essential components for TGF-β signal transduction in T cells, yet the mechanisms by which the receptors are regulated remain poorly understood. We show here that Poly(ADP-ribose) polymerase-1 (PARP-1) regulates TGF-β receptor I (TβRI) and II (TβRII) expression in CD4+ T cells and subsequently affects Smad2/3-mediated TGF-β signal transduction. Inhibition of PARP-1 led to the upregulation of both TβRI and TβRII, yet the underlying molecular mechanisms were distinct. PARP-1 selectively bound to the promoter of TβRII, whereas the enzymatic activity of PARP-1 was responsible for the inhibition of TβRI expression. Importantly, inhibition of PARP-1 also enhanced expression of TβRs in human CD4+ T cells. Thus, PARP-1 regulates TβR expression and TGF-β signaling in T cells.
Introduction
Transforming growth factor-β (TGF-β) receptor I (TβRI) and II (TβRII) are essential components of TGF-β signaling1and play an indispensable role in generation of regulatory T cells (Tregs). In mice, selective deletion of TβRI2or TβRII3,4 in T cells results in a severe defect in Treg generation. However, the underlying mechanisms are poorly understood. The expression of TβRs in T cells determines TGF-β signal strength, which has profound effects on T-cell responses and differentiation.5,6 Thus, insights into the mechanisms that regulate TβR expression are not only essential for understanding Treg generation, but also important for treatment of autoimmune diseases, transplant rejection, cancer, and infection.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme that is conventionally linked to DNA repair.7-9 However, PARP-1 has also been shown to function as a transcription factor involved in the transcription of many genes.10,11 Inhibition of PARP-1 activity by inhibitors or gene mutation has been shown to lead to both suppression12-15 and exacerbation16 of chronic inflammation and autoimmune disease models. Recently, it was shown that deletion of PARP-1 inhibited nuclear factor–κB (NF-κB) activation and decreased tumor necrosis factor-α (TNF-α) and inducible nitric oxide synthesis in macrophages.14,17 However, the role of PARP-1 in T-cell–mediated immune responses remains elusive.
Here, we show that PARP-1 regulates the expression of TβRs and thereby controls Treg generation in T cells. Deletion of PARP-1 in mice (PARP-1−/−) results in a T-cell–intrinsic preference to generate more thymic Tregs and convert more naive T cells into induced Tregs in vitro and in vivo. Treg increase was attributed to enhanced sensitivity of CD4+ T cells to TGF-β signals by upregulation of both TβRI and II, and subsequent Smad2/3 activation in PARP-1−/− T cells. We show that PARP-1 inhibits TβRI expression through its enzymatic function, and modulates TβRII by directly binding to TβRII gene (Tgfbr2). In addition, PARP-1 deficiency enriched the binding of Smad3 at the enhancer of the forkhead box p3 (Foxp3) gene. Importantly, inhibition of PARP-1 enzyme activity resulted in increased FOXP3 and TGFBR genes expression in human CD4+ T cells. Together, these data reveal an unrecognized role for PARP-1 in the regulation of TβR expression.
Materials and methods
Mice
Generation of PARP-1−/− (sv/129 × C57BL/6 background) mice was previously described.9 PARP-1−/− mice on a C57BL/6 background were obtained by backcrossing with C57BL/6 mice for at least 6 generations and used in the experiments unless otherwise stated. Rag-1−/− and C57BL/6 (CD45.2+ or CD45.1+) mice were from The Jackson Laboratory. Mice were used per National Institutes of Health (NIH) guidelines for use and care of live animals and approved by the Animal Care and Use Committee of the National Institute of Dental and Craniofacial Research (NIDCR).
Antibodies and reagents
Mouse anti-CD3 (clone 145-2C11), anti-CD28 (clone 37.51), anti-CD16/CD32 (clone 93), phycoerythrin (PE)– or allophycocyanin-conjugated anti-CD25 (clone PC61.5), fluorescein isothiocyanate (FITC)– or peridinin chlorophyll protein complex (PerCP)–conjugated anti-CD4 (clone GK1.5), FITC- or PerCP-conjugated anti-CD8 (clone 53-6.7) monoclonal antibodies (mAbs) were from BD Biosciences. Allophycocyanin-conjugated anti-TβRI and PE- or allophycocyanin-conjugated anti-TβRII and anti–TGF-β1, 2, 3 mAbs were from R&D Systems. Anti–PARP-1 (B-10) mAb was from Santa Cruz Biotechnology. Anti-Smad3 (ab28379) and rabbit control immunoglobulin G (IgG) chromatin immunoprecipitation (ChIP) grade antibodies were from Abcam. Phospho-Smad2 (S465/467), Smad2 (L16D3) antibodies were from Cell Signaling Technology. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was from Imgenex. The mouse and human CD4+CD25+ T isolation kit were from Miltenyi Biotec. Allophycocyanin- or PE-conjugated anti-Foxp3 (clone FJK-16s) and rat IgG2a isotype control, IL-6 enzyme-linked immunosorbent assay kits were from eBioscience. TβRI kinase inhibitor II was from Calbiochem.
Cell isolation, cell-culture experiments, mixed bone marrow chimeras, flow cytometry analysis, ChIP assay, luciferase assay, and house dust mites–induced asthma, real-time polymerase chain reaction (PCR), oral tolerance, immunoblot analysis, and isolation of subsets of human CD4+ T cells and cell culture are described in supplemental Methods (available on the Blood website).
Statistical analysis
Statistical significance of differences was determined by the unpaired 2-tailed Student t test unless otherwise stated.
Results
Deletion of PARP-1 results in enhanced sensitivity to TGF-β1 in CD4+ T cells
To study the role of PARP-1 in T cells in response to TGF-β signaling, we first investigated Treg generation in PARP-1−/− mice, as TGF-β signaling is crucial in Foxp3+ Treg generation.18,19 We observed that PARP-1−/− mice had significantly higher frequencies of CD4+Foxp3+ Tregs in the spleen, thymus, and peripheral lymph nodes, compared with wild-type (PARP-1+/+) littermates (data not shown). PARP-1−/− Tregs exhibited similar levels of apoptosis (supplemental Figure 1A-B), cell proliferation (supplemental Figure 1C), and activation markers CD44, CD45RB, CD62L, and CD69 compared with PARP-1+/+ Tregs (data not shown). These results suggest a role for PARP-1 in controlling Treg generation, which is consistent with a recent report.20 However, whether the increase in Tregs in PARP-1−/− mice was due to a T-cell–intrinsic mechanism was undetermined in the recent report. Therefore, we generated bone marrow chimeras to address this question. We injected a mixture of C57BL/6 (CD45.1+) and PARP-1−/− (CD45.2+) bone marrow or a mixture of C57BL/6 (CD45.1+) and PARP-1+/+ (CD45.2+) bone marrow at 1:1 ratio, into sublethally irradiated recipient Rag1−/− mice. Both spleen and thymus reconstituted with PARP-1−/− (CD45.2+) bone marrow contained a higher percentage of CD4+Foxp3+ Tregs than did those transplanted with PARP-1+/+ (CD45.2+) bone marrow (Figure 1A-C). There was no significant increase of total Treg numbers in PARP-1−/− bone marrow chimers since the number of total splenocytes in PARP-1−/− bone marrow chimers was lower (data not shown). These data confirm that PARP-1 deficiency confers T cells with an increased capacity to differentiate toward Treg fate in vivo.
Figure 1.
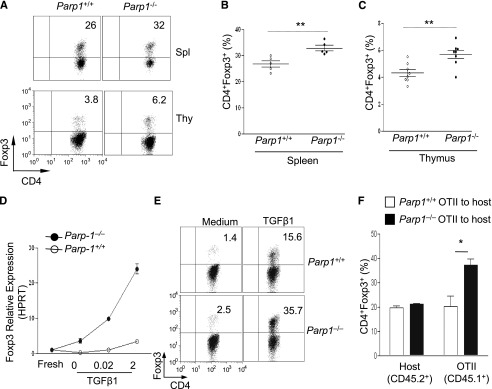
Mice deficient in PARP-1 gene expression have more Treg cells. (A-C) Analysis of PARP-1−/− (CD45.2+) and PARP-1+/+ (CD45.2+) CD4+ T cells in mixed bone marrow chimeras; representative staining of CD4 vs Foxp3 in CD4+ cells in the spleen and thymus (A). Frequency of CD4+Foxp3+ Tregs in spleen (B) and thymus (C) (mean ± SEM, PARP-1+/+ chimeras, n = 5; PARP-1−/− chimeras, n = 5). (D) Quantitative PCR analysis of the expression of Foxp3 (mean ± SEM) in freshly isolated (fresh) and CD4+CD25− T cells stimulated with anti-CD3– (5µg/ml) and CD28– (2 µg/mL) specific antibodies in the absence and presence of indicated concentrations of TGF-β1 for 24 hours. (E) CD4+CD25− T cells were cultured in the presence of anti-CD3 antibody (0.5 μg/mL) and wild-type APCs with (TGF-β1) or without TGF-β1 (Med) for 2 days. FACS plots show representative staining of CD4 vs Foxp3. (F) Treg induction in PARP-1−/− T cells in vivo. CD4+CD25– T cells were sorted from the spleens of CD45.1+ OTII transgenic PARP-1−/− or PARP-1+/+ littermate control mice and 1 × 106 sorted cells were transferred into CD45.2+ recipient mice. Recipient mice were received OVA protein in the drinking water for 5 days. On day 6, the small intestine LP lymphocytes (LPLs) were isolated and stained with Foxp3 to assess Treg induction in vivo by flow cytometry analysis. Bar graph shows the frequency of CD45.2+ (Host) and CD45.1+ (OT-II) CD4+Foxp3+ T cells in the LPLs of recipient mice. Data are representative of 2 (A-D,F) or 3 (E) independent experiments. *P < .05; **P < .01 (unpaired 2-tailed Student t test). FACS, fluorescence-activated cell sorter; LPL, LP lymphocyte.
Furthermore, we determined whether PARP-1 deficiency affected Treg generation in culture. We induced Foxp3 expression in naive CD4+CD25– T cells by culturing cells with anti-CD3 antibody, antigen-presenting cells (APCs), and TGF-β1.18 There were higher amounts of Foxp3 messenger RNA (mRNA) (Figure 1D; supplemental Figure 2A) and protein (Figure 1E; supplemental Figure 2B-C) in PARP-1−/− CD4+ T cells compared with PARP-1+/+CD4+ T cells. PARP-1−/−CD4+ T cells also expressed higher levels of Foxp3 mRNA and protein in response to lower concentrations of TGF-β1, suggesting that PARP-1−/− T cells are more sensitive to TGF-β1 stimulation (Figure 1D; supplemental Figure 2B). In addition, PARP-1−/− T cells converted to Foxp3-expressing cells much faster than PARP-1+/+ T cells in response to 2 ng/mL TGF-β1 (supplemental Figure 2C). A similar increase in Tregs was seen in PARP-1−/− T cells when they were cultured with anti-CD3 and CD28 antibodies and TGF-β1 (data not shown).
Enhanced Treg generation in PARP-1−/− T cells in vitro prompted us to explore whether this phenomenon would also occur in vivo. We addressed this in an oral tolerance model.21-23 We transferred PARP-1−/− or PARP-1+/+ CD4+CD25– T cells from OTII T-cell receptor (TCR) transgenic mice (CD45.1+) into C57BL/6 (CD45.2+) mice and fed the mice with chicken ovalbumin (OVA) protein (1.5%) in drinking water for 5 constitutive days. The mice were sacrificed to assess Treg induction in the lamina propria (LP). We analyzed the Foxp3 expression in the CD4+CD45.1+ OTII TCR transgenic T cells and observed that PARP-1−/− CD4+ OTII T cells showed significantly higher frequencies of Foxp3+ Tregs than PARP-1+/+ OTII T cells (Figure 1F). As expected, there was no significant difference in Tregs in the spleens and peripheral lymph nodes in CD45.2+ host CD4+ T cells. Collectively, these data indicate that PARP-1 expression in CD4+ T cells affects Treg generation.
Higher expression of TβRs in PARP-1−/− CD4+CD25– T cells
As PARP-1−/− T cells showed an increased sensitivity to TGF-β1, we hypothesized that PARP-1 could affect TGF-β signaling by altering TβR expression. To test this hypothesis, we first examined the mRNA expression of Tgfbr1 and Tgfbr2 in CD4+ T cells. Freshly isolated PARP-1−/− CD4+CD25– T cells expressed higher levels of Tgfbr1 and Tgfbr2 mRNA compared with PARP-1+/+ CD4+CD25– T cells (Figure 2A; supplemental Figure 3). TCR stimulation of PARP-1−/− CD4+CD25– T cells with anti-CD3 and CD28 antibodies for 15 to 90 minutes also resulted in higher Tgfbr1 and Tgfbr2 mRNA expression compared with PARP-1+/+ CD4+CD25– T cells (Figure 2B). Stimulation of PARP-1−/− CD4+CD25– T cells with TGF-β1 (2 ng/mL) plus anti-CD3 (0.5 μg/mL) and CD28 (0.2 μg/mL) antibodies also led to higher levels of Tgfbr1 and Tgfbr2 mRNA compared with PARP-1+/+ CD4+CD25– T cells (Figure 2C). Intriguingly, freshly isolated PARP-1+/+ CD4+CD8–CD25– thymocytes showed much higher expression (twofold to threefold) of Tgfbr1 and Tgfbr2 mRNA than splenic CD4+CD25– T cells from the same mice (Figure 2D). Again, deletion of PARP-1 led to slightly, but reproducibly, higher expression levels of Tgfbr1 and Tgfbr2 mRNA on PARP-1−/− CD4+CD8–CD25– thymocytes compared with PARP-1+/+ cells (Figure 2D). In contrast to CD4+CD25– T cells, freshly isolated PARP-1−/− Tregs from the spleen (Figure 2E) and thymus showed similar levels of Tgfbr1 and Tgfbr2 mRNA to Tregs from PARP-1+/+ mice. We also found that PARP-1 deficiency upregulated the surface protein expression of TβR I and II on splenic CD4+CD25+ T cells, as well as CD4+CD25− T cells (Figure 2F-H). Together, these data demonstrate that a deficiency in PARP-1 led to increased expression of TβRI and TβRII in CD4+ T cells, suggesting that PARP-1 may have an inhibitory role in the control of TβR expression.
Figure 2.

Increased expression of TβRI and TβRII in CD4+CD25– T cells of PARP-1−/− mice. (A-C) Quantitative PCR analysis of the expression of Tgfbr1 and Tgfbr2 mRNA in (A) freshly isolated splenic CD4+CD25- T cells from PARP-1−/− mice and PARP-1+/+ littermate controls, in (B) CD4+CD25– T cells were stimulated for 15 minutes with CD3 (5 µg/mL) and CD28 antibodies (2 µg/mL) or in (C) CD4+CD25− T cells were stimulated for 15 minutes with CD3 (0.5 µg/mL), and CD28 (0.2 µg/mL) specific antibodies in the presence of TGF-β1 (2 ng/mL). (D-E) Quantitative PCR analysis of the expression of Tgfbr1 and Tgfbr2 mRNA in CD4+CD25– thymocytes and splenocytes (D) and in freshly isolated CD4+CD25+ Tregs (E). Data are representative of at least 3 independent experiments (A-E) (mean ± SEM of the duplicate measurements). (F-H) Flow cytometry analysis of the expression of TβRI (G) and TβRII (H) in splenocytes which were from both PARP-1−/− and PARP-1+/+ mice. (F) A representative dot plot showing cells analyzed for CD4 and TβRI or TβRII expressions in CD4+CD25− T cells. (G) TβRI expression in CD4+CD25+ (left 2 bars) and CD4+CD25− (right 2 bars) cells. (H)TβRII expression in CD4+CD25+ (left 2 bars) and CD4+CD25− (right 2 bars) cells. Graphs show mean ± SD. ***P < .002. (I) ChIP-coupled quantitative PCR analysis of PARP-1 enrichment around 500 bp up/downstream (primers no. 3) from TSS of Tgfbr1 and Tgfbr2 genes (mean ± SEM of duplicate wells). Data are representative of 2 independent experiments. (J) Quantitative PCR analysis of the expression of Tgfbr1 and Tgfbr2 mRNA in 5-AIQ–treated CD4+CD25– T cells. Data are representative of 4 independent experiments (mean ± SEM) *P < .05 (unpaired 2-tailed Student t test).
PARP-1 binds to Tgfbr2 but not to Tgfbr1 gene
We next determined how PARP-1 regulated TβR expression. We first determined whether PARP-1 could directly bind to Tgfbr1 and Tgfbr2 genes and thereby regulate their transcription in CD4+ T cells. Using freshly isolated CD4+CD25– T cells from the spleen of wild-type mice, we assessed the binding of PARP-1 to Tgfbr1 and Tgfbr2 genes by ChIP-coupled quantitative PCR. We designed a series of primers which covered −2 kb to +1 kb from the transcriptional starting site (TSS) of Tgfbr1 and Tgfbr2 genes (supplemental Figure 4A). We observed binding of PARP-1 to the regions close to the TSS and +1 kb from the TSS at the Tgfbr2 gene (Figure 2I; supplemental Figure 4B). However, we did not detect enrichment of PARP-1 binding to the same regions of the Tgfbr1 gene. The data suggest that PARP-1 differentially regulates Tgfbr1 and Tgfbr2 gene expression, directly interacting with the promoter of the former, but not the latter.
To further confirm the hypothesis that PARP-1 proteins control TβR gene expression, we overexpressed PARP-1 protein and cotransfected it with pGL4-based constructs which contained a fragment of murine TβR promoter to the EL4/LAF cell lines. We found that overexpression of PARP-1 protein reduced the activity of both TβRI and II in the presence of TGF-β1 (supplemental Figure 5).These data together further support our hypothesis that PARP-1 negatively regulates TβR signaling.
Inhibition of PARP-1 enzymatic activity increases Tgfbr1 but not Tgfbr2 expression
We next investigated the mechanisms by which PARP-1 regulated TβRI expression. Because PARP-1 possesses enzymatic activity in addition to its transcriptional function, we determined whether PARP-1 could influence TβRI expression through its catalytic function. We pretreated wild-type (C57BL/6) CD4+CD25– T cells with PARP-1–specific inhibitor 5-aminoisoquinoline (5-AIQ),24 followed by stimulation of these T cells with anti-CD3 and CD28 antibodies for 30 minutes. 5-AIQ treatment significantly upregulated Tgfbr1 expression in wild-type CD4+CD25– T cells (Figure 2J). Surprisingly, 5-AIQ–treated CD4+CD25– T cells showed no significant changes in Tgfbr2 expression compared with untreated CD4+CD25– T cells (Figure 2J). These data reveal that PARP-1 controls TβRI expression through its catalytic activity.
PARP-1 deletion leads to increased Smad2/3 phosphorylation
As PARP-1−/− CD4+ T cells showed increased TβR expression, and therefore an increased sensitivity to TGF-β signaling, we next determined the effect of PARP-1 deficiency on the phosphorylation and function of Smad2/3 (Smad2 and/or Smad3). Smad2/3 are immediate downstream mediators of TβRs and key factors in mediating TGF-β–induced Foxp3 gene transcription.2,25-27 We used an anti-phosphorylated Smad2 (P-Smad2) as an indicator for Smad2/3 phosphorylation,2 and observed that even freshly isolated PARP-1−/− CD4+CD25– T cells showed substantially higher amounts of P-Smad2 than PARP-1+/+ CD4+CD25– T cells (Figure 3A-B). This corresponded to the higher levels of TβRs in PARP-1−/− T cells (Figure 2A). Stimulation of PARP-1−/− CD4+CD25– T cells with anti-CD3 and CD28 antibodies for 20 to 30 minutes substantially enhanced P-Smad2 compared with that of PARP-1+/+CD4+CD25– T cells (Figure 3A-B). As expected, addition of exogenous TGF-β1 increased P-Smad2 in both PARP-1−/− and PARP-1+/+ CD4+ T cells compared with stimulation with TCR alone, but the level of P-Smad2 in PARP-1−/− T cells was still higher than that in PARP-1+/+ T cells (Figure 3A-B). Total Smad2 levels were similar between the PARP-1−/− and PARP-1+/+ CD4+ T cells irrespective of treatment (Figure 3A-B). These data suggest PARP-1 regulates Smad2/3 phosphorylation, which is likely due to increased expression of TβR.
Figure 3.

PARP-1 regulates Smad3 phosphorylation and Smad3 binding at the Foxp3 enhancer. (A) Western blot of P-Smad2, Smad2 and GAPDH expressions in freshly isolated CD4+CD25− T cells and CD4+CD25− T cells stimulated for 15 minutes with CD3 (5 µg/mL) plus CD28 (2 µg/mL) antibodies in the absence or presence of TGF-β1 (2 ng/mL). Data are representative of 3 independent experiments. (B) Summarization of 3× P-Smad2 western blot analysis. Western blot bands were analyzed by Odyssey software, the PARP-1+/+ P-Smad2 expression was set as 1, PARP-1−/− P-Smad2 expression was presented relative to PARP-1+/+. *P < .05, unpaired 1-tailed Student t test. (C) ChIP-coupled quantitative PCR analysis of Smad3 enrichment in the enhancer region of Foxp3 gene assessed using an antibody to Smad3 and presented relative to input and compared with a control IgG. Data are representative of 4 independent experiments (mean ± SEM); ***P < .001 (unpaired 2-tailed Student t test).
PARP-1 regulates Smad3 binding to the Foxp3 transcriptional enhancer
Smad3 has been shown to preferentially bind to the enhancer region of Foxp3 gene,27 and plays a critical role in the initiation and mediation of the cascade of transcription factors which drive Foxp3 gene activation.25,27 We next investigated whether PARP-1 affected Smad3 binding to the enhancer region of the Foxp3 gene.27 It has been reported that PARP-1 is a Smad-interacting partner and can attenuate Smad-mediated transcription by blocking the binding of Smad3 to its target gene and/or dissolving the bound Smad3 from its target gene in tumor cells.28 We hypothesized that PARP-1 might also interfere with Smad3 binding to the Foxp3 gene enhancer in naive CD4+ T cells and that a deficiency in PARP-1 protein would increase Smad3 binding at the enhancer of the Foxp3 gene and induce more Foxp3 transcription. We cultured PARP-1−/− and PARP-1+/+ CD4+CD25– T cells with anti-CD3 and CD28 antibodies plus TGF-β1 for 30 minutes (an optimal time for Smad3 binding at the Foxp3 enhancer)25 and examined the binding of Smad3 to the enhancer region (+2085 to +2231) of Foxp3 gene by ChIP-coupled quantitative PCR. PARP-1 deficiency significantly enhanced the Smad3 binding to the enhancer of Foxp3 gene in naive CD4+CD25– T cells in response to TCR and TGF-β1 treatment (Figure 3C).
More interleukin-17 (IL-17)–producing T helper (Th17) cells in PARP-1−/− T cells in response to TGF-β1 and IL-6 in vitro
Because TGF-β1 is also required for Th17 cell differentiation in the presence of proinflammatory cytokine IL-6,6,29-31 we next examined whether deletion of PARP-1 affected Th17 cell differentiation. We found that treatment with TGF-β1 and IL-6 in the presence of anti-CD3 antibody plus wild-type APCs resulted in significantly more IL-17 production from PARP-1−/− than PARP-1+/+ CD4+CD25– T cells (Figure 4A-B; supplemental Figure 6B). Stimulation with TGF-β1 plus IL-6 in the presence of immobilized anti-CD3 and CD28 antibodies increased IL-17 producing as well, although the overall frequency was lower than that in the cultures with APCs (data not shown). Consistent with the enhanced sensitivity to TGF-β1 treatment in Treg generation, PARP-1−/− CD4+CD25– T cells also showed significantly higher Th17 cell polarization over a wide range of TGF-β1 concentrations (supplemental Figure 6A). In line with IL-17 production, treatment with TGF-β1 plus IL-6 also induced higher levels of Rorc expression and RORγt protein in PARP-1−/− T cells (Figure 4C-D). Together, our data indicate that in the absence of PARP-1, CD4+ T cells have an increased ability to differentiate into the Th17 fate in vitro, although this is unlikely to occur in naive PARP-1−/− mice in the steady state because naive PARP-1−/− mice are unlikely to show increased IL-6 production in the absence of exogenous inflammatory stimuli. Indeed, naive PARP-1−/− splenocytes and purified CD4+CD25– T cells secreted substantially lower amounts of IL-6 in response to TCR stimulation in vitro compared with PARP-1+/+ control mice (supplemental Figure 6C).
Figure 4.

Greater differentiation of Th17 in PARP-1−/− T cells in vitro. (A) Scatterplot (left) of staining of IL-17 vs IFNγ on sorted CD4+ T cells from PARP-1−/− or PARP-1+/+ littermate controls stimulated with anti-CD3 (0.5 µg/mL) antibody and PARP-1+/+ APCs in the absence (Med) or presence of TGF-β1 plus IL6 (TGFβ1+IL6) for 4 days. Data are representative of 2 independent experiments. The histogram (right) is summarized analysis of 2 experiments. *P < .05 (unpaired 1-tailed Student t test). (B) IL-17 levels in supernatants from cultures described in panel A (mean ± SEM of duplicate measurements). (C) Quantitative PCR analysis of Rorc expression in CD4+CD25− T cells from PARP-1−/− or PARP-1+/+ littermate controls stimulated with an anti-CD3 (0.5 µg/mL) antibody and PARP-1+/+ APCs in the absence (Med) or presence of TGF-β1 plus IL6 (TGFβ-1+IL6) for 2 days (mean ± SEM of duplicate measurements). (D) Staining of RORγt in CD4+T cells from PARP-1−/− or age-matched PARP-1+/+ control littermates which were stimulated with anti-CD3 (0.5 µg/mL) antibody and PARP-1+/+ APCs in the absence (Med) or presence of TGF-β1 and IL6 (TGFβ1+IL6) for 4 days. *P < .05; **P < .01 (unpaired 2-tailed Student t test).
Increased expression of Foxp3 and TβRI in human CD4+ T cells following PARP-1 inhibition
Finally, to determine whether the pathways we identified in mouse are present in human, we assessed whether reduction of PARP-1 activity in human CD4+ T cells affected Foxp3 expression, as TGF-β1 signaling is also required for Foxp3 induction in human CD4+CD25– T cells.32,33 We pretreated CD4+CD25– human T cells with the PARP-1 inhibitor 5-AIQ and then cultured with anti-CD3and CD28 antibodies overnight, which was documented to induce FOXP3 transcription by an endogenous TGF-β1–dependent mechanism.32 Inhibition of PARP-1 activity with 5-AIQ significantly enhanced FOXP3 expression in human CD4+CD25– T cells (Figure 5A), suggesting PARP-1 controls the sensitivity of human T cells to TGF-β signaling.
Figure 5.

Inhibition of PARP-1 in human CD4+CD25– T cells increases the expression of FOXP3 and TGFBR1. CD4+CD25– T cells were isolated from human peripheral blood and preincubated with 5-AIQ for 20 minutes, then cells were stimulated with human CD3- and CD28-specific antibodies for the indicated time. Quantitative PCR analysis showed the expression of FOXP3, TGFBR1, and TGFBR2. (A) FOXP3 expression was determined after 12 hours of culture. Data are mean ± SEM of 3 independent experiments. (B) TGFBR1 and TGFBR2 mRNA expression was determined after 90 minutes of culture. Data are mean ± SEM of 3 independent experiments. *P < .05 (unpaired 2-tailed Student t test).
We next investigated whether inhibition of PARP-1 activity affected the expression of TβRs. We cultured human CD4+CD25– T cells with anti-CD3 and CD28 antibodies in the absence and presence of 5-AIQ for 90 minutes. Analysis of TβR expression revealed that the PARP-1 inhibitor significantly increased TGFBR1 gene expression in 5-AIQ–treated resting CD4+CD25– T cells compared with untreated controls (Figure 5B), which confirmed our results from mice (Figure 2J). In contrast to Tgfbr2 gene expression in mouse T cells, inhibition of PARP-1 with 5-AIQ in TCR-stimulated human CD4+CD25– T cells resulted in higher levels of TGFBR2 gene expression compared with control T cells receiving TCR stimulation alone (Figure 5B). Nevertheless, the data indicate that PARP-1 also controls the expression of TβRs in human T cells, and therefore could also potentially influence Foxp3 expression in human T cells.
Discussion
ΤβRs are essential components in transducing TGF-β signal that is required for Treg differentiation,2,3,34 yet the molecular mechanisms is poorly understood. In this article, we have shown that PARP-1 can control expression of TβRs and therefore affect Smad2/3-mediated signaling in CD4+ T cells. We show that PARP-1 regulates TβRI expression by its catalytic activity, yet regulates TβRII expression by its transcriptional function. We also demonstrate that there is increased binding of Smad3 to the Foxp3 enhancer in PARP-1−/− CD4+ T cells; a critical step in initiating Foxp3 gene transcription in response to TGF-β1.5 Our data also show that PARP-1 regulates the expression of TβRs in human CD4+ T cells.
Several conclusions can be drawn from the present study. First, PARP-1 controls TGF-β signaling by restraining TβR expression in CD4+ T cells on mRNA level. We also observed an increased TβR staining on the cell surface in the absence of PARP-1 by flow cytometry. However, these data need to be confirmed by more experiments because the reliability of antibodies to detect cell-surface ΤβRs is still controversial. The upregulation of TβRI and TβRII was evident in fresh and stimulated PARP1−/− CD4+CD25– T cells, which is in line with the enhanced sensitivity of these knockout T cells to TGF-β1. Likewise, PARP-1−/− CD4+CD8–CD25– thymocytes also showed increased expression of TβRs compared with those from PARP-1+/+ mice. Intriguingly, CD4+CD25+Foxp3+ Tregs from PARP-1−/− mice exhibited no increase in TβRI or TβRII expression compared with PARP-1+/+ Tregs. The reason why PARP-1 is able to control TβR expression in naive CD4+CD25– T cells but not in CD4+CD25+Foxp3+ Tregs remain unknown. However, it could be because CD4+CD25+Foxp3+ Tregs already express optimal levels of TβRs. This is in line with data showing that TGF-β1 is unable to further upregulate Foxp3 expression in isolated CD4+CD25+Foxp3+ Tregs.18 Consistent with the increase in TβR expression, PARP-1−/− T cells show increased levels of activated Smad2/3. Short TCR stimulation alone (15-90 minutes) without TGF-β1 substantially increased phosphorylated Samd2/3 (P-Smad2/3) in PARP-1−/− T cells.
Our surprising findings are that PARP-1 regulates TβRI and TβRII through different molecular mechanisms and reveals an unrecognized regulation pathway for TβRs. We show that PARP-1 functions as both a transcription factor and an enzyme in controlling TβR expression. These data are in line with the reported functions of PARP-1.35 Intriguingly, PARP-1 binds directly to the Tgfbr2 gene, but not to the Tgfbr1 gene. This indicates that PARP-1 could directly limit Tgfbr2 gene activation through transcriptional regulation which is consistent with a previous report of PARP-1 regulation of TβRII in cancer cells.36 The lack of binding of PARP-1 at the Tgfbr1 gene prompted us to investigate a possible role of the enzymatic activity of PARP-1 in regulating TβRI expression. Indeed, blockade of PARP-1 enzymatic activity with inhibitor 5-AIQ upregulated TβRI expression in CD4+CD25– T cells, but had less impact on TβRII expression. Although it remains to be determined how PARP-1 executes this dual function, it is not without precedence that an enzyme can also be transcription factor; it was recently reported that indoleamine 2,3-dioxygenase also functions as a signal protein in plasmacytoid dendritic cells.37
We have shown that Smad2/3 proteins are constitutively phosphorylated. In addition, the deficiency of PARP-1 leads to the enhanced binding of Smad3 at the enhancer of the Foxp3 gene.5,25,27 The enhanced binding of Smad3 at the enhancer may be partially responsible for the increased Foxp3 expression in PARP-1−/− T cells. Enhanced Treg generation in PARP-1−/−T cells was also confirmed in a model of oral tolerance in mice.21,22
The increased differentiation of Th17 cells in PARP-1−/− T cells in vitro further validates the role for PARP-1 in controlling sensitivity to TGF-β signals. However, this upregulation of Th17 cells is unlikely to occur in the steady state in PARP-1−/− mice as PARP-1−/− mice showed decreased levels of IL-6 compared with PARP-1+/+ mice. However, increased Th17 differentiation could occur where mice encounter infections and/or inflammatory stimuli that trigger IL-6 production from APCs. Indeed, PARP-1 deficiency in mice showed no protective effect upon immunization to induce experimental autoimmune encephalomyelitis despite PARP-1−/− mice having elevated Tregs, as PARP-1−/− mice showed elevated levels of Th17 cells (data not shown). We would like to explore the function of PARP-1 in Th17 differentiation in the future and this might help to resolve a controversial issue of whether responsiveness to TGF-β makes more or less pathogenic Th17 cells.38
We also show that PARP-1 affects expression in human T cells and thereby regulates FOXP3 gene expression. Our data show that inhibition of PARP-1 activity in human T cells substantially enhanced FOXP3 induction and this was likely attributable to enhanced TGF-β1 signaling. Indeed, treatment of human CD4+CD25– resting T cells with 5-AIQ significantly enhanced TGFBR1 expression, consistent with mouse CD4+ T cells. Intriguingly, in contrast to mice, inhibition of PARP-1 with 5-AIQ also caused upregulation of TGFBR2 expression. It should be noted that the effect results from the prevention of TCR-mediated downregulation of TGFBR2 mRNA, as TCR stimulation for 90 minutes was shown to cause downregulation of TβRII in human CD4+ T cells.
Our data show that PARP-1 restrains TGF-β signaling in T cells by decreasing TβR expression. Except for responding to DNA damage, PARP-1 has been shown to regulate both chromatin structure and gene transcription.39 Here, we add to these studies and show that in the absence of PARP-1 TGF-β signaling is enhanced. Our data therefore suggest that PARP-1 functions to dampen regulatory signals in T cells. Although considered to be activated following DNA damage, PARP-1 has been shown to be active in resting cells and by both ERK1/2 and Notch signaling. Interestingly, PARP-1 has been shown to be activated during T-cell stimulation.40 As such, it is possible that PARP-1 plays a role in directly promoting immune responses. This would be achieved at 2 discreet points, first at sites of inflammation where reactive oxidant species are present, and secondly in T cells activated in lymph nodes. Thus, PARP-1 could function to dampen immunoregulatory pathways and promote T-cell activation in the lymph node and further protect T cells from any regulatory mechanisms at the site of inflammation. This fits with previous data showing that PARP-1 inhibition reduces cytokine production14,17 and allergic asthma.41 Further suggesting a role for PARP-1 in promoting immune responses are data that show increased tumor formation in PARP-1−/− mice.39 PARP-1 has also been shown to act as a transcriptional coregulator of NF-κB and is important for activation of this vital mediator of inflammatory response.39
Our data have uncovered an unrecognized role for PARP-1 in regulating Treg differentiation and development through the regulation of TβR expression. This provides a starting point to further understand the network of factors regulating TβRs and their expression. Our data also have implications for exploring the possibility of blocking PARP-1 as a potential therapy in human autoimmune diseases. Collectively, we show a vital role for PARP-1 in restraining TGF-β signaling in T cells.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Dental and Craniofacial Research (National Institutes of Health).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: W.J.C. designed research; P.Z., H.N., E.T., S.K., K.C., T.M., G.W., J.E.K., and B.A. performed research; Z.-Q.W. and K.Z. provided new reagents and analytic tools and crucial input; M.I. provided crucial input on the PARP-1 overexpression assay; and P.Z., H.N., E.T., T.M., J.E.K., and W.J.C. analyzed data and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: WanJun Chen, Mucosal Immunology Section, OPCB, NIDCR, NIH, Building 30, Room 304, Bethesda, MD 20892; e-mail: wchen@dir.nidcr.nih.gov.
References
- 1.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9(6):632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 3.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25(3):455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25(3):441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Maruyama T, Li J, Vaque JP, et al. Control of the differentiation of regulatory T cells and T(H)17 cells by the DNA-binding inhibitor Id3. Nat Immunol. 2011;12(1):86–95. doi: 10.1038/ni.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou L, Lopes JE, Chong MM, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453(7192):236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113(6):677–683. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 8.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286(5446):1897-1905. [DOI] [PubMed]
- 9.Wang ZQ, Auer B, Stingl L, et al. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9(5):509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 10.Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J Biol Chem. 2003;278(46):45145–45153. doi: 10.1074/jbc.M307957200. [DOI] [PubMed] [Google Scholar]
- 11.Abramson J, Giraud M, Benoist C, Mathis D. Aire’s partners in the molecular control of immunological tolerance. Cell. 2010;140(1):123–135. doi: 10.1016/j.cell.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 12.Burkart V, Wang ZQ, Radons J, et al. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat Med. 1999;5(3):314–319. doi: 10.1038/6535. [DOI] [PubMed] [Google Scholar]
- 13.Eliasson MJ, Sampei K, Mandir AS, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3(10):1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 14.Farez MF, Quintana FJ, Gandhi R, Izquierdo G, Lucas M, Weiner HL. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol. 2009;10(9):958–964. doi: 10.1038/ni.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Virág L, Jagtap P, Szabó E, et al. Garcia Soriano F. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7(1):108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 16.Selvaraj V, Soundarapandian MM, Chechneva O, et al. PARP-1 deficiency increases the severity of disease in a mouse model of multiple sclerosis. J Biol Chem. 2009;284(38):26070–26084. doi: 10.1074/jbc.M109.013474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pétrilli V, Herceg Z, Hassa PO, et al. Noncleavable poly(ADP-ribose) polymerase-1 regulates the inflammation response in mice. J Clin Invest. 2004;114(8):1072–1081. doi: 10.1172/JCI21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140(6):845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 20.Nasta F, Laudisi F, Sambucci M, Rosado MM, Pioli C. Increased Foxp3+ regulatory T cells in poly(ADP-ribose) polymerase-1 deficiency. J Immunol. 2010;184(7):3470–3477. doi: 10.4049/jimmunol.0901568. [DOI] [PubMed] [Google Scholar]
- 21.Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204(8):1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun CM, Hall JA, Blank RB, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204(8):1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konkel JE, Chen W. Balancing acts: the role of TGF-β in the mucosal immune system. Trends Mol Med. 2011;17(11):668–676. doi: 10.1016/j.molmed.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald MC, Mota-Filipe H, Wright JA, et al. Effects of 5-aminoisoquinolinone, a water-soluble, potent inhibitor of the activity of poly (ADP-ribose) polymerase on the organ injury and dysfunction caused by haemorrhagic shock. Br J Pharmacol. 2000;130(4):843–850. doi: 10.1038/sj.bjp.0703391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruan Q, Kameswaran V, Tone Y, et al. Development of Foxp3(+) regulatory T cells is driven by the c-Rel enhanceosome. Immunity. 2009;31(6):932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takimoto T, Wakabayashi Y, Sekiya T, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development [published correction appears in J Immunol. 2011;186(1):632]. J Immunol. 2010;185(2):842–855. doi: 10.4049/jimmunol.0904100. [DOI] [PubMed] [Google Scholar]
- 27.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9(2):194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 28.Lönn P, van der Heide LP, Dahl M, Hellman U, Heldin CH, Moustakas A. PARP-1 attenuates Smad-mediated transcription. Mol Cell. 2010;40(4):521–532. doi: 10.1016/j.molcel.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 29.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 30.Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 31.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Amarnath S, Dong L, Li J, Wu Y, Chen W. Endogenous TGF-beta activation by reactive oxygen species is key to Foxp3 induction in TCR-stimulated and HIV-1-infected human CD4+CD25- T cells. Retrovirology. 2007;4:57. doi: 10.1186/1742-4690-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110(8):2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463(7282):808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hakmé A, Wong HK, Dantzer F, Schreiber V. The expanding field of poly(ADP-ribosyl)ation reactions. ‘Protein Modifications: Beyond the Usual Suspects’ Review Series. EMBO Rep. 2008;9(11):1094–1100. doi: 10.1038/embor.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sterling JA, Wu L, Banerji SS. PARP regulates TGF-beta receptor type II expression in estrogen receptor-positive breast cancer cell lines. Anticancer Res. 2006;26(3A):1893–1901. [PubMed] [Google Scholar]
- 37.Pallotta MT, Orabona C, Volpi C, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12(9):870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 38.Ghoreschi K, Laurence A, Yang X-P, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 2010;467(7318):967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valdor R, Schreiber V, Saenz L, et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol Immunol. 2008;45(7):1863–1871. doi: 10.1016/j.molimm.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 41.Oumouna M, Datta R, Oumouna-Benachour K, et al. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5 [published correction appears in J Immunol. 2007;179(8):5604]. J Immunol. 2006;177(9):6489–6496. doi: 10.4049/jimmunol.177.9.6489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.