

Abstract
Many individuals with multiple or large colorectal adenomas, or early-onset colorectal cancer (CRC), have no detectable germline mutations in the known cancer predisposition genes. Using whole-genome sequencing, supplemented by linkage and association analysis, we identified specific heterozygous POLE or POLD1 germline variants in several multiple adenoma and/or CRC cases, but in no controls. The susceptibility variants appear to have high penetrance. POLD1 is also associated with endometrial cancer predisposition. The mutations map to equivalent sites in the proof-reading (exonuclease) domain of DNA polymerases ε and δ, and are predicted to impair correction of mispaired bases inserted during DNA replication. In agreement with this prediction, mutation carriers’ tumours were microsatellite-stable, but tended to acquire base substitution mutations, as confirmed by yeast functional assays. Further analysis of published data showed that the recently-described group of hypermutant, microsatellite-stable CRCs is likely to be caused by somatic POLE exonuclease domain mutations.
Mutations in at least 10 genes are responsible for Mendelian syndromes associated with colorectal cancer. For some of these genes (APC and MUTYH), the primary predisposition is to multiple adenomas, the benign precursors of many colorectal cancers (CRCs). For other genes (STK11/LKB1, SMAD4, BMPR1A and GREM1), CRC risk is mediated through the development of hamartomas or mixed polyps; and for yet other genes (MSH2, MLH1, MSH6 and PMS2), there is usually no great number of polyps and early-onset CRC or endometrial cancer is the usual presentation. The functions of these 10 genes vary, although it is notable that four encode (MSH2, MLH1, MSH6 and PMS2) encode DNA mismatch repair proteins and a fifth (MUTYH) codes for a glycolsylase that effects base excision repair (MUTYH). There exists a set of patients who appear a priori to have a strong chance of carrying a high-penetrance CRC predisposition allele, but who have no mutations in these genes. One such group of patients is characterised by multiple adenomas, typically 10-100 in number, often presenting before the age of 60 and frequently progressing to CRC unless treated. Some of these individuals come from extensive CRC pedigrees, but many have no significant family history of colorectal tumours.
As part of the Oxford-Illumina WGS500 project, we undertook whole-genome sequencing (WGS) of 15 probands who had been diagnosed with at least 10 colorectal adenomas by 60 years of age (Supplementary Table 1). For three probands, a relative with more than 5 adenomas was also sequenced, and for one proband two additional affected relatives were sequenced. Eight of the 20 sequenced individuals had developed colorectal carcinoma (CRC) and all of the remainder had a first-degree relative with CRC. Known Mendelian cancer syndromes had reportedly been excluded previously in the clinical diagnostic setting, by mutation screening for APC, MUTYH and the mismatch repair genes, together with microsatellite instability (MSI) testing and immunohistochemistry for mismatch repair proteins in some cases. The pedigrees of the 15 families showed a degree of heterogeneity in their phenotypes and patterns of inheritance, and we anticipated that this might reflect underlying genetic heterogeneity.
RESULTS
Identification of colorectal tumour susceptibility variants
Following standard quality assurance procedures, whole-genome sequencing reads were mapped and called using the established local pipeline based on the STAMPY 1 and PLATYPUS programs (further details in Methods). We initially excluded two individuals who were found, despite previous testing, to have pathogenic heterozygous germline mutations in genes known to cause high-penetrance predisposition to colorectal tumours (Supplementary Table 1). One of these individuals had attenuated familial adenomatous polyposis (FAP) owing to a splice site germline mutation in the APC gene (c.423 −2 A>G) and the other had Lynch syndrome caused by an MSH6 mutation (R911X).
In the absence of information regarding the underlying genetic architecture of the multiple adenoma phenotype, including the mode of inheritance in our families, we treated analysis of the remaining 13 WGS families as the Discovery Phase of our project. We then used a pragmatic approach to prioritise genes for further investigation in a larger Validation Phase of individuals with a similar phenotype. Based on the assumption that at least some individuals would carry mutations with direct, strong effects on protein function, we filtered the data so as to examine only protein-coding and splice site sequence. We filtered variants further using the algorithm shown in Supplementary Figure 1 and then searched for genes that harboured probably pathogenic variants across 4 or more families. However, no genes matched these criteria.
We therefore switched focus to single families. We initially examined the pedigree (SM2702, Figure 1) from which 3 individuals had been sequenced. We used pre-existing linkage data from 5 SM2702 individuals with ≥5 early-onset colorectal adenomas 2,3 to restrict our search to 8 shared regions of the genome. There were 6 non-silent coding variants present in 4 different chromosomal regions that were shared by the 3 sequenced members of SM2702 (Supplementary Table 2). Further genotyping determined that the additional affected members of SM2702 (Figure 1) only shared one of these variants, POLE L424V (NM_006231:c.C1270G). POLE encodes the catalytic and proof-reading sub-unit of polymerase ε. The L424V amino acid is highly conserved (Supplementary Figure 2a) and the L>V change has predicted severe functional consequences for protein function (SIFT score=0.01, Polyphen2=0.993, Phylop=1.000, cons46=607, LOD=390 and MutationTaster=0.987).
Figure 1. Pedigrees of the POLE L424V and POLD1 S478N families in the Discovery Phase.

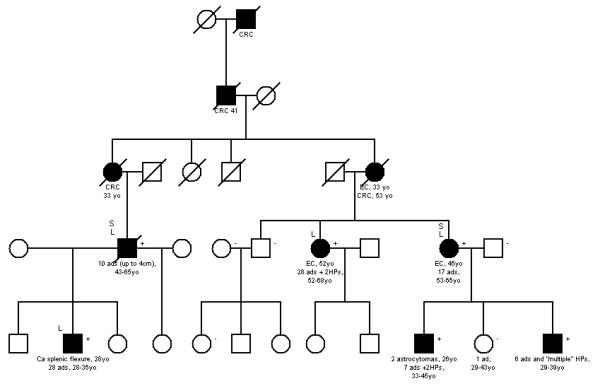

Families (a) SM2702 (POLE), (b) SM1645 (POLD1) and (c) SM1412 (POLD1) are shown. [•]=affected, [+]=mutation carrier and [−]=wildtype. S=whole genome-sequenced, L=genome-wide linkage analysis. For colorectal adenomas (ads), we show the cumulative tumour numbers from age at first presentation or screening colonoscopy to age at last contact. Diameter of the largest adenoma is also given where reported. Hyperplastic polyp (HP) numbers are also shown. For colorectal carcinomas (CRCs), endometrial carcinomas (ECs) and brain tumours, age at first presentation is given. Location of the CRC (colon, caecum, rectum) is also given where reported.
We genotyped the POLE L424V variant in a set of Validation Phase samples. These comprised 3,805 white UK CRC patients enriched for family history of colorectal tumours, multiple adenomas and early-onset disease (see Methods). For comparison, we genotyped 6,721 white UK controls, of whom about two-thirds were population-based and one-third, selected for absent personal history of colorectal tumours. We found 12 additional unrelated cases and no controls who were L424V heterozygotes (P=2.46×10−6, Fisher’s exact test, one-tailed). In addition, L424V was not present in 10,755 controls sequenced as part of the NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), despite >50X median read depth at this site in the genome. We confirmed that L424V mRNA is expressed, without any evidence of allele-specific instability (details not shown).
The pedigrees of the 12 Validation Phase cases with POLE L424V are shown in Supplementary Figure 3. They are compatible with a dominantly-inherited trait. All pedigrees except one contained at least one individual with multiple or very large adenomas, multiple CRCs, or early-onset CRC. Microsatellite marker analysis provided no evidence of a recent common ancestor in these families (data not shown). All the L424V mutation carriers were found in our highly-selected groups of patients from the CORGI, multiple adenoma and NSCCG studies (see Methods) rather than in our CRC clinical trial patients (VICTOR and QUASAR2). This suggests that the relatively severe phenotypes of our L424V carriers do not simply reflect ascertainment criteria and also shows that these mutations are likely to be a rare cause of CRC in the general population. Genotyping of additional members of these 12 families showed that all L424V carriers developed colorectal tumour(s) and that no tumour-free blood relatives of cases carried L424V. One non-carrier, from a kindred whose other affected members developed tens of adenomas or multiple carcinomas, had developed a single tiny (~0.5mm diameter) colorectal polyp by 33 years of age, a phenotype compatible with a sporadic occurrence or an additional “complex” genetic predisposition in the family. Based on a relatively stringent definition of affection status – ≥5 colorectal adenomas, ≥1 large (>2cm diameter) adenoma, ≥2 CRCs, or CRC at ≤40 years of age – and on the reasonable assumption that all L424V alleles within each family were identical by descent, the probability of each affected non-proband sharing L424V in the 12 Validation Phase families was 1.22×10−4 after ascertainment correction.
Overall, the phenotypes of our L424V pedigrees were consistent with dominantly-inherited, high-penetrance predisposition to colorectal adenoma and carcinoma, with a variable tendency to develop multiple and large tumours. Therapeutic or prophylactic colectomy had occurred in several cases, thus attenuating the phenotype, and it is also likely that some tumours were under-reported, especially in early generations. However, even allowing for these factors, tumour multiplicity appeared to vary among individuals (Figure 1a, Supplementary Figure 3). Moreover, adenomas did not, in general appear to progress rapidly to carcinoma in all individuals. The histological features of the L424V carriers’ colorectal tumours were unremarkable, mostly comprising conventional adenomas and carcinomas without specific morphology or obvious site predilection in the large bowel. No conclusive evidence of a predisposition to extra-colonic tumours or non-tumour phenotypes was found in the L424V-mutant families (Figure 1a, Supplementary Figure 3).
We examined 39 tumours from 11 POLE L424V carriers for somatic mutations. “Second hits” by LOH involving the germline wildtype allele were found in some tumours, including adenomas (Supplementary Figure 4). In contrast to Lynch syndrome, all tumours were microsatellite-stable (MS-stable), and the two carcinomas analysed had chromosomal instability. Most tumours were screened for KRAS and BRAF driver mutations, and a sub-set of tumours was screened for somatic mutations of established pathogenicity in APC, CTNNB1, PIK3CA and FBXW7 (Table 1, Supplementary Table 3). Mutations were found in all genes apart from CTNNB1, suggesting that the tumours at least partly followed the classical pathway of colorectal tumorigenesis. Mutations were all base substitutions. For APC, this was a notable difference from sporadic tumours in which ~60% of mutations are frameshifts 4. Moreover, certain sites not commonly mutated in sporadic colorectal tumours seemed to be mutation hotspots, for example codons 1114 and 1338 of APC and codon 146 of KRAS. Unlike in MUTYH-associated polyposis, where somatic driver mutations are almost all G:C>T:A changes, our patients’ tumours showed no evidence of a predominant type of substitution in the 6 genes analysed, although at this stage, we could not exclude more subtle biases in this regard (see below).
Table 1. Summary of somatic mutation spectra in colorectal cancer driver genes in tumours from POLE L424V patients.
For each patient, the data from several tumours are combined. The position of the somatic mutation and the base change are shown for all detected changes. Full details, including numbers of tumours tested, are shown in Supplementary Table 3.
| Family | Patient | Somatic mutations |
|---|---|---|
| 2702 | 1 | APC Q1338X (C>T); FBXW7 R465C (C>T) |
| 2702 | 2 | None found |
| 2702 | 3 | APC R1114X (C>T), Q1338X (C>T); KRAS G12D (G>A), A146T (A>C) |
| A | 1 | None found |
| I | 1 | APC Q1338X (C>T); KRAS A146T (A>C); FBXW7 R479Q (G>A) |
| I | 2 | APC Q1338X (C>T); KRAS G12D (G>A) × 3 |
| F | 1 | None found |
| K | 1 | APC R1114X (C>T) |
| C | 1 | None found |
| H | 1 | None found |
We then considered variants shared by the two affected individuals sequenced in our other large family, SM1645. Using the same strategy as used for SM2702 – searching for shared functionally important variants and filtering using linkage analysis (Supplementary Table 4) – we identified two SM1645 cases who shared a heterozygous POLD1 mutation (S478N; NM_002691:c.G1433A). We also found the same variant in another proband (SM1412) who had undergone whole-genome sequencing in the WGS500. This variant was stably expressed in mRNA (details not shown) and was predicted to have a severe effect on protein function (SIFT score=0.01, Polyphen2=0.992), Phylop=0.987, cons46 (607, LOD=390) and MutationTaster=1.000). Moreover, the equivalent of POLD1 478 is POLE 428, very close to the site of the L424V mutation (Supplementary Figure 5).
We typed S478N in all available members of SM1645 and SM1412 (Figure 1b,c). All S478N carriers developed colorectal tumour(s) and all affected individuals (see above for criteria) carried S478N (P=0.008 after ascertainment correction). Affected members of SM1645 and SM1412 shared alleles at microsatellites D19S867 and D19S904, consistent with a common ancestor. No tumour-free blood relatives of cases carried the mutation. We noted that a formally unaffected non-carrier from SM1645, whose other members had multiple polyps (Figure 1b), had developed one, very small (<1mm diameter) colorectal adenoma by 43 years of age. We then genotyped the POLD1 S478N variant in the Validation Phase and found one additional carrier, a case diagnosed with colorectal carcinoma at age 28 whose father also had colorectal cancer at 44 years of age. No control carried POLD1 S478N, and the variant was also absent from the NHLBI GO Exome Sequencing Project database.
The colorectal phenotypes of POLD1 S478N mutation carriers were generally similar to those of the POLE L424V mutants (Figure 1, Supplementary Figure 3). However, 7 patients with POLD1 mutations had developed endometrial carcinoma, some at relatively young ages, and one had developed two primary brain tumours. No cases of either tumour type were present in the 13 families with POLE mutations. As a result of this finding, we screened 386 early-onset endometrial cancer cases without a reported family history of CRC for POLD1 S478N, but did not find any further mutation carriers.
We examined 23 tumours from POLD1 S478N carriers for LOH and for the somatic mutations that had been tested in the POLE-mutant tumours (Table 2, Supplementary Table 5). In most respects, the POLD1-mutant tumours resembled POLE-mutant tumours: “second hits” occurred at POLD1 by LOH involving the germline wildtype allele (Supplementary Figure 4), tumours were all MS-stable, carcinomas had chromosomal instability, C>T substitution mutations were most common, and the APC mutation spectrum was atypical compared with sporadic CRCs. However, there were also some possible differences: KRAS mutations tended to target codons 13 and 61 in the POLD1 mutants, as compared with codons 12 and 146 in POLE mutants, and some POLD1 tumours had BRAF mutations.
Table 2. Summary of somatic mutation spectra in colorectal cancer driver genes in tumours from POLD1 S478N patients.
For each patient, the data from several tumours are combined. The position of the somatic mutation and the base change are shown for all detected changes. Full details, including numbers of tumours tested, are shown in Supplementary Table 5.
| Family | Patient | Somatic mutations |
|---|---|---|
| 1412 | 1 | None found |
| 1645 | 1 | APC E1309X (C>T); KRAS Q61H (A>C) |
| 1645 | 2 |
APC R1114X (C>T), Q1338X (C>T) × 3, Q1367X (C>T); KRAS G13D (G>A) × 4; BRAF V600E (T>A) × 2; FBXW7 R465H (G>A), R465C (C>T) |
| 1645 | 3 | None found |
| 1645 | 4 | None found |
| 1645 | 5 | None found |
Functional and structural assessment of POLE L424V and POLD1 S478N
Human POLE is the catalytic subunit of the polymerase epsilon (Polε) enzyme complex 5. Polε is responsible for synthesis of the leading strand during DNA replication. In addition to DNA-binding and polymerase domains, Polε has proof-reading capacity through the POLE exonuclease domain. This capacity is essential for the maintenance of replication fidelity and may act not only on newly misincorporated bases, but also on mismatches produced by non-proof reading polymerases such as Polα. L424 is conserved in the exonuclease domain of class B DNA polymerases (Supplementary Figure 2a) and a Blast search against the Protein Data Bank (www.pdb.org) revealed that L424 maps directly to the active site of the exonuclease domain (L479 in yeast DNA polymerase [PDB ID: 3IAY]) 6. The importance of leucine 424 for proof-reading has been shown by mutation of the equivalent residue (L479S) in S. cerevisiae Polδ/Pol3. This results in a mutator phenotype similar to that of the pol3-01 mutation, which has less than 0.5% wild-type exonuclease activity 7,8.
POLD1 encodes the catalytic and proof-reading sub-unit of Polδ, the equivalent lagging strand polymerase to Polε 9,10, and Polδ may also participate in the mismatch and base excision repair pathways 11,12. POLD1 serine 478 is highly evolutionarily conserved in eukaryotes including S. cerevisiae, although in S. pombe, the homologous amino acid is the structurally very similar cysteine (Supplementary Figure 2b). S478N lies close to other mutations that have a mutator phenotype in S. cerevisiae Pol3 8, but mutation of this specific residue has not been reported. We therefore constructed the equivalent mutation in fission yeast and determined its effect on reversion of the ade6-485 allele, which is a C to G transversion that reverts by base substitution 13. The Pol3-C462N strain (equivalent to human POLD1 S478N) had a mutation rate 12-fold higher than a wild-type strain that was constructed using the same strategy (Table 3, Supplementary Table 6). We also examined a Pol3-C462S strain, equivalent to the human wild-type, and this mutation did not have a significant effect on the ade6 reversion rate.
Table 3. Assessment of the functional effects of the S. pombe equivalent of human POLD1 S478N.
Human S478 is partially conserved in S. pombe (C462). The cysteine was therefore mutated to both the human wildtype (S, wt) and mutant (N) alleles. The Table shows reversion rates of the ade6-485 allele in the genetic backgrounds listed, with standard deviations in parentheses. Numbers are mean values of Ade+ revertants per 109 divisions with standard deviations in parentheses. Reversion rates were determined from three experiments. Strains used were 3176 (wt), 3177 (C462N) and 3178 (C462S).
| Site mutated | Mutation rate (x109) | Fold increase (mutant/wt) |
|---|---|---|
| C462N | 72 (9.6) | 12 |
| C462S | 4.2 (2.1) | 0.7 |
| Wildtype | 6.0 (4.2) | 1 |
POLE and POLD1 are related B family polymerases that have greatest homology (23% identity, 37% similarity) over the exonuclease domain (residues 223-517 of POLE and 245-571 of POLD1). Mapping these two mutations onto the structure of yeast DNA polymerase reveals that they pack together at the interface between two helices that form the base of the exonuclease active site. Modelling the ssDNA component of T4 DNA polymerase/exonuclease complex (1NOY) reveals that POLE L424 is only ~5Å from the ssDNA, and mutations of POLE 424 or POLD1 478 will distort the packing of the helices which will be propagated through to the active site, affecting nuclease activity (Figure 2).
Figure 2. Modelling of the germline and exonuclease domain mutations.

a) Composite model of the catalytic subunit of the yeast DNA polymerase δ (in ternary complex with DNA and an incoming nucleotide [PDB ID: 3IAY]) and the ssDNA component of the T4 polymerase complex [PDBID: 1NOY], modeled into the exonuclease active site. The polymerase is coloured blue, the exonuclease domain is shown in green, the dsDNA in orange and magenta, and the ssDNA in the exonulease active site in yellow. Mutations map to the active site of the exonuclease domain.
b) Germline mutations POLE L424V (in red) and POLD1 S478N (magenta) map to a helix (478-487) and pack against another helix, forming part of the base of the exonuclease active site. Mutations will disrupt this packing of helices and distort the active site. The active site is defined by the ssDNA substrate shown in yellow.
c) Mapping of the possibly pathogenic germline and somatic mutations to the exonuclease domain. All the POLE mutations (exonuclease domain somatic changes from the TCGA colorectal cancer data), and POLD1 P327L (germline variant from our patient, same location as POLE P286H) cluster around the active site (in red), whilst the POLD1 mutations S370R and G426S (germline variants from two other patients, shown in magenta) are more peripheral.
Search for additional POLE and POLD1 predisposition variants in the exonuclease domain
We wondered whether additional germline POLE and POLD1 exonuclease domain variants might predispose to colorectal tumours. We screened these regions (codons 268-471of POLE, codons 304-517 of POLD1) for germline variants in 469 cases with multiple colorectal adenomas and/or familial CRC (sample sets (i) and (iv) in Methods). We found three heterozygous POLD1 variants, each in a single case, that were absent from public control databases and from the non-cancer cases in the WGS500: P327L (c.C981G), S370R (c.C1110G) and G426S (c.G1276A). Structural studies showed that both S370R and G426S were unlikely to be involved in direct DNA binding and were predicted to have only moderate functional effects. By contrast, for P327L, which occurred in a 70 year-old with 10 colorectal adenomas, there was good evidence of functional consequences. The residue is fully evolutionarily conserved and lies on the DNA interface of the POLD1 exonuclease domain. Moreover, POLD1 327 is analogous to POLE 286, an amino acid that lies at the DNA-binding interface and is somatically mutated in sporadic CRCs (Figure 2 and Supplementary Table 7).
Assessment of POLE and POLD1 somatic mutations and their consequences in sporadic colorectal cancers
There is currently little evidence of pathogenic somatic POLD1 mutations in sporadic CRCs (Supplementary Table 7), but there have been some reports of somatic POLE mutations in sporadic CRCs 14-16. Of particular interest is the recent TCGA sequencing of 224 CRC exomes 17. The authors highlighted POLE as being an important somatic change, but did not demonstrate a significant excess of mutations over background. Moreover, the importance of individual mutations had not yet been assessed and we therefore examined the TCGA POLE data in the light of our own findings above (Table 4, Supplementary Table 7). Fifteen TCGA CRCs had one or more POLE mutations at locations throughout the gene. Six of these cancers were hypermutated and/or MS-unstable owing to MLH1 methylation. A further two CRCs were MS-stable without hypermutation, and one of these had an exonuclease domain mutation. Strikingly, all of the remaining 7 POLE-mutant cancers were hypermutated and MS-stable or -low, and all of these had a mutation in the POLE exonuclease domain (P<0.001, Fisher’s exact test; Supplementary Table 7). In some cancers, the exonuclease mutation was accompanied by a second nonsense (sample 17) or missense mutation elsewhere in the protein (Table 4). Structural analysis showed that the exonuclease domain mutations cluster around the DNA-binding site (Figure 2), including one mutation, P436R (P327L in POLD1), that lies in a disordered loop which becomes ordered on DNA binding (sample #15, Supplementary Table 7). All of these mutations will have a direct effect on either DNA binding and/or activity. The 2 exceptions are S370R and G426S that lie on the periphery of the domain, and mutations at these sites must act indirectly, if they are pathogenic.
Table 4. Summary of somatic POLE and POLD1 mutations and mutations in known colorectal cancer driver genes from the TCGA colorectal cancer sequencing data.
For the POLE and POLD1 mutations: Cat=catalytic domain; Exo=exonuclease domain; DFU-putative C-terminal DNA-binding domain; None=no specific domain; and Loss=protein-truncating mutation presumed to be null. The positions of the driver mutations in APC, KRAS, BRAF, PIK3CA and FBXW7, and the specific base change, are shown. Full results are shown in Supplementary Table 7.
| Tumour | Hyper- mutated? |
MSI | MLH1 methylated? |
Gene | AA Change |
Domain | Somatic mutations |
|---|---|---|---|---|---|---|---|
| TCGA-AA-3516 | Yes | MSI-H | Yes | POLD1 | p.A864T | Cat | BRAF V600E (T>A) |
| TCGA-AA-3710 | Yes | MSI-H | Yes | POLD1 | p.A145D | None | None found |
| TCGA-AA-3947 | Yes | MSI-H | Yes | POLD1 | p.P787L | Cat | APC E874fs; BRAF V600E (T>A), PIK3CA (H1074R (A>G) |
| TCGA-AA-3949 | Yes | MSI-H | Yes | POLD1 | p.Q461H | Exo | BRAF V600E (T>A); FBXW7 R367X (C>T) |
| TCGA-AA-A00J | Yes | MSI-H | Yes | POLD1 | p.R808H | Cat | APC R554X (C>T); BRAF V600E (T>A) |
| POLE | p.A2056T | None | |||||
| TCGA-AA-3518 | Yes | MSI-H | Yes | POLE | p.V1368M | None | FBXW7 R465C (C>T) |
| TCGA-AA-3525 | Yes | MSI-H | Yes | POLE | p.A2213V | None | BRAF V600E (T>A) |
| POLE | p.K1008N | Cat | |||||
| POLE | p.R762W | Cat | |||||
| TCGA-AA-3555 | Yes | MSS | No | POLE | p.P286H | Exo | APC R499X (C>T), R1450X (C>T); KRAS A146T (G>A) |
| TCGA-AA-3678 | No | MSS | No | POLE | p.D1752N | DFU | APC E1309fs |
| TCGA-AA-3710 | Yes | MSI-H | Yes | POLE | p.P1421S | None | None found |
| TCGA-AA-3864 | Yes | MSI-H | No | POLE | p.R231H | None | APC R348X (C>T), R564X (C>T), R1432X (C>T) |
| TCGA-AA-3977 | Yes | MSS | No | POLE | p.K777N | Cat | APC R1114X (C>T), E1309X (G>T) |
| POLE | p.F367S | Exo | |||||
| TCGA-AA-3984 | Yes | MSS | No | POLE | p.V411L | Exo | APC R1114X (C>T) |
| TCGA-AA-A00N | Yes | MSI-L | No | POLE | p.L1255V | None | APC S1281X (C>A), E1408X (G>T); KRAS G13D (G>A) |
| POLE | p.V411L | Exo | |||||
| TCGA-AA-A010 | Yes | MSI-L | No | POLE | p.P436R | Exo | APC R1450X (C>T) |
| POLE | p.A189T | None | |||||
| TCGA-AG-3892 | Yes | MSS | No | POLE | p.S459F | Exo | APC R1114X (C>T) |
| TCGA-AG-A002 | Yes | MSS | No | POLE | p.S459F | Exo | APC R1450X (C>T), E1538X (G>T) |
| POLE | p.R150X | Loss | |||||
| TCGA-AG-A01W | No | MSS | Not reported | POLE | p.D2013N | None | APC R876X (C>T); KRAS G13D (G>A) |
Exonuclease domain-mutant (EDM) CRCs in the TCGA data set tended to have nonsense mutations in the APC gene, including some of the same hotspots that we observed in the patients with germline POLE or POLD1 mutations (Table 4, Supplementary Table 7). For example the APC R1114X mutation was found in 6 of 224 TCGA CRCs, 3 of which had POLE exonuclease domain mutations (P<0.001, Fisher’s exact test). Genome-wide analysis showed that the set of 7 POLE EDM CRCs from the TCGA data had an increased tendency to acquire all types of base substitution compared with cancers that had POLE mutations outside the exonuclease domain and with all the other TCGA cancers. Indeed, non-EDM POLE-mutant cancers were MS-unstable and showed no difference in mutation spectrum or frequency from MS-unstable cancers without POLE mutations (Supplementary Table 8).The EDM-associated mutator phenotype did not, however, affect all substitutions equally. Hence, whilst C:G>T:A changes remained the most common type of mutation in EDM tumours, with an ~30-fold excess over other cancers, there was a particularly large (~100-fold) relative excess of G:C>T:A changes (Supplementary Table 8). An analysis of individual EDM cancers (details not shown) demonstrated that they all showed this mutation spectrum, further suggesting that each of these exonuclease domain changes was functionally deleterious. However, within the EDM group, two CRCs had acquired strikingly large numbers of mutations (over 6,000 each), these being the tumour with the mutation in the loop that became ordered on DNA binding and the tumour that had both an exonuclease domain mutation and a truncating mutation in POLE. We therefore speculate that these cancers may have had particularly deficient proof-reading.
A very recent study by Seshagiri et al 18 analysed 74 CRC exomes. Although a direct comparison between large-scale sequencing studies using non-identical platforms and analysis pipelines must be performed with caution, we found that the data of Seshagiri et al provide support for our conclusions from the TCGA data. Although Seshagiri et al did not highlight POLE in their manuscript, their data set included two hypermutated MS-stable cancers, both of which were EDM (P286R in both cases) and had an genome-wide mutation spectrum similar to that of the EDM TCGA cancers (details not shown).
We examined four tumours from our POLE L424V and POLD1 S478N carriers using an extended Ion Torrent panel based on 150 cancer gene exomes (see Methods). These tumours all showed mutation spectra similar to the POLE EDM cancers (Supplementary Table 8).
DISCUSSION
We have shown that the germline POLE variant L424V predisposes to multiple colorectal adenomas and carcinomas. The allele shows dominant inheritance, apparently with high penetrance, and tumours occur predominantly or exclusively in the large bowel. Some individuals have a predominantly multiple adenoma phenotype, similar to MUTYH-associated polyposis (MAP) that results from defective base excision repair. In other patients, the phenotype is one of large adenomas or early-onset carcinoma, thus resembling Lynch syndrome that results from defective DNA mismatch repair. This phenotypic variation is found in both MAP 19 and Lynch syndrome 20, and is consistent with the action of genetic modifiers. However, it may have other causes (for example, random mutation, different environments, reduction in risk owing to therapeutic surgery or other tumour prophylaxis, under-reporting of adenomas especially in older family members, or premature death). The evidence strongly suggests that the mechanism of tumorigenesis is decreased fidelity of replication-associated polymerase proof-reading, leading to an increased mutation rate by base substitution.
We have also shown that POLD1 S478N predisposes to colorectal tumours, endometrial cancer and, perhaps, to brain tumours. The colorectal phenotype cannot be distinguished from that of POLE L424V mutants in the current data set. The apparently different tumour spectrum between POLE 424V and POLD1 S478N carriers is unexplained, but similar differences are found in Lynch syndrome, in which carriers of MSH6 mutations, for example, seem relatively prone to endometrial cancer 21. One possible explanation is that a mutator phenotype is not the only tumour-promoting consequence of POLE and POLD1 – and indeed, mismatch repair gene – mutation.
It is highly plausible, based on theoretical considerations and the TCGA data in sporadic CRCs, that a variety of germline POLE or POLD1 variants in the exonuclease domain can influence CRC risk. A strong candidate from our own analysis is POLD1 P327L. The effects of these mutations might vary depending on the degree to which they impair proof reading. Other pathogenic mutations might occur in the non-proof-reading domains of POLE or POLD1 or in genes that encode the other components of Polε and Polδ (POLE2, POLE3, POLE4, POLD2, POLD3 and POLD4). Such mutations might act a little differently from those in the proof-reading domain, for example if they caused a tendency for the polymerase to incorporate mispaired bases into the growing strand. Of note, we have previously shown that common variation at the POLD3 locus is associated with CRC risk in the general population 22.
Mice that carry double Pole mutations (D272A;E274A) close to the exonuclease active site impair proof reading and cause a mutator phenotype in both the heterozygous and homozygous states 23,24. The proof-reading mutations cause a tendency to base substitutions, but not to MSI. Pole homozygote mutants are specifically predisposed to developing intestinal adenomas and carcinomas. Mice with germline Pold1 D400A mutations (equivalent to human D380A) had a similar mutator phenotype in both the heterozygous and homozygous states, and homozygous mutant animals had a tendency to develop a variety of cancers, although, intriguingly, the tumours spectrum differed from that of the Pole mutants, with most tumours being of cutaneous, rather than intestinal, origin 23,25-28. There was also evidence of different, but overlapping mutation spectra in the Pole- and Pold1-mutant animals.
The question remains as to whether POLE and POLD1 mutations act as classical tumour suppressors, with “second hits”, as haploinsufficient alleles, or through some hybrid mechanism, such as loss of a specific protein function. We cannot resolve this issue yet from our own data or the TCGA findings, but we note the following: (i) “second hits” do occur in some of our patients’ tumours, including early lesions, at frequencies greater than the background expected from other studies 29, but many POLE- or POLD1 EDM tumours do not have identified “second hits” (Supplementary Tables 3, 5 and 7); (ii) the L424V and S478N alleles produce stable mutant mRNA, and there is no evidence from our data that protein-truncating or null POLE or POLD1 mutations predispose to tumours; and (iii) bi-allelic Pole or Pold1 mutations that cause complete loss of enzyme function are probably cell-lethal in null mice. One intriguing mechanism to consider is that mutations are only pathogenic when they couple functional polymerase activity to non-functional proof-reading in the same molecule.
When added to the DNA mismatch repair defects that underlie Lynch syndrome and the base excision repair defects that cause MAP, the mutations in POLE and POLD1 emphasise the critical role of replication errors and coupled repair of base pair-level mutations in predisposition to colorectal and endometrial cancer. This is in contrast to cancers of the breast and ovary, in which double strand break repair is more important in predisposition. POLE L424V and POLD1 S478N mutations can be easily tested and should be considered in any individual with an unexplained personal or family history of multiple or large colorectal adenomas, and/or multiple or early-onset colorectal (or endometrial) carcinoma. Whilst further data are required, it may be prudent in the short term to manage mutation carriers in a fashion intermediate between Lynch syndrome and MAP, with regular and frequent colonoscopic polypectomy and consideration of prophylactic surgery.
Methods
Samples
All cases and controls were UK residents and had self-reported European ancestry. For most individuals, large-scale genotyping data were available that allowed ethnic clustering with HapMap CEU samples to be checked. None of the groups described below overlapped. All studies were performed with UK national ethical committee approval (MREC/06/Q1702/99).
Discovery Phase cases
These were obtained from the COloRectal tumour Gene Identification (CORGI) study 30. The study has recruited through Clinical Genetics Departments from throughout the United Kingdom since 1999 and has to date enrolled 1,698 families. General eligibility criteria for the study are given below. Discovery Phase cases were selected from among the set of CORGI probands for a disease phenotype that was most strongly reminiscent of a high-penetrance predisposition. We specifically enriched for multiple tumours and/or an early age of presentation and/or multiple family members with CRC or adenomas. Formal criteria were not set owing to the impossibility of weighing these different features against each other and the heterogeneity of clinical data (for example comparing an individual who had had fewer adenomas but also a colectomy against one who had more adenomas but only polypectomies). Our prioritisation resulted in the identification of a set of 15 unrelated individuals who had been diagnosed with ≥10 colorectal tumours before the age of 60 years (Supplementary Table 1). All individuals had CRC or a first- or second-degree relative with CRC, but they were not otherwise selected for family or personal history of other tumour types. All individuals had been screened for APC and MUTYH mutations by their local Genetics service and Lynch syndrome had been screened by microsatellite instability and/or mismatch repair protein expression in tumours and/or direct MSH2, MLH1, MSH6 and PMS2 mutation testing. DNA from these samples and 5 additional family members was sequenced across the entire genome as part of the Oxford University-Illumina WGS500 collaboration.
Validation Phase cases
These samples comprised the following non-overlapping collections, all comprising white individuals from the United Kingdom.
Multiple adenoma cases comprised 271 individuals who had developed ≥5 colorectal adenomas at any age, irrespective of family history of colorectal tumours. Germline APC and MUTYH mutations had been excluded. These individuals were recruited principally from St Mark’s Hospital, London and were included in the study irrespective of other personal or family history. The mean number of adenomas was 16 (range 4-65), mean age of presentation was 54 (range 23-80); 21% had had CRC and 31% reported a family history of CRC.
CORGI cases comprised 1,476 individuals from the COloRectal tumour Gene Identification study 30. All recruits had developed CRC at age ≤80, and/or “significant” colorectal adenoma(s) that fulfilled one or more of the following criteria: ≥3 in number; severe dysplasia; villous histology; and presentation at <45 years of age. All adenoma-only cases (N=503) had a first or second degree relative with CRC. Mean age of participants was 58 years (range 18-80).
National Study of Colorectal Cancer Genetics (NSCCG) CRC cases (N=300) were selected from the main NSCCG study to be aged <55 at diagnosis and to have at least one first-degree relative affected with CRC. The main NSCCG study recruited any individual with CRC from Oncology clinics throughout the United Kingdom 31.
Whole-genome sequencing case data were obtained using the Complete Genomics platform 32 from 198 CRC cases previously selected for early age of presentation and family history from the CORGI and NSCCG studies.
VICTOR/QUASAR2 samples comprised 1,560 cases from the VICTOR and QUASAR2 clinical trials of stage II/III CRC treated with curative intent 33. VICTOR was a post-primary therapy randomised controlled trial of rofecoxib and QUASAR2, a randomised trial of adjuvant 5-fluorouracil±bevacizumab. The mean age of patients derived from these trials was 61 years (range 44-70).
Validation Phase controls
These comprised the following collections, all comprising white individuals from the United Kingdom
CORGI controls were 1,666 spouses/partners of CORGI cases and reported no personal or family history of colorectal tumours.
1958 Birth Cohort sequencing set controls 34 (N=300) had undergone whole-exome sequencing.
Population controls (N=3,812) were obtained from the population-based, publicly-available 1958 Birth Cohort and National Blood Service 35 sample sets.
GLACIER controls without cancer (N=943) were derived from the GLACIER study of lobular breast cancer predisposition and were cancer-free spouses, partners or friends (E. Sawyer, I. Tomlinson, R.Roylance, unpubl.).
Although not used for primary statistical comparisons, additional population-based control data were derived from the Exome variant Server (NHLBI GO Exome Sequencing Project).
Endometrial cancer cases
386 cases with primary carcinoma of the endometrium and without a family history of CRC were derived from the National Study of Endometrial Cancer Genetics (NSECG) that recruited white UK cases with endometrial cancer under the age of 70 years old 36.
Germline genotyping and mutation screening
WGS data for POLE, POLD1 and other variants within regions of linkage 2,3 were validated by bi-directional Sanger sequencing using standard methods (details available from authors) and variants identified both automatically and manually in the Mutation Surveyor (Soft Genetics) program. For screening the Validation Phase samples, we designed KASPar 37 allele-specific SNV primers for POLE L424V and POLD1 S478N (Supplementary Figure 6), and included two known variant samples in each run in order to facilitate genotype clustering (details available from authors). All potential L424V and S478N variants detected were subsequently checked by bi-directional Sanger sequencing. For screening the full exonuclease domains of POLE (codons 269-471) and POLD1 (codons 304-517), bi-directional Sanger sequencing was used (details available from authors).
Expression of the mutant POLD1 S478N and POLE L424V alleles
mRNA was extracted from pelleted untransformed lymphocytes from one patient from SM1645 and two patients from SM 2702 using the RNeasy kit (Qiagen) following manufacturer’s instructions. RNA was treated with DNaseI (Fermentas) and converted into cDNA using the High Capacity cDNA reverse transcription kit (Applied Biosystems). cDNA was tested in triplicate using the KASPar genotyping assays described above. Heterozygous gDNA samples were run alongside these samples together with cloned alleles in various mixtures to determine whether the mRNA expression of the two alleles was balanced.
Loss of heterozygosity (LOH) and common ancestor analysis
Microsatellites mapping close to POLE (D12S1723, D12S1628, D12S357, D12S1638) or POLD1 (D19S867, D19S904, D19S907) were analysed in the Gene Marker (Soft Genetics) program and used to assess whether the L424V or S478N alleles were derived from a common ancestor (PCR conditions available from authors). LOH was assessed using the same set of microsatellites. LOH was scored if the intensity of any allele was reduced by ≥50% relative to the other allele after taking account of the relative allelic intensities in paired constitutional DNA. In the case of discordance between microsatellites, precedence was given to the one closest to POLE or POLD1. For a small sub-set of POLD1-mutant tumours, LOH analysis was performed genome-wide using the Illumina Goldengate HumanLinkage system (Panel V). Briefly, ~1μg of tumour DNA and paired constitutional DNA were amplified as per the manufacturer’s instructions and hybridised separately to Sentrix BeadArrays overnight. Each chip was then washed and stained, and scanned immediately using an Illumina BeadArray reader. GenomeStudio software was used to identify regions with loss of heterozygosity (LOH) and/or copy number changes. It should be noted that some tumours in the set analysed were very small and contamination by normal cells may have obscured LOH in some cases. Similarly, it was not possible to screen the large POLE and POLD1 genes for “second hits” other than LOH owing to limiting tumour material.
Somatic CRC driver mutation screening: Ion Torrent cancer hotspot panel
Using the Ion Torrent cancer hotspot panel, we screened CRC-associated mutations for somatic changes in tumours from POLE L242V and POLD1 S478N carriers using direct sequencing. Owing to limited quantities of DNA from the formalin-fixed, paraffin-embedded tumours available from our patients, we focussed on relatively common somatic mutations for which there was excellent pre-existing evidence of pathogenicity. Specifically, based on the existing literature and databases (http:// http://www.sanger.ac.uk), we regarded the following as pathogenic: APC (reference sequence NM_000038) – protein-truncating mutations between codons 167 and 1580; CTNNB1 (NM_001904) – missense mutations at codons 32, 33, 34, 37, 41 or 45; KRAS (NM_004985) – missense mutations at codons 12, 13, 61 or 146; BRAF (NM_004333) – V600E; PIK3CA (NM_006218) missense mutations at codons 345, 420, 542, 545, 546, 1025 or 1047; and FBXW7 (NM_033632) – protein-truncating mutations and missense mutations at codons 465, 479, 505 or 582. TP53 was not analysed because of the uncertain pathogenicity of some missense variants. Mutations were called using Ion Reporter (Invitrogen) and bam files were additionally inspected manually in order to remove likely artefacts and to detect any mutations - especially insertions and deletions in APC - that were not called by the Ion Reporter software. KRAS and BRAF mutations were additionally screened in an extended sample set using Sanger sequencing.
Somatic mutation spectrum screening: Ion Torrent cancer gene custom panel
One tumour a from POLE L424V carrier (family SM2702) and three tumours from POLD1 S478N carriers (two from a member of SM1645 and one from a member of SM1412) were screened for exonic variants in a custom panel of 150 cancer-related genes using the Ion Torrent Ampliseq method (details available on request). After excluding the above known CRC driver genes and known polymorphisms, the somatic mutation spectrum was determined. All but two in-del mutations were substitutions.
Fission yeast strain construction and mutation rate assays
Strains used in this study are listed in Supplementary Table 6. Standard genetic methods were used for strain construction 38. The pol3-C462N mutant strain (equivalent to human POLD1 S478N) was constructed by amplifying pol3 segments with primer combinations 1075 & 1077; 1076 and 1078 (see Supplementary Table 6). The products were purified, annealed and amplified using primers 1075 & 1076. The product was digested with BamHI and AscI and inserted into pFA6a-kanMX6 39. The pol3-C462S (equivalent to human POLD1 wild type) strain was constructed in the same way using initial primer combinations 1075 & 1079; 1080 & 1076. As a control, a pol3+ (wild-type) strain was also constructed using the same procedure, using primer combinations 1075 & 1076 to amplify the wt sequence. Plasmids were integrated into the pol3 locus after linearization with CspCI and selecting for G418-resistance. Constructs were verified by sequencing. Mutation rates of the ade6-485 allele in different strain backgrounds were determined by fluctuation analysis as previously described 40. Eleven cultures were used for each experiment and plates were scored after 7 days. Mutation rates were calculated using the MSS-Maximum likelihood estimator 41.
Structural analysis
Human mutations in both POLE and POLD1 were visualised in PyMOL (http://pymol.org) on the catalytic subunit of the yeast DNA polymerase δ [PDB ID: 3IAY]), with the ssDNA component of the T4 polymerase complex [PDBID: 1NOY], modelled into the exonuclease active site.
Other methods are provided in the Supplementary Material.
Supplementary Material
Acknowledgements
We are grateful to all the patients and their relatives from the families studied and to those who have provided their medical care. We acknowledge the help of many colleagues who are parts of the teams who work on the CORGI study. This work was principally funded by Cancer Research UK and the Oxford NIHR Comprehensive Biomedical Research Centre (to IT). We also acknowledge core funding to the Wellcome Trust Centre for Human Genetics from the Wellcome Trust (090532/Z/09/Z). Work in the RSH laboratory is supported by funding from Cancer Research UK (C1298/A8362 supported by the Bobby Moore Fund). Work in SEK’s lab was supported by project grants from Cancer Research UK and the John Fell Oxford University Press (OUP) Research Fund; we thank Oliver Fleck for fission yeast strains. IS is supported by a fellowship from the Junta de Extremadura, Spain (Consejería de Economía, Comercio e Innovación). RH and IT acknowledge funding from the European Union Seventh Framework Programme (FP7/207-2013) under grant no. 258236, FP7 collaborative project SYSCOL.
Footnotes
Accession numbers APC: NM_000038; CTNNB1: NM_001904; KRAS: NM_004985; BRAF: NM_004333; PIK3CA: NM_006218; FBXW7: NM_033632.
Competing financial interests None declared.
URLs PLATYPUS program: http://www.well.ox.ac.uk/platypus
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP
HapMap: http://www.hapmap.org
1000Genomes: http://www.1000genomes.org
Wellcome Trust Case Control Consortium: http://www.wtccc.org.uk/
Exome Variant Server: http://evs.gs.washington.edu/EVS/
COSMIC: http://www.sanger.ac.uk/COSMIC/
SIFT: http://sift.jcvi.org/
PolyPhen: http://genetics.bwh.harvard.edu/pph
Mutation taster: http://www.mutationtaster.org/
Cancer Genome Atlas project: http://cancergenome.nih.gov
OMIM: http://www.omim.org/
HGMD Professional: http://www.hgmd.org/
GeneCards: http://www.genecards.org/
Clustal: http://www.ebi.ac.uk/Tools/msa/clustalo/
Pymol: http://www.pymol.org
NCBI Structure server: http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi
Life Technologies: http://tools.invitrogen.com/content/sfs/brochures/IonAmpliseq_CancerPanel_AppNote_CO 32199_06042012.pdf
References
- 1.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–9. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kemp Z, et al. Evidence for a colorectal cancer susceptibility locus on chromosome 3q21-q24 from a high-density SNP genome-wide linkage scan. Hum Mol Genet. 2006;15:2903–10. doi: 10.1093/hmg/ddl231. [DOI] [PubMed] [Google Scholar]
- 3.Papaemmanuil E, et al. Deciphering the genetics of hereditary non-syndromic colorectal cancer. Eur J Hum Genet. 2008;16:1477–86. doi: 10.1038/ejhg.2008.129. [DOI] [PubMed] [Google Scholar]
- 4.Laurent-Puig P, Béroud C, Soussi T. APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1998;26:269–270. doi: 10.1093/nar/26.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struct Mol Biol. 2009;16:979–86. doi: 10.1038/nsmb.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM. Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′-5′-exonuclease activities of Pol delta in the creation of a ligatable nick. The Journal of biological chemistry. 2003;278:1626–33. doi: 10.1074/jbc.M209803200. [DOI] [PubMed] [Google Scholar]
- 8.Murphy K, Darmawan H, Schultz A, Fidalgo da Silva E, Reha-Krantz LJ. A method to select for mutator DNA polymerase deltas in Saccharomyces cerevisiae. Genome. 2006;49:403–10. doi: 10.1139/g05-106. [DOI] [PubMed] [Google Scholar]
- 9.Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–5. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hindges R, Hubscher U. DNA polymerase delta, an essential enzyme for DNA transactions. Biol Chem. 1997;378:345–62. doi: 10.1515/bchm.1997.378.5.345. [DOI] [PubMed] [Google Scholar]
- 11.Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001;8:1076–92. doi: 10.1038/sj.cdd.4400948. [DOI] [PubMed] [Google Scholar]
- 12.Mitra S, Boldogh I, Izumi T, Hazra TK. Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ Mol Mutagen. 2001;38:180–90. doi: 10.1002/em.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleck O, Lehmann E, Schar P, Kohli J. Involvement of nucleotide-excision repair in msh2 pms1-independent mismatch repair. Nature genetics. 1999;21:314–7. doi: 10.1038/6838. [DOI] [PubMed] [Google Scholar]
- 14.da Costa LT, et al. Polymerase delta variants in RER colorectal tumours. Nat Genet. 1995;9:10–1. doi: 10.1038/ng0195-10. [DOI] [PubMed] [Google Scholar]
- 15.Flohr T, et al. Detection of mutations in the DNA polymerase delta gene of human sporadic colorectal cancers and colon cancer cell lines. Int J Cancer. 1999;80:919–29. doi: 10.1002/(sici)1097-0215(19990315)80:6<919::aid-ijc19>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida R, et al. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. Eur J Hum Genet. 2011;19:320–5. doi: 10.1038/ejhg.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seshagiri S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, et al. MYH mutations in patients with attenuated and classic polyposis and with young-onset colorectal cancer without polyps. Gastroenterology. 2004;127:9–16. doi: 10.1053/j.gastro.2004.03.070. [DOI] [PubMed] [Google Scholar]
- 20.Lanspa SJ, et al. Colorectal adenomas in the Lynch syndromes. Results of a colonoscopy screening program. Gastroenterology. 1990;98:1117–22. doi: 10.1016/0016-5085(90)90323-s. [DOI] [PubMed] [Google Scholar]
- 21.Wijnen J, et al. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet. 1999;23:142–4. doi: 10.1038/13773. [DOI] [PubMed] [Google Scholar]
- 22.Dunlop MG, et al. Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet. 2012;44:770–776. doi: 10.1038/ng.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albertson TM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106:17101–4. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Preston BD, Albertson TM, Herr AJ. DNA replication fidelity and cancer. Semin Cancer Biol. 2010;20:281–93. doi: 10.1016/j.semcancer.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldsby RE, et al. Defective DNA polymerase-delta proofreading causes cancer susceptibility in mice. Nat Med. 2001;7:638–9. doi: 10.1038/88963. [DOI] [PubMed] [Google Scholar]
- 26.Goldsby RE, et al. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc Natl Acad Sci U S A. 2002;99:15560–5. doi: 10.1073/pnas.232340999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkatesan RN, Hsu JJ, Lawrence NA, Preston BD, Loeb LA. Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase delta. J Biol Chem. 2006;281:4486–94. doi: 10.1074/jbc.M510245200. [DOI] [PubMed] [Google Scholar]
- 28.Venkatesan RN, et al. Mutation at the polymerase active site of mouse DNA polymerase delta increases genomic instability and accelerates tumorigenesis. Mol Cell Biol. 2007;27:7669–82. doi: 10.1128/MCB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones AM, et al. Analysis of copy number changes suggests chromosomal instability in a minority of large colorectal adenomas. J Pathol. 2007;213:249–56. doi: 10.1002/path.2234. [DOI] [PubMed] [Google Scholar]
- 30.Tomlinson I, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–8. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 31.Penegar S, et al. National study of colorectal cancer genetics. Br J Cancer. 2007;97:1305–9. doi: 10.1038/sj.bjc.6603997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drmanac R, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 33.Pendlebury S, Duchesne F, Reed KA, Smith JL, Kerr DJ. A trial of adjuvant therapy in colorectal cancer: the VICTOR trial. Clin Colorectal Cancer. 2003;3:58–60. doi: 10.3816/CCC.2003.n.013. [DOI] [PubMed] [Google Scholar]
- 34.Power C, Elliott J. Cohort profile: 1958 British birth cohort (National Child Development Study) Int J Epidemiol. 2006;35:34–41. doi: 10.1093/ije/dyi183. [DOI] [PubMed] [Google Scholar]
- 35.Ouwehand WH. Platelet genomics and the risk of atherothrombosis. J Thromb Haemost. 2007;5(Suppl 1):188–95. doi: 10.1111/j.1538-7836.2007.02550.x. [DOI] [PubMed] [Google Scholar]
- 36.Spurdle AB, et al. Genome-wide association study identifies a common variant associated with risk of endometrial cancer. Nat Genet. 2011;43:451–4. doi: 10.1038/ng.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuppen E. Genotyping by Allele-Specific Amplification (KASPar) CSH Protoc. 2007;2007 doi: 10.1101/pdb.prot4841. pdb prot4841. [DOI] [PubMed] [Google Scholar]
- 38.Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods in enzymology. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 39.Bahler J, et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–51. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 40.Marti TM, Mansour AA, Lehmann E, Fleck O. Different frameshift mutation spectra in non-repetitive DNA of MutSalpha- and MutLalpha-deficient fission yeast cells. DNA Repair (Amst) 2003;2:571–80. doi: 10.1016/s1568-7864(03)00023-5. [DOI] [PubMed] [Google Scholar]
- 41.Hall BM, Ma CX, Liang P, Singh KK. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics. 2009;25:1564–5. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.