

Abstract
Antigen-binding Fc fragments (Fcab) are generated by engineering the C-terminal loop regions in the CH3 domain of human immunoglobulin G class 1-crystallizable fragment (IgG1-Fc). For an optimum library design with high percentage of well-folded clones for efficient binder selection, information about the correlation between primary structure and stability is needed. Here, we present a rapid method that allows determination of the overall stability of whole libraries of IgG1-Fc on the surface of yeast by flow cytometry. Libraries of IgG1-Fc mutants with distinct regions in AB-, CD- and EF-loops of the CH3 domains randomized or carrying therein insertions of five additional residues were constructed, incubated at increasing temperatures and probed for residual binding of generic Fc ligands. Calculated temperatures of half-maximal irreversible denaturation of the libraries gave a clear hierarchy of tolerance to randomization of distinct loop positions. Experimental data were evaluated by a computational approach and are discussed with respect to the structure of IgG1-Fc and variation in sequence and length of these loops in homologous Fc proteins. Generally, the described method allows for quick assessment of the effects of randomization of distinct regions on the foldability and stability of a yeast-displayed protein library.
Keywords: Fcab, IgG1-Fc, library design, loop engineering, therapeutic antibody, thermal denaturation, yeast surface display
Introduction
Monoclonal antibodies (mAbs) are the most rapidly growing and successful drug class in the clinic today. They are important therapeutics for the treatment of cancer as well as immunological and infectious diseases (Nelson et al., 2010) due to high specificity and potency in binding virtually any antigen. The first generation of antibody drugs simply mimicked the natural immunoglobulin G class 1 (IgG1) topology with two antigen-binding fragments (Fabs) and one crystallizable fragment (Fc). The latter interacts with the neonatal Fc receptor (FcRn) resulting in a prolonged in vivo half-life, binds to Fcγ-receptors (e.g. FcγRIIIa, CD16a) triggering antibody-dependent cell-mediated cytotoxicity (ADCC) and interacts with C1q initiating the classical pathway of the complement system, resulting in complement-dependent cytotoxicity (CDC) (Kubota et al., 2009).
The next generation of therapeutic mAbs focused on making smaller fragments of the full IgG (150 kDa), including single-chain variable fragments (scFvs) (27 kDa), Fabs (50 kDa), minibodies (80 kDa) and various scFv- and Fab-based multimers (Holliger and Hudson, 2005). Recently, even smaller alternative binding domains have been engineered (e.g. DARPins or affibodies) (Orlova et al., 2006; Zahnd et al., 2006) and more recently there has been a rapid increase in design of multifunctional antibodies with multiple binding scaffolds. However, many of these new formats suffer from the absence of binding sites for ligands that trigger ADCC, CDC or that mediate long half-life.
One interesting development is the use of the Fc fragment of IgG1 as starting scaffold for engineering. The Fc protein has—with the exception of an antigen-binding site—all the properties of a full-size IgG, i.e. the ability to bind Fcγ-receptors, the complement activator C1q and FcRn. However, since the immunoglobulin fold is known to tolerate variability in both the length and sequence of structural loops that connect the β-strands (Halaby et al., 1999), these loops can be used for the generation of novel binding sites. Upon engineering the C-terminal loops in the CH3 domains of human IgG1-Fc (Fig. 1A and B), antigen-binding Fc fragments (i.e. Fcab) could be selected that were able to bind to HER2/neu (Wozniak-Knopp et al., 2010) or αvβ3-integrin (Traxlmayr et al., 2012c). A HER2/neu-binding Fcab has been demonstrated to elicit ADCC and to have a long half-life in vivo at only one-third of the molar mass (∼50 kDa) of a full-size IgG1 molecule (Wozniak-Knopp et al., 2010; Kainer et al., 2012).
Fig. 1.

(A) Crystal structure of the CH3 domain of human IgG1 (PDB code 1OQO), showing the typical immunoglobulin fold composed of two β-sheets formed by three and four β-strands, respectively. The loops connecting these β-strands at the C-terminus of the CH3 domain are shown in blue (AB-loop), red (CD-loop) and yellow (EF-loop). (B) Multiple sequence alignment of IgG1-CH3 of 12 species. Loop regions are colored according to the ClustalW color code in order to emphasize possible conservation of amino acid side chain character (Larkin et al., 2007).
However, introduction of novel binding sites might impair the immunoglobulin fold thereby decreasing the conformational and thermal stability of the respective proteins. Successful stability engineering of an Fcab by rational mutagenesis (Wozniak-Knopp and Rüker, 2012) and a directed evolution strategy based on the work of Shusta et al. (2000) have been reported recently. The latter approach included soft randomization of distinct loop regions of the parental binder followed by heat incubation of the yeast-displayed protein library and selection for retained antigen binding (Traxlmayr et al., 2013). Nevertheless, the systematic usage of C-terminal AB-, CD- and EF-loops of the IgG1-Fc CH3 domains (Fig. 1A and B) for the design of novel therapeutic mAbs needs a more systematic evaluation of (i) those amino acid positions that can be randomized without significantly decreasing the thermal stability of the resulting variants as well as of (ii) those sites in loops that allow insertion of additional amino acid residues in order to increase loop length and thus the potential interaction surface with antigens. Since these questions cannot be answered by expression and biophysical characterization of individual variants, a more rapid and comprehensive method had to be applied.
Here, we have determined the relative thermal stability of distinct IgG1-Fc libraries displayed on the surface of yeast by using flow cytometry. The applied protocol is based on work by Orr et al. (2003) where a clear correlation was reported between the denaturation temperature of the solubly expressed and the respective yeast displayed form of individual single-chain variable fragment (scFv) and single-chain T cell receptor mutants. This allowed rapid comparison of primary structural information and stability. We adapted this method for the determination of half-maximum irreversible denaturation temperatures of entire IgG1-Fc libraries having either distinct (overlapping) regions in the AB-, CD- and/or EF-loop randomized or carrying insertions at certain positions. Additionally, using in silico methods the medians of change in free energy of unfolding of each individual loop position being replaced by the other 19 amino acids as well as of all library designs (i.e. sum of medians of change in free energy of unfolding calculated for the randomized residues according to the different library designs) have been calculated and compared with the wild-type protein. Experimental and computational data correlate strongly and are discussed with respect to the structure of IgG1-Fc. Our findings provide important criteria for the design of Fcab libraries with a high percentage of well-folded mutants with high thermostability. In general, the described method allows for quick assessment of potential destabilization upon randomization of distinct regions of a protein that can be displayed on yeast.
Materials and methods
Cloning and library construction
The gene coding for human IgG1-Fc (Hinge region, CH2 and CH3 domains) was codon-optimized for the expression in yeast and cloned into pYD1 (Invitrogen, Carlsbad, CA, USA) for Saccharomyces cerevisiae surface expression using the restriction sites for BamHI and NotI (Boder and Wittrup, 1997). A stop codon was introduced at the 3′ end of the region coding for the CH3 domain to exclude C-terminal tags present on pYD1. For the use in the construction of CD- and EF-loop libraries, two novel BsmBI restriction sites were introduced upstream of the region coding for the CD-loop of the CH3 domain and downstream of the EF-loop (QuikChange Lightning Site-Directed Mutagenesis Kit, Agilent Technologies, Santa Clara, CA, USA). Accordingly, a non-coding stuffer fragment was modified by introducing two BsmBI restriction sites. Both plasmid and stuffer fragment were then digested with BsmBI, followed by treatment of the linearized vector with CIAP and ligation using the T4 DNA ligase, yielding the vector pYD1-2BN-CDEF, which would not lead to surface display of wild-type IgG1-Fc as a consequence of incomplete BsmBI digest or spontaneous religation during transformation. Likewise, for the construction of AB-loop libraries the region coding for AB- to EF-loop was replaced by a non-coding stuffer fragment carrying two BsmBI restriction sites at the respective positions to yield the vector pYD1-2BN-AB upon restriction digest with BsmBI.
The introduction of randomized loop sequences was done by saturated mutagenesis using NNK oligonucleotides (N codes for a mixture of all four nucleotides, whereas K represents a mix of G and T; ordered from Sigma, St. Louis, MO, USA) in a multi-step polymerase chain reaction, yielding fragments comprising upstream and downstream regions of homology to the linearized vector backbone for homologous recombination in yeast and (partially) randomized sequences in the regions coding for AB-, CD- or EF-loop.
Saccharomyces cerevisiae EBY100 (Invitrogen) were transformed with the gel-purified library inserts together with BsmBI-digested pYD1-2BN using the lithium acetate method (Gietz and Schiestl, 2007). Plasmids were reconstituted by gap repair driven homologous recombination in S.cerevisiae due to the presence of homologous regions in the insert and the BsmBI-digested pYD1-2BN. The S.cerevisiae libraries were grown in SD-CAA medium [20 g/l glucose, 0.1 M KH2PO4/K2HPO4, pH 6, 10 g/l (NH4)2SO4, 0.1 g/l l-leucine (all Sigma), 3.4 g/l yeast nitrogen base, 10 g/l bacto casamino acids (all Difco, BD, Franklin Lakes, NJ, USA)] at 28°C for 48 h. The Zymoprep Yeast Plasmid Miniprep Kit II (Zymo Research, Orange, CA, USA) was used to isolate pYD1-2BN vector DNA, which was then used for Escherichia coli transformation and ensuing sequencing.
Expression of libraries in the yeast surface display format
Saccharomyces cerevisiae libraries were grown over night in SD-CAA medium, passaged to an OD600 of 1, incubated at 28°C for 4 h while shaking and centrifuged and resuspended in SGR-CAA-medium [20 g/l galactose, 10 g/l raffinose, 0.1 M KH2PO4/K2HPO4, pH 6, 10 g/l (NH4)2SO4, 0.1 g/l l-leucine (all Sigma), 3.4 g/l yeast nitrogen base, 10 g/l bacto casamino acids (all Difco, BD)] to an OD600 of 1. The SGR-CAA cultures were induced for surface expression at 20°C for 18–20 h, centrifuged and resuspended in phosphate-buffered saline (PBS)/bovine serum albumin (BSA) [0.2 g/l KCl, 0.2 g/l KH2PO4, 8 g/l NaCl, 1.15 g/l Na2HPO4 (anhydrous), 20 g/l bovine serum albumin (Sigma)] to an OD600 of 3. An induction temperature of 20°C was chosen in order to avoid a pre-selection by the protein quality control system of yeast (Traxlmayr and Obinger, 2012). The cell suspensions were aliquoted, transferred to microcentrifuge tubes and incubated for 30 min on ice or at 50, 55, 60, 65, 70 or 75°C, respectively, in thermomixers (Eppendorf, Hamburg, Germany) upon shaking at 300 rpm. After cooling on ice, 50 µl of each aliquot were transferred to a round-bottom 96-well-plate (TPP, Trasadingen, Switzerland) followed by incubation at 4°C for 30 min. After centrifugation, the cells were stained with 2.5 µg/ml anti-Xpress antibody (Invitrogen) conjugated to allophycocyanin (APC) using the LYNX Rapid APC Antibody Conjugation Kit (AbD Serotec, Kidlington, UK) and with either 2 μg/ml fluorescein isothiocyanate isomer 1-labeled anti-human IgG CH2-domain antibody (anti-CH2-FITC, clone MK 1 A6, AbD Serotec) or 1 μg/ml His-tagged Fcγ-receptor I (FcγRI) (R&D Systems, Abingdon, UK). For staining with FcγRI, cells were washed with PBS/BSA and stained with 1 μg/ml anti-His antibody conjugated to Alexa Fluor 488 (QIAGEN, Venlo, Netherlands). All stainings were done in PBS/BSA at 4°C for 30 min, shaking. Following a terminal washing step, cells were resuspended in PBS/BSA and analyzed on a FACSCanto II (BD).
Flow cytometric measurements and data analysis
During flow cytometric measurements, 10 000 events were measured and gated for the (i) desired morphology, (ii) presence as singlets, (iii) display of the expression tag (Xpress epitope at the N-terminus of the Fc) and, finally, (iv) the fluorescence signal of the respective fluorescence-labeled ligand bound to the displayed Fc protein was measured. Resulting data were analyzed using the FACSDiva software (BD). To derive a measure of overall library stability from flow cytometric data, temperatures of half-maximal irreversible denaturation (T1/2) were calculated for each distinct library as previously described by Orr et al. (2003) for individually displayed scFvs. The mean fluorescence intensity (MFI) of anti-Xpress positive yeast cells displaying wild-type Fc on the cell surface, stained with fluorescence-labeled FcγRI and incubated at 0°C was set to 100% relative MFI, while the lowest MFI of a measurement series was set to 0%. Resulting data were fitted according to 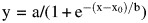 , where x corresponds to the respective incubation temperatures, y corresponds to the relative MFI after heat incubation, a and b correspond to the maximum and minimum y values as defined by the model and x0 corresponds to T1/2.
, where x corresponds to the respective incubation temperatures, y corresponds to the relative MFI after heat incubation, a and b correspond to the maximum and minimum y values as defined by the model and x0 corresponds to T1/2.
For validation of the method described above midpoints of thermal denaturation of soluble wild-type and mutant IgG1-Fc determined by differential scanning calorimetry (DSC) (Traxlmayr et al., 2013) were compared with the T1/2 values obtained from flow cytometric analysis of the corresponding yeast-displayed mutants incubated at increasing temperatures.
Prediction of ΔΔG values using FoldX
We used the computer algorithm FoldX (Guerois et al., 2002; Schymkowitz et al., 2005) to mutate each loop residue to the other 19 amino acids. FoldX predicts the effect of point mutations on protein stability in terms of change in free energy of unfolding. In a first step, the FoldX command <RepairPDB> was used to repair the crystal structure of human IgG1-Fc (PDB ID 1OQO), identifying residues with bad torsion angles or van der Waals clashes. Following this, we applied <BuildModel> to the C-terminal loop residues of chains A and B of the repaired structure. Using this function, every amino acid of the CH3 domain was exchanged by the other possible 19 amino acids and values for the change in free energy of unfolding (ΔΔG = ΔGmutant− ΔGwild-type) for each mutation at each position were predicted. We calculated the median of all determined ΔΔG values at each loop position to estimate the overall destabilization upon mutation of each particular residue. Median values of loop positions were then summed up according to the respective library designs to predict the effect of randomization of more than one residue.
Results
Validation of the method
In the Fc fragment of IgG1, the main contributions to the formation of the homodimeric structure stem from the two disulfide bonds in the hinge region as well as from extensive interactions between residues of the two CH3 domains and to a lesser degree from contacts between the carbohydrate moieties linked to residue N297 in the CH2 domains (Dall'Acqua and Carter, 1998). Recently, it has been demonstrated that mutation of the C-terminal loops of the CH3 domains of human IgG1-Fc can impair the folding and in consequence the conformational and thermal stability of these proteins. The decrease in thermal stability strongly depended on the actual position being altered (Traxlmayr et al., 2012a,b,c; Traxlmayr et al., 2013). This clearly underlined the need of a method for quick assessment of destabilization effects due to randomization of distinct loop regions in order to design Fc libraries with a high percentage of well-folded clones.
Initially, based on the work by Orr et al. (2003) on scFv and scTCR, we validated the use of yeast display for quantitation of thermal stability of Fc variants by using flow cytometry. Wild-type Fc, an Fcab (H10-03-06) binding to HER2/neu (Wozniak-Knopp et al., 2010) and variants stabilized by in vitro directed evolution (STAB1, STAB5, STAB11, STAB14, STAB15 and STAB19, respectively) (Traxlmayr et al., 2013) were expressed in Pichia pastoris and HEK-295 cells and evaluated for thermal stability by DSC. Table I summarizes the observed endothermic midpoint transition temperatures of the CH3 domain of the soluble proteins. As previously reported, wild-type Fc heterologously expressed in P.pastoris shows the first Tm1 at 66°C representing the unfolding of the CH2 domains, and Tm2/Tm3 values at 78.0 and 82.6°C, respectively, reflecting the thermal denaturation of the CH3 domains. By contrast, during unfolding of the HEK-293 expressed protein only two endothermic transitions occur corresponding to melting of CH2 (71°C) followed by CH3 domains (82.4°C). In the parental HER2/neu binder (H10-03-06) only one broad transition at ∼65.9°C (P.pastoris) or 69.5°C (HEK-293) was seen, whereas in the STAB variants the endothermic transitions shifted to higher temperatures (Table I) covering a range of Tm values from 66 to 82°C (for P.pastoris produced proteins) and 69–82°C (for HEK-293 produced proteins). The same set of proteins was displayed on the surface of S.cerevisiae by expressing them as fusions to the yeast cell wall protein Aga2p. Yeast suspensions were incubated at increasing temperatures (0, 50, 55, 60, 65, 70 and 75°C) for 30 min, cooled and analyzed for binding to the fluorescently labeled structural markers FcγRI (CD64) and CH2 domain-specific antibody (aCH2). Residual binding after incubation at each temperature with respect to the binding of non-heat-incubated wild-type protein was plotted against the respective heat shock temperature (Fig. 2A). From the sigmoidal fit of the resulting data points T1/2 values representing the temperatures of half-maximum irreversible denaturation could be calculated as described in the Materials and methods Section (Orr et al., 2003; Table I).
Table I.
Comparison of the thermal stability of solubly expressed wild-type human IgG1-Fc and Her2/neu-binding Fc variants (Fcabs) with the corresponding yeast surface displayed forms.
| P.pastoris | HEK-293 | FcγRI | aCH2 | |
|---|---|---|---|---|
| IgG1-Fc variant | Tm (°C) | Tm (°C) | T1/2 (°C) | T1/2 (°C) |
| Wild-type human IgG1-Fc | 78.0, 82.6 | 82.4 | 64.5 ± 0.8 | 66.1 ± 0.8 |
| H10-03-06 | 65.9 | 69.5 | 56.4 ± 0.1 | 55.9 ± 0.1 |
| STAB1 | 68.8 | 70.0 | 58.0 ± 0.2 | 58.1 ± 0.2 |
| STAB5 | 68.9 | 71.4 | 59.0 ± 0.3 | 59.4 ± 0.1 |
| STAB11 | 69.9 | n.d. | 59.7 ± 0.2 | 59.9 ± 0.3 |
| STAB14 | 70.2 | n.d. | 59.1 ± 0.2 | 59.3 ± 0.1 |
| STAB15 | 68.1 | 70.0 | 57.1 ± 0.2 | 57.5 ± 0.3 |
| STAB19 | 74.5 | 75.3 | 60.7 ± 0.4 | 61.4 ± 0.3 |
Soluble proteins were heterologously expressed in P.pastoris and HEK-293 cells and characterized by DSC (Traxlmayr et al., 2013). The displayed formats were characterized by flow cytometry for residual binding to either FcγRI or an antibody that recognizes intact CH2 domain (aCH2) after heat incubation of the yeast cells. Resulting temperatures of half-maximal irreversible denaturation (T1/2) of the displayed variants were calculated and compared with midpoints of denaturation (Tm) measured for solubly expressed Fcabs by DSC. Only thermal unfolding data of the CH3 domain are depicted.
n.d., not determined.
Fig. 2.

Comparison of thermal stability of soluble HER2/neu-binding human IgG1-Fc variants and wild-type protein with that of the yeast surface-displayed formats. (A) Temperature exposure of wild-type IgG1-Fc and two representative variants expressed on the surface of yeast cells. The residual binding of the Fc proteins to FcγRI is plotted versus the incubation temperature (100% corresponds to non-heat treated wild-type IgG1-Fc binding to the ligand). Resulting data were fitted according to the depicted equation yielding T1/2 values, which correspond to the irreversible denaturation of the CH3 domain of the displayed protein. These values were used to calculate ΔT1/2 that relates the stability of respective protein to that of the wild-type IgG1-Fc. (B) Correlation between midpoint temperatures of denaturation (Tm) as determined for soluble proteins by DSC with flow cytometrically measured T1/2 values of the yeast-displayed formats (see also Table I). T1/2 values were determined either by assessing residual binding to FcγRI or to an anti-CH2-antibody (aCH2).
Comparison of DSC-derived Tm values for solubly expressed proteins with flow cytometrically determined T1/2 values for the respective yeast surface displayed proteins showed a strong correlation as demonstrated in Fig. 2B. This is valid for all possible combinations of data, i.e. correlation of T1/2 values from flow cytometric analyses that probed the residual FcγRI binding with Tm values for P.pastoris or HEK-293 produced soluble proteins as well as correlation of T1/2 values calculated from flow cytometric analyses that probed CH2 structure using binding of the specific antibody with the Tm values of the soluble counterparts. Evaluating the irreversible denaturation of IgG1-Fc fusions on the yeast surface by flow cytometry was highly reproducible and thus allowed rapid comparison of primary structural information of these libraries with their overall stability.
Design, construction and characterization of model and insertion libraries
In the next step, we determined the T1/2 values of distinct Fc loop libraries aiming at analyzing the destabilizing effect of randomization of amino acids in varying sequence context in the three relevant C-terminal loops. For this purpose we designed ‘model’ libraries as summarized in Table II. Additionally, we planned to identify suitable sites for insertion of five additional residues, which would increase the number of available topologies to select for binding to antigens of different sizes and geometries.
Table II.
Determination of the effect of engineering of the C-terminal AB-, CD- and EF-loops of the CH3 domains (Fig. 1) of IgG1-Fc on the structural fitness of the resulting libraries
| AB-loop libraries (RDELTKNQ) | ΔT1/2 (°C) | CD-loop libraries (SNGQPENNY) | ΔT1/2 (°C) | EF-loop libraries (DKSRWQQGNV) | ΔT1/2 (°C) |
|---|---|---|---|---|---|
| LT | −2.8 ± 0.7 | SN | −2.1 ± 0.8 | QQGNV | −3.3 ± 0.1 |
| LTK | −3.9 ± 0.5 | NG | −2.8 ± 0.9 | RWQQGNV | −5.0 ± 0.2 |
| LTKN | −3.5 ± 0.7 | GQ | −3.0 ± 0.6 | DKSRWQQGNV | −8.0 ± 0.6 |
| TK | −1.1 ± 0.4 | QP | −1.8 ± 0.7 | DK | −2.1 ± 0.3 |
| TKN | −1.1 ± 0.7 | PE | −2.9 ± 0.6 | KS | −2.4 ± 0.2 |
| KN | −1.7 ± 0.5 | EN | −3.1 ± 0.9 | DKS | −2.4 ± 0.3 |
| RDELTKN | −9.7 ± 1.0 | NN | −1.4 ± 0.8 | DKSRW | −5.3 ± 0.3 |
| NY | −4.4 ± 0.7 | RW | −3.6 ± 0.2 | ||
| DKSrwQQGNV | −6.3 ± 0.4 | ||||
| AB insertion libraries |
CD insertion libraries |
EF insertion libraries |
|||
| LT5 | −5.7 ± 0.9 | SN5 | −4.4 ± 0.4 | DK5 | −8.7 ± 0.5 |
| LTK5 | −6.0 ± 0.8 | NG5 | −3.7 ± 0.3 | KS5 | −8.3 ± 0.7 |
| LTKN5 | −5.2 ± 0.8 | GQ5 | −4.4 ± 0.2 | ||
| TK5 | −3.8 ± 0.7 | QP5 | −4.5 ± 0.1 | ||
| TKN5 | −4.0 ± 0.9 | PE5 | −5.4 ± 0.2 | ||
| KN5 | −3.9 ± 0.7 | EN5 | −5.3 ± 0.1 | ||
| NN5 | −5.1 ± 0.1 | ||||
| NY5 | −7.0 ± 0.1 |
The randomized residues in the respective loops are indicated. Furthermore, insertion libraries with five additional residues inserted after the (also randomized) amino acid positions were designed. Constructed libraries were expressed on the surface of yeast and cell suspensions were incubated at increasing temperatures and (after cooling) were probed for binding to FcγRI using flow cytometry. Obtained ΔT1/2 values (for calculation see Fig. 2 and the Materials and methods Section) describe the overall destabilization of a distinct library as a consequence of randomization with respect to the displayed wild-type protein.
In detail, library designs were based on the definition of loop regions according to information drawn from (i) visual inspection of the crystal structure of IgG1-Fc, (ii) secondary structure prediction using DSSP (Kabsch and Sander, 1983), (iii) previous work (Traxlmayr et al., 2012b; Wozniak-Knopp et al., 2010) and (iv) sequence alignment of the IgG1-CH3 domains from 12 species as depicted in Fig. 1 (Larkin et al., 2007). Residues R355 to N361 were defined as the loop connecting strands A and B of the CH3 domain (AB-loop), residues S383 to Y391 as CD-loop and residues D413 to V422 as EF-loop (Fig. 1 and Table II) (Eu numbering system according to Kabat et al., 1991). For AB- and CD-loop libraries a sliding-window design was applied (Table II). Since the EF-loop contains a stabilizing motif (R416, W417) as determined in previous experiments (Wozniak-Knopp et al., 2010; Traxlmayr et al., 2012a,b,c), in EF-loop libraries residues N- and C-terminal of R416-W417 were randomized. Additionally, insertion libraries were created according to a sliding-window design with five additional residues inserted after the indicated (also randomized) amino acids (Table II).
These libraries were constructed to a size of 106 and expressed on the surface of yeast. The cell suspensions were incubated at increasing temperatures for 30 min, cooled and then probed for binding to FcγRI or aCH2 using flow cytometry. From the resulting sigmoidal curves, T1/2 values were calculated as described above. Data are presented as ΔT1/2 values that describe the destabilization of a library as a consequence of randomization of distinct loop regions with respect to the wild-type protein (T1/2 = 64.5°C). Table II as well as Fig. 3 summarizes those ΔT1/2 values for all constructed libraries.
Fig. 3.

ΔT1/2 values for model and insertion libraries with respect to the wild-type protein displayed on the yeast cell surface. Respective loop sequences are shown on the horizontal axes. Black horizontal bars represent the residues that were randomized in each model library (see also Table II). Gray horizontal bars represent randomized residues with five additional residues introduced at this site in each insertion library.
Randomization of the whole AB-loop significantly destabilized the resulting library (ΔT1/2 = −9.7°C). AB-loop libraries having L358 randomized (LT, LTK, LTKN) were destabilized by −2.8 to −3.5°C, whereas those that did not include this position in the design (TK, TKN, KN) were less affected (−1.1 to −1.7°C). A similar relation could be observed for AB-loop insertion libraries, where including L358 in the design resulted in ΔT1/2 values being significantly more negative compared with those that did not include this position (Table II).
Evaluation of optimum positions for randomization in the CD-loop showed that randomizations at S383, N384, Q386, P387, N389 and N390 were well tolerated, whereas including G385, E388 and Y391 in the design destabilized the libraries (Table II). Insertion of five additional residues in the CD-loop is best tolerated in the region after N384-G385, whereas insertion in the C-terminal part CD-loop significantly destabilized the library (Table II).
Randomization of the EF-loop positions gave ΔT1/2 values of −8.0°C. All designs that included R416-W417 exhibited the most pronounced destabilization effects (Table II), thereby confirming the important role of these two residues in the conformational stability of IgG1-Fc (Wozniak-Knopp et al., 2010; Traxlmayr et al., 2012b). By contrast, randomization N- and C-terminal of this motif (i.e. designs including D413, K414, S415 as well as Q418, Q419,G420, N421, V422) was tolerated. Insertion of five residues N-terminal of the RW motif drastically decreased the structural fitness of the resulting libraries.
We also hypothesized, that in addition to using the MFI from the fluorescence-activated cell sorting (FACS) histograms for calculation of T1/2 values, further information could be obtained from the variance of the fluorescence intensities across the library population, i.e. the width and shape of the histogram. Thus, we have looked at the respective FACS histograms of the displayed wild-type protein and model libraries incubated at distinct temperatures and calculated the variance of the fluorescence intensities across the library population that still bound to CD64. However, no correlation between incubation temperatures or respective positions of randomization and the coefficients of variance could be observed. The coefficients of variance of the population of wild-type IgG1-Fc still binding to CD64 after incubation at increasing temperatures was more or less constant around 50%, whereas for the libraries the coefficients varied between 50 and 70%. Thus, the variance of the fluorescence intensities across populations cannot be used as an additional measure of quality. Apparently, the potential differences are not observable due to intrinsic dispersion of the fluorescent signals.
Evaluation of experimental data
Supplementary Fig. S1 shows that the determined ΔT1/2 values correlate roughly with the residual average binding of the respective libraries to a structural marker at room temperature (i.e. without heat incubation). The higher the ΔT1/2 values were, the lower was the percentage of residual binding to CD64 at 25°C in comparison with the wild-type protein.
For further validation of the experimental data, we performed FoldX calculations to investigate whether the observed destabilization of the described libraries could be predicted computationally. Each loop residue was replaced by the other 19 amino acids and the ΔΔG value for each single mutant was calculated. The median of all these ΔΔG values at one position served as a prediction value for a more general effect of randomization at this position. Median values were than summed up according to the designs of the model libraries (Table II) and plotted against the respective T1/2 values. Figure 4 clearly demonstrates that there is a correlation between high ΔΔG values and the experimentally determined destabilization of libraries represented by the T1/2 values. However, it must be emphasized that the computational method cannot substitute the experimental work as is evident by closer inspection of Fig. 4. For some library designs the correlation between ΔΔG values and T1/2 values was weak.
Fig. 4.
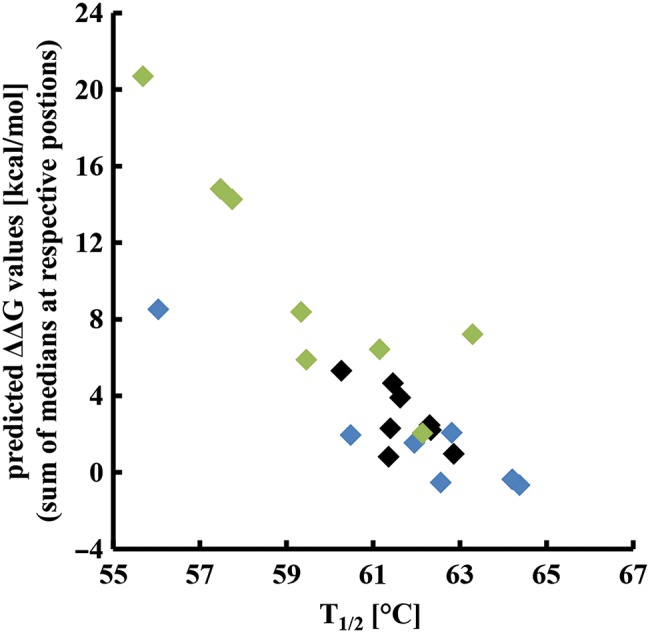
Correlation between computationally predicted ΔΔG values and experimentally determined T1/2 values. Using the FoldX function <BuildModel>, ΔΔG values determined for all possible mutations at the respective loop positions were calculated and median values for each position were determined. To estimate the additive effect of randomization of more than one residue, ΔΔG medians were summed up according to the different library designs and plotted against the experimentally obtained T1/2 values (Table II).
Discussion
Insertion of new functions (e.g. antigen-binding sites) into proteins often decreases the conformational and thermal stability in comparison with that of the parental scaffold. For an optimum library design, the availability of detailed information on the contribution of distinct amino acid positions or sections to the foldability and stability of the entire protein is crucial. Recently, we published a method that allows the construction of a stability landscape of a protein at single residue resolution by combining directed evolution with high-throughput sequencing (Traxlmayr et al., 2012b). Thereby, a clear picture of the contribution of individual residues to the overall stability of the CH3 domain was obtained. However, this method is not suitable for routinely probing and evaluating library designs since it accounts only for the contribution of single residues, whereas for the design of novel binding sites the knowledge about the potentially destabilizing effect of randomization of different loop residues at the same time is needed.
In this paper, we have introduced a new method that rapidly and reproducibly allows the comparison of different library designs, thereby further developing the work done by Orr et al. (2003). These authors have demonstrated that the hierarchy in thermal stability of soluble scFvs and scTCR as determined by various biophysical techniques is very well reflected by the hierarchy of thermal stability of the same proteins immobilized on the surface of yeast cells. This made it possible to eliminate the time-consuming purification and final characterization of large numbers of individual scFvs and scTCRs (Orr et al., 2003). We could show that this fact also holds true for more complex proteins like IgG1-Fc variants and, finally, have adapted this method to evaluate the overall relative thermal stability of entire IgG1-Fc libraries.
We have focused on libraries in which the C-terminal loops of the CH3 domains of IgG1-Fc were randomized. The knowledge about residues in the AB-, CD- and EF-loops (Fig. 1) that tolerate randomization is important in the design of Fcabs. In total, 24 libraries and 16 insertion libraries were constructed, displayed on yeast and probed for residual ligand binding after heat incubation. From the sigmoidal denaturation curves temperatures of half-maximum denaturation of the entire library pool were calculated, reflecting the average of thermal unfolding of thousands of individual clones.
As expected, randomization of the entire AB-loop resulted in strong destabilization of the library (−9.7°C) mainly due to disruption of the intra-loop 310 helix formed by R355-D356-E357 (Fig. 1). Additionally, introduction of L358 in the design destabilized the libraries, whereas the sequence T359–K360–N361 tolerates randomization better and thus should be included in library design for Fcab selection. Insertion of five additional residues is also better tolerated if the wild-type residue is kept constant at position 358. These findings nicely correlate with the published stability landscape of the CH3 domain of human IgG1 (Traxlmayr et al., 2012b) showing highest tolerance to mutation for residues R355, T359, K360 and N361 and strongly decreased tolerance for residues D356, E357 and L358.
The CD-loop connects the two β-sheets of the CH3 domain and is therefore supposed to be relatively flexible. The N-terminus of the loop is formed by a β-bend type II, consisting of S383, N384 and G385. The importance of glycine in bend formation is underlined by its destabilizing effect when it was randomized. By contrast, destabilization was less pronounced upon diversification of S383, N384 as well as Q386, P387, N389 and N390. Our findings suggest that E388 should not be included in the library design as glutamate 388 forms an ionic interaction with the guanidinium group of R416 in the EF-loop. Furthermore, Y391 should not be mutated and the four N-terminal positions of the CD-loop are significantly more tolerant to insertion than the C-terminal positions. Again, these results are in accordance with the stability landscape of the CH3 domain of IgG1 (Traxlmayr et al., 2012b).
As the EF-loop has a peculiar symmetry, we applied a different strategy during library design. We chose R416 and W417 as a central stabilizing motif, which was suggested by previous experiments and inspection of the crystal structure of IgG1-Fc. Arginine 416 forms an ionic interaction with E388 in the CD-loop, whereas the indole ring of W417 points to the hydrophobic core of the β-barrel of the CH3 domain thereby contributing to its hydrophobic packing (Fig. 1). Wozniak-Knopp et al. (2010) showed that—after randomizing R416 and W417 by parsimonious mutagenesis during affinity maturation of an Fcab binding to HER2/neu—both residues were recovered in the matured Fcab. Also, the stability landscape and the evolutionary conservation of these residues of the CH3 domain (Fig. 1B) clearly demonstrated that they are intolerant to mutation (Traxlmayr et al., 2012b). Moreover, the overall half-maximum denaturation temperatures of all libraries that included R416 and W417 were significantly decreased underlining the validity of the applied method. The other residues of the EF-loop can be included in the library design.
In summary, the presented method allows rapid evaluation of the correlation between primary structural information and stability of whole libraries. As model system we have investigated the optimum design of loop libraries of IgG1-Fc, suggesting three suitable positions (359–361) in the AB-loop, six in the CD-loop (383, 384, 386, 387, 389 and 390) and eight in the EF-loop (413–415, 418–422). These data will enable us to construct Fcab libraries containing a higher fraction of stable and well-folded protein variants. This, in turn, will result in increased functional library sizes for the selection of high affinity and stable Fcab clones.
Similar information can be obtained for all other engineering aspects of proteins that can be displayed on the surface of yeast cells. Flow cytometry allows fast analysis of thousands of clones within a distinct library, whereas cloning, expression, purification and characterization of a representative number of individual clones are not feasible. Of particular importance for the presented approach is the choice of the range of heat incubation temperatures as well as of the length of incubation. This must be established for each protein of interest and needs the investigation of the (un)folding pathway as well as its reversibility. If, for example, the temperature or the incubation time is too low and the library pool as well as the parental protein remains in the native fold, it will not be possible to detect a difference in the binding intensity to a structurally specific ligand between the more stable parental protein and the destabilized library pool. However, having established this knowledge, the presented approach will facilitate the identification of regions within a protein that are particularly tolerant or intolerant to mutation and therefore better suitable for insertion of new protein functions such as antigen binding.
Supplementary data
Funding
Funding to pay the Open Access publication charges for this article was provided by the Austrian Science Fund (FWF): W1224, Doctoral Program on Biomolecular Technology of Proteins, BioToP.
Supplementary Material
Acknowledgements
This work was supported by the Christian Doppler Research Association (Christian Doppler Laboratory for Antibody Engineering), the company F-star, as well as the “Austrian Science Fund” (FWF W1224—Doctoral Program on Biomolecular Technology of Proteins—BioToP). The authors want to thank Irene Schaffner and Elisabeth Lobner for excellent technical assistance.
Conflict of interest: G.W.-K. and F.R. hold stock in F-star. P.C.J. was an employee of F-star. No disclosures by M.W.T., C.H., G.S. and C.O.
References
- Boder E.T., Wittrup K.D. Nat. Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- Dall'Acqua W., Carter P. Curr. Opin. Struct. Biol. 1998;8:443–450. doi: 10.1016/s0959-440x(98)80121-8. [DOI] [PubMed] [Google Scholar]
- Gietz R.D., Schiestl R.H. Nat. Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- Guerois R., Nielsen J.E., Serrano L. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Halaby D.M., Poupon A., Mornon J. Protein Eng. 1999;12:563–571. doi: 10.1093/protein/12.7.563. [DOI] [PubMed] [Google Scholar]
- Holliger P., Hudson P.J. Nat. Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- Kabat E.A., Wu T.T., Perry H.M., Gottesman K.S., Foeller C. Sequences of Proteins of Immunological Interest. 5th edn. National Institutes of Health; 1991. pp. 688–696. [Google Scholar]
- Kabsch W., Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kainer M., Antes B., Wiederkum S., Wozniak-Knopp G., Bauer A., Rüker F., Woisetschläger M. Arch. Biochem. Biophys. 2012;526:154–158. doi: 10.1016/j.abb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Kubota T., Niwa R., Satoh M., Akinaga S., Shitara K., Hanai N. Cancer Sci. 2009;100:1566–1572. doi: 10.1111/j.1349-7006.2009.01222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M.A., Blackshields G., Brown N.P., et al. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Nelson A.L., Dhimolea E., Reichert J.M. Nat. Rev. Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- Orlova A., Magnusson M., Eriksson T.L., et al. Cancer Res. 2006;66:4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- Orr B.A., Carr L.M., Wittrup K.D., Roy E.J., Kranz D.M. Biotechnol. Prog. 2003;19:631–638. doi: 10.1021/bp0200797. [DOI] [PubMed] [Google Scholar]
- Schymkowitz J.W., Rousseau F., Martins I.C., Ferkinghoff-Borg J., Stricher F., Serrano L. Proc. Natl Acad. Sci. USA. 2005;102:10147–10152. doi: 10.1073/pnas.0501980102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shusta E.V., Holler P.D., Kieke M.C., Kranz D.M., Wittrup K.D. Nat. Biotechnol. 2000;18:754–759. doi: 10.1038/77325. [DOI] [PubMed] [Google Scholar]
- Traxlmayr M.W., Faissner M., Stadlmayr G., Hasenhindl C., Antes B., Rüker F., Obinger C. Biochim. Biophys. Acta. 2012a;1824:542–549. doi: 10.1016/j.bbapap.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxlmayr M.W., Hasenhindl C., Hackl M., Stadlmayr G., Rybka J.D., Borth N., Grillari J., Rüker F., Obinger C. J. Mol. Biol. 2012b;423:397–412. doi: 10.1016/j.jmb.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxlmayr M.W., Lobner E., Antes B., et al. Protein Eng. Des. Sel. 2013;26:255–265. doi: 10.1093/protein/gzs102. [DOI] [PubMed] [Google Scholar]
- Traxlmayr M.W., Obinger C. Arch. Biochem. Biophys. 2012;526:174–180. doi: 10.1016/j.abb.2012.04.022. [DOI] [PubMed] [Google Scholar]
- Traxlmayr M.W., Wozniak-Knopp G., Antes B., Stadlmayr G., Rüker F., Obinger C. J. Biotechnol. 2012c;155:193–202. doi: 10.1016/j.jbiotec.2011.06.042. [DOI] [PubMed] [Google Scholar]
- Wozniak-Knopp G., Bartl S., Bauer A., et al. Protein Eng. Des. Sel. 2010;23:289–297. doi: 10.1093/protein/gzq005. [DOI] [PubMed] [Google Scholar]
- Wozniak-Knopp G., Rüker F. Arch. Biochem. Biophys. 2012;526:181–187. doi: 10.1016/j.abb.2012.03.024. [DOI] [PubMed] [Google Scholar]
- Zahnd C., Pecorari F., Straumann N., Wyler E., Plückthun A. J. Biol. Chem. 2006;281:35167–35175. doi: 10.1074/jbc.M602547200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.