

Abstract
Heterologous immunologic memory has been considered a potent barrier to tolerance induction in primates. Induction of such tolerance for a previously transplanted organ may be more difficult, because specific memory cells can be induced and activated by a transplanted organ. In the current study, we attempted to induce tolerance to a previously transplanted kidney allograft in nonhuman primates. The conditioning regimen consisted of low dose total body irradiation, thymic irradiation, antithymocyte globulin, and anti- CD154 antibody followed by a brief course of a calcineurin inhibitor. This regimen had been shown to induce mixed chimerism and allograft tolerance when kidney transplantation (KTx) and donor bone marrow transplantation (DBMT) were simultaneously performed. However, the same regimen failed to induce mixed chimerism when delayed DBMT was performed after KTx. We found that significant levels of memory T cells remained after conditioning, despite effective depletion of naïve T cells. By adding humanized anti-CD8 monoclonal antibody (cM-T807), CD8 memory T cells were effectively depleted and these recipients successfully achieved mixed chimerism and tolerance. The current studies provide ‘proof of principle’ that the mixed chimerism approach can induce renal allograft tolerance, even late after organ transplantation if memory T-cell function is adequately controlled.
Keywords: Kidney transplantation, memory T cell, mixed chimerism, tolerance
Introduction
For induction of allograft tolerance, numerous strategies have been developed in rodents. However, only a few strategies have been successfully translated to nonhuman primate models (1–5). Heterologous immunologic memory induced by exposure to environmental pathogens in primates, has been suggested as one explanation for this critical discrepancy (6–10).
We have previously reported a nonmyeloablative conditioning regimen that can induce mixed chimerism and renal allograft tolerance in nonhuman primates (1,11–13). However, these regimens require treatment of subjects beginning 6 days prior to organ transplantation, which limits their applicability to only living donor transplant recipients. In an effort to extend the applicability of this approach, we evaluated a ‘delayed tolerance’ protocol, in which kidney transplantation is performed first with conventional immunosuppressive therapy, which was followed by conditioning and donor bone marrow transplantation at a later time. To our knowledge, delayed induction of such allograft tolerance has never been achieved in large animal or rodent models. If this approach is feasible, any stable recipient of either living donor or deceased donor kidney transplantation (KTx) could be a potential candidate, if either fresh (living donor) or cryopreserved (deceased donor) DBM cells are available. However, induction of such tolerance for a previously transplanted organ may be more difficult, because specific memory cells can be activated by a transplanted organ even under immunosuppression. We show here that significant levels of memory T cells remained after conditioning, in spite of effective deletion of naïve T cells. Addition of anti-CD8 monoclonal antibody, cM-T807, to suppress post-conditioning expansion of CD8+ memory T cells is shown to improve chimerism induction and renal allograft survival.
Materials and Methods
Animals
Twenty-four male cynomolgus monkeys that weighed 3–5 kg were used (Charles River Primates, Wilmington, MA). Recipient and donor pairs were selected for compatible ABO blood types and mismatched cynomolgus leukocyte (CyLA)-MHC antigens. CyLA class I antigenic disparity was determined by serologic typing with allele-specific anti-HLA class I monoclonal antibodies (mAbs) that cross-react with cynomolgus alleles. CyLA class II antigenic disparity was confirmed by a positive mixed lymphocyte response and polymerase chain reaction (PCR) assay using primers specific to cynomolgus DR. All surgical procedures and postoperative care of animals were performed in accordance with National Institute of Health guidelines for the care and use of primates and were approved by the Massachusetts General Hospital Subcommittee on Animal Research.
Conditioning regimens
The original regimen consisted of nonmyeloablative total body irradiation (TBI, 1.5 GyX2) on days −6 and −5, thymic irradiation (TI, 7 Gy) on day −1, equine ATG (ATGAM, Pharmacia and Upjohn, Kalamazoo, MI, 50 mg/kg/day on days −2,−1 and 0) and anti-CD154 monoclonal antibody (anti-CD154, American Type Culture Collection catalog number 5c8.33, 20 mg/kg on days 0 and +2). The recipients underwent simultaneous kidney and bone marrow transplantation (SKBMT) on day 0 and treated with a 1-month course of cyclosporine (CyA) (Novartis, Basel, Switzerland) (tapered from an initial dose of 15 mg/kg/day) to maintain therapeutic serum levels (>300 ng/mL). CyA was discontinued on day 28 posttransplant, after which serum CyA levels typically became undetectable by day 60.
Group A: The recipients underwent kidney transplantation alone, which followed by CyA administration, TBI (1.5 Gy on days 1 and 2, relative to the date of KTx), TI (7 Gy on day 5) and ATGAM (days 4, 5 and 6). DBMT was performed on day 6 and anti-CD154 was administered on days 6 and 8. CyA was discontinued at 1 month after DBMT.
In Groups B and C, recipients underwent KTx alone with a conventional triple drug immunosuppressive regimen. The recipients then received a 6-day conditioning regimen and DBMT 4 months later. The triple immunosuppressive regimen consisted of tacrolimus (Astellas Pharma Inc. Osaka, Japan, intramuscular injection, starting from 0.1 mg/kg to maintain trough levels of 15 to 25 ng/mL), mycophenolate mofetil (Roche Inc., Nutley, NJ, 50 mg/kg/day for 2 weeks, followed by 3 days/week until DBMT) and prednisone (starting from 10 mg/kg, which was tapered over a week to maintenance dose of 0.3 mg/kg/day).
Group B: The recipients received a 6-day conditioning regimen 4 months after KTx. The conditioning regimen consisted of TBI 1.5 Gy on days −6 and −5 relative to the day of DBMT, followed by ATGAM (days −2, −1 and 0), TI (day −1), DBMT (day 0), anti-CD154 (days 0 and +2). Tacrolimus was discontinued 1 month after DBMT.
Group C: A 1-month course of humanized anti-CD8 mAb administration (cM-T807 provided by Centocor Inc. Horsham, PA) was begun at 5 mg/kg on days −1, 0 and 2 and continued at 1 mg/kg weekly for 1 month after DBMT in addition to the Group B regimen.
Except for M6601 (Group B) and M2402 (Group C), all recipients treated with anti-CD154 were pretreated with Ketorolac (1 mg/kg intravenous injections on days −1 and 0) to prevent thrombosis, as previously described (14).
Renal transplantation
Monkeys underwent heterotopic KTx and bilateral nephrectomies under general anesthesia as previously reported (15). Post-conditioning support with irradiated blood products (washed red cells and/or platelets) from ABO-matched animals were administered as required during the period of aplasia.
Bone marrow transplantation
Bone marrow was harvested from donor iliac bones by multiple percutaneous aspirations. If the donor animal was sacrificed, bone marrow cells were also harvested from the vertebral bones after the euthanasia. These cells (0.5 − 3.0 × 108 mononuclear cells/kg) were infused intravenously.
Detection of chimerism
After standard water shock treatment, peripheral blood cells were first stained with donor-specific mAbs chosen from a panel of mouse anti-human HLA class I mAbs that cross-react with cynomolgus monkeys. Cells were incubated for 30 min at 4°C, and then washed twice. Cell-bound mAb was detected with fluorescein isothiothianate (FITC)-conjugated rat anti-mouse immunoglobulin IgG2a mAb (Pharmingen, San Diego, CA), which was incubated for 30 min at 4°C, followed by two washes and analysis on FACScan (Becton Dickinson, Mountain View, CA). In all experiments, the percentage of cells that stained with each mAb was determined from one color fluorescence histogram and compared with those obtained from donor and pretreatment frozen recipient cells, which were used as positive and negative controls. The percentage of cells considered positive was determined with a cutoff chosen as the fluorescence level at the beginning of the positive peak for the positive control stain and by subtracting the percentage of cells stained with an isotype control. By using forward and 90° light scatter (FSC and SSC, respectively) dot plots, lymphocyte (FSC- and SSC-low), granulocyte (SSC-high), and monocyte (FSC-high but SSC-low) populations were gated, and chimerism was determined separately for each population. Nonviable cells were excluded by propidium iodide staining.
Flow cytometric analysis
Cell surface antigens were analyzed by multicolor flow cytometry. Phenotypic markers for memory and naïve T cells in cynomolgus monkeys were chosen based on the studies by Pitcher et al. (16), as well as studies in our laboratory (Nadazdin O and Benichou G. et al., manuscript submitted). PBMC were directly labeled with a combination of the following monoclonal antibodies: CD3 PerCP (SP 34-2), CD4 PerCP (L-200), CD8 PerCP (RPA-T8), CD8 APC (RPA-T8), CD16 (NKP15), CD20 PE (2H7), CD95 FITC (DX2), CD95 APC (DX2) CD28 PE (CD28.2) and CD28 FITC (CD28.2) purchased from BD Pharmingen, (San Jose, CA). In Group C, to avoid competitive binding with cM-T807, CD8 RPE (DK25, DAKO, Glostrup, Denmark) was used to detect CD8+ cells. The fluorescence of the stained samples was analyzed using FACS Calibur and FACS Scan flow cytometers and Cell Quest Software (BD Immunocytometry Systems, San Jose, CA). Lymphocytes were gated on the forward and side light scatter and 3000-5000 events were collected.
Pathology studies
Sequential renal biopsies were obtained at 2- to 4-month intervals in animals with stable function and whenever a rise in serum creatinine occurred in any recipient. In Groups B and C, recipients underwent a protocol biopsy 1 week prior to the DBMT conditioning. Tissue was processed for routine microscopy and a portion frozen for immunoperoxidase staining.
Statistical analysis
We used as a summary for each animal the average value of the absolute cell counts or the percentage of donor cells between days 0 and 28. A comparison of animals treated with the original regimen (Groups A and B) and the modified regimen with cM-T807 (Group C) was done using the Kruskal-Wallace test.
Results
Induction of chimerism and renal allograft survival
The original regimen consisted of low-dose TBI on days −6 and −5, TI on day −1, ATGAM, anti-CD154 and a 1-month course of CyA. In SKBMT, the recipients underwent simultaneous kidney and bone marrow transplantation on day 0 (Table 1). All eight recipients of SKBMT consistently developed mixed chimerism (Table 1) and seven of eight recipients survived long term (Figure 1), with four of them showing no evidence of either acute or chronic rejection.
Table 1. Conditioning regimens and induction of multilineage chimerism.
| Regimen | Conditioning and DBMT1 | Multilineage Chimerism | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| N | TBI (1.5Gy × 2) | TI (7Gy) | DBMT | cM-T807 | |||
| SKBMT | 8 | Days −5 and −62 | Day −1 | Day 0 | − | 8/8 | |
| Delayed | A | 4 | Day +1, +2 | Day +5 | Day +6 | − | 0/4 |
| B | 3 | +4 Months | +4 Months | +4 Months | − | 0/3 | |
| C | 5 | +4 Months | +4 Months | +4 Months | + | 3/5 | |
All recipients were also treated with ATGAM and anti-CD154 administration, followed by a 1-month course of calcineurin inhibitor after DBMT.
Relative to the time of kidney translplantation.
Figure 1. Posttransplant course (serum creatinine levels) of recipients in Groups SKBMT, A, B and C.

Red solid lines and black dashed lines indicate creatinine changes in the tolerant recipients and rejectors, respectively. SKBMT: All eight recipients of SKBMT developed mixed chimerism and 7 of 8 survived long term without acute rejection with four of them acquired allograft tolerance. Group A: All four recipients in Group A failed to develop mixed chimerism and rejected their kidney allografts. Group B: One recipient that developed limited chimerism survived long term, but eventually developed chronic rejection and was euthanized due to kidney failure on day 703 after DBMT. Two recipients failed to develop chimerism and rejected their kidney allograft on days 64 and 16. Group C: Three recipients (red lines) in Group C successfully developed mixed chimerism and have never developed rejection with two recipients currently surviving at 1.8 and 2.8 years after DBMT with normal kidney function. The other (red squares) died on day 67 due to refractory ascites caused by right atrial thrombus, which may be attributed to side effects of anti-CD154 antibody at the time of DBMT. Two recipients failed to develop mixed chimerism and rejected their allograft on days 61 and 35.
In Group A, four recipients initially underwent KTx alone, which was immediately followed by initiation of the conditioning regimen and DBMT on day 6 (Table 1). All four recipients in this group failed to develop mixed chimerism and rejected their kidney allografts soon after DBMT (Figure 1). In Group B, kidney allograft recipients were initially treated with a conventional immunosuppressive regimen and received conditioning and DBM 4 months after KTx. In this group, two of three recipients failed to develop chimerism and rejected their allografts soon after discontinuation of their immunosuppressive medication (Figure 1). One recipient developed limited chimerism only in the myeloid lineage but developed chronic rejection (Figures 2A,B) after discontinuation of immunosuppression. Thus, in contrast to the results of SKBMT, in which the original regimen consistently induced mixed chimerism and long-term allograft survival (Table 1), the same regimen failed to induce multilineage chimerism and renal allograft tolerance, if DBMT was administered separately after KTx. However, by adding anti-CD8 mAb (cM-T807) to the original regimen, three of five recipients successfully developed multilineage chimerism (Table 1) with two of them surviving longer than 500 days with no evidence of either acute or chronic rejection (Figures 2C,D). The other recipient who developed mixed chimerism was euthanized on day 67 without rejection due to a thromboembolic complication presumably caused by anti-CD154 mAb (14,17). The fourth recipient in Group C failed to develop mixed chimerism and developed acute rejection requiring euthanasia on day 61. The last recipient in this group had suffered an acute rejection episode 1 week prior to DBMT while being treated with conventional immunosuppression. Although this rejection episode was successfully reversed with steroid pulse therapy and he underwent conditioning and DBMT on day 119, this recipient failed to develop chimerism. Anti-donor alloantibody production and terminal kidney allograft rejection occurred within 35 days after DBMT (Figure 1).
Figure 2. Renal allograft biopsies obtained from recipients in Groups B and C.
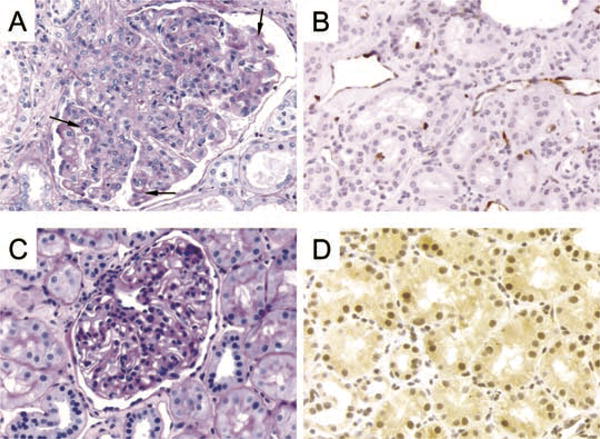
(A) Renal allograft from autopsy of a Group B long-term survivor (M6601) on day 702 after DBMT. An enlarged glomerulus shows mesangial and endocapillary hypercellularity with duplication of the basement membranes (arrows), PAS stain, 400×. (B) Renal allograft (M6601 on day 702) showing capillary staining for C4d, 400×. (C) Biopsy taken on day 629 after DBMT from M 1902 (Group C) showing a normal glomerulus without hypercellularity and with a normal basement membrane, PAS stain, 400×. (D) C4d staining of the biopsy taken from M 1902 on day 629. No C4d staining of the peritubular capillaries, 400×.
Lymphocyte subsets after conditioning
In Groups A and B, a significant number of both CD4+ or CD8+ T cells still remained in the peripheral blood. By adding humanized anti-CD8 monoclonal antibody (cM-T807) to the original regimen (Group C), total circulating CD8+ T-cell counts were significantly suppressed for 3 weeks (p < 0.05). There was no significant difference in CD4+CD3+, CD3–CD16+ (NK cells) or CD3–CD20+ (B cells) among Groups A–C (Figure 3).
Figure 3. Lymphocyte subsets after DBMT.

After DBMT, there is no significant difference observed in the absolute counts of CD3+CD4+, CD3-CD16+ (NK cell) and CD3-CD20+ (B cell) cells in the peripheral blood in Groups A–C. The CD8+CD3+ cells were significantly suppressed (p < 0.05) for 3 weeks in Group C. Statistical analysis was performed by comparing the average of cell counts between days 0–16 in Groups A (n = 4), B (n = 3) and C (n = 5) using Kruskal–Wallace test. N.S.; statistically not significant.
Naïve and memory T-cell subsets after conditioning
Further analyses of naïve and memory T-cell subsets were performed in Group B and Group C recipients. In both groups, significant numbers of CD4+ central memory T cells (CD4+CD95+CD28+,CD4 TCM) remained after conditioning (Figure 4C), in spite of effective depletion of naïve CD4 and CD8 T cells (CD95−) (Figures 4A,D). In Group B, although effective depletion of CD8 effector memory T cells (CD8 +CD95+CD28−, TEM) was initially observed until day 5, rapid expansion of CD8 TEM ensued thereafter. In contrast, post-conditioning expansion of CD8 TEM was effectively inhibited by adding cM-T807 and CD8 TEM remained suppressed more than a month in Group C (Figure 4E). CD8 TCM were also more suppressed in Group C than those in Group B (Figure 2F). In summary, effective depletion of CD8 memory T cells was associated with improved induction of mixed chimerism and renal allograft survival in the ‘delayed tolerance’ protocol.
Figure 4. Absolute counts of naïve and memory T-cell subsets after DBMT in Group B (n = 2) and Group C recipients (n = 3).

(A) CD4 naïve T cells were effectively depleted and remained suppressed. (B) The absolute counts of CD4 effector memory T cells (CD95+CD28 & minus; CD4Tem remained low in the peripheral blood. (C) CD4 central memory T cells (CD95+CD28+, CD4Tcm were resistant to the conditioning regimen and their absolute counts remained >200/mm3 in both Groups B and C. (D) CD8 naïve T cells (CD95−) were effectively depleted and remained suppressed after DBMT in both Groups B and C, regardless of cM-T807 treatment. (E) CD8 effector memory T cells (CD95+CD28−, CD8 TEM) were effectively depleted until day 5 in both groups. However, rapid expansion of CD8 TEM ensued thereafter in Group B, while such rapid expansion of CD8 TEM was effectively inhibited by cM-T807 treatment in Group C. (F) Absolute counts of CD8 TCM were also more suppressed in Group C than those in Group B.
Discussion
In order to extend the clinical applicability of our tolerance-inducing non-myeloablative regimen to recipients of deceased donor transplants, we initially evaluated conditioning regimens in which treatment was begun within 24-h of KTx. However, simple compression of the previously effective 6-day therapeutic protocol into a 24-h period not only failed to induce chimerism but also led to unacceptable toxicity (data not shown). Therefore, in the current study, we evaluated a post-kidney transplant (delayed tolerance) protocol, in which recipients initially underwent KTx alone with conventional immunosuppression, followed by a 6-day preparative conditioning and DBMT at a later time. This approach would potentially extend the applicability of our regimen to any current recipient of previously transplanted allografts from either living or deceased donors, if DBM is available.
However, in the current study, we found that using the ‘delayed tolerance’ protocol is more difficult to induce mixed chimerism and allograft tolerance than in SKBMT. This could be explained by specific activation of memory T cells by the initially transplanted organ, even without clinical evidence of rejection. Extensive FACS analyses of T-cell subsets in our recipients revealed that a substantial number of memory T cells remained after the conditioning regimen, in spite of effective depletion of naïve T cells. To further analyze subsets of these residual memory T cells, we used CD95 and CD28 to define memory T cells, based on the studies by Pitcher et al. in rhesus monkeys (16), as well as studies on cynomolgus monkeys performed in our laboratory (Nadazdin and Benichou et al., manuscript submitted). These analyses revealed that CD4 TCM were resistant to the conditioning regimen and remained more than 200/mm3. Both CD8 TEM and CD8 TCM were initially depleted effectively but rapid post-conditioning expansion of CD8 TEM was observed after day 5, while CD8 TCM counts remained unchanged. This was an unexpected in vivo finding, since TEM have been reported to be terminally differentiated cells with limited replicative capacity in vitro(18,19). It is possible that these vigorously proliferating CD8 TEM were differentiated from CD8 TCM(20), resulting in no apparent expansion of CD8 TCM. Neujahr et al. demonstrated that partial T-cell depletion resulted in homeostatic proliferation of the residual lymphocytes, which lead to tolerance resistance in their mice cardiac transplant model. They proposed two different strategies to block homeostatic proliferation of memory T cells after T-cell depletion; first, by the adoptive transfer of additional unprimed regulatory cells at the time of transplantation, and second, by the adjunctive use of nondepleting anti-CD4 and anti-CD8 mAbs (10). In our studies, a humanized anti-CD8 mAb, cM-T807, effectively inhibited expansion of CD8 TEM, which resulted in improved chimerism and tolerance induction. The antibody cM-T807 is a strong depleting antibody, which effectively depletes CD8+ cells from the peripheral blood and lymph nodes (21). These results may indicate that a powerful depleting antibody can also be a tool to overcome homeostatic proliferation of memory T cells.
Among memory T cells, donor-specific CD8 memory Tcells may be particularly important in DBMT. In a murine model, adoptively transferred donor-specific CD8 memory T cells consistently prevented induction of mixed chimerism and skin allograft tolerance, while CD4 memory T cell had no effects on tolerance induction (7). In other studies, adoptively transferred anti-donor CD4 memory T cells prevented induction of cardiac allograft tolerance after treatments with CD154 blockade and donor-specific transfusion. However, in that model, depletion of effector CD8 T cells resulted in significant prolongation of heart graft survival even in the presence of anti-donor CD4 memory T cells (22). In the current study, we only targeted CD8 memory T cells, with no extensive depletion being made against residual CD4 memory T cells. In targeting CD4 memory T cells, it may be important not to interfere with CD4+ T regulatory cells (23), which bear some similarities to memory T cells in their phenotype and function (10,24–26). In a mouse model, low-dose TBI, anti-CD154 and DBMT reliably induced mixed chimerism and allograft tolerance. However, CD4+ T-cell depletion prevented the development of mixed chimerism and tolerance due to a lack of regulation over donor-reactive CD8+ T cells (27). In a clinical trial for tolerance induction using Campath antibody, profound depletion of both CD4+ and CD8+ cells resulted in an increased incidence of acute rejection (28,29). Nevertheless, intervention of residual CD4 memory T cells may be necessary to further improve the consistency of the tolerance induction.
In conclusion, the current studies provide ‘proof of principle’ that the mixed chimerism approach can induce tolerance even several months after organ transplantation by additional intervention against CD8 memory T cells. Monoclonal antibody cM-T807 effectively inhibited expansion of CD8 memory T cells and improved induction of chimerism and renal allograft survival.
Acknowledgments
This study was supported in part by NHL-BI, POI-HL18646, NIH-NIAID, ROI A137692 and NIH/NIAID 5R01 AI50987-03.
We thank Dr. David Schoenfeld for statistical analysis, Patricia Della Pelle and Joanne Phelan for technical assistance and Diann Funk Flavin for anesthesia and postoperative care.
cM-T807 used in this study was provided by the NIH Nonhuman Primate Reagent Resource (RR016001 andAI040101).
Abbreviations
- CI
calcineurin inhibitor
- DBMT
donor bone marrow transplantation
- KTx
Kidney transplantation
- SKBMT
simultaneous kidney and bone marrow transplantation
- TBI
total body irradiation
- TCM
central memory cells
- TEM
effector memory T cells
References
- 1.Kawai T, Cosimi AB, Colvin RB, et al. Mixed allogeneic chimerism and renal allograft tolerance in cynomolgus monkeys. Transplantation. 1995;59:256–262. [PubMed] [Google Scholar]
- 2.Preston EH, Xu H, Dhanireddy KK, et al. IDEC-131 (anti-CD154), sirolimus and donor-specific transfusion facilitate operational tolerance in non-human primates. Am J Transplant. 2005;5:1032–1041. doi: 10.1111/j.1600-6143.2005.00796.x. [DOI] [PubMed] [Google Scholar]
- 3.Thomas JM, Carver FM, Kasten-Jolly J, et al. Further studies of veto activity in rhesus monkey bone marrow in relation to allograft tolerance and chimerism. Transplantation. 1994;57:101–115. doi: 10.1097/00007890-199401000-00018. [DOI] [PubMed] [Google Scholar]
- 4.Kirk AD, Burkly LC, Batty DS, et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med. 1999;5:686–693. doi: 10.1038/9536. [DOI] [PubMed] [Google Scholar]
- 5.Knechtle SJ, Vargo D, Fechner J, et al. FN18-CRM9 immunotoxin promotes tolerance in primate renal allografts. Transplantation. 1997;63:1–6. doi: 10.1097/00007890-199701150-00002. [DOI] [PubMed] [Google Scholar]
- 6.Adams AB, Pearson TC, Larsen CP. Heterologous immunity: An overlooked barrier to tolerance. Immunol Rev. 2003;196:147–160. doi: 10.1046/j.1600-065x.2003.00082.x. [DOI] [PubMed] [Google Scholar]
- 7.Adams AB, Williams MA, Jones TR, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Z, Bensinger SJ, Zhang J, et al. Homeostatic proliferation is a barrier to transplantation tolerance. Nat Med. 2004;10:87–92. doi: 10.1038/nm965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valujskikh A. The challenge of inhibiting alloreactive T-cell memory. Am J Transplant. 2006;6:647–651. doi: 10.1111/j.1600-6143.2005.01215.x. [DOI] [PubMed] [Google Scholar]
- 10.Neujahr DC, Chen C, Huang X, et al. Accelerated memory cell homeostasis during T cell depletion and approaches to overcome it. J Immunol. 2006;176:4632–4639. doi: 10.4049/jimmunol.176.8.4632. [DOI] [PubMed] [Google Scholar]
- 11.Kimikawa M, Sachs DH, Colvin RB, Bartholomew A, Kawai T, Cosimi AB. Modifications of the conditioning regimen for achieving mixed chimerism and donor-specific tolerance in cynomolgus monkeys. Transplantation. 1997;64:709–716. doi: 10.1097/00007890-199709150-00008. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Poncelet A, Sachs DH, et al. Long-term outcome and alloantibody production in a non-myeloablative regimen for induction of renal allograft tolerance. Transplantation. 1999;68:1767–1775. doi: 10.1097/00007890-199912150-00022. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Sogawa H, Boskovic S, et al. CD154 blockade for induction of mixed chimerism and prolonged renal allograft survival in nonhuman primates. Am J Transplant. 2004;4:1391–1398. doi: 10.1111/j.1600-6143.2004.00523.x. [DOI] [PubMed] [Google Scholar]
- 14.Koyama I, Kawai T, Andrews D, et al. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation. 2004;77:460–462. doi: 10.1097/01.TP.0000110291.29370.C0. [DOI] [PubMed] [Google Scholar]
- 15.Cosimi AB, Delmonico FL, Wright JK, et al. Prolonged survival of nonhuman primate renal allograft recipients treated only with anti-CD4 monoclonal antibody. Surgery. 1990;108:406–413. discussion 413–414. [PubMed] [Google Scholar]
- 16.Pitcher CJ, Hagen SI, Walker JM, et al. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168:29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6:114. doi: 10.1038/72162. [DOI] [PubMed] [Google Scholar]
- 18.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 19.Hamann D, Baars PA, Rep MH, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labalette M, Leteurtre E, Thumerelle C, Grutzmacher C, Tourvieille B, Dessaint JP. Peripheral human CD8(+)CD28(+)T lymphocytes give rise to CD28(-)progeny, but IL-4 prevents loss of CD28 expression. Int Immunol. 1999;11:1327–1336. doi: 10.1093/intimm/11.8.1327. [DOI] [PubMed] [Google Scholar]
- 21.Schmitz JE, Simon MA, Kuroda MJ, et al. A nonhuman primate model for the selective elimination of CD8+ lymphocytes using a mouse-human chimeric monoclonal antibody. Am J Pathol. 1999;154:1923–1932. doi: 10.1016/S0002-9440(10)65450-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Heeger PS, Valujskikh A. In vivo helper functions of alloreactive memory CD4+ T cells remain intact despite donor-specific transfusion and anti-CD40 ligand therapy. J Immunol. 2004;172:5456–5466. doi: 10.4049/jimmunol.172.9.5456. [DOI] [PubMed] [Google Scholar]
- 23.Zheng XX, Sanchez-Fueyo A, Domenig C, Strom TB. The balance of deletion and regulation in allograft tolerance. Immunol Rev. 2003;196:75–84. doi: 10.1046/j.1600-065x.2003.00089.x. [DOI] [PubMed] [Google Scholar]
- 24.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: Contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–1786. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- 25.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation of regulatory T cells in the human fetus. Eur J Immunol. 2005;35:383–390. doi: 10.1002/eji.200425763. [DOI] [PubMed] [Google Scholar]
- 27.Fehr T, Sykes M. Tolerance induction in clinical transplantation. Transpl Immunol. 2004;13:117–130. doi: 10.1016/j.trim.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Kirk AD, Hale DA, Mannon RB, et al. Results from a human renal allograft tolerance trial evaluating the humanized CD52-specific monoclonal antibody alemtuzumab (CAMPATH-1H) Transplantation. 2003;76:120–129. doi: 10.1097/01.TP.0000071362.99021.D9. [DOI] [PubMed] [Google Scholar]
- 29.Kirk AD, Mannon RB, Kleiner DE, et al. Results from a human renal allograft tolerance trial evaluating T-cell depletion with alemtuzumab combined with deoxyspergualin. Transplantation. 2005;80:1051–1059. doi: 10.1097/01.tp.0000174341.49741.8f. [DOI] [PubMed] [Google Scholar]