

Abstract
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with profound effects on multiple organ systems. In patients with SLE, the immune system is subverted to target numerous self-antigens and the ensuing inflammatory response elicits a vicious cycle of immune cell activation and tissue damage. Both genetic and environmental factors are essential for the development of this debilitating condition, although the exact cause remains unclear. Early studies on the pathogenesis of lupus centered on the adaptive immune system as lymphocyte abnormalities were thought to be primary cause of autoimmunity. In the past decade, however, this paradigm has shifted with rapid advances in the field of innate immunity. These developments have yielded important insights to how the autoimmune response in SLE is initiated and maintained. Monocytes and macrophages represent an essential arm of the innate immune system with a multitude of immunological functions including antigen presentation, phagocytosis, and cytokine production. Aberrations of monocyte / macrophage phenotype and function are increasingly recognized in SLE and animal models of the disease. In this review, we summarize the current knowledge of monocyte / macrophage abnormalities in human SLE and discuss their implications for understanding the pathogenesis of lupus.
Keywords: systemic lupus erythematosus, monocytes, macrophages
I. Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by autoantibody production and chronic inflammation targeting multiple organs (Reeves 2004). In healthy individuals, the immune system defends against microbes by distinguishing self- from foreign-antigens. For reasons not completely understood, immune tolerance is breached in SLE and the immune system actively responds to a wide-array of autoantigens (Hanh 2005). The resulting immune cell activation leads to autoantibody production and establishes a vicious cycle of chronic inflammation and tissue destruction.
Although the etiology of SLE remains unknown, early studies focused on the adaptive immune system as primary abnormalities of B and T lymphocyte functions were considered the likely basis of the disease (Hanh 2005). This paradigm has shifted with recent advances in the field of innate immunity. It is now increasingly recognized that components of the innate immune system, which normally function to detect invasion by microbial pathogens, play an essential role in the recognition of self antigens in SLE (Theofilopoulos et al. 2005; Marshak-Rothstein 2006).
Monocytes are a key component of the innate immune system involved in the regulation of the adaptive immune response (Unanue 1978). These bone marrow-derived myeloid cells and their derivatives, including tissue macrophages, Kupffer cells, and conventional dendritic cells (CDCs), are equipped with an arsenal of conserved innate sensors designed to recognize pathogen-associated molecular patterns (PAMPs) (Takeda et al. 2003). Activation of these receptors leads to the production of a wide-spectrum of cytokines and chemokines. This capacity of monocytes to initiate inflammation and recruit other immune cells is coupled to their ability to present antigens in the context of products of the major histocompatibility complexes (MHC), making them an important link between the innate and adaptive immune systems. Monocytes also serve as direct precursors to tissue macrophages found in almost every organ in the body (Gordon et al. 2005). Tissue macrophages can be further divided into different subsets based on their specialized functions in immune regulation and wound healing (Mosser et al. 2008). Importantly, both monocytes and tissue macrophages also possess potent phagocytic activity essential for the clearance of dead or dying cells, cellular debris, microbes, and other foreign material (Aderem et al. 1999). Current knowledge in these areas is summarized in several excellent reviews of monocyte biology (Grage-Griebenow et al. 2001; Gordon et al. 2005; Mosser et al. 2008; Auffray et al. 2009).
Abnormalities in monocyte phenotype and functions have been associated with a variety of autoimmune disorders, including SLE (Katsiari et al. 2009). While animal models have significantly enhanced our understanding of monocyte / macrophage involvement in lupus, this review will mainly focus on findings in human SLE. We will briefly review the classification of monocyte subsets and the current understanding of monocyte defects associated with human SLE including abnormalities in 1) surface marker expression, 2) phagocytosis of cellular debris, and 3) cytokine production.
II. Monocyte subsets and SLE
The heterogeneity of human peripheral blood monocytes has been known for decades and early studies distinguished subsets of regular monocytes and intermediate monocytes based on phenotypic and functional characteristics (Akiyama et al. 1983; Grage-Griebenow et al. 2001). Regular monocytes were defined by their larger size, significant peroxidase activity, and enhanced capacity to mediate antibody-dependent cellular cytotoxicity (Figdor et al. 1982; Grage-Griebenow et al. 2001). The intermediate monocyte subset, on the other hand, possesses a greater ability to secrete inflammatory cytokines (Akiyama et al. 1983).
Subsequently, it was found that human monocyte subsets also can be distinguished based on the surface expression of CD14 and CD16 (Passlick et al. 1989). A population that expresses high levels of CD14, but not CD16, accounts for the majority of circulating monocytes (Ziegler-Heitbrock et al. 1988; Ziegler-Heitbrock 1996). These CD14+CD16- monocytes express the chemokine receptor CCR2 and may correspond to the Ly6Chi inflammatory monocyte subset in mice (Passlick et al. 1989). However, unlike their murine counterparts, CD14+CD16- monocytes do not seem to be the predominant cytokine-producing monocyte subset in human. A small population of circulating monocytes with strong surface expression of CD16, on the other hand, is responsible for the inducible production of inflammatory cytokines upon stimulation (Grage-Griebenow et al. 2001). Based on the prominent surface expression of CX3CR1 (CX3CL1 / fractalkine receptor), CD14+CD16+ monocytes are thought to be the human equivalent of the Ly6Clo residential monocytes in mice (Passlick et al. 1989). A third subset of human monocytes characterized as CD14dimCD16+ has also been described although its functional significance is less clear (Skrzeczynska-Moncznik et al. 2008).
The homeostasis of monocyte subsets is often altered in various disease states. In patients with rheumatoid arthritis (RA), an increased number of CD14+CD16+ monocytes correlates with elevated erythrocyte sedimentation rate and serum C-reactive protein levels (Kawanaka et al. 2002; Wijngaarden et al. 2003). In addition, expansion of the CD14+CD16+ monocyte population is described in patients with coronary artery disease and is positively associated with the severity of hypercholesterolemia (Hornung et al. 2002). This population also is expanded in patients with sepsis (Skrzeczynska et al. 2002), inflammatory bowel disease (Hanai et al. 2004), and hemolytic uremic syndrome in children (Berland et al. 2006).
Monocyte subsets also have been examined in SLE but the findings vary among several studies. Two groups reported that the percentages of CD14+CD16+ monocytes in SLE vs. healthy subjects were not significantly different (Asahara et al. 1997; Cairns et al. 2002). In contrast, an expansion of CD14+CD16+ monocytes was reported in another study in which it was shown that steroid treatment decreased the number of these monocytes in a dose-related manner (Sumegi et al. 2005). In the largest study to date with 205 SLE patients and 74 normal healthy controls, we recently demonstrated that although the absolute monocyte count is decreased in SLE patients, there is no relative difference in proportion of CD14+CD16-, CD14+CD16+, or CD14dimCD16+ monocytes between the groups (Li et al. 2009). Neither the percentage nor the absolute number of monocytes in the different subsets correlates with disease activity or manifestations, however. It remains unclear whether the reduction in total monocyte count in SLE is due to a defect in monocyte development or active recruitment of these cells to sites of inflammation such as the kidneys. The latter view is supported by increased expression of CX3CL1 and accumulation of CD16+ monocytes in glomeruli of active lupus nephritis, which correlate with impaired renal function and the presence of anti-dsDNA autoantibodies.
III. Aberrant monocyte surface marker expression
Monocytes express a variety of surface molecules that confer the ability to recognize the environment and respond to changes induced by exogenous as well as endogenous stimuli. Aberrant expression of these surface markers, therefore, may skew the immune response. A number of abnormalities have been described in monocytes from SLE patients and may contribute to disease pathogenesis (Figure 1A).
Figure 1. Abnormal monocyte surface marker expression and cytokine / chemokine production in SLE.
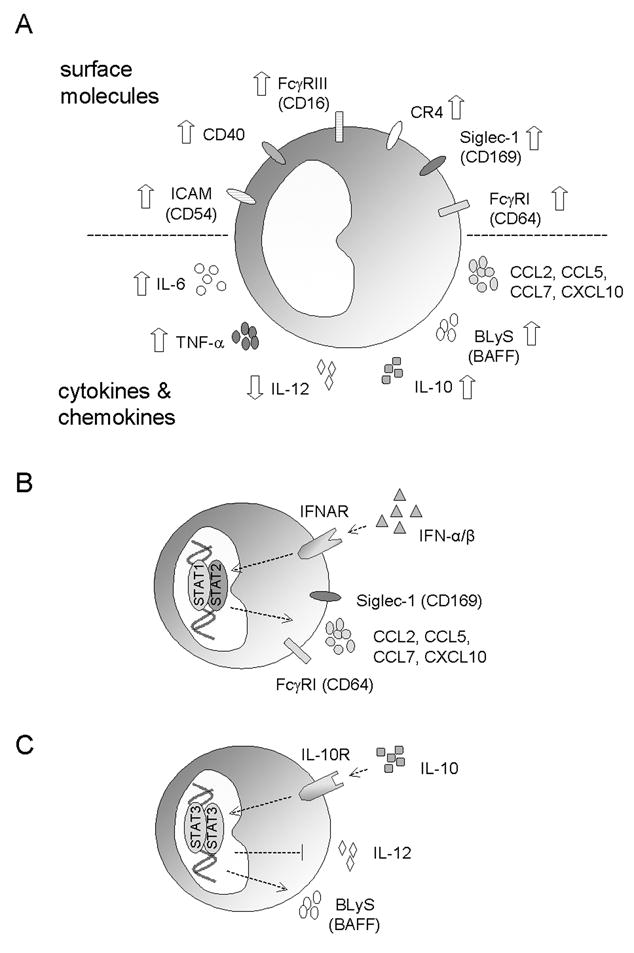
A) Illustration of surface marker abnormalities (top) and excess cytokine / chemokine production (bottom) associated with SLE. B) Elevated serum levels of IFN-I in SLE impacts the expression surface markers (FcγRI and Siglec-1) and chemokines (CCL2, CCL5, CCL7, and CXCL10) in monocytes. C) Increased IL-10 production in SLE modulates the expression of IL-12 and BlyS. Abbreviations: BLyS, B-lymphocyte stimulator; CR, complement receptor; ICAM, intercellular adhesion molecule-1; IFN, interferon; IFNAR, IFN-α/β receptor; STAT, signal transducers and activators of transcription; TNF, tumor necrosis factor.
Fc gamma receptors (FcγRs), which are involved in phagocytosis, cytolysis, degranulation, and induction of inflammatory cytokines (Nimmerjahn et al. 2008), are among the surface molecules dysregulated in SLE. The outcome of interactions with immune complexes, which are produced abundantly in SLE, is determined by the balance of activating and inhibitory FcγRs on the cell surface of APCs such as monocytes. Deletion of Fc receptor γ-chain, an essential signaling component of activating FcγRs, is sufficient to inhibit the development of glomerulonephritis lupus-prone mice (Clynes et al. 1998). A subsequent study showed that the presence of Fc γ-chain on monocytes, but not renal mesangial cells, is required for this effect (Bergtold et al. 2006). In contrast, the absence of the inhibitory FcγRIIb induces autoantibody production and accelerates disease progression in experimental models (Bolland et al. 2002).
In humans, polymorphisms of both activating and inhibitory FcγRs are implicated in the development of lupus and the surface expression of these molecules is altered in SLE (Li et al. 2009). Several single nucleotide polymorphisms of FcγRII and FcγRIII affecting protein expression, ligand interaction, and transcription factor binding have been established as risk alleles for SLE (Blank et al. 2005; Floto et al. 2005; Su et al. 2007; Li et al. 2009). Copy number polymorphisms of FcγRIII also influence the risk for SLE. A low copy number predisposes to the development of lupus while a high copy number protects against the disease (Aitman et al. 2006; Fanciulli et al. 2007).
Whereas decreased expression of the inhibitory FcγRIIb (CD32) is found on certain B lymphocyte subsets (Mackay et al. 2006), the activating receptor FcγRI (CD64) is over-expressed in monocytes from SLE patients and is expressed at even higher levels in the subset of patients with renal disease (Hepburn et al. 2004; Li et al. 2009). FcγRI expression positively correlates with markers of renal dysfunction (blood urea nitrogen, serum creatinine, microalbumin / creatinine ratio) and parameters of ongoing systemic inflammation (C3, C-reactive protein) but is largely unaffected by conventional medications used in SLE (Li et al. 2009). In the presence of immune complexes, the predominance of activating FcγRs may further augment the inflammatory response in SLE (Kavai et al. 2007).
Over-expression of adhesion molecules and costimulatory molecules may lead to aberrant monocyte migration and lymphocyte activation. Monocytes from SLE patients with active disease display elevated surface levels of ICAM-1 (Intercellular Adhesion Molecule-1, CD54), a transmembrane protein involved in endothelial transmigration and inflammatory cytokine production (Funauchi et al. 1993), an effect that may be partially offset by the inhibitory effect of corticosteroids on ICAM-1 expression (Hepburn et al. 2004). Compared with healthy controls, lupus patients’ monocytes also express greater levels of the costimulatory molecule CD40 and this feature is most pronounced in patients with active disease and hypergammaglobulinemia (Katsiari et al. 2002). Interaction between CD40 and its ligand (CD40L) triggers lymphocyte activation, proliferation, and initiation of immunoglobulin isotype switch (Banchereau et al. 1994). Interestingly, CD40L expression is elevated concomitantly on T and B lymphocytes from lupus patients with active disease (Desai-Mehta et al. 1996; Koshy et al. 1996). Aberrant expression of CD40 on monocytes, therefore, may promote the proliferation of autoreactive lymphocytes and generation of autoantibodies through excess CD40-CD40L interactions.
Monocytes from lupus patients also exhibit abnormalities in antigen presentation. Decreased expression of class II MHC on monocytes is documented in patients with active disease (Shirakawa et al. 1985) and impaired antigen presentation is associated with defective upregulation of CD80 on activated APCs (Tsokos et al. 1996). However, monocytes from lupus patients have a propensity to differentiate into dendritic cells with potent capacity to present self-antigens (Blanco et al. 2001). Monocyte-derived dendritic cells from lupus patients also appear to exhibit higher CD86 expression compared to those from healthy individuals (Decker et al. 2006; Ding et al. 2006).
The mechanisms underlying these phenotypic changes on monocytes are largely unknown, although contributions from both genetic and environmental factors have been described. The involvement of genetic factors is best illustrated by natural polymorphisms and copy number variations found in FcγRs (as discussed above) and complement receptors (Dykman et al. 1984; Yang et al. 2007; Li et al. 2009). Recent studies demonstrate that the serum cytokine milieu in SLE may also play a role. Several groups have identified a panel of type-I interferon (IFN-I)-stimulated genes (ISGs) highly expressed in peripheral blood mononuclear cells (PBMC) from lupus patients (Baechler et al. 2003; Bennett et al. 2003; Lee et al. 2007). This interferon signature is associated with a wide spectrum of clinical manifestations and with autoantibody production. Dysregulated IFN-I production also may account for the over-expression of FcγRI in monocytes from lupus patients, as IFN-I signaling induces the expression of FcγRI (but not other FcγRs) in monocytes and elevated levels of FcγRI transcripts in PBMCs from lupus patients are noted in microarray studies (Bennett et al. 2003; Biesen et al. 2008; Li et al. 2009). Sera from lupus patients can also induce FcγRI expression in monocytes form healthy controls in an IFN-I-dependently manner (Li et.al. unpublished data). In addition, the expression of sialoadhesin (Siglec-1, CD169) on monocytes from lupus patients is also increased in SLE due to dysregulated IFN-I production (Biesen et al. 2008). Both CD14+CD16- and CD14+CD16+ monocyte subsets display elevated surface levels of sialoadhesin, a feature that correlates with disease activity and the of anti-dsDNA autoantibody levels, but inversely correlates with the complement levels (Biesen et al. 2008). Interestingly, excess production of IFN-I and expression of sialoadhesin are also present in patients with systemic sclerosis (York et al. 2007).
The elevated levels of IFN-I found in lupus patients also may contribute to the accumulation of inflammatory phenotype of monocytes at the sites of tissue damage (Figure 1B). Excess expression of IFN-I and ISGs has been described at sites of inflammation including the skin and kidneys of lupus patients (Farkas et al. 2001; Peterson et al. 2004). Among the genes induced by IFN-I are the potent monocyte chemoattractants CCL2 and CCL7 (Bauer et al. 2006). As illustrated recently in a mouse model of SLE, recruitment of inflammatory monocytes in response to local secretion of these IFN-inducible chemokines may perpetuate the chronic inflammatory responses associated with lupus (Lee et al. 2008; 2009). Supporting a pathogenic role of IFN-I, early clinical data evaluating the utility of anti-IFN-I antibodies in SLE have suggests therapeutic efficacy (Liao et al. 2004).
IV. Abnormalities in cytokine production
Aberrant activation of the adaptive immune system in SLE is augmented by the dysregulated production of cytokines (Kyttaris et al. 2005). In many instances, monocytes / macrophages are a major source of these immunomodulators, which may promote the activation of autoreactive T and B cells (Figure 1A).
In line with excess generation of immunoglobulins, elevated serum levels of the T helper 2 (Th2) cytokines IL-6 and IL-10 are found in lupus patients (Linker-Israeli et al. 1991; Llorente et al. 1993). Both of these cytokines promote the production of IgG and IL-6 also is required (along with IFN-I) for the differentiation of B cells into antibody-secreting plasma cells (Jego et al. 2003). Monocytes are the primary source of both IL-6 and IL-10 in the peripheral blood (Hagiwara et al. 1996), although production by lymphocytes also is reported (Linker-Israeli et al. 1991; Llorente et al. 1993; Mellor-Pita et al. 2009). Monocyte are a significant source of B-lymphocyte stimulator (BLyS; also known as BAFF), which promotes the survival and proliferation factor for B lymphocytes (Moore et al. 1999; Nardelli et al. 2001). Lupus patients over-produce BLyS/BAFF and its levels correlate with the levels of autoantibodies against dsDNA (Zhang et al. 2001).
The cytokine dysregulation in lupus is further complicated by interactions among the different cytokines (Figure 1C). For example, the secretion of BLyS/BAFF can be further enhanced by IL-10 and IFN-γ (Nardelli et al. 2001). The elevated levels of IL-10, on the other hand, down-regulates the production of IL-12 (Liu et al. 1998)and reduce the inhibitory effects of IL-12 on antibody production (Houssiau et al. 1997). IFN-I, a family of cytokines overproduced in the majority of lupus patients (as described above), act synergistically with IL-6 to induce plasma cell differentiation (Jego et al. 2003) while priming with IFN-I unleashes the proinflammatory functions of IL-10 (Sharif et al. 2004). IFN-I also primes the IFN-γ signaling cascade and hyper-responsiveness of monocytes from lupus patients to IFN-γ stimulation has been reported recently (Karonitsch et al. 2009).
One explanation for the excess cytokine production is the presence of nucleic acid-containing immune complexes in the serum of lupus patients. Human monocytes express the endosomal nucleic acid sensors TLR-7/TLR-8 and TLR9, which mediate the inflammatory response to viral ssRNA and bacterial CpG DNA, respectively (Klinman et al. 2002; Gorden et al. 2005). However, endogenous nucleic acids are also capable of activating these receptors (Marshak-Rothstein 2006). Normally the immune system is safeguarded from endogenous RNA and DNA by rapid degradation of nucleic acids during apoptosis. Grouping of these endosomal TLRs in the endoplasmic reticulum and late endosomes provides further insurance against unwanted responses to endogenous nucleic acids (Barton et al. 2006). These endogenous TLR ligands are normally restricted from entering the endosomal compartments where the nucleic acid-sensing TLRs are located. In SLE, however, autoantibodies against DNA and RNA-associated antigens promote the formation of nucleic acid-containing immune complexes (Barrat et al. 2005; Vollmer et al. 2005). FcγR mediated uptake of these complexes may interfere with the actions of endogenous nucleases, resulting in translocation of the endogenous nucleic acids into the endosomal compartment containing TLR 7/8 and TLR9. This mechanism induces maturation of monocytes (Vollmer et al. 2005) and may be responsible for the excess production of inflammatory cytokines such as tumor necrosis factor α (TNF-α) by lupus monocytes (Steinbach et al. 2000). The same mechanism is believed to be responsible for the aberrant production of IFN-I by plasmacytoid DCs in lupus (Barrat et al. 2005; Vollmer et al. 2005; Kelly et al. 2006). Furthermore, these effects on cytokine production may be augmented by the increased ratio of activating to inhibitory FcγRs (Kavai et al. 2007) and/or delayed removal of endogenous nucleic acids secondary to deficiency of serum nucleases in lupus patients (Lee-Kirsch et al. 2007).
V. Defects in phagocytosis and clearance of cellular debris
The phagocytic capacity of monocytes / macrophages is essential for host defense against pathogens and homeostatic clearance of dead or dying cells (Aderem et al. 1999). Upon activation of the immune system through the detection of PAMPs, peripheral blood monocytes and tissue macrophages migrate to the site of infection and initiate phagocytosis of the invading organisms through several mechanisms. Cellular recognition of pathogen components by surface molecules such as mannose receptors, integrins, and scavenger receptors can directly induce the uptake of microbes. Moreover, opsonization by antibodies and complement potently stimulates phagocytosis via complement receptors and Fc receptors on the surface of phagocytic cells (Aderem et al. 1999).
Similarly, apoptotic and necrotic cells are removed by phagocytosis (Birge et al. 2008). During necrosis, contents of the dying cell are released and may subsequently trigger an inflammatory response through innate sensors such as TLRs and inflammasome components (Cavassani et al. 2008; Iyer et al. 2009; Li et al. 2009). Despite the continuous turnover of aging or injured cells, apoptotic cell debris normally does not activate the inflammatory response because the receptors responsible for its uptake by macrophages are linked to anti-inflammatory signal pathways (Fadok et al. 1998; Fadok et al. 2001; Chung et al. 2006). Recognition of apoptotic cells through a receptor for phosphotidylserine on macrophages inhibits the production of inflammatory mediators including IL-1β, IL-8, IL-10, TNF-α, leukotriene C4 and thromboxane B2 (Fadok et al. 1998; Hoffmann et al. 2001). The anti-inflammatory effects are mediated by the synthesis of transforming growth factor (TGF)-beta1, prostaglandin E2, and platelet-activating factor (Fadok et al. 1998). Accumulating data from animal models and in vitro studies illustrate that removal of apoptotic cell by macrophages is likely mediated by multiple pathways in addition to phosphotidylserine receptor. The involvement of CD14, c-Mer, liver receptor X, and vitronectin receptor is well documented and deficiency of some of these components is associated with the development of autoimmune manifestations in mice (Fadok et al. 1992; Devitt et al. 1998; Fadok et al. 1998; Scott et al. 2001; A-Gonzalez et al. 2009), although their role on human SLE is less clear at this time.
Ineffective clearance of dying cells and debris may provide a source of autoantigens for the development of an autoimmune response (Figure 2). Indeed, abnormal clearance of apoptotic cells by macrophages from patients with SLE was demonstrated more than a decade ago (Herrmann et al. 1998). In vitro phagocytosis of autologous apoptotic cells is significantly impaired in monocyte-derived macrophages from SLE patients compared to healthy controls. Supporting these findings, examination of lymph node biopsy samples from SLE patients revealed an accumulation of apoptotic cells near germinal centers and a decreased number of phagocytic tingible macrophages (Baumann et al. 2002). The clearance defect is compounded by the burden of chronic inflammation and increased rate of apoptosis in SLE (Ren et al. 2003). Moreover, sera from lupus patients possess enhanced capacity to induce apoptosis (Bengtsson et al. 2004). An important question raised by these findings is whether the aberrant uptake of apoptotic cells represents an inherent defect of macrophage function or a secondary phenomenon driven by serum abnormalities associated with the disease (e.g. low complement levels and presence of autoantibodies).
Figure 2. Defects in phagocytosis and apoptotic cell clearance in SLE.
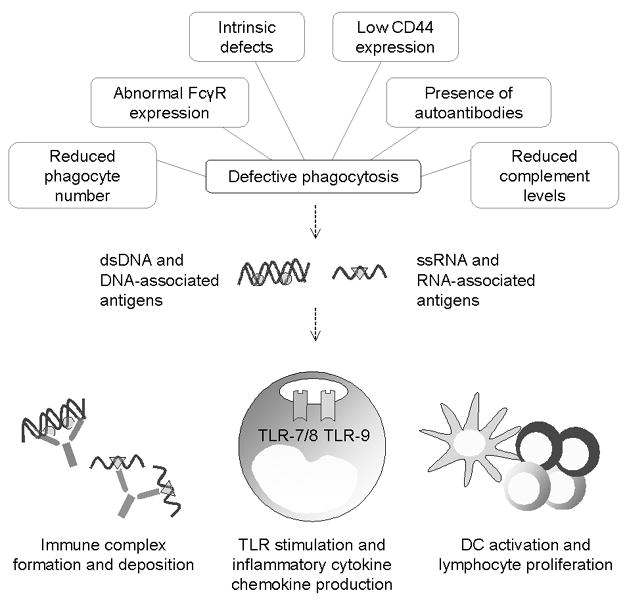
A number of intrinsic and extrinsic factors contribute to the phagocytic defects associated with SLE (top). Impaired clearance of apoptotic cells and debris results in the accumulation of endogenous nucleic acids and nucleic acid-associated antigens (e.g. histones bound to DNA and components of the small ribonuclear protein complex associated with RNA). Upon on translocation of these autoantigens into endosomes via FcγR-dependent uptake, endogenous nucleic acids stimulate the endosomal Toll-like receptors on monocytes and dendritic cells, triggering the production of inflammatory cytokines / chemokines and proliferation of lymphocytes. The presence of autoantibodies further perpetuates the inflammatory response due to immune complex formation and deposition within tissues.
The presence of an intrinsic defect in the clearance of dying cells is supported by several lines of evidence. Despite normal surface binding of apoptotic cells, macrophages from lupus patients display reduced ability to internalize the targets compared to those from healthy controls or patients with RA (Tas et al. 2006). This defect may be partially explained by reduced surface expression of the glycoprotein receptor CD44 on monocytes. CD44 mediates the clearance of apoptotic neutrophils by monocytes and decreased expression of this molecule is found in lupus, but not RA patients (Cairns et al. 2001). An intrinsic defect of phagocytosis also is revealed by a study comparing CD34+ hematopoietic stem cell (HSC)-derived macrophages from lupus patients and healthy controls (Gaipl et al. 2005). Similar to monocyte freshly isolated from the peripheral blood, macrophages derived from CD34+ HSCs of SLE patients demonstrated a reduced phagocytic capacity. This problem is compounded by the low number of CD34+ HSCs in SLE patients and their ineffective differentiation into macrophages (Papadaki et al. 2001; Gaipl et al. 2005). Freshly isolated monocytes and cultured macrophages from SLE patients also display increased rates of spontaneous cell death due to fas-mediated apoptosis (Shoshan et al. 2001). Thus, both quantitative and qualitative (functional) defects of the monocyte / macrophage lineage may contribute to the impaired apoptotic cell uptake in SLE. However, the phagocytic defect in macrophages from lupus patients can be partially reversed by sera from healthy controls (Ren et al. 2003). Conversely, addition of serum from lupus patients to macrophages from healthy controls reduces the uptake of apoptotic cells (Ren et al. 2003). These findings suggest that humoral mediators of phagocytosis may be dysregulated in SLE. The deficiency of complement in SLE perhaps provides the best supporting evidence for this hypothesis.
The complement system is comprised of a cascade of self-regulated proteins that directs bacteriolysis, antigen opsonization, neutrophil chemotaxis, and immune complex clearance (Carroll 1998). Complement components also bind to apoptotic cells and promote phagocytosis by macrophages (Mevorach et al. 1998). This system is critical to the prevention of autoimmunity as genetic deficiencies of the early classical complement components (e.g. C1q, C4) are associated with the development of SLE in humans as well as mouse models (Manderson et al. 2004). Autoantobodies to complement components such as C1q provides another mechanism to suppress the clearance of apoptotic cell (Uwatoko et al. 1984). Increased levels of anti-C1q antibodies are frequently found in lupus patients with renal disease (Coremans et al. 1995). Indeed, reduced serum levels of C1q, C3 and C4 in SLE may partially explain the inhibitory effect of lupus sera on the uptake of apoptotic cells by macrophages (Bijl et al. 2006). However, other factors are likely involved as sera from inactive lupus patients do not suppress phagocytosis despite the presence of decreased complement levels (Grevink et al. 2005).
Factors capable of inhibiting phagocytosis may provide an alternative explanation for the effect of lupus sera. Indeed, autoantibodies targeting nuclear antigens in SLE have been linked to the phagocytic abnormalities in macrophages. Opsonization by IgG from lupus patients, but not healthy controls, inhibits the phagocytosis of late apoptotic cells by macrophages (Reefman et al. 2007). Both FcγRI and FcγRII are required for this inhibitory effect, although the exact mechanism remains to be elucidated. Conversely, autoantibodies promote the uptake of necrotic cells (Grossmayer et al. 2008) and secondary necrotic cell-derived material, which induces the production of pro-inflammatory cytokines (Munoz et al. 2009).
In addition to these mechanisms, autoantibodies may also act directly on macrophages. Autoantibodies targeting surface molecules involved in the phagocytosis of apoptotic cells, such as scavenger receptors, have been demonstrated in sera from lupus patients and lupus-prone mice (Wermeling et al. 2007).
Conclusion
SLE is a chronic autoimmune disorder associated with a plethora of immunological abnormalities. Phenotypic and functional abnormalities in monocytes / macrophages increasingly are recognized to play a role in this complex disease. Recent studies in lupus patients have identified numerous monocyte / macrophage defects involving surface protein expression, cytokine production, and phagocytic capacity. However, we are only beginning to understand the mechanisms underlying these abnormalities. While further studies are needed to define the genetic and environmental factors involved, abnormalities of immunomodulation also warrant exploration. With monocyte depletion therapy already being assessed in inflammatory diseases such as RA, a better understanding of the causes and sequelae of the monocyte / macrophage abnormalities may aid in the design of novel therapeutic approaches in SLE.
Acknowledgments
This work was supported by R01-AR44731 from the US Public Health Service and by generous gifts from Lupus Link, Inc. (Daytona Beach, FL) and Mr. Lewis M. Schott to the UF Center for Autoimmune Disease.
Abbreviations
- SLE
systemic lupus erythematosus
- DC
dendritic cells
- PAMPs
pathogen-associated molecular patterns
- TLR
Toll-like receptor
- MHC
major histocompatibility complexes
- FcγR
Fc gamma receptor
- ICAM-1
intercellular ahesion molecule-1
- IFN-I
type-I interferon
- ISG
interferon-stimulated gene
- ds
double-stranded
- ss
single-stranded
- Th
T helper
- BLyS
B-lymphocyte stimulator
- RA
rheumatoid arthritis
- HSC
hematopoietic stem cell
References
- A-Gonzalez N, Bensinger SJ, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–58. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Aitman TJ, Dong R, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- Akiyama Y, Miller PJ, et al. Characterization of a human blood monocyte subset with low peroxidase activity. J Clin Invest. 1983;72:1093–105. doi: 10.1172/JCI111034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Murohara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Auffray C, Sieweke MH, et al. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–92. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Bazan F, et al. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- Barrat FJ, Meeker T, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Kagan JC, et al. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- Bauer JW, Baechler EC, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3:e491. doi: 10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann I, Kolowos W, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Bengtsson AA, Sturfelt G, et al. Induction of apoptosis in monocytes and lymphocytes by serum from patients with systemic lupus erythematosus - an additional mechanism to increased autoantigen load? Clin Exp Immunol. 2004;135:535–43. doi: 10.1111/j.1365-2249.2003.02386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L, Palucka AK, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–23. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergtold A, Gavhane A, et al. FcR-bearing myeloid cells are responsible for triggering murine lupus nephritis. J Immunol. 2006;177:7287–95. doi: 10.4049/jimmunol.177.10.7287. [DOI] [PubMed] [Google Scholar]
- Berland R, Fernandez L, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–40. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Biesen R, Demir C, et al. Sialic acid-binding Ig-like lectin 1 expression in inflammatory and resident monocytes is a potential biomarker for monitoring disease activity and success of therapy in systemic lupus erythematosus. Arthritis Rheum. 2008;58:1136–45. doi: 10.1002/art.23404. [DOI] [PubMed] [Google Scholar]
- Bijl M, Reefman E, et al. Reduced uptake of apoptotic cells by macrophages in systemic lupus erythematosus: correlates with decreased serum levels of complement. Ann Rheum Dis. 2006;65:57–63. doi: 10.1136/ard.2005.035733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birge RB, Ucker DS. Innate apoptotic immunity: the calming touch of death. Cell Death Differ. 2008;15:1096–102. doi: 10.1038/cdd.2008.58. [DOI] [PubMed] [Google Scholar]
- Blanco P, Palucka AK, et al. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–3. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- Blank MC, Stefanescu RN, et al. Decreased transcription of the human FCGR2B gene mediated by the -343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum Genet. 2005;117:220–7. doi: 10.1007/s00439-005-1302-3. [DOI] [PubMed] [Google Scholar]
- Bolland S, Yim YS, et al. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(-/-) mice. J Exp Med. 2002;195:1167–74. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns AP, Crockard AD, et al. The CD14+ CD16+ monocyte subset in rheumatoid arthritis and systemic lupus erythematosus. Rheumatol Int. 2002;21:189–92. doi: 10.1007/s00296-001-0165-8. [DOI] [PubMed] [Google Scholar]
- Cairns AP, Crockard AD, et al. Reduced expression of CD44 on monocytes and neutrophils in systemic lupus erythematosus: relations with apoptotic neutrophils and disease activity. Ann Rheum Dis. 2001;60:950–5. doi: 10.1136/ard.60.10.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol. 1998;16:545–68. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- Cavassani KA, Ishii M, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–21. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung EY, Kim SJ, et al. Regulation of cytokine production during phagocytosis of apoptotic cells. Cell Res. 2006;16:154–61. doi: 10.1038/sj.cr.7310021. [DOI] [PubMed] [Google Scholar]
- Clynes R, Dumitru C, et al. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–4. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- Coremans IE, Spronk PE, et al. Changes in antibodies to C1q predict renal relapses in systemic lupus erythematosus. Am J Kidney Dis. 1995;26:595–601. doi: 10.1016/0272-6386(95)90595-2. [DOI] [PubMed] [Google Scholar]
- Decker P, Kotter I, et al. Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2006;45:1087–95. doi: 10.1093/rheumatology/kel061. [DOI] [PubMed] [Google Scholar]
- Desai-Mehta A, Lu L, et al. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996;97:2063–73. doi: 10.1172/JCI118643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devitt A, Moffatt OD, et al. Human CD14 mediates recognition and phagocytosis of apoptotic cells. Nature. 1998;392:505–9. doi: 10.1038/33169. [DOI] [PubMed] [Google Scholar]
- Ding D, Mehta H, et al. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J Immunol. 2006;177:5878–89. doi: 10.4049/jimmunol.177.9.5878. [DOI] [PubMed] [Google Scholar]
- Dykman TR, Hatch JA, et al. Polymorphism of the human C3b/C4b receptor. Identification of a third allele and analysis of receptor phenotypes in families and patients with systemic lupus erythematosus. J Exp Med. 1984;159:691–703. doi: 10.1084/jem.159.3.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, et al. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest. 2001;108:957–62. doi: 10.1172/JCI14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Savill JS, et al. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol. 1992;149:4029–35. [PubMed] [Google Scholar]
- Fadok VA, Warner ML, et al. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3) J Immunol. 1998;161:6250–7. [PubMed] [Google Scholar]
- Fanciulli M, Norsworthy PJ, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–3. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas L, Beiske K, et al. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159:237–43. doi: 10.1016/s0002-9440(10)61689-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figdor CG, Bont WS, et al. Isolation of functionally different human monocytes by counterflow centrifugation elutriation. Blood. 1982;60:46–53. [PubMed] [Google Scholar]
- Floto RA, Clatworthy MR, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. 2005;11:1056–8. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- Funauchi M, Ohno M, et al. Abnormal expression of intercellular adhesion molecule-1 on peripheral blood mononuclear cells from patients with systemic lupus erythematosus. J Clin Lab Immunol. 1993;40:115–24. [PubMed] [Google Scholar]
- Gaipl US, Voll RE, et al. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev. 2005;4:189–94. doi: 10.1016/j.autrev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Gorden KB, Gorski KS, et al. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174:1259–68. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Grage-Griebenow E, Flad HD, et al. Heterogeneity of human peripheral blood monocyte subsets. J Leukoc Biol. 2001;69:11–20. [PubMed] [Google Scholar]
- Grage-Griebenow E, Zawatzky R, et al. Identification of a novel dendritic cell-like subset of CD64(+) / CD16(+) blood monocytes. Eur J Immunol. 2001;31:48–56. doi: 10.1002/1521-4141(200101)31:1<48::aid-immu48>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Grevink ME, Horst G, et al. Levels of complement in sera from inactive SLE patients, although decreased, do not influence in vitro uptake of apoptotic cells. J Autoimmun. 2005;24:329–36. doi: 10.1016/j.jaut.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Grossmayer GE, Munoz LE, et al. IgG autoantibodies bound to surfaces of necrotic cells and complement C4 comprise the phagocytosis promoting activity for necrotic cells of systemic lupus erythaematosus sera. Ann Rheum Dis. 2008;67:1626–32. doi: 10.1136/ard.2007.081828. [DOI] [PubMed] [Google Scholar]
- Hagiwara E, Gourley MF, et al. Disease severity in patients with systemic lupus erythematosus correlates with an increased ratio of interleukin-10:interferon-gamma-secreting cells in the peripheral blood. Arthritis Rheum. 1996;39:379–85. doi: 10.1002/art.1780390305. [DOI] [PubMed] [Google Scholar]
- Hanai H, Watanabe F, et al. Correlation of serum soluble TNF-alpha receptors I and II levels with disease activity in patients with ulcerative colitis. Am J Gastroenterol. 2004;99:1532–8. doi: 10.1111/j.1572-0241.2004.30432.x. [DOI] [PubMed] [Google Scholar]
- Hanh BH. Systemic Lupus Erythematosus. In: Kasper DLea., editor. Harrison’s Principles of Internal Medicine. New York City: McGraw-Hill Medical; 2005. pp. 1956–67. [Google Scholar]
- Hepburn AL, Mason JC, et al. Expression of Fcgamma and complement receptors on peripheral blood monocytes in systemic lupus erythematosus and rheumatoid arthritis. Rheumatology (Oxford) 2004;43:547–54. doi: 10.1093/rheumatology/keh112. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Voll RE, et al. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–50. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hoffmann PR, deCathelineau AM, et al. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol. 2001;155:649–59. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Rothenfusser S, et al. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- Houssiau FA, Mascart-Lemone F, et al. IL-12 inhibits in vitro immunoglobulin production by human lupus peripheral blood mononuclear cells (PBMC) Clin Exp Immunol. 1997;108:375–80. doi: 10.1046/j.1365-2249.1997.d01-1009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, Pulskens WP, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:20388–93. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego G, Palucka AK, et al. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–34. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- Karonitsch T, Feierl E, et al. Activation of the interferon-gamma signaling pathway in systemic lupus erythematosus peripheral blood mononuclear cells. Arthritis Rheum. 2009;60:1463–71. doi: 10.1002/art.24449. [DOI] [PubMed] [Google Scholar]
- Katsiari CG, Liossis SN, et al. The Pathophysiologic Role of Monocytes and Macrophages in Systemic Lupus Erythematosus: A Reappraisal. Semin Arthritis Rheum. 2009 doi: 10.1016/j.semarthrit.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Katsiari CG, Liossis SN, et al. Aberrant expression of the costimulatory molecule CD40 ligand on monocytes from patients with systemic lupus erythematosus. Clin Immunol. 2002;103:54–62. doi: 10.1006/clim.2001.5172. [DOI] [PubMed] [Google Scholar]
- Kavai M, Szegedi G. Immune complex clearance by monocytes and macrophages in systemic lupus erythematosus. Autoimmun Rev. 2007;6:497–502. doi: 10.1016/j.autrev.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Kawanaka N, Yamamura M, et al. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–86. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- Kelly KM, Zhuang H, et al. “Endogenous adjuvant” activity of the RNA components of lupus autoantigens Sm/RNP and Ro 60. Arthritis Rheum. 2006;54:1557–67. doi: 10.1002/art.21819. [DOI] [PubMed] [Google Scholar]
- Klinman DM, Takeshita F, et al. CpG DNA: recognition by and activation of monocytes. Microbes Infect. 2002;4:897–901. doi: 10.1016/s1286-4579(02)01614-3. [DOI] [PubMed] [Google Scholar]
- Koshy M, Berger D, et al. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J Clin Invest. 1996;98:826–37. doi: 10.1172/JCI118855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttaris VC, Juang YT, et al. Immune cells and cytokines in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2005;17:518–22. doi: 10.1097/01.bor.0000170479.01451.ab. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Gong M, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–7. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- Lee PY, Kumagai Y, et al. TLR7-dependent and Fc{gamma}R-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PY, Li Y, et al. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. 2009;175:2023–33. doi: 10.2353/ajpath.2009.090328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PY, Li Y, et al. Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum. 2007;56:3759–69. doi: 10.1002/art.23035. [DOI] [PubMed] [Google Scholar]
- Li H, Ambade A, et al. Cutting edge: Necrosis activates the NLRP3 inflammasome. J Immunol. 2009;183:1528–32. doi: 10.4049/jimmunol.0901080. [DOI] [PubMed] [Google Scholar]
- Li X, Ptacek TS, et al. Fcgamma receptors: structure, function and role as genetic risk factors in SLE. Genes Immun. 2009;10:380–9. doi: 10.1038/gene.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lee PY, et al. Increased expression of FcgammaRI/CD64 on circulating monocytes parallels ongoing inflammation and nephritis in lupus. Arthritis Res Ther. 2009;11:R6. doi: 10.1186/ar2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CH, Yao TC, et al. Polymorphisms in the promoter region of RANTES and the regulatory region of monocyte chemoattractant protein-1 among Chinese children with systemic lupus erythematosus. J Rheumatol. 2004;31:2062–7. [PubMed] [Google Scholar]
- Linker-Israeli M, Deans RJ, et al. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol. 1991;147:117–23. [PubMed] [Google Scholar]
- Liu TF, Jones BM. Impaired production of IL-12 in systemic lupus erythematosus. I. Excessive production of IL-10 suppresses production of IL-12 by monocytes. Cytokine. 1998;10:140–7. doi: 10.1006/cyto.1997.0268. [DOI] [PubMed] [Google Scholar]
- Llorente L, Richaud-Patin Y, et al. Spontaneous production of interleukin-10 by B lymphocytes and monocytes in systemic lupus erythematosus. Eur Cytokine Netw. 1993;4:421–7. [PubMed] [Google Scholar]
- Mackay M, Stanevsky A, et al. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med. 2006;203:2157–64. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderson AP, Botto M, et al. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431–56. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–35. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor-Pita S, Citores MJ, et al. Monocytes and T lymphocytes contribute to a predominance of interleukin 6 and interleukin 10 in systemic lupus erythematosus. Cytometry B Clin Cytom. 2009;76B:261–70. doi: 10.1002/cyto.b.20468. [DOI] [PubMed] [Google Scholar]
- Mevorach D, Mascarenhas JO, et al. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. 1998;188:2313–20. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PA, Belvedere O, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–3. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz LE, Janko C, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1733–42. doi: 10.1002/art.24535. [DOI] [PubMed] [Google Scholar]
- Nardelli B, Belvedere O, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- Papadaki HA, Boumpas DT, et al. Increased apoptosis of bone marrow CD34 (+) cells and impaired function of bone marrow stromal cells in patients with systemic lupus erythematosus. Br J Haematol. 2001;115:167–74. doi: 10.1046/j.1365-2141.2001.03076.x. [DOI] [PubMed] [Google Scholar]
- Passlick B, Flieger D, et al. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- Peterson KS, Huang JF, et al. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J Clin Invest. 2004;113:1722–33. doi: 10.1172/JCI19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reefman E, Horst G, et al. Opsonization of late apoptotic cells by systemic lupus erythematosus autoantibodies inhibits their uptake via an Fcgamma receptor-dependent mechanism. Arthritis Rheum. 2007;56:3399–411. doi: 10.1002/art.22947. [DOI] [PubMed] [Google Scholar]
- Reeves WH, Narain S, Satoh M. Autoantibodies in systemic lupus erythematosus. Philadelphia: Lippincott Williams & Wilkins; 2004. [Google Scholar]
- Ren Y, Tang J, et al. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 2003;48:2888–97. doi: 10.1002/art.11237. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–11. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- Sharif MN, Tassiulas I, et al. IFN-alpha priming results in a gain of proinflammatory function by IL-10: implications for systemic lupus erythematosus pathogenesis. J Immunol. 2004;172:6476–81. doi: 10.4049/jimmunol.172.10.6476. [DOI] [PubMed] [Google Scholar]
- Shirakawa F, Yamashita U, et al. Reduced function of HLA-DR-positive monocytes in patients with systemic lupus erythematosus (SLE) J Clin Immunol. 1985;5:396–403. doi: 10.1007/BF00915337. [DOI] [PubMed] [Google Scholar]
- Shoshan Y, Shapira I, et al. Accelerated Fas-mediated apoptosis of monocytes and maturing macrophages from patients with systemic lupus erythematosus: relevance to in vitro impairment of interaction with iC3b-opsonized apoptotic cells. J Immunol. 2001;167:5963–9. doi: 10.4049/jimmunol.167.10.5963. [DOI] [PubMed] [Google Scholar]
- Skrzeczynska-Moncznik J, Bzowska M, et al. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand J Immunol. 2008;67:152–9. doi: 10.1111/j.1365-3083.2007.02051.x. [DOI] [PubMed] [Google Scholar]
- Skrzeczynska J, Kobylarz K, et al. CD14+CD16+ monocytes in the course of sepsis in neonates and small children: monitoring and functional studies. Scand J Immunol. 2002;55:629–38. doi: 10.1046/j.1365-3083.2002.01092.x. [DOI] [PubMed] [Google Scholar]
- Steinbach F, Henke F, et al. Monocytes from systemic lupus erythematous patients are severely altered in phenotype and lineage flexibility. Ann Rheum Dis. 2000;59:283–8. doi: 10.1136/ard.59.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su K, Yang H, et al. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. 2007;178:3272–80. doi: 10.4049/jimmunol.178.5.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumegi A, Antal-Szalmas P, et al. Glucocorticosteroid therapy decreases CD14-expression and CD14-mediated LPS-binding and activation of monocytes in patients suffering from systemic lupus erythematosus. Clin Immunol. 2005;117:271–9. doi: 10.1016/j.clim.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, et al. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Tas SW, Quartier P, et al. Macrophages from patients with SLE and rheumatoid arthritis have defective adhesion in vitro, while only SLE macrophages have impaired uptake of apoptotic cells. Ann Rheum Dis. 2006;65:216–21. doi: 10.1136/ard.2005.037143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Baccala R, et al. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- Tsokos GC, Kovacs B, et al. Defective antigen-presenting cell function in patients with systemic lupus erythematosus. Arthritis Rheum. 1996;39:600–9. doi: 10.1002/art.1780390409. [DOI] [PubMed] [Google Scholar]
- Unanue ER. The regulation of lymphocyte functions by the macrophage. Immunol Rev. 1978;40:227–55. doi: 10.1111/j.1600-065x.1978.tb00408.x. [DOI] [PubMed] [Google Scholar]
- Uwatoko S, Aotsuka S, et al. Characterization of C1q-binding IgG complexes in systemic lupus erythematosus. Clin Immunol Immunopathol. 1984;30:104–16. doi: 10.1016/0090-1229(84)90011-4. [DOI] [PubMed] [Google Scholar]
- Vollmer J, Tluk S, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. 2005;202:1575–85. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermeling F, Chen Y, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. 2007;204:2259–65. doi: 10.1084/jem.20070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijngaarden S, van Roon JA, et al. Fcgamma receptor expression levels on monocytes are elevated in rheumatoid arthritis patients with high erythrocyte sedimentation rate who do not use anti-rheumatic drugs. Rheumatology (Oxford) 2003;42:681–8. doi: 10.1093/rheumatology/keg174. [DOI] [PubMed] [Google Scholar]
- Yang Y, Chung EK, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–54. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York MR, Nagai T, et al. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum. 2007;56:1010–20. doi: 10.1002/art.22382. [DOI] [PubMed] [Google Scholar]
- Zhang J, Roschke V, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166:6–10. doi: 10.4049/jimmunol.166.1.6. [DOI] [PubMed] [Google Scholar]
- Ziegler-Heitbrock HW. Heterogeneity of human blood monocytes: the CD14+ CD16+ subpopulation. Immunol Today. 1996;17:424–8. doi: 10.1016/0167-5699(96)10029-3. [DOI] [PubMed] [Google Scholar]
- Ziegler-Heitbrock HW, Passlick B, et al. The monoclonal antimonocyte antibody My4 stains B lymphocytes and two distinct monocyte subsets in human peripheral blood. Hybridoma. 1988;7:521–7. doi: 10.1089/hyb.1988.7.521. [DOI] [PubMed] [Google Scholar]