

Abstract
Reverse signaling via members of the tumor necrosis factor (TNF) superfamily is increasingly recognized among cells of the immune system where it controls multiple aspects of immune function. Here we document TNFα reverse signaling in the nervous system for the first time and show that it plays a crucial role in establishing sympathetic innervation. During postnatal development, sympathetic axons express TNFα as they grow and branch in their target tissues which in turn express TNFR1. In culture, soluble forms of TNFR1 act directly on postnatal sympathetic axons to promote growth and branching by a mechanism that depends on membrane integrated TNFα and downstream MEK/ERK activation. Sympathetic innervation density is significantly reduced in several tissues in postnatal and adult mice lacking either TNFα or TNFR1. These findings reveal that target-derived TNFR1 acts as a reverse signaling ligand for membrane-integrated TNFα to promote sympathetic axon growth and branching.
Keywords: reverse signaling, sympathetic neuron, neurite growth, TNFα, TNFR1, development
The growth, guidance and the terminal arborization of axons are regulated by numerous extracellular signals that either promote or inhibit axon extension and influence axon trajectory by acting as either attractants or repellents at the growth cone 1. The sympathetic neurons of the mouse superior cervical ganglion (SCG) comprise an extensively studied, experimentally tractable population of neurons for investigating the regulation of axonal growth and target field innervation in the developing peripheral nervous system 2, 3. A variety of secreted proteins promote sympathetic axon growth at different stages of development, including nerve growth factor (NGF), neurotrophin-3 (NT-3), glial cell-derived neurotrophic factor (GDNF), artemin and hepatocyte growth factor (HGF). Of these, the most extensively studied and best understood is NGF, which is produced in target tissues and promotes the growth and branching of sympathetic axons within many of these tissues 4. Target-derived NGF is also required for and regulates the survival of developing sympathetic neurons 2, 3.
Recent work has shown that several members of the TNF superfamily (TNFSF) are capable of modulating neurite growth in the developing nervous system. For example, in certain populations of neurons, FasL and GITRL enhance neurite growth 5-7 whereas TNFα, LIGHT and RANKL reduce neurite growth 8-11. TNFSF members are type II transmembrane glycoproteins that are active both as membrane-integrated ligands and as soluble ligands following cleavage from the cell membrane, and exert their effects by binding to members of the TNF receptor superfamily (TNFRSF) 12. In addition to functioning as ligands, several membrane-integrated TNFSF members have been reported to act as reverse signaling receptors for their respective TNFRSF partners in cell lines and several cell types of the immune system 13. Here we report the occurrence of TNFSF reverse signaling in the nervous system. We demonstrate that TNFR1 acts as a ligand for membrane-integrated TNFα expressed along the axons of postnatal SCG neurons and that TNFR1-activated TNFα reverse signaling promotes axon growth and tissue innervation by an ERK1/ERK2-dependent mechanism.
RESULTS
TNFα and its receptors in SCG neurons and targets
As the starting point of an investigation to clarify the role of TNFα in sympathetic neuron development, we carried out a detailed study of the expression of TNFα and its receptors, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2), in SCG neurons and tissues innervated by these neurons. In low-density dissociated cultures of P0 mouse SCG neurons, we observed intense TNFα immunoreactivity in the cell bodies and throughout the neurite arbors of the neurons. In marked contrast, intense TNFR1 immunoreactivity was restricted to the neuron cell bodies, and the neurites were completely unlabeled (Fig. 1a). Accordingly, in histological sections of the SCG, neuron cell bodies were labeled by both anti-TNFα and anti-TNFR1 antibodies, and post-ganglionic sympathetic fibre bundles, identified by tyrosine hydroxylase immunostaining, were labeled with anti-TNFα antibodies but not with anti-TNFR1 antibodies (Fig. 1b). We observed weak TNFR2 immunoreactivity in the cell bodies and neurites of the neurons (not shown). Quantification of TNFα and TNFR1 transcripts in dissected SCG revealed a gradual increase from embryonic day 13 (E13) to reach a peak in expression by postnatal day 5 (P5), followed by a decline to low or negligible levels in the adult (Figs. 1c and 1d).
Figure 1.
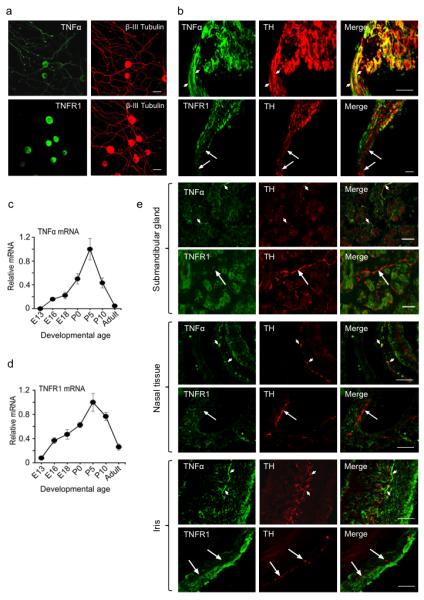
Expression of TNFα and TNFR1 in developing SCG neurons and their targets. (a) Photomicrographs of representative P0 SCG neurons double labeled for β-III tubulin and either TNFα or TNFR1 after being cultured for 24 h in the presence of 10 ng/ml NGF. (b) Sections passing through P0 SCG with an attached nerve double labeled with anti-tyrosine hydroxlyase (TH) and either anti-TNFα or anti-TNFR1. Small arrows indicate examples of TH-positive fibre bundles double-labeled with anti-TNFα, and large arrows indicate examples of TH-positive fibre bundles that are not stained with anti-TNFR1. (c-d) Levels of TNFα mRNA (c) and TNFR1 mRNA (d) relative to reference mRNAs in SCG of different ages. The data are normalized to a value of 1.0 at the peak of expression of TNFα and TNFR1 mRNAs at P5. Mean ± s.e.m. of data from three separate sets of ganglia at each age are shown. (e) Sections of the submandibular gland, nasal turbinate tissue and iris of P0 mice double labeled with anti-TH and either anti-TNFα or anti-TNFR1. Small arrows indicate examples of tyrosine hydroxlyase-positive sympathetic fibres that are double-labeled with anti-TNFα, and large arrows indicate examples of tyrosine hydroxlyase-positive sympathetic fibres that are not stained with anti-TNFR1. Scale bars = 50 μm.
In the SCG target tissues selected for detailed analysis in this study (submandibular salivary gland, nasal turbinate tissue and iris), tyrosine hydoxylase-positive sympathetic fibres were clearly labeled by anti-TNFα antibodies, but were not labeled by anti-TNFR1 antibodies (Fig. 1e). Submandibular gland tubules exhibited clear TNFR1 immunostaining and a low level of TNFα immunostaining. The connective tissue in the core of nasal turbinates, where blood vessels and sympathetic fibres are concentrated, was immunostained with anti-TNFR1 antibodies, whereas the mucosa was labeled with anti-TNFα antibodies. The connective tissue of the iris was labeled with both anti-TNFα and anti-TNFR1 antibodies (Fig. 1e). Sections incubated with secondary antibody alone exhibited no background immunofluorescence, and sections of tissues obtained from tnf−/− and tnfr1−/− mice were not labeled by anti-TNFα and anti-TNFR1 antibodies, respectively (not shown).
TNFα reverse signaling promotes sympathetic axon growth
Our finding that TNFα is expressed on postnatal sympathetic axons ramifying in TNFR1-expressing targets raised the possibility that target-derived TNFR1 might act as a reverse signaling ligand for TNFα. To examine the possibility that TNFα reverse signaling might influence sympathetic axon growth, P0 SCG neurons were cultured with NGF to sustain their survival with and without a divalent TNFR1-Fc chimera that has been shown to be a potent reverse signaling ligand for TNFα 14. After 24 hours incubation, the neurite arbors of neurons treated with the TNFR1-Fc were larger than those grown with NGF alone (Fig. 2a,b). Quantification of the size and complexity of the neurite arbors showed that the TNFR1-Fc chimera caused highly significant increases in neurite length (Fig. 2c) and branch point number (Fig. 2d), and the Sholl profiles, which plot neurite branching with distance from the cell body, were clearly larger in the presence of TNFR1-Fc (Fig. 2e). NGF supplemented neurons treated with an Fc protein fragment did not have significantly larger neurite arbors than neurons growth with NGF alone (Figs. 2c-e), indicating that the Fc fragment of the TNFR1-Fc chimera did not affect neurite growth. Detailed dose response analysis (not shown) revealed that the effect of the TNFR1-Fc chimera on neurite growth reached a plateau at a concentration of 10 ng/ml. Soluble recombinant monovalent TNFR1 (sTNFR1), which activites TNFα reverse signaling at much higher concentrations 15-17, also significantly enhanced neurite growth and branching (Figs. 2c-e). Cell counts showed that neither the TNFR1-Fc chimera nor sTNFR1 significantly affected neuron survival in these NGF-supplemented cultures (NGF alone 79.4 ± 8.4%, NGF + TNFR1-Fc 83.9 ± 7.8%, NGF + sTNFR1 97.2 ± 9.4%, number of neurons surviving 24 hr after plating expressed as a percentage of number plated, mean ± s.e.m.).
Figure 2.
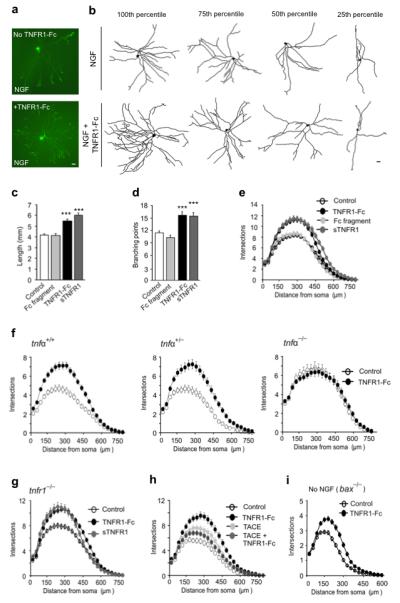
TNFR1-promoted TNFα reverse signaling enhances neurite growth from SCG P0 neurons. (a) Representative neurons cultured for 24 h in medium containing 10 ng/ml NGF, with or without 10 ng/ml TNFR1-Fc. (b) Representative camera lucida drawings of neurons corresponding to the 100th, 75th, 50th and 25th percentiles in terms of total neurite length. Scale bars = 20 μm. Length (c), branch point number (d) and Sholl profiles (e) of neurons cultured for 24 h with either NGF alone (control, n = 342) or NGF plus 10 ng/ml of human Fc fragment (n = 228), 10 ng/ml TNFR1-Fc (n = 171) or 5 μg/ml sTNFR1 (n = 180). (f) Sholl plots of neurons from P0 tnfα+/+, tnfα+/− and tnfα−/− littermates cultured for 24 h with NGF alone (control) or NGF plus TNFR1-Fc (n = 150). (g) Sholl plots of neurons from tnfr1−/− mice grown for 24 h with either NGF alone (control) or NGF plus TNFR1-Fc or sTNFR1 (n = 150). (h) Sholl plots of neurons cultured for 24 h with either NGF alone (control, n = 234) or NGF plus TNFR1-Fc (n = 243), 200 ng/ml TACE (n = 247) and TNFR1-Fc plus TACE (n = 248). (i) Sholl plots of the neurite arbors of neurons from bax−/− mice grown for 24 h in the presence (n = 174) or absence (n= 196) of TNFR1-Fc in medium lacking NGF. Mean ± s.e.m of data from 3 to 5 separate experiments of each type (*** indicates P <0.001, statistical comparison with control).
To verify that TNFα is essential for neurite growth enhancement by TNFR1-Fc, we compared the effects of TNFR1-Fc on cultures of SCG neurons obtained from tnfα+/+, tnfα+/− and tnfα−/− mice. For these studies, we crossed tnfα+/− mice and established separate SCG neuron cultures from each littermate. The Sholl profiles of SCG neurons of newborn tnfα+/+ and tnfα+/− mice grown with TNFR1-Fc plus NGF were markedly larger than those grown with NGF alone, whereas the Sholl profiles of NGF supplemented SCG neurons of newborn tnfα−/− mice grown with and without TNFR1-Fc were very similar (Fig. 2f). Statistical analysis revealed that whereas TNFR1-Fc caused highly significant increases neurite length and branch point number in the neurite arbors of neurons cultured from newborn tnfα+/+ and tnfα+/− mice (P<0.001, data not shown), TNFR1-Fc had no significant effect on neurite arbor size in cultures established from newborn tnfα−/− mice. These results demonstrate that TNFα is essential for the effect of TNFR1-Fc on neurite growth. In contrast to TNFα-deficient neurons, TNFR1-deficient neurons responded, just like wild types neurons, to TNFR1-Fc and sTNFR1 with enhanced neurite growth (Fig. 2g).
To determine whether membrane-integrated TNFα is required for neurite growth enhancement by TNFR1-Fc, we pre-treated P0 SCG neurons with TNFα-converting enzyme (TACE, ADAM17), a metalloproteinase that cleavages and releases the ectodomain of membrane-integrated TNFα 18, thereby preventing reverse signaling. The Sholl analysis revealed that neurite arbors of neurons cultured with TACE plus TNFR1-Fc were smaller than those cultured with TNFR1-Fc alone (Fig. 2h). Statistical analysis revealed that TACEcaused significant reductions in neurite length and branch point number from TNFR1-Fc treated neurons (P<0.01, data not shown). These findings are consistent with transduction of the TNFR1-Fc signal by intact membrane integrated TNFα.
To examine whether TNFR1 is able to enhance neurite growth independently of NGF, we cultured Bax-deficient neonatal SCG neurons, which fail to undergo apoptosis in the absence of NGF 19, without NGF in the presence and absence of TNFR1-Fc. Although the neurite arbors of these neurons were very much smaller than neurons grown with NGF, Sholl analysis revealed that TNFR1-Fc increased neurite arbor size and complexity (Fig. 2i). We carried out similar experiments using SCG neurons from wild type neurons treated with the cell permeable broad-spectrum caspase inhibitor Q-VD-OPh to prevent apoptosis in the absence of NGF. In these experiments, TNFR1-Fc likewise significantly increased neurite arbor size and complexity (not shown).
To determine whether TNFR1-Fc enhances neurite growth during a particular phase of sympathetic neuron development, we examined the effect of TNFR1-Fc on SCG neuron cultures established over a range of ages. Sholl analysis revealed that TNFR1-Fc did not enhance neurite growth from either E18 or P10 SCG neurons, but enhanced neurite arbor size and complexity from P0 and P5 neurons (Fig. 3a). This indicates that TNFα reverse signaling enhances neurite growth from SCG neurons during a restricted period of postnatal development when the axons of these neurons are ramifying in their target tissues.
Figure 3.
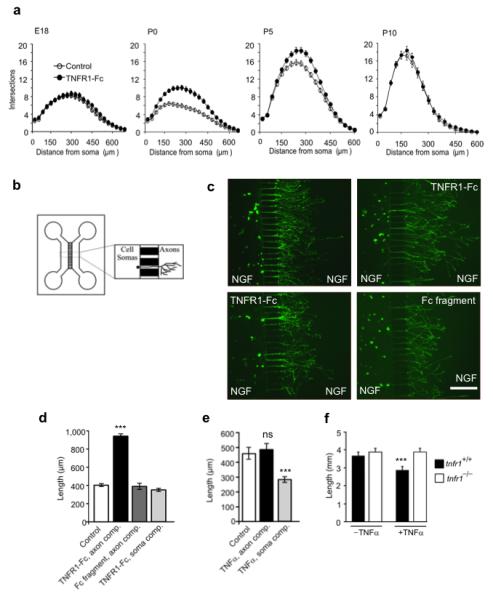
TNFR1-Fc acts locally on SCG axons to enhance growth over a window of postnatal development. (a) Sholl plots of the neurite arbors of E18, P0, P5 and P10 SCG neurons cultured for 24 h with or without 10 ng/ml TNFR1-Fc in presence of 10 ng/ml NGF. (b) Schematic illustration of the two-chamber microfluidic device. (c) Representative images of calcein-AM labeled P0 SCG neurons that were cultured for 24 h in a two-compartment microfluidic device containing 10 ng/ml NGF in both compartments, with either 10 ng/ml TNFR1-Fc or 10 ng/ml Fc fragment in the axon compartment or TNFR1-Fc in the soma compartment. Scale bar = 100 μm. (d,e) Bar charts of mean axon length of neurons projecting axons into the axon compartment under the experimental conditions indicated. The data represent the mean ± s.e.m of 9 independent experiments. (f) Plots of neurite arbor length of P0 SCG neurons from tnfr1+/+ and tnfr1−/− mice cultured for 24 h with NGF in the presence and absence of 10 ng/ml TNFα. The data shown represent the mean ± s.e.m of neurite arbor data 150 neurons per condition combined from 3 to 5 separate experiments of each type (*** indicates P<0.001, statistical comparison with control).
TNFR1-Fc acts locally on axons to enhance their growth
The expression of TNFR1 within sympathetic target tissues raises the possibility that it acts as a reverse signaling ligand on the TNFα-expressing sympathetic axon terminals to promote their growth locally as they ramify within these tissues. To test whether TNFR1 can act locally on sympathetic axon terminals to promote growth, we cultured sympathetic neurons in microfluidic devices in which the cell soma and growing axon terminals are cultured in different compartments separated by a barrier (Fig. 3b). We seeded P0 SCG neurons into one compartment (the soma compartment) of a two-compartment device that contained NGF in both compartments to sustain neuronal survival and encourage axon growth from the soma compartment into the axon compartment. We added TNFR1-Fc to either the soma or axon compartment and used an Fc protein fragment as control. After 24 hours incubation, we labeled the axons in the axon compartment with the fluorescent vital dye calcein-AM which also retrogradely labeled the cell bodies of the neurons projecting axons into the axon compartment. We used a stereological method to quantify the extent of axon growth in the axon compartment relative to the number of neurons projecting axons into this compartment. Addition of TNFR1-Fc to the axon compartment resulted in a marked and highly significant increase in axon growth within this compartment compared with Fc-treated controls. In contrast, addition of TNFR1-Fc to the soma compartment had no significant effect on axon growth in the axon compartment (Figs. 3c and 3d). These results suggest that TNFR1 expressed in sympathetic targets is capable of acting locally to enhance the growth of sympathetic axon terminals ramifying within these tissues.
We have previously shown that soluble TNFα significantly reduces the extent of NGF-promoted neurite growth from cultured neonatal SCG neurons 9. Additional compartment culture experiments showed that TNFα only impaired axon growth if it was added to the soma compartment, not the axon compartment (Fig. 3e). Our demonstration that the neurite growth inhibitory effect of soluble TNFα is completely eliminated by deletion of the tnfr1 gene (Fig. 3f), suggests that soluble TNFα exerts this effect via TNFR1. Our finding that neurite growth inhibition is observed when TNFα is applied to the cell soma, not axon terminals, of the neurons is consistent with the restriction of TNFR1 expression to the cell soma (Fig. 1), and suggests that TNFα expressed within target tissues in vivo plays no role in regulating sympathetic innervation density.
Reduced innervation density in tnfα−/− and tnfr1−/− mice
To ascertain whether the increase in sympathetic axon growth and branching brought about by TNFR1-activated TNFα reverse signaling in vitro is physiologically relevant for the establishment of sympathetic innervation in vivo, we used tyrosine hydroxylase immunofluorescence to identify sympathetic fibres and quantify sympathetic innervation density 20 in mice that are homozygous or heterozygous for targeted deletions of the tnfα 21 and tnfr1 22 genes and wild type littermates. We chose iris, nasal turbinate tissue and submandibular gland for this analysis because they receive a dense innervation of sympathetic fibres from the SCG. We initially carried out this analysis at P10, which is immediately after the period of development when TNFR1-activated TNFα reverse signaling enhances neurite growth in vitro and is at a stage in vivo when the sympathetic innervation of these tissues has become well established.
We crossed tnfα+/− mice to generate litters of tnfα+/+, tnfα+/− and tnfα−/− pups and and crossed tnfr1+/− mice to generate litters of tnfr1+/+, tnfr1+/− and tnfr1−/− pups. Sections of the iris, nasal turbinate tissue and submandibular gland of tnfα−/− and tnfr1−/− mice revealed reductions in tyrosine hydroxylase immunofluorescence compared with wild type littermates (Figs. 4a and 4e). Quantification of the level of tyrosine hydroxylase immunofluorescence revealed highly significant reductions in these tissues in both tnfα−/− mice (Figs. 4b, 4c and 4d) and tnfr1−/− mice (Figs. 4f, 4g and 4h) compared to wild type littermates. The levels oftyrosine hydroxylase immunofluorescence were also lower in the tissues of tnfα+/− and tnfr1+/− mice compared with wild type mice, although these reductions were only statistically significant in the nasal turbinate tissue of tnfα+/− mice (Fig. 4c) and the submandibular gland of tnfr1+/− mice (Fig. 4h).
Figure 4.
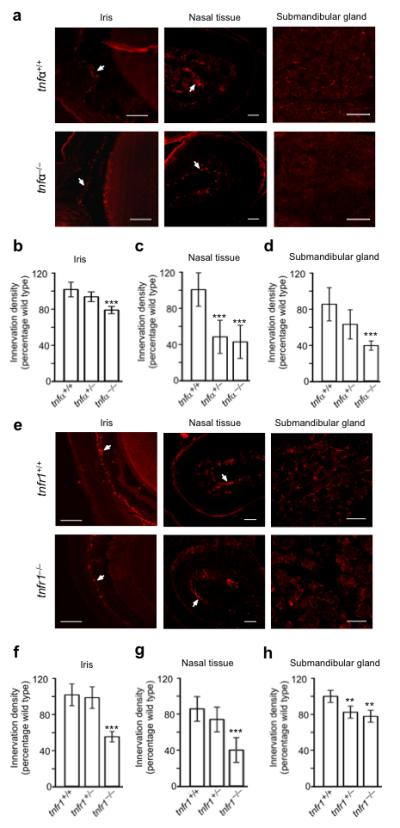
Reduced sympathetic innervation density in tnfα−/− (a-d) and tnfr1−/− mice (e-h). (a) Representative photomicrographs of TH labeled sections of the iris, nasal turbinate tissue and submandibular gland parenchyma of P10 tnfα+/+ and tnfα−/− mice. Arrows indicate the major location of TH-positive fibres: in the part of the section passing through the iris and the central core of connective tissue of a nasal turbinate where blood vessels and sympathetic fibres are concentrated. Scale bar = 100 μm. Bar charts of the relative levels of TH immunofluorescence in the iris. (b), nasal turbinate tissue (c) and submandibular gland (d) of P10 tnfα+/+, tnfα+/− and tnfα−/− mice expressed as a percentage of the mean level in tnfα+/+ mice. (e) Representative photomicrographs of TH labeled sections of the iris, nasal turbinate tissue and submandibular gland parenchyma of P10 tnfr1+/+ and tnfr1−/− mice. Scale bar, 100 μm. Bar charts of the relative levels of TH immunofluorescence in the iris. (f), nasal turbinate tissue (g) and submandibular gland (h) of P10 tnfr1+/+, tnfr1+/− and tnfr1−/− mice expressed as a percentage of the mean level in P10 tnfr1+/+ mice. Mean ± s.e.m of data from 5 × tnfα+/+, 6 × tnfα+/−, 7 × tnfα−/−, 7 × tnfr1+/+, 6 × tnfr1+/− and 6 × tnfr1−/− (** indicates P<0.01 and *** indicates P<0.001, statistical comparison with control).
To exclude the possibility that the decreases in tyrosine hydroxylase immunofluorescence in the tissues of tnfα−/− and tnfr1−/− mice were secondary to down-regulation of tyrosine hydroxylase expression in the innervating neurons, we used Western blotting to quantify the levels of tyrosine hydroxylase protein in the SCG of these mice and the corresponding wild type mice. This analysis revealed no significant differences in the levels of tyrosine hydroxylase protein relative to the level of the neuron-specific β-III tubulin protein in the SCG of tnfα−/− mice (Supplementary Fig. 1a-b) mice and tnfr1−/− mice (Supplementary Fig. 1d-e) compared with the ganglia of wild type mice at P10.
To determine if reduction in sympathetic innervation density observed in the tissues of tnfα−/− and tnfr1−/− mice was secondary to a decrease in the size of the innervating population of neurons, we counted the number of neurons in the SCG of tnfα−/− and tnfr−/− mice and their corresponding wild type littermates at P10. These counts revealed no significant differences in the numbers of SCG neurons between tnfα−/− and tnfα+/+ mice (Supplementary Fig. 1c) and between tnfr1−/− and tnfr1+/+ mice (Supplementary Fig. 1f). Taken together, these results demonstrate that TNFα and TNFR1 have a crucial role in establishing sympathetic innervation in vivo and suggest that the influence of TNFR1-activated TNFα reverse signaling on axon growth and branching is physiologically relevant.
To ascertain whether the significant reductions in sympathetic innervation observed in tnfα−/− and tnfr1−/− mice at P10 are maintained in the mature nervous system, we quantifiedtyrosine hydroxylase immunofluorescence in sections of the anatomically circumscribed tissues of the iris and submandibular salivary gland of adult mice. This analysis not only revealed highly significant reductions in tyrosine hydroxylase immunofluorescence in both tissues in tnfα−/− and tnfr1−/− adults compared with age-matched wild type animals (Figs. 5a-f), but the proportional reductions were greater than those observed at P10, especially in the submandibular gland, where the level of tyrosine hydroxylase immunofluorescence was reduced by between 70 and 80% in tnfα−/− and tnfr1−/− mice (Fig. 5c and 5f). These findings suggest that TNFα and TNFR1 play an ongoing role in maintaining sympathetic innervation in vivo.
Figure 5.
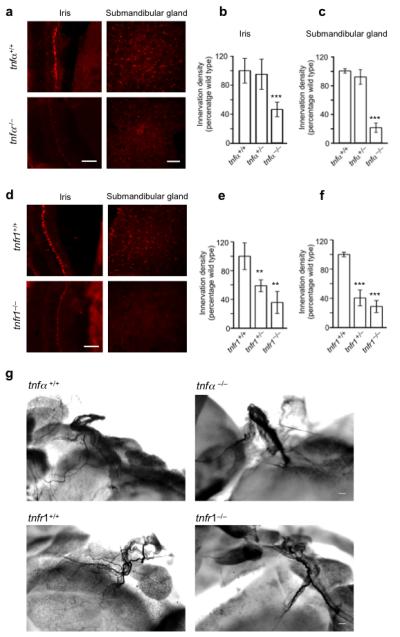
Reduced sympathetic innervation density in adult tnfα−/− (a-c) and tnfr1−/− mice (d-f) and reduced nerve fibre branching in P10 tnfα−/− and tnfr1−/− mice (g-i). (a) Representative photomicrographs of TH labeled sections of the iris and submandibular gland parenchyma ofadult tnfα+/+ and tnfα−/− mice. Bar charts of the relative levels of TH immunofluorescence in the iris (b) and submandibular gland (c) of adult tnfα+/+, tnfα+/− and tnfα−/− mice expressed as a percentage of the mean level in tnfα+/+ mice. (d) Representative photomicrographs of TH labeled sections of the iris and submandibular gland parenchyma of adult tnfr1+/+ and tnfr1−/− mice. Bar charts of the relative levels of TH immunofluorescence in the iris (e) and submandibular gland (f) of adult tnfr1+/+, tnfr1+/− and tnfr1−/− mice expressed as a percentage of the mean level in tnfr1+/+ mice. Mean ± s.e.m of data from three animals of each genotype are shown (** indicates P<0.01 and *** indicates P<0.001, statistical comparison with wild type). (g) Representative images of TH labeled whole mount preparations of the submandibular gland hilus of P10 tnfα+/+, tnfα−/−, tnfr1+/+ and tnfr1−/− mice. Scale bars = 100 μm.
We also employed tyrosine hydroxylase staining in whole mount tissue preparations to visualize sympathetic innervation in TNFα-deficient and TNFR1-deficient mice. This technique was especially suitable for the soft tissue of the submandibular gland. Here sympathetic nerves enter the gland at the hilus and subsequently split into numerous branches that ramify extensively within the gland. These preparations, carried out on tissue from P10 littermates, revealed very clear differences in sympathetic fibre branching between wild type and knockout animals. Whereas tyrosine hydroxylase-positive nerves branched into multiple finer branches near the hilus in wild type mice, tyrosine hydroxylase-positive nerves at the glandular hilus of tnfα−/− mice and tnfr1−/− mice did not give off as many branches, and the main nerve trunk had a tapped appearance, suggesting that whereas sympathetic nerves reach this gland, many fibres failed to grow into and ramify within this tissue (Fig. 5g). Using a stereological method to estimate the extent of branching near the hilus, we found highly significantly reductions in the number of nerve branches in tnfα−/− mice (37.3 ± 7.0% reduction, mean ± s.e.m., P<0.00001, n = 7) and tnfr1−/− mice (17.9 ± 5.1%, mean ± s.e.m., P<0.003, n = 13).
To corroborate our conclusion based on tyrosine hydroxylase immunohistochemistry that tnfα−/− and tnfr1−/− mice have reduced sympathetic innervation density compared with wild type mice, we used dopamine β-hydroxylase (DBH) immunohistochemistry as an alternative marker of sympathetic fibres in tissue sections of separate sets of P10 mutant and wild type mice. Although this analysis was based on a smaller number of animals, quantification of the level of DBH immunofluorescence nonetheless revealed highly significant reductions in the iris, nasal turbinate tissue and submandibular gland of both tnfα−/− mice and tnfr1−/− mice compared to wild type (Supplementary Fig. 2). Furthermore, we also observed significant decreases levels of DBH immunofluorescence in several tissues of heterozygous mice.
TNFR1-Fc enhances axonal growth by activating ERK1/ERK2
To elucidate the molecular mechanism underlying the enhancement of neurite growth from SCG neurons by TNFα reverse signaling, we explored a common link in intracellular signaling between the control of neurite growth and TNFα reverse signaling in the immune system. The MEK/ERK pathway has been shown to be activated both by TNFα reverse signaling in monocytes 23 and by NGF in PC12 cells and SCG neurons and to contribute to neurite growth in response to NGF 7, 24-26.
To investigate whether MEK/ERK signaling contributes to TNFR1-promoted neurite growth from SCG neurons, we first tested whether TNFR1-Fc activates ERK1 and ERK2 in these neurons. In these experiments, we initially cultured P0 SCG neurons for 12 hours with NGF before treating them with TNFR1-Fc. The initial period of culture permitted the levels of phospho-ERK1 and phospho-ERK2 to fall to basal levels so that any subsequent increase could be observed more easily. Treatment with TNFR1-Fc after 12 hours caused rapid increases in the levels phospho-ERK1 and phospho-ERK2 within 5 minutes, returning to basal levels within 30 to 45 minutes (Fig. 6a).
Figure 6.
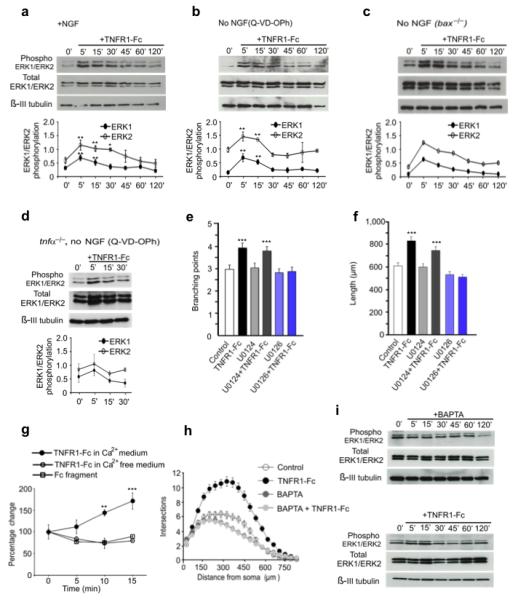
Cytosolic Ca2+ elevation and ERK1/ERK2 activation are required for TNFR1-Fc-promoted neurite growth. (a-c) Representative Western blots probed for phospho-ERK1/ERK2, total ERK1/ERK2 and β-III tubulin of lysates of P0 SCG neurons grown for 12 h with either 10 ng/ml NGF (a), 10 μM Q-VD-OPh without NGF (b) or BAX-deficient neurons without NGF (c) prior to treatment with 10 ng/ml TNFR1-Fc. (mean ± s.e.m of densitometry from 4 experiments. (d) Representative Western blot of lysates of P0 TNFα-deficient neurons grown for 12 h with Q-VD-OPh without NGF prior to TNFR1-Fctreatment. Mean ± s.e.m of densitometry from 3 experiments Branch point number (e) and length (f) of neurons pretreated for 2 h with either 10 μM U0126 or U0124 prior to TNFR1fcaddition and incubation for 24 h in medium containing Q-VD-OPh (mean ± s.e.m., >200 neurons per condition from four independent experiments). (g) Percentage change in cytosolic free-Ca2+ after addition of either Fc fragment or TNFR1-Fc in either Ca2+-free medium or medium containing 1.2 mM Ca2+ (mean ± s.e.m. of 3 experiments, >20 neurons imaged per condition per experiment). (h) Sholl profiles of P0 SCG neurons cultured for 24 h with either NGF (control) or NGF plus 1 μM BAPTA-AM,, TNFR1-Fc, or BAPTA-AM plus TNFR1-Fc (mean ± s.e.m. >150 neurons per condition from three independent experiments). (i) Representative Western blots of lysates of P0 SCG neurons grown for 12 h with Q-VD-OPh without NGF prior to treatment with either 1 μM BAPTA-AM or BAPTA-AM plus TNFR1-Fc (** P<0.01 and *** P<0.001, comparison with control).
To determine whether TNFR1-Fc treatment is able to activate ERK1 and ERK2 independently of NGF, we initially cultured P0 SCG neurons for 12 hours with the caspase inhibitor Q-VD-OPh to prevent apoptosis in the absence of NGF prior to TNFR1-Fc treatment. As with neurons maintained with NGF, TNFR-Fc caused a rapid transient increase in the levels phospho-ERK1 and phospho-ERK2 (Fig. 6b). Likewise, Bax-deficient SCG neurons cultured in the absence of NGF exhibited very similar changes in the levels of phospho-ERK1 and phospho-ERK2 following TNFR1-Fc treatment after 12 hours of culture (Fig. 6c). To confirm that TNFα is needed for the activation of ERK1/ERK2 by TNFR1-Fc, we treated SCG neurons obtained from tnfα−/− mice with TNFR1-Fc. We observed no significant changes in phospho-ERK1/phospho-ERK2 relative to total ERK1/ERK2 protein in TNFα-deficient neurons following TNFR1-Fc treatment (Fig. 6d). Taken together, these results show that TNFα reverse signaling causes rapid, transient ERK1/ERK2 activation independently of NGF.
To investigate if ERK1/ERK2 activation is responsible for the enhanced neurite growth brought about by TNFR1-Fc, we examined whether U0126, a selective MEK1/MEK2 inhibitor that interferes with MEK1/MEK2 dependent activation of ERK1/ERK2 27, could prevent the increase in neurite growth. In these experiments, we plated P0 SCG neurons in NGF-free medium containing Q-VD-OPh and were pretreated for 2 hours with either U0126 or the inactive analog U0124 before adding TNFR1-Fc and imaging and quantifying the neurite arbors 24 hours later. U0126, but not U0124, completely prevented TNFR1-Fc enhanced neurite growth, as shown by quantification of branch point number (Fig. 6e) and neurite length (Fig. 6f). This suggests that MEK/ERK signaling plays a crucial role in mediating the effect of TNFα reverse signaling on neurite growth.
Previous studies in a macrophage cell line have shown that activation of TNFα reverse signaling leads to Ca2+ influx and rapid elevation of cytosolic Ca2+ 28. Because elevated cytosolic Ca2+ can trigger ERK1/ERK2 activation by a variety of mechanisms in neurons 29, we asked whether stimulating TNFα reverse signaling also leads to elevation of cytosolic Ca2+ in SCG neurons and whether this is a necessary step in ERK1/ERK2 activation and enhanced neurite growth. Ca2+ imaging studies showed that treating SCG neurons with TNFR1-Fc, but not with a control Fc fragment, caused a rapid, significant increase in cytosolic Ca2+ (Fig. 6g). This elevation failed to take place if we treated the neurons with TNFR1-Fc in Ca2+-free medium (Fig. 6g), suggesting that it was due to Ca2+ influx. Treating neurons with 1 μM BAPTA-AM, a membrane permeable acetomethyl ester that chelates cytosolic Ca2+ when hydrolyzed in the cytoplasm, prevented activation of ERK1/ERK2 by TNFR1-Fc without affecting the levels of phospho-ERK1 and phospho-ERK2 on its own (Fig. 6i). 1 μM BAPTA-AM also eliminated the enhanced neurite growth from SCG neurons grown with TNFR1-Fc, while having negligible effect on NGF-promoted neurite growth on its own (Fig. 6h). These results suggest that elevation of cytosolic Ca2+ is a necessary step in ERK1/ERK2 activation and enhanced neurite growth brought about by TNFα reverse signaling.
DISCUSSION
Reverse signaling via membrane-integrated members of the TNFSF is becoming increasingly recognized among cells of the immune system where it controls multiple aspects of immune function 13. While there is a burgeoning literature on the roles of the TNFSF in the nervous system in development, physiology and pathology 30, 31, this has been investigated and interpreted within the framework of conventional forward signaling. Here we report for the first time TNFSF reverse signaling in the nervous system. We show that soluble monovalent TNFR1 and divalent TNFR1-Fc chimera, proteins that initiate TNFα reverse signaling in a variety of TNFα-expressing cell lines and cells of the immune system 14-17, enhance axonal growth and branching from TNFα-expressing postnatal SCG in culture. The expression of membrane-integrated TNFα is essential for the effect of TNFR1-Fc on neurite growth because TNFα converting enzyme, which cleaves the extracellular domain of TNFα, significantly impairs the ability of TNFR1-Fc to enhance neurite growth.
The neurite growth enhancing effect of TNFR1-Fc is restricted to a period of postnatal development from P0 to P5 when sympathetic axons are growing and ramifying within their targets in vivo under the influence of target-derived NGF 2, 3. TNFR1-Fc not only significantly enhances the size and complexity of the neurite arbors of postnatal SCG neurons beyond that seen with maximally effective concentrations of NGF, but promotes neurite growth and branching in the absence NGF in cultures in which neuronal apoptosis is prevented by caspase inhibition or deletion of the pro-apoptotic Bax protein. This indicates that TNFα reverse signaling enhances neurite growth and branching independently of NGF and is therefore capable of affecting axonal growth throughout the range of NGF concentrations developing SCG neurons encounter in vivo.
While SCG neurons co-express TNFα and TNFR1, these proteins exhibit different spatial patterns of expression in the neurons. Whereas TNFα is distributed along the axons both in vitro and in vivo, TNFR1 expression is restricted to the soma. The expression of TNFR1 by cells in tissues innervated by SCG neurons, together with our demonstration in compartment cultures that TNFR1-Fc enhances axon growth when applied to the axon terminals, but not to the cell soma, suggests that target-derived TNFR1 acts locally on axons to promote growth. TNFR1 expressed in target tissues could act on the membrane-integrated TNFα of the innervating axons either as a soluble, diffusible protein following its release from the cells that synthesize it 32, 33 or as a membrane integrated protein 34. Although we do know whether either or both of these alternatives pertain in vivo, the restriction of TNFR1 to the cells that produce it as membrane-integrated protein could potentially have a more precise influence on regulating the local growth, branching and disposition of axons in target tissues. Although SCG neurons co-express TNFR1 and TNFα, our observation that the magnitude of NGF-promoted neurite growth from cultured postnatal SCG neurons lacking either TNFR1 or TNFα is not significantly different from that of wild type neurons suggests that any potential autocrine signaling between these membrane proteins has no significant influence on neurite growth.
The physiological significance of the axon growth-promoting effect of target-derived TNFR1 on TNFα-expressing sympathetic axons in vivo is amply demonstrated by extensive, blind quantification of the sympathetic innervation density of several tissues of tnfα and tnfr1 mutant mice and wild type littermates. This analysis revealed highly statistically significant reductions in the levels of TH immunofluorescence in the irides, nasal tissue and submandibular gland of tnfα−/− and tnfr1−/− mice at P10 compared wild type littermates. There were also smaller, statistically significant reductions in the nasal tissue of tnfα+/− mice and the submandibular gland of tnfr1+/− mice compared to wild type littermates, hinting at a gene dosage effect in heterozygous mice. Quantification of levels of tyrosine hydroxylase protein and numbers of neurons in the SCG of tnfα−/− and tnfr1−/− mice and corresponding wild type mice indicated that the significant decreases in sympathetic innervation density were not secondary to decreases in either tyrosine hydroxylase expression or the size of the innervating population of sympathetic neurons. Quantification of sympathetic innervation density using DBH immunofluorescence as an alternative marker of sympathetic fibres, likewise revealed highly significant reductions in the iris, nasal turbinate tissue and submandibular gland of both tnfα−/− mice and tnfr1−/− mice compared to wild type. Furthermore, analysis of whole mount preparations suggests that whereas sympathetic nerves reach their targets in tnfα−/− and tnfr1−/− mice, many fibres fail to grow into and ramify within these targets. Quantification of sympathetic innervation density in adult mice revealed even greater decreases in tnfα−/− and tnfr1−/− mice than those observed at P10. Taken together, these studies suggest that TNFR1-activated TNFα reverse signaling makes a significant contribution to the establishment of sympathetic innervation of several cranial tissues in postnatal mice and the maintenance of sympathetic innervation in throughout life.
In common with TNFα reverse signaling in monocytes 23, we show that TNFα reverse signaling in postnatal SCG neurons leads to a rapid, pronounced and transient activation of ERK1/ERK2. Like the enhanced neurite growth promoted by TNFR1-Fc, the activation of ERK1/ERK2 by TNFR1-Fc is dependent on the expression of TNFα and occurs independently of NGF. Several studies have shown that activation of ERK1/ERK2 by NGF in SCG neurons contributes to its ability to promote neurite growth 7, 25, 26. Our demonstration that pharmacological inhibition of MEK1/MEK2, the kinases that phosphorylate and activate ERK1/ERK2, completely prevents neurite growth enhancing action of TNFR1-Fc suggests that MEK/ERK signaling mediates the effect of TNFα reverse signaling on neurite growth. As in macrophages 28, activation of TNFα reverse signaling in SCG neurons causes Ca2+ influx and rapid elevation of cytosolic Ca2+. Our demonstration that the intracellular Ca2+ chelator BAPTA prevents both ERK1/ERK2 activation and enhanced neurite growth in response to TNFR1-Fc, suggests that elevation of cytosolic Ca2+ is a necessary step in mediating the effect of TNFα reverse signaling on neurite growth. The identity of the channels that mediate Ca2+ influx and how TNFα reverse signaling gates these channels remain intriguing questions.
We previously reported that soluble TNFα reduces the extent of NGF-promoted neurite growth from cultured newborn SCG neurons by an IKKβ/NF-κB dependent mechanism 9. Our demonstration that SCG neurons cultured from tnfr1−/− mice lack this inhibitory response suggests that it is mediated by TNFR1. In accordance with the restriction of TNFR1 to the soma of SCG neurons, the growth inhibitory effect of soluble TNFα in compartment cultures was only observed when TNFα was added to the soma compartment, not the axon compartment. This suggests that target-derived TNFα is unlikely to play a role in regulating axon growth in vivo. Moreover, if the growth inhibitory effect of soluble TNFα observed in vitro were the predominant, physiologically relevant influence of TNFα in vivo, one might predict an increase in sympathetic innervation density in tnfα−/− and tnfr1−/− mice, not the decrease we observe.
The expression of TNFR1 and TNFR2 by neurons has been implicated in the regulation of neuronal death and survival. There is evidence that TNFα/TNFR1 signaling in fetal mouse SCG and trigeminal sensory neurons plays a role in accelerating neuronal apoptosis following NGF deprivation 35. Conversely, TNFα/TNFR2 signaling has been implicated in the survival effects of NGF on P5 rat DRG sensory neurons 36. However, by P10 we observe no significant differences in the number of SCG neurons between tnfα−/−, tnfr1−/− and wild type littermates, indicating that by this late stage of development compensatory have taken place in these mice for any effects of TNFα on neuronal survival and the timing and magnitude of naturally occurring neuronal death observed earlier in development.
Our discovery of TNF reverse signaling in the nervous system increases our appreciation of the diversity and complexity of signaling between cells in the nervous system and may necessitate a re-evaluation of the mechanistic explanation for previous in vivo and in vitro studies of TNFα function in the nervous system. For example, the phenotypic consequences of deleting the tnfα gene may result from eliminating either forward or reverse signaling. Likewise, the addition of soluble TNFα to cultured cells could either activate forward signaling or interfere with potential endogenous reverse signaling by competing for TNFR1 binding. In addition to providing important new insights into the regulation of the growth of sympathetic axons and the establishment of sympathetic innervation in vivo, our findings highlight the importance of evaluating the relative contributions of forward and reverse signaling in different developmental systems and experimental paradigms.
METHODS
Real-time QPCR
The levels of TNFα and TNFR1 mRNAs were quantified by RT-QPCR relative to a geometric mean of mRNAs for glyceraldehyde phosphate dehydrogenase (GAPDH) and succinate dehydrogenase (SDHA). Total RNA was extracted from SCG with the RNeasy Mini extraction kit (Qiagen, Crawley, UK), and 5 μl was reverse transcribed for 1 h at 45°C using the AffinityScript kit (Agilent, Berkshire, UK) in a 25 μl reaction according to the manufacturer’s instructions. 2 μl of cDNA was amplified in a 20 μl reaction volume using Brilliant III ultrafast QPCR master mix reagents (Agilent, Berkshire, UK). QPCR products were detected using dual-labeled (FAM/BHQ1) hybridization probes specific to each of the cDNAs (MWG/Eurofins, Ebersberg, Germany). The PCR primers were: TNFα forward, 5′-TAC TTA GAC TTT GCG GAG-3′ and reverse, 5′-AGA GTA AAG GGG TCA GAG-3′; TNFR1 forward, 5′-TTC CCA GAA TTA CCT CAG-3′ and reverse, 5′-AAC TGG TTC TCC TTA CAG-3′; GAPDH forward, 5′-GAG AAA CCT GCC AAG TAT G-3′ and reverse, 5′-GGA GTT GCT GTT GAA GTC-3′; SDHA forward, 5′-GGA ACA CTC CAA AAA CAG-3′ and reverse, 5′-CCA CAG CAT CAA ATT CAT-3′. Dual labeled probes were: TNFα, FAM-CAG GTC TAC TTT GGA GTC ATT GCT C-BHQ1; TNFR1, FAM-CAC CGT GTC CTT GTC AGC-BHQ1; GAPDH, FAM-AGA CAA CCT GGT CCT CAG TGT-BHQ1; SDHA, FAM-CCT GCG GCT TTC ACT TCT CT-BHQ1. Forward and reverse primers were used at a concentration of 150 nM each and dual-labeled probes were used at a concentration of 300 nM. PCR was performed using the M×3000P platform (Agilent, Berkshire, UK) using the following conditions: 45 cycles of 95°C for 12 seconds and 60°C for 35 seconds. Standard curves were generated in every 96-well plate, for each cDNA for every real time PCR run, by using serial three-fold dilutions of reverse transcribed spleen total RNA (Ambion, Paisley, UK). Three separate dissections were performed for each age.
Immunocytochemistry and immunohistochemistry
For immunocytochemistry, the cultures were fixed in ice-cold methanol for 5 min and were washed with phosphate-buffered saline (PBS) before blocking nonspecific binding and permeablizing the cells with 5% bovine serum albumin (BSA) plus 0.1% Triton X-100 (Sigma, Dorset, UK) in PBS for 1 h at room temperature. Neurons were incubated overnight with primary antibody in 1% blocking solution at 4°C. After washing with PBS, the cultures were incubated with the appropriate secondary antibody.
For immunohistochemistry, tissue was fixed in 4% paraformaldehyde for 24 h and was cryoprotected in 30% sucrose before being frozen. OCT embedded tissues were snap frozen in isopentene (Sigma, Dorset, UK) cooled with dry ice and serially sectioned at 14 μm. Eyes were sectioned at right angles to the visual axis and the entire nasal tissue was sectioned in the coronal plane. The submandibular gland was sectioned in the coronal plane. The sections were mounted onto electrostatic charged slides (Leica Microsystems, Peterborough, UK), blocked with 5% BSA containing 0.1% Triton X-100 in PBS for 1 h at room temperature, and then incubated for 18 h at 4°C with primary antibodies. The sections were washed in PBS before being incubated with an appropriate secondary antibody.
The primary antibodies were: monoclonal anti-β-III tubulin antibody (1:10000, R&D systems, Abingdon, UK, catalogue number MAB1195), polyclonal anti-TNFα antibody (1:200, Abcam, Cambridge, UK, catalogue number ab34674), polyclonal anti-TNFR1 antibody (1:200, Abcam, Cambridge, UK, catalogue number ab19139), polyclonal anti-tyrosine hydroxylase antibody (1:200, Millipore, Watford, UK, catalogue numbers AB1542 and AB152) and polyclonal anti-DBH antibody (1:100, Abcam, Cambridge, UK, catalogue number ab43868). Secondary antibodies were alexa fluor conjugated anti-Ig antibodies fromLife Technologies, Invitrogen, Paisley, UK used at 1:500 (catalogue numbers: donkey anti-rabbit IgG alexa fluor 488, A21206, goat anti-rabbit IgG alexa fluor 546, A11035, goat anti-mouse IgG alexa fluor 546, A11005, donkey anti-sheep IgG alexa fluor 546, A11016) and horse raddish peroxidase-conjugated anti-IgG antibodies from Progema, Southampton, UK, used at 1:1000 (catalogue numbers: anti-mouse IgG, W4021 and anti-rabbit IgG, W4011). Images were obtained using a Zeiss Axioplan confocal microscope (Zeiss, Cambridge, UK).
Neuron culture
SCG were trypsinized and plated at very low density (~ 200 neurons per dish/well) in poly-ornithine and laminin-coated 35 mm tissue culture dishes (Greiner, Gloucestershire, UK) or 4-well dishes (Starlab, Milton Keynes, UK) in serum-free Hams F14 medium 37 supplemented with 0.25% Albumax I (Invitrogen, Paisley, UK). Neuronal survival was estimated by counting the number of neurons in 4-well dishes 2 h after plating and again at 24 h. All neurons in each well were counted. The number of neurons surviving at 24 h was expressed as a percentage of the number at 2 h. Analysis of the size and complexity of neurite arbors was carried out in 35 mm dishes 24 h after plating. The neurite arbors were labeled by incubating the neurons with the fluorescent vital dye calcein-AM (1:1000, Invitrogen, Paisley, UK) at the end of the experiment. Images of neurite arbors were acquired by fluorescence microscopy and analyzed to obtain total neurite length, number of branch points and Sholl profiles 38.
For compartment cultures two-compartment microfluidic devices were used (Xona microfludics, CA, USA). P0 SCG neurons were plated on one compartment and both compartments received NGF. The TNFR1-Fc chimera was added to either compartment and a human Fc fragment was used as control. After 24 h incubation, the axons in the axon compartment and the cell bodies that project axons into this compartment were labeled by adding the fluorescent vital dye calcein-AM to the axon compartment. Axon length was quantified by a modification of a previously described method 39. Briefly, using NIH ImageJ, a grid of vertical lines was constructed with an interline interval of 200 μm. Total intersections between neurites and the grid were counted and normalized against the number of labeled somas in the cell body compartment. Average neurite length per projecting cell body was calculated using the formula L= πDI/2, where L is the estimated length, D is the interline interval and I the average number of intersections per projecting cell body. Measurements were independently carried out in all fields along the microfluidic barrier.
For harvesting protein for Western blotting, neurons were cultured at a high density (~85 000 neurons per well) in a 96-well plate for 12 h in medium containing NGF, caspase inhibitor or no factors in case of SCG neurons obtained from bax−/− mice. The neurons were then treated with TNFR1-Fc for times ranging from 5 to 120 minutes.
The majority of cultures were established from CD1 mice. Bax-deficient neurons were obtained from bax−/− mice back-crossed into a CD1 background. Tnfα and tnfr1 mutant mice were maintained in a c57bl6 background. Neonates of different genotypes were generated by crossing heterozygous mice. Separate cultures were established from each littermate resulting from these crosses, and the genotypes were only determined after the cultures had been analyzed by a PCR based approach using tissue samples obtained at the time the cultures were set up. All animal experiments were conducted in accordance with the 1986 Animal Procedures Act approved by the Home Office (UK).
Purified recombinant NGF, TNFR1-Fc, soluble TNFR1, TNFα and caspase inhibitor Q-VD-OPh were obtained from R&D Systems and the human Fc fragment was obtained from Abcam.
Quantification of the sympathetic innervation of SCG targets
Batches of tissue from littermates of all three genotypes of each mouse mutant were processed at the same time to ensure they were stained in an identical manner. For the iris, all sections were imaged. For the nasal turbinate tissue and submandibular gland, every fifth section was imaged. The outline of the iris and the core tissue of the nasal turbinates (i.e., turbinate tissue excluding the nasal mucosa, which displays some non-specific staining) in these images was traced using Adobe Photoshop CS. Total iris and core nasal turbinate area and the area containing intense immunoreactive tyrosine hydroxylase-positive fibers were estimated by automated pixel counts using identical settings for all sections and all genotypes, and the ratio tyrosine hydroxylase-positive area to total iris area and core turbinate area was calculated. For the submandibular gland, multiple random images were analyzed in which the ratio of immunoreactive tyrosine hydroxylase-positive fibers to total image area was estimated. Background staining was subtracted from all images prior to quantification. Background staining was obtained by imaging sections of the tissues that were incubated with secondary antibody alone. The data are expressed as a percentage of the mean wild type data for each tissue. This analysis was carried out by multiple authors and was done blind.
For whole mount studies, the submandibular glands of P10 of tnfα+/+, tnfα−/−, tnfr1+/+ and tnfr1−/− pups were fixed in 4% paraformaldehyde for at least 24 h. The tissue was dehydrated in 50% methanol for 1h at room temperature and 80% methanol for a further 1 h. Endogenous peroxidase activity was quenched by placing tissue in a solution of 80% methanol, 20% DMSO and 3% H2O2 overnight at 4°C. Tissue was rehydrated by placing in 50% methanol for 1h, 30% methanol for 1h, PBS for 1h, all at room temperature, and was blocked overnight at 4°C with 4% BSA containing 1% Triton X-100 in PBS. The tissue was then incubated with polyclonal anti-tyrosine hydroxylase antibody (1:200, Millipore, Watford, UK) in blocking solution for 72 h at 4°C. After washing 3 × 2 h in 1% Triton X-100 in PBS at room temperature, the tissue was kept at 4°C overnight in a fourth wash before being incubated with anti-rabbit HRP conjugated antibody (1:300, Promega, Southampton, UK) in blocking solution at 4°C overnight. The tissue was then washed for 2 h at room temperature with PBS containing 1% Triton X-100. Tyrosine hydroxylase-positive fibres were visualized by DAB-HRP staining: the tissue was incubated with 1×DAB for 20 min atroom temperature and then with 1×DAB containing 0.006% H2O2 for 2-5 min to develop the staining. After washing with PBS, the tissue was and incubated at 4°C overnight in PBS. BABB (1 part benzyl alcohol: 2 parts benzyl benzoate) was used as a clearing solution. The tissue was placed in 50% methanol for 10 min, washed 3 times with 100% methanol (1 × 30 min, 2 × 15 min) at RT and was incubated in 50% BABB for 5 min before being placed in BABB. To compare the extent of sympathetic nerve branching near the gland hilus, a modified line-intercept method was used. Using ImageJ, a grid of 24 squares (4 × 6 squares) of side length 158 μm per square was aligned in a standard orientation next to the hilus of each gland. The number of fibre bundles intersecting the sides of squares in the grid was scored blind for the glands from each animal. Fibre density was estimated using the formula πDI/2, where D is the interline interval (158) and I the mean number of intersections along one side of each square in the grid. The data are expressed as a percentage of the mean wild type data.
Quantification of neuron numbers in SCG
Estimates of the numbers of neurons in the SCG of P10 tnfr1+/+, tnfr1−/−, tnfα+/+ and tnfα−/− pups were carried out by stereological analysis of ganglia serially sectioned at 8 μm and immunolabeled for β-III tubulin, as described previously 35. The analysis was done blind to avoid any observer bias.
Immunoblotting
Immunoblotting was carried out using the BioRad TransBlot (BioRad, Hertfordshire, UK) as previously described 40. The blots were probed with antibodies to phospho-ERK1/ERK2 (1:1,000, Cell Signaling, Hertfordshire, UK, catalogue number), total ERK1/ERK2 (1:1,000, Cell Signaling, Hertfordshire, UK, catalogue number) or β-III tubulin (1:10,000, R&D systems, Abingdon, UK, catalogue number). Binding of the primary antibodies was visualized with an HRP-conjugated secondary antibody (1:2,000; Promega, Southampton, UK, catalogue number) and ECL-plus (Amersham, Buckinghamshire, UK). Densitometry was carried out using Gel-Pro Analyzer 32 program (Media Cybernetics, USA). The levels of phospho-ERK1 and phospho-ERK2 were normalized to the levels of total ERK1 and ERK2.
Calcium imaging
SCG neurons were cultured at high density (50,000 cells per 35 mm dish) in medium containing 10 ng/ml NGF plus an inhibitor of TNFα processing (TAPI-0, 300 nM, Enzo Life Sciences) to inhibit cleavage of membrane integrated TNFα. Twelve hours after plating, the medium was changed to Ringer’s solution (125 mM NaCl, 4 mM KCl, 1.2 mM CaCl2, 0.5 mM MgCl2, 10 mM Glucose and 10 mM HEPES, pH 7.4) or containing 0.1% BSA and Fura-2 AM (1 mg/ml, Invitrogen). After a further 30 min at room temperature, the cells were washed 2 × 10 min with Ringer’s solution or Ca2+-free Ringer’s solution (Ca2+-free Ringer’s solution (125 mM NaCl, 4 mM KCl, 0.5 mM MgCl2, 10 mM Glucose and 10 mM HEPES, pH 7.4.) and were treated with either 5 μg/ml TNFR1-Fc in Ringers solution or Ca2+-free Ringers solution or 5 μg/ml Fc fragment in Ringers solution. Time lapse imaging of the 340/380 nm ratio was carried out at intervals using a Zeiss Axiovert 200 fluorescence microscope. At least 20 neurons were imaged per condition in each experiment, and the mean percentage change in the 340/380 nm ratio (minus background) was calculated.
Statistical analysis
Data are expressed as mean and standard errors. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those generally employed in the field.. Following normality test and homogeneity variance (F-test), group comparison were made using a t-test or one-way ANOVA as appropriate followed by Fisher’s post hoc test for normal distributed data. Nonparamentric test, Kruskal-Wallis test was performed for data that was not normal distributed. Differences were considered significant for p < 0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank Matthew White for providing RNA samples and Kevin Fox, Andrew Dick and Stanley Korsmeyer for providing tnfα, tnfr1 and bax mutant mice. This work was supported by grant from the Welcome Trust.
REFERENCES
- 1.Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Ann. Rev. Neurosci. 2003;26:509–563. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- 2.Glebova NO, Ginty DD. Growth and survival signals controlling sympathetic nervous system development. Ann. Rev. Neurosci. 2005;28:191–222. doi: 10.1146/annurev.neuro.28.061604.135659. [DOI] [PubMed] [Google Scholar]
- 3.Davies AM. Extracellular signals regulating sympathetic neuron survival and target innervation during development. Auto.n Neurosci. 2009;151:39–45. doi: 10.1016/j.autneu.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Glebova NO, Ginty DD. Heterogeneous requirement of NGF for sympathetic target innervation in vivo. J. Neurosci. 2004;24:743–751. doi: 10.1523/JNEUROSCI.4523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desbarats J, et al. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat. Cell Biol. 2003;5:118–125. doi: 10.1038/ncb916. [DOI] [PubMed] [Google Scholar]
- 6.Zuliani C, et al. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1) Cell Death Diff. 2006;13:31–40. doi: 10.1038/sj.cdd.4401720. [DOI] [PubMed] [Google Scholar]
- 7.O’Keeffe GW, Gutierrez H, Pandolfi PP, Riccardi C, Davies AM. NGF-promoted axon growth and target innervation requires GITRL-GITR signaling. Nat. Neurosci. 2008;11:135–142. doi: 10.1038/nn2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neumann H, et al. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J. Neurosci. 2002;22:854–862. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutierrez H, O’Keeffe GW, Gavalda N, Gallagher D, Davies AM. Nuclear factor kappa B signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J. Neurosci. 2008;28:8246–8256. doi: 10.1523/JNEUROSCI.1941-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavalda N, Gutierrez H, Davies AM. Developmental regulation of sensory neurite growth by the tumor necrosis factor superfamily member LIGHT. J. Neurosci. 2009;29:1599–1607. doi: 10.1523/JNEUROSCI.3566-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez H, Kisiswa L, O’Keeffe GW, Smithen MJ, Wyatt S, Davies AM. Regulation of neurite growth by tumour necrosis superfamily member RANKL. Open Biol. 2013;3:120150. doi: 10.1098/rsob.120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun M, Fink PJ. A new class of reverse signaling costimulators belongs to the TNF family. J. Immunol. 2007;179:4307–4312. doi: 10.4049/jimmunol.179.7.4307. [DOI] [PubMed] [Google Scholar]
- 14.Eissner G, et al. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J. Immunol. 2000;164:6193–6198. doi: 10.4049/jimmunol.164.12.6193. [DOI] [PubMed] [Google Scholar]
- 15.Waetzig GH, et al. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. Faseb J. 2005;19:91–93. doi: 10.1096/fj.04-2073fje. [DOI] [PubMed] [Google Scholar]
- 16.Xin L, et al. Dual regulation of soluble tumor necrosis factor-alpha induced activation of human monocytic cells via modulating transmembrane TNF-alpha-mediated ‘reverse signaling’. Int. J. Mol. Med. 2006;18:885–892. [PubMed] [Google Scholar]
- 17.Yu M, et al. Influence of reverse signaling via membrane TNF-alpha on cytotoxicity of NK92 cells. Euro. J. Cell Biol. 2009;88:181–191. doi: 10.1016/j.ejcb.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Black RA, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 19.Deckwerth TL, et al. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 20.Vizard TN, et al. Regulation of axonal and dendritic growth by the extracellular calcium-sensing receptor. Nat. Neurosci. 2008;11:285–291. doi: 10.1038/nn2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korner H, et al. Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Euro. J. Immunol. 1997;27:2600–2609. doi: 10.1002/eji.1830271020. [DOI] [PubMed] [Google Scholar]
- 22.Pfeffer K, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 23.Kirchner S, et al. LPS resistance in monocytic cells caused by reverse signaling through transmembrane TNF (mTNF) is mediated by the MAPK/ERK pathway. J. Leuco. Biol. 2004;75:324–331. doi: 10.1189/jlb.0703343. [DOI] [PubMed] [Google Scholar]
- 24.Gomez N, Cohen P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature. 1991;353:170–173. doi: 10.1038/353170a0. [DOI] [PubMed] [Google Scholar]
- 25.Thompson J, Dolcet X, Hilton M, Tolcos M, Davies AM. HGF promotes survival and growth of maturing sympathetic neurons by PI-3 kinase- and MAP kinase-dependent mechanisms. Mol. Cell Neurosci. 2004;27:441–452. doi: 10.1016/j.mcn.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Goold RG, Gordon-Weeks PR. The MAP kinase pathway is upstream of the activation of GSK3beta that enables it to phosphorylate MAP1B and contributes to the stimulation of axon growth. Mol. Cell Neurosci. 2005;28:524–534. doi: 10.1016/j.mcn.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Favata MF, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 28.Watts AD, et al. A casein kinase I motif present in the cytoplasmic domain of members of the tumour necrosis factor ligand family is implicated in ‘reverse signalling’. EMBO J. 1999;18:2119–2126. doi: 10.1093/emboj/18.8.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiegert JS, Bading H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: A link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium. 49:296–305. doi: 10.1016/j.ceca.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Park KM, Bowers WJ. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell. Sig. 2010;22:977–983. doi: 10.1016/j.cellsig.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reich A, Spering C, Schulz JB. Death receptor Fas (CD95) signaling in the central nervous system: tuning neuroplasticity? Trends Neurosci. 2008;31:478–486. doi: 10.1016/j.tins.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Xanthoulea S, et al. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004;200:367–376. doi: 10.1084/jem.20040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Islam A, et al. Extracellular TNFR1 release requires the calcium-dependent formation of a nucleobindin 2-ARTS-1 complex. J. Biol. Chem. 2006;281:6860–6873. doi: 10.1074/jbc.M509397200. [DOI] [PubMed] [Google Scholar]
- 34.Harashima S, et al. Outside-to-inside signal through the membrane TNF-alpha induces E-selectin (CD62E) expression on activated human CD4+ T cells. J. Immunol. 2001;166:130–136. doi: 10.4049/jimmunol.166.1.130. [DOI] [PubMed] [Google Scholar]
- 35.Barker V, Middleton G, Davey F, Davies AM. TNFalpha contributes to the death of NGF-dependent neurons during development. Nat. Neurosci. 2001;4:1194–1198. doi: 10.1038/nn755. [DOI] [PubMed] [Google Scholar]
- 36.Takei Y, Laskey R. Tumor necrosis factor alpha regulates responses to nerve growth factor, promoting neural cell survival but suppressing differentiation of neuroblastoma cells. Mol. Biol. Cell. 2008;19:855–864. doi: 10.1091/mbc.E07-06-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies AM, Lee KF, Jaenisch R. p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron. 1993;11:565–574. doi: 10.1016/0896-6273(93)90069-4. [DOI] [PubMed] [Google Scholar]
- 38.Gutierrez H, Davies AM. A fast and accurate procedure for deriving the Sholl profile in quantitative studies of neuronal morphology. J. Neurosci. Meth. 2007;163:24–30. doi: 10.1016/j.jneumeth.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Ronn LC, et al. A simple procedure for quantification of neurite outgrowth based on stereological principles. J. Neurosci. Meth. 2000;100:25–32. doi: 10.1016/s0165-0270(00)00228-4. [DOI] [PubMed] [Google Scholar]
- 40.Gallagher D, et al. Nuclear factor-kappaB activation via tyrosine phosphorylation of inhibitor kappaB-alpha is crucial for ciliary neurotrophic factor-promoted neurite growth from developing neurons. J. Neurosci. 2007;27:9664–9669. doi: 10.1523/JNEUROSCI.0608-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.